
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAcid-catalyzed transformation of cassane diterpenoids from Caesalpinia bonduc to aromatic derivatives†
Ting Liuab,
Zunxi Ningab,
Yan Yinab,
Shizhou Qiab and
Huiyuan Gao *ab
*ab
aSchool of Traditional Chinese Materia Medica, Shenyang Pharmaceutical University, Shenyang 110016, People's Republic of China. E-mail: sypugaohy@163.com; Tel: +86-024-43520781
bKey Laboratory of Structure-Based Drug Design & Discovery of Ministry of Education, Shenyang Pharmaceutical University, Shenyang 110016, People's Republic of China
First published on 22nd June 2021
Abstract
Transformations of cassane diterpenoids from Caesalpinia bonduc into aromatic derivatives, either in CDCl3 or in CHCl3 irradiated with UV light or catalyzed by AlCl3, were described. Caesalmin C (2) was hydrolyzed with Na2CO3 upon refluxing in MeOH to yield compound 1. Dissolving compound 1 with CDCl3 resulted in an unexpected aromatization process of a C ring to obtain 1a, and aromatic derivatives 6-acetoxy-3-deacetoxycaesaldekarin e (2a), caesall A (3a), caesaldekarin e (5a), caesalpinin MC (5b), 2-acetoxycaesaldekarin e (6a) and new compound 6b could be obtained from corresponding cassane diterpenoids (2–8) under the same conditions. Furthermore, the photochemical reactions of cassane diterpenoids 1–8 occurring in CHCl3 also yielded aromatic derivatives 1a, 2a, 3a, 5a, 6a, new compounds 2b and 3b, and 17-norcassane diterpenoids norcaesalpinin MC (2c) and caesalmin J (3c). In addition, cassane diterpenoids 1–8, treated with AlCl3 in CHCl3 or CH2Cl2, gave the same results in CDCl3 and with even shorter reaction time. The role of AlCl3 in the aromatization of 1 has been explained by DFT calculations.
1 Introduction
Cassane diterpenoids, a group of rearranged abietane metabolites, have been isolated from different species of medicinal plants of the Caesalpinia genus in recent years, and have received much attention for their wide range of pharmacological activities including anti-inflammatory, antitumor, antimalarial, antimicrobial, antiviral, antioxidant and antinociceptive properties.1 Among these compounds with great structural diversity, some of these bearing an aromatic C ring have earned special attention due to their significant biological activities. Researchers have synthesized some of these interesting diterpenoids including benthaminin 1,2 taepeenin D,3 taepeenin F,4 (5α)-vouacapane-8(14),9(11)-diene,5 sucutinirane C and sucutiniran D6 from commercially available materials. In this paper the transformation of cassane-type diterpenoids with a fused furan ring into aromatic derivatives in CDCl3, or upon irradiation with UV light in CHCl3, or catalyzed by AlCl3 in CHCl3 or CH2Cl2 at room temperature, was described.2 Experimental
2.1 General experimental procedures
Optical rotation was measured in MeOH solution on an Anton-Paar MCP 200 polarimeter at room temperature. NMR spectra were recorded on Bruker ARX-400 and AV-600 instruments (Bruker Corporation, Bremen, Germany) with tetramethylsilane as the internal standard. HRESIMS spectra were recorded on a Bruker micro-TOF-Q mass spectrometer. Silica gel (100–200 mesh, and 200–300 mesh) for open-column chromatography and TLC plates (GF254) was purchased from Qingdao Marine Chemical Co. Ltd. (Qingdao, China). The HPLC system consisted of a Shimadzu LC-20AR instrument equipped with a SPD-20A UV detector (Shimadzu, Kyoto, Japan) and a YMC C-18 reversed-phase column (5 μm, 20 × 250 mm) (YMC, Kyoto, Japan).2.2 Plant material
The seeds of Caesalpinia bonduc (L.) Roxb. (a synonym for Guilandina bonduc L.) were purchased from An Guo Medicinal Material Corporation (Hebei Province, China), and authenticated by Associated Prof. Jiuzhi Yuan, Shenyang Pharmaceutical University, Shenyang, China. A voucher specimen (No. SY-2018-01A) was deposited in the Department of Natural Products Chemistry, Shenyang Pharmaceutical University, Shenyang, China.2.3 Extraction and isolation
The powdered air-dried seed kernels (12.0 kg) of C. bonduc were defatted three times with petroleum ether, and then were extracted with 75% ethanol for three times. The mixture was filtered and the filtrate concentrated in vacuo in a rotary evaporator. The concentrated extract was suspended in water and partitioned with CHCl3 and n-BuOH, successively. The residue of the CHCl3 (214.0 g) was subjected to silica gel column chromatography eluted with CH2Cl2–MeOH (100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0 to 1:1) to yield ten constituents (constituents 1–10). Constituent 2 (18.8 g) (CH2Cl2–MeOH = 50:1) was isolated by silica gel column chromatography using gradient solvent system of petroleum ether–EtOAc (15:1 to 2:1) to give five constituents (constituents 2.1–2.5). Constituent 2.2 (petroleum ether–EtOAc = 10:1) was subjected to preparative RP-HPLC with MeOH–H2O (65:35) to afford compounds 2 (600.0 mg), 6 (125.5 mg) and 9 (45.5 mg). Constituent 2.3 (petroleum ether–EtOAc = 5:1) was applied to RP-HPLC with MeOH–H2O (65:35) to get compounds 5 (35.2 mg) and 11 (29.5 mg), and further purified using preparative TLC plate with CH2Cl2–acetone (25:1) to obtain compound 10 (38.5 mg). Constituent 2.4 (petroleum ether–EtOAc = 3:1) was purified by RP-HPLC with MeOH–H2O (55:45) to yield compounds 7 (30.2 mg) and 8 (93.0 mg). Constituent 3 (38.3 g) (CH2Cl2–MeOH = 25:1) was subjected to silica gel column chromatography eluted with petroleum ether–EtOAc gradient (10:1–1:1) to give five constituents (constituents 3.1–3.5). Constituent 3.1 (petroleum ether–EtOAc = 10:1) was further applied to silica gel column with petroleum ether–EtOAc (7:1–1:1) and isolated by RP-HPLC with MeOH–H2O (65:35) to furnish compounds 3 (85.5 mg) and 4 (65.0 mg).
:0 to 1:1) to yield ten constituents (constituents 1–10). Constituent 2 (18.8 g) (CH2Cl2–MeOH = 50:1) was isolated by silica gel column chromatography using gradient solvent system of petroleum ether–EtOAc (15:1 to 2:1) to give five constituents (constituents 2.1–2.5). Constituent 2.2 (petroleum ether–EtOAc = 10:1) was subjected to preparative RP-HPLC with MeOH–H2O (65:35) to afford compounds 2 (600.0 mg), 6 (125.5 mg) and 9 (45.5 mg). Constituent 2.3 (petroleum ether–EtOAc = 5:1) was applied to RP-HPLC with MeOH–H2O (65:35) to get compounds 5 (35.2 mg) and 11 (29.5 mg), and further purified using preparative TLC plate with CH2Cl2–acetone (25:1) to obtain compound 10 (38.5 mg). Constituent 2.4 (petroleum ether–EtOAc = 3:1) was purified by RP-HPLC with MeOH–H2O (55:45) to yield compounds 7 (30.2 mg) and 8 (93.0 mg). Constituent 3 (38.3 g) (CH2Cl2–MeOH = 25:1) was subjected to silica gel column chromatography eluted with petroleum ether–EtOAc gradient (10:1–1:1) to give five constituents (constituents 3.1–3.5). Constituent 3.1 (petroleum ether–EtOAc = 10:1) was further applied to silica gel column with petroleum ether–EtOAc (7:1–1:1) and isolated by RP-HPLC with MeOH–H2O (65:35) to furnish compounds 3 (85.5 mg) and 4 (65.0 mg).
2.4 Deacetylation of caesalmin C (2)
To a solution of compound 2 (25.0 mg, 0.053 mmol) in methanol (10 mL) was added Na2CO3 (28 mg, 0.264 mmol). After refluxing for 1 h, the solvent was removed under reduced pressure and CH2Cl2 (20 mL) was added. The organic extract was washed successively with 2 M HCl (5 mL × 2) and saturated NaCl (5 mL × 2) and dried with anhydrous Na2SO4. The solution was filtrated and the solvent was removed under reduced pressure, and 18.0 mg (72%) of compound 1 was obtained as a white amorphous powder.2.5 Aromatization of compound 1 in CDCl3
Compound 1 (1.0 mg) was dissolved in deuterated chloroform (CDCl3, 0.6 mL) at room temperature, and the solution instantly turned pink and quickly became canary yellow, resulting in the formation of compound 1a as evidenced by TLC, which showed a blue spot after being visualized with vanillin–sulfuric acid while compound 1 dissolved in methanol showed an aubergine spot with different Rf values under the same separating condition. Vaporizing CDCl3 and compound 1a was obtained.2.6 Aromatization of compounds 2–11 in CDCl3
Compounds 2–11 (0.0025 mmol) were dissolved in CDCl3 (0.6 mL) at room temperature, and the formation of aromatic derivatives was monitored by TLC. When the reactant disappeared, the reaction was quenched by evaporating the CDCl3, and the residue was purified by HPLC (C18 column) eluted with MeOH–H2O (65:35) or silica gel column chromatography eluted with petroleum ether–EtOAc.
2.7 Photochemical reaction of compounds 1–11 in CHCl3
A solution of cassane diterpenoid (0.01 mmol) in CHCl3 (5.0 mL) was irradiated with a UV lamp (254 nm) at room temperature. After the starting material was consumed, the solvent was removed under reduced pressure and the residue was subjected to HPLC (C18 column) eluted with MeOH–H2O (65:35) or silica gel column chromatography eluted with petroleum ether–EtOAc.
2.8 Aromatization of compounds 1–11 catalyzed by AlCl3
AlCl3 (0.012 mmol) was added to a solution of reactant (0.012 mmol) in CHCl3 (5 mL) and stirred at room temperature until the reactant couldn't be detected by TLC. The mixture was filtrated and then washed with water (2 mL × 3) and dried with anhydrous Na2SO4. The solvent of the organic layer was evaporated under reduced pressure, and aromatic derivative was obtained by silica gel column chromatography eluted with petroleum ether–EtOAc.![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) OCH3), 172.5 (C, 7-OOCH3), 171.2 (C, 1-OOCH3), 155.9 (C, C-12), 146.7 (CH, C-16), 141.6 (C, C-9), 131.8 (C, C-14), 128.0 (C, C-13), 127.7 (C, C-8), 106.5 (CH, C-15), 105.1 (CH, C-11), 80.9 (C, C-5), 78.3 (CH, C-6), 76.8 (CH, C-1), 76.7 (CH, C-7), 50.1 (C, C-10), 39.7 (C, C-4), 33.6 (CH2, C-3), 30.8 (CH3, C-18), 29.3 (CH3, C-20), 25.2 (CH3, C-19), 23.4 (CH2, C-2), 22.0 (CH3, 6-OCOH3), 21.1 (CH3, 7-OCOH3), 21.0 (CH3, 1-OCOH3), 16.1 (CH3, C-17); HRESIMS m/z 495.1989 [M + Na]+ (calcd for C26H32O8Na, 495.1995).OCH3), 155.8 (C, C-12), 146.2 (CH, C-16), 141.5 (C, C-9), 132.5 (C, C-14), 130.5 (C, C-8), 127.7 (C, C-13), 106.2 (CH, C-15), 104.6 (CH, C-11), 86.1 (CH, C-7), 81.0 (C, C-5), 77.3 (CH, C-1), 76.9 (CH, C-6), 57.5 (CH3, 7-OCH3), 49.1 (C, C-10), 40.0 (C, C-4), 33.9 (CH2, C-3), 31.3 (CH3, C-18), 29.0 (CH3, C-20), 25.9 (CH3, C-19), 23.6 (CH2, C-2), 21.1, (CH3, 1-OCOH3), 16.2 (CH3, C-17); HRESIMS m/z 425.1935 [M + Na]+ (calcd for C23H30O6Na, 425.1940).OCH3), 171.5 (C, 2-OOCH3), 171.3 (C, 1-OOCH3), 159.2 (C, C-12), 143.5 (C, C-9), 133.9 (C, C-14), 126.6 (C, C-8), 125.2 (C, C-13), 102.8 (CH, C-11), 78.2 (CH, C-3), 77.3 (C, C-5), 74.9 (CH, C-1), 71.8 (CH2, C-16), 68.0 (CH, C-2), 49.3 (C, C-10), 44.0 (C, C-4), 31.1 (CH3, C-20), 29.9 (CH2, C-15), 25.4 (CH3, C-19), 25.3 (CH2, C-6), 24.2 (CH2, C-7), 23.5 (CH3, C-18), 21.0, (CH3, 1-OCOH3), 20.9, (CH3, 3-OCOH3), 20.6, (CH3, 2-OOCH3), 16.3 (CH3, C-17); HRESIMS m/z 497.2146 [M + Na]+ (calcd for C26H34O8Na, 497.2151).
OCH3), 172.5 (C, 7-OOCH3), 171.2 (C, 1-OOCH3), 155.9 (C, C-12), 146.7 (CH, C-16), 141.6 (C, C-9), 131.8 (C, C-14), 128.0 (C, C-13), 127.7 (C, C-8), 106.5 (CH, C-15), 105.1 (CH, C-11), 80.9 (C, C-5), 78.3 (CH, C-6), 76.8 (CH, C-1), 76.7 (CH, C-7), 50.1 (C, C-10), 39.7 (C, C-4), 33.6 (CH2, C-3), 30.8 (CH3, C-18), 29.3 (CH3, C-20), 25.2 (CH3, C-19), 23.4 (CH2, C-2), 22.0 (CH3, 6-OCOH3), 21.1 (CH3, 7-OCOH3), 21.0 (CH3, 1-OCOH3), 16.1 (CH3, C-17); HRESIMS m/z 495.1989 [M + Na]+ (calcd for C26H32O8Na, 495.1995).OCH3), 155.8 (C, C-12), 146.2 (CH, C-16), 141.5 (C, C-9), 132.5 (C, C-14), 130.5 (C, C-8), 127.7 (C, C-13), 106.2 (CH, C-15), 104.6 (CH, C-11), 86.1 (CH, C-7), 81.0 (C, C-5), 77.3 (CH, C-1), 76.9 (CH, C-6), 57.5 (CH3, 7-OCH3), 49.1 (C, C-10), 40.0 (C, C-4), 33.9 (CH2, C-3), 31.3 (CH3, C-18), 29.0 (CH3, C-20), 25.9 (CH3, C-19), 23.6 (CH2, C-2), 21.1, (CH3, 1-OCOH3), 16.2 (CH3, C-17); HRESIMS m/z 425.1935 [M + Na]+ (calcd for C23H30O6Na, 425.1940).OCH3), 171.5 (C, 2-OOCH3), 171.3 (C, 1-OOCH3), 159.2 (C, C-12), 143.5 (C, C-9), 133.9 (C, C-14), 126.6 (C, C-8), 125.2 (C, C-13), 102.8 (CH, C-11), 78.2 (CH, C-3), 77.3 (C, C-5), 74.9 (CH, C-1), 71.8 (CH2, C-16), 68.0 (CH, C-2), 49.3 (C, C-10), 44.0 (C, C-4), 31.1 (CH3, C-20), 29.9 (CH2, C-15), 25.4 (CH3, C-19), 25.3 (CH2, C-6), 24.2 (CH2, C-7), 23.5 (CH3, C-18), 21.0, (CH3, 1-OCOH3), 20.9, (CH3, 3-OCOH3), 20.6, (CH3, 2-OOCH3), 16.3 (CH3, C-17); HRESIMS m/z 497.2146 [M + Na]+ (calcd for C26H34O8Na, 497.2151).2.9 Computational methods
The B3LYP density functional method was employed in this work to carry out all the computations. Empirical dispersion GD3BJ was also considered. The 6-31G (d,p) basis set was used for the atoms in geometry optimizations. Vibrational frequency analyses at the same level of theory were performed on all optimized structures to characterize stationary points as local minima or transition states. Furthermore, intrinsic reaction coordinate (IRC) computations were carried out to confirm that transition states connect to the appropriate reactants and products. The Gaussian 09 program package was used throughout.3 Results and discussion
The fast facile aromatization process in CDCl3 occurred when we attempted to obtain the NMR spectra of hydrolysate of caesalmin C (2),7 a cassane-type diterpenoid isolated from both Caesalpinia Minax Hance and Caesalpinia bonduc (L.) Roxb. in our previous work. Compared with other cassane diterpenoids isolated from seeds of above two plants of Caesalpinia genus, caesalmin C (2) has relatively high abundance but no distinct bioactivity in our previous activity screening. For this reason, caesalmin C (2) was deacetylated with Na2CO3 by refluxing in MeOH for 1 h. Upon completion of the deacetylation, product 1 dissolved in CDCl3 could quickly transformed to aromatic derivative 1a, as evidenced by thin layer chromatography (TLC) behavior and NMR data. Aromatization reactions of caesalmin C (2),7 bonducellpin G (4),8 caesalmin E (7)7 and caesalmin F (8)7 to 6-acetoxy-3-deacetoxycaesaldekarin e (2a),9 ζ-caesalpin (3)10 to caesall A (3a),11 caesalpinin C (5)12 to caesaldekarin e (5a),13 and 14(17)-dehydrocaesalpin F (6)14 to 2-acetoxycaesaldekarin e (6a)14 also been observed under this condition with excellent yields. Caesalpinin MC (5b)15 and new compound 6b were observed as the main byproducts in the aromatization processes of 5 and 6, respectively. When compounds 1–8 were dissolved in CHCl3 and irradiated with UV light, aforementioned aromatic derivatives 2a, 3a, 5a and 6a could be produced from corresponding substrates, accompanied by the byproducts of new compound 2b and norcaesalpinin MC (2c),15 and new compound 3b and caesalmin J (3c)16 from 2 and 3, respectively. Furthermore, compounds 1–8 treated with AlCl3 in CHCl3 or CH2Cl2 could give to corresponding aromatic derivatives same as observed in CDCl3 with excellent yields and even shorter reaction time, and the role of AlCl3 in the aromatization of 1 has been explained by density functional theory (DFT) calculations.Caesalmin C (2) was deacetylated with Na2CO3 by refluxing in MeOH for 1 h to generate 1, for which the formation was confirmed by the disappearance of acetoxy methyl signals (δH 1.97, 2.08, 2.11), carbonyl carbons (δC 169.3, 171.0, 171.0) and methyl carbons (δC 21.5, 21.6, 21.9), and proton signals of three oxygenated methines all shifted to higher fields in the NMR spectra. When 1 was dissolved in CDCl3, it quickly converted to 1a at room temperature (Fig. 1), as evidenced by TLC behavior and NMR data. Nevertheless, this aromatization reaction couldn't proceed in other deuterated solvents including DMSO, CD3OD, (CD3)2CO and C5D5N. Compound 1 was obtained as white amorphous powder, when it was dissolved in CDCl3, the solution instantly turned pink and them became canary yellow. Compound 1a was obtained as a white amorphous power, and its molecular formula was deduced to be C20H26O4 from the HRESIMS (m/z 353.1723 [M + Na]+, calcd 353.1729). The 1H NMR spectrum of 1a displayed three methyl signals (δH 1.27, s; 1.33, s; 1.34, s), a benzylic methyl signal (δH 2.41, s), two oxygenated methine signals (δH 4.52, t, J = 7.8 Hz; 4.55, br s), one aromatic proton (δH 7.42, s) and two protons (δH 6.75, d, J = 2.3 Hz; 7.55, d, J = 2.3 Hz) of a 1,2-disubstituted furan ring, which revealed the existence of aromatic ring. The 13C NMR displayed the presence of 20 carbons including four methyls, three methylenes, five methines (three olefinic at δC 104.6, 105.2, 144.7 and two oxygenated at δC 69.7, 73.6) and eight quaternary carbons (five olefinc at δC 126.1, 128.1, 128.8, 140.0, 154.0 and one oxygenated at δC 78.5), which further supported the formation of benzene ring. The direct connections between the protons and carbons were identified by the HSQC spectrum. In the HMBC spectrum (Fig. 2), H-6 showed long-range correlation with C-7, and H2-7 showed correlations with C-6, C-9 and C-14, indicating that the hydroxy group at C-7 in 1 has disappeared in the aromatization process. The orientation of hydroxy groups at C-1 and C-6 remained unchanged as evidenced by the correlations of H-1 with H-2α and H-2β, and H-6 with H3-20 in the NOESY spectrum (Fig. 3). In order to determine the absolute configuration of 1a, the calculated ECD spectra were compared with experimental spe-ctrum (Fig. 4), and the result showed that the calculated ECD of 1S,5R,6S,10S was consistent with experimental data of 1a. Thus, compound 1a was identified as 1α,5α,6α-trihydroxy-14-methylvoucapane-8(14),9(11)-diene.
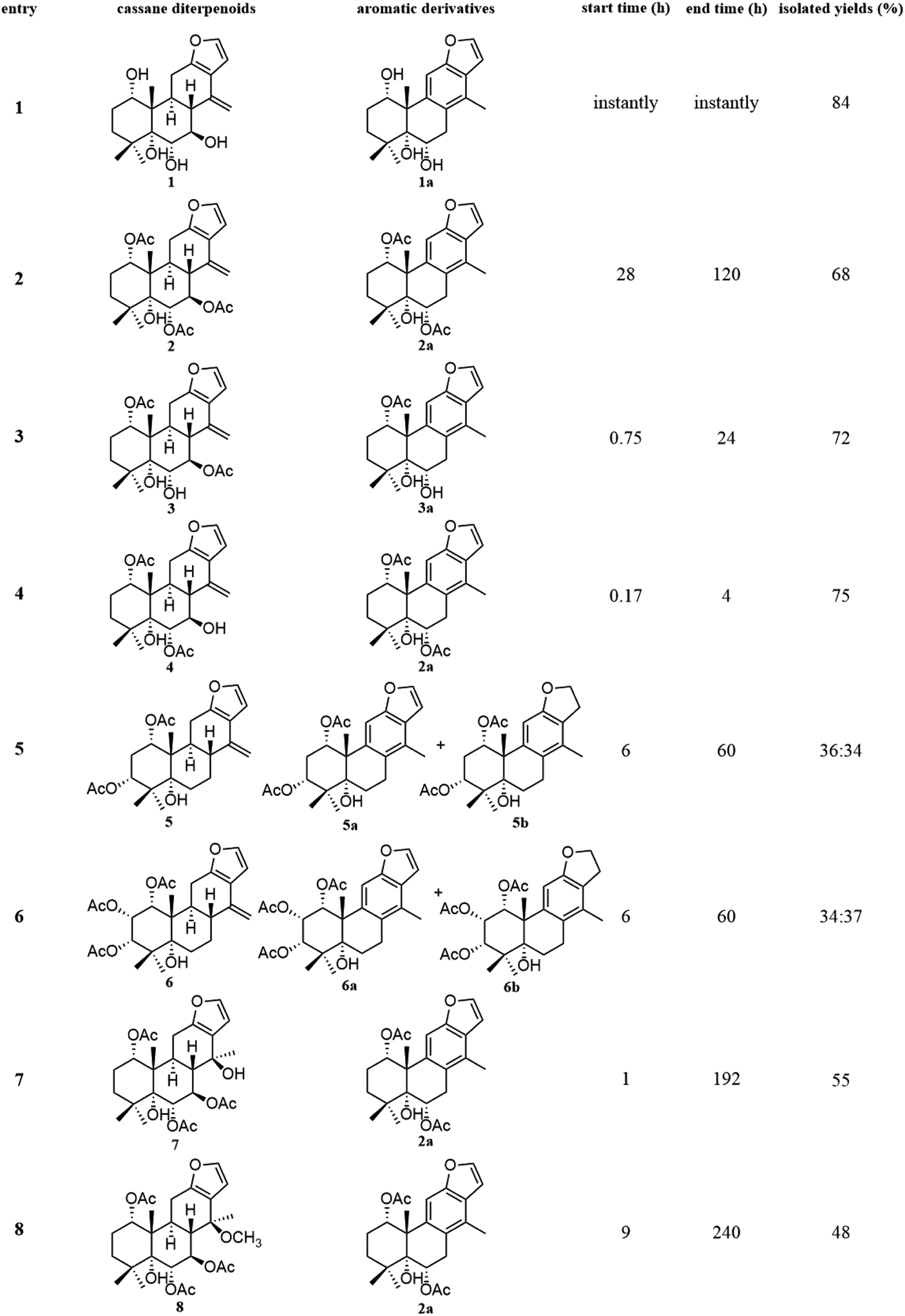 | ||
| Fig. 1 Scope of aromatization of cassane diterpenoids in CDCl3. | ||
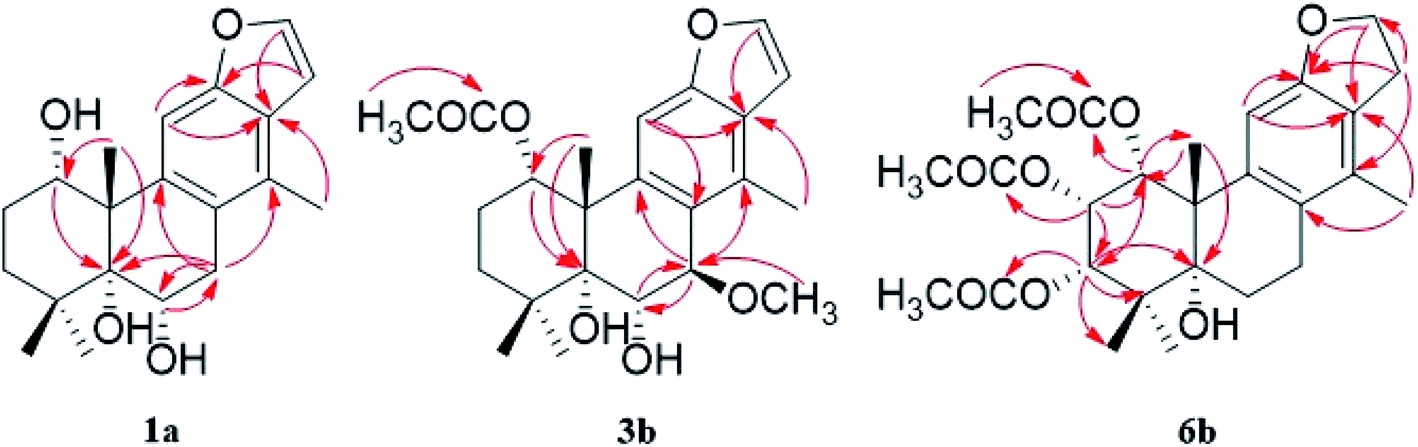 | ||
| Fig. 2 Key HMBC correlations of compounds 1a, 3b and 6b. | ||
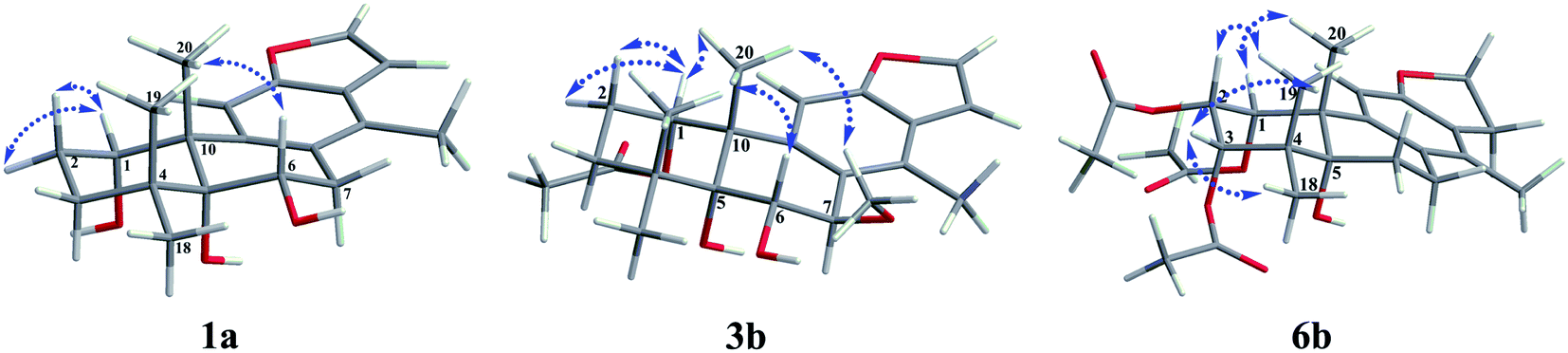 | ||
| Fig. 3 Key NOESY correlations of compounds 1a, 3b and 6b. | ||
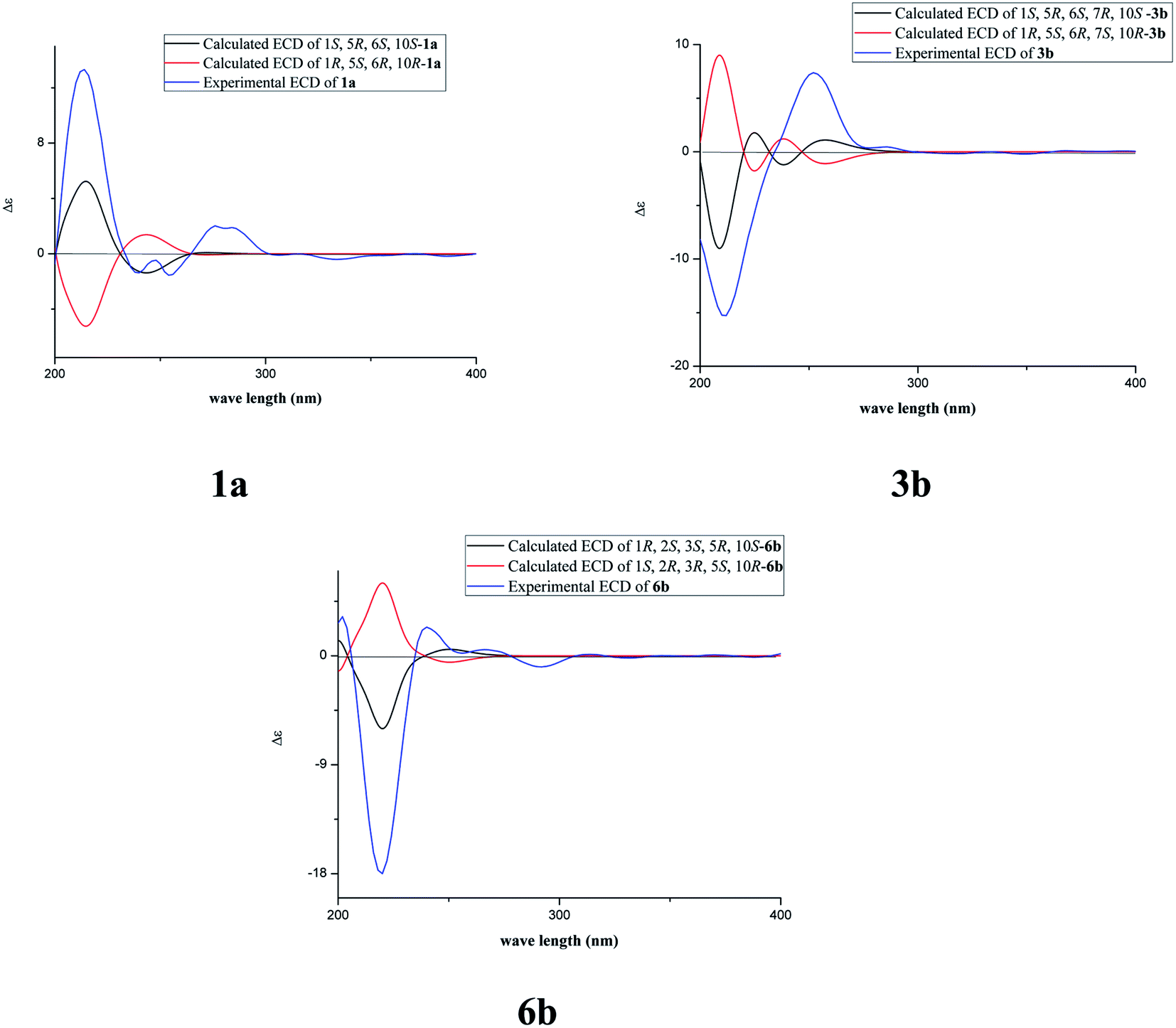 | ||
| Fig. 4 Experimental and calculated ECD spectra of compounds 1a, 3b and 6b. | ||
Inspired by this phenomenon, compounds 2–11, isolated from seed kernels of C. bonduc, were individually dissolved in CDCl3 to test the scope and limitations of the method. Consequently, as shown in Fig. 1, aromatic derivatives 6-acetoxy-3-deacteoxycaesaldekarin e (2a)9 from caesalmin C (2),7 bonducellpin G (4),8 caesalmin E (7)7 and caesalmin F (8),7 caesall A (3a)11 from ζ-caesalpin (3),10 caesaldekarin e (5a)13 and caesalpinin MC (5b)15 from caesalpinin C (5),12 and 2-acetoxycaesaldekarin e (6a)14 and 6b from 14(17)-dehydrocaesalpin F (6)14 were obtained with excellent yields but the aromatization of δ-caesalpin (9),9 bonducellpin C acetate (10)17 and norcaesalpinin E (11)12 (Fig. S1†) were not achieved, indicating that diene system or allylic hydroxy group and allylic methoxy group in C ring might be indispensable to the aromatization conditions. Furthermore, among diene systems, compounds decorated with homoallylic hydroxy group (1 and 4) were more susceptible to the aromatization process than acetyl derivatives (2 and 3) of the homoallylic alcohol and free counterparts (5 and 6). Additionally, 5 dissolved in CDCl3 yielded a mixture of 5a and 5b in almost 5:5 ratio, similar to the case of 6. When 7 or 8 was subjected to the same condition, the first step was the elimination of hydroxy or methoxy at C-14 to give 2 as an intermediate, and subsequently generate 2a.
Compound 6b was assigned as a new cassane diterpenoid. 6b was obtained as a white amorphous powder, and its molecular formula was established to be C26H34O8 by HRESIMS analysis (m/z 497.2146 [M + Na]+, calcd 497.2151). The 1H NMR spectrum displayed three methyl signals (δH 1.18, s; 1.29, s; 1.44, s), three acetoxy methyl signals (δH 1.96, s; 1.97, s; 2.11, s), a benzylic methyl signal (δH 2.13, s), two dihydrofuran methylene signals (δH 3.10, t, J = 8.6 Hz, 2H; 4.45, dt, J = 16.9, 8.6 Hz; 4.47, dt, J = 16.9, 8.6 Hz), three oxygenated methine signals (δH 5.23, d, J = 3.8 Hz; 5.69, t, J = 3.6 Hz; 5.90, d, J = 3.2 Hz) and one aromatic proton (δH 6.24, s). The 13C NMR spectrum revealed the presence of 26 carbons including seven methyls, four methylenes (one oxygenated at δC 71.8), four methines (one olefinic at δC 102.8 and three oxygenated at δC 68.0, 74.9, 78.2) and eleven quaternary carbons (three ester carbonyls at δC 171.4, 171.5, 171.7, five olefinic at δC 125.2, 126.6, 133.9, 143.5, 159.2 and one oxygenated at δC 77.3). The 1H and 13C NMR spectra were similar to those of 5b, except for an extra acetoxy group in 6b, suggesting the presence of a dihydrobenzofuran fragment. The correlations of H-1 with C-10, C-20 and δC 171.4, H-2 with C-1, C-3 and δC 171.5, and H-3 with C-4, C-5, C-19 and δC 171.7 in the HMBC spectrum (Fig. 2) indicated that the acetoxy groups were attached at C-1, C-2 and C-3, respectively. The relative configuration of 6b was determined on the basis of coupling constants and the NOESY spectrum (Fig. 3). The small coupling constants between H-1 and H-2 (3.2 Hz), and between H-2 and H-3 (3.8 Hz), and NOE correlations from H-1 to H3-20, from H-2 to H3-19 and H3-20, and from H-3 to H3-18 and H3-19 suggested that acetoxy groups at C-1, C-2 and C-3 were all α-orientated. The absolute configuration of 6b was determined to be 1R,2S,3S,5R,10S by comparing experimental ECD spectrum with calculated spectra of a pair of enantiomers (Fig. 4). Thus, the structure of 6b was established as 1α,2α,3α-triacetoxy-5α-hydroxy-15,16-dihydro-14-methylvoucapane-8(14),9(11)-diene.
A previous work has reported the transformation of certain biomedical compounds such as bilirubin, thymine, uracil, dehydrocholesterol and vitamin D3 observed in deuterated solvents such as heavy water and CDCl3, and the chemical reaction of bilirubin in CDCl3 exhibited a striking resemblance to the photochemical reaction in CHCl3.18 Inspired by this research, the photochemical reactions of cassane diterpenoids in CHCl3 were explored. Gratifyingly, the photochemical reactions of cassane diterpenoids 1–8 in CHCl3 under laboratory conditions (room temperature and exposure to UV light, 254 nm) could produce corresponding aromatic derivatives similar in CDCl3 and other related compounds (Fig. 5). When 2 was dissolved in CHCl3 and irradiated with UV light, aromatic derivatives 2a, 2b and 17-norcassane diterpenoid norcaesalpinin MC (2c)15 were produced. The 1H NMR and 13C NMR spectra of 2b were similar to those of caesalpine D,19 except for the orientation of acetoxy group at C-7. Influenced by the rigid plane structure of benzene ring, the coupling constant between H-6 and H-7 is relatively small when the substituent groups at C-6 and C-7 were α- and β-oriented, respectively. Published compound caesalpin E20 and the following 3b have proved this point. Moreover, the orientation of substituent at C-7 remained unchanged in the photochemical reactions of 2 and 3 as evidenced by the products 2c, 3b and 3c. Hence the structure of 2b was established as 1α,6α,7β-triacetoxy-5α-hydroxy-14-methylvoucapane-8(14),9(11)-diene. As shown in Fig. 5, when 3 was subjected to the same condition, 3a, 3b and 17-norcassane diterpenoid caesalmin J (3c)16 were produced. The structure of 3b was assigned as a new cassane diterpenoid, and its molecular formula was established to be C23H30O6 by HRESIMS (m/z 425.1935 [M + Na]+, calcd 425.1940). The 1H NMR spectrum showed three methyl signals (δH 1.36, s; 1.39, s; 1.48, s), an acetoxy methyl signal (δH 1.90, s), a benzylic methyl signal (δH 2.49, s), a methoxy signal (δH 3.62, s), three oxygenated methine signals (δH 4.51, d, J = 4.3 Hz; 4.67, d, J = 4.3 Hz; 5.59, t, J = 3.7 Hz), one aromatic proton (δH 6.93, s) and two protons (δH 6.87, d, J = 2.2 Hz; 7.66, d, J = 2.2 Hz) of a 1,2-disubstituted furan ring. The 13C NMR spectrum displayed the presence of 23 carbons signals, including six methyls (one methoxy at δC 57.5), two methylenes, six methines (three olefinic at δC 104.6, 106.2, 146.2 and three oxygenated at δC 76.9, 77.3, 86.1) and nine quaternary carbons (one ester carbonyl at δC 171.5, five olefinic at δC 127.7, 130.5, 132.5, 141.5, 155.8 and one oxygenated at δC 81.0). The proton signals were assigned to the corresponding carbons through direct 1H and 13C correlations in the HSQC spectrum. In the HMBC spectrum (Fig. 2), the correlations of H-1 with C-5 and δC 171.5, H-6 with C-7, H-7 with C-6, C-9 and C-14, and –OCH3 with C-7 indicated that acetoxy, hydroxy and methoxy groups were attached to C-1, C-6 and C-7, respectively. The relative configuration of 3b was established through the NOESY spectrum (Fig. 3). The NOE correlations of H-1 with H-2α, H-2β and H3-20, H-6 with H3-20, and OCH3-7 with H3-20 suggested that acetoxy at C-1, hydroxy at C-6 and methoxy at C-7 were α-, α-, and β-oriented, respectively. Comparing the experimental ECD spectrum with calculated ECD spectra (Fig. 4) of two enantiomers, the absolute configuration of compounds 3b were determined to be 1S,5R,6S,7R,10S. Thus, compound 3b was identified as 1α-acetoxy-5α,6α-dihydroxy-7β-methoxy-14-methylvoucapane-8(14),9(11)-diene. As for the photochemical reactions of 7 and 8, the first step was the elimination of hydroxy or methoxy at C-14 to yield 2, followed by the reaction same as entry 2 (Fig. 5).
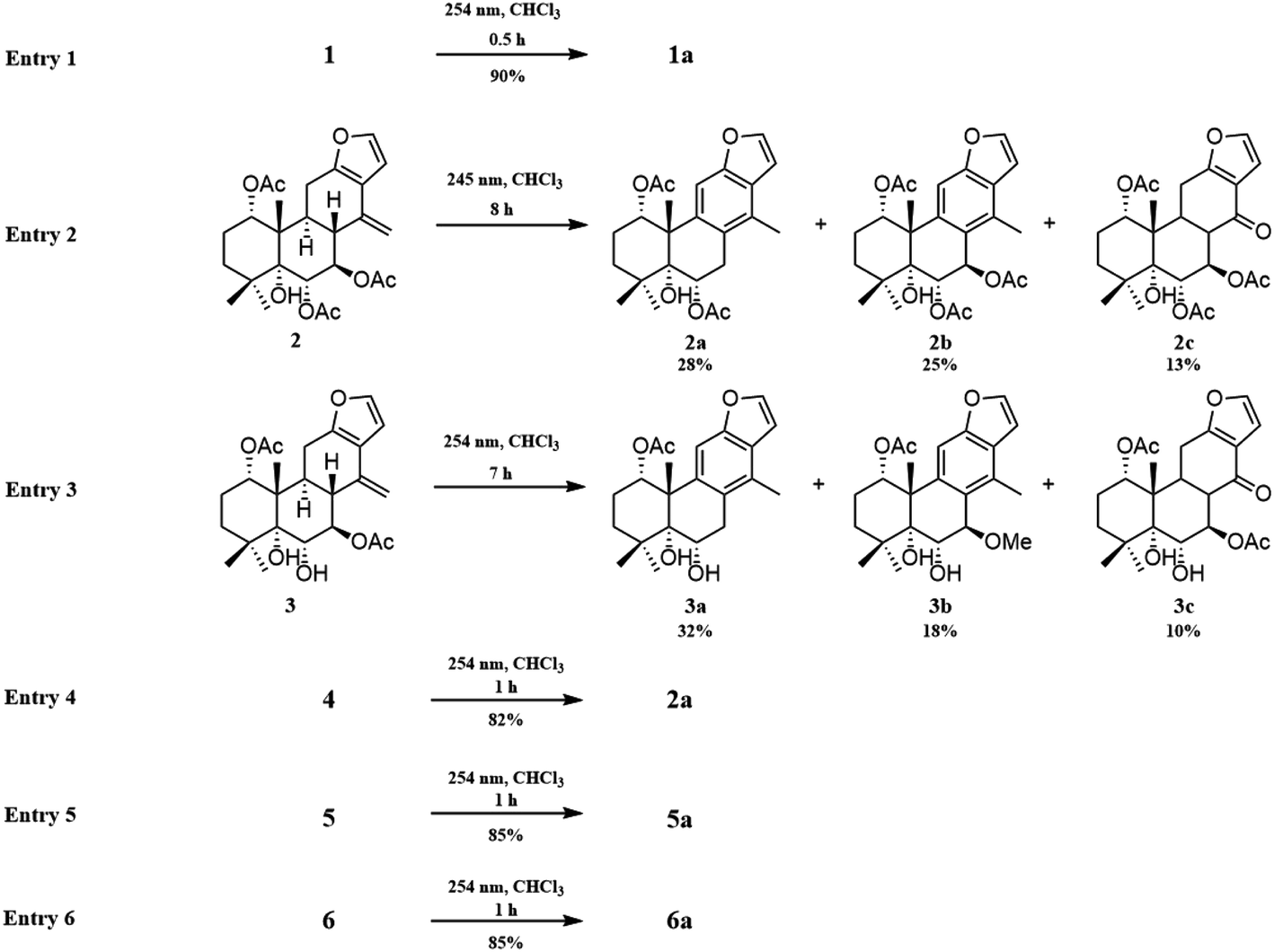 | ||
| Fig. 5 Scope of aromatization of cassane diterpenoids in CHCl3 irradiated with UV light. | ||
Given that CHCl3 might have some acidic impurities such as HCl upon storage and therefore promote acid-catalyzed aromatization of these compounds, so we anticipated that some Lewis acid might involve in the aforementioned aromatization reaction. To explore this idea the compound 1 was treated with a few Lewis acids (AlCl3, FeCl3, ZnCl2) in CHCl3 at room temperature, and fortunately, the use of AlCl3 generated the desired aromatic derivative 1a with excellent yield. Of the solvents screened (CH2Cl2, CHCl3, acetone, MeOH and CH3CN), both CHCl3 and CH2Cl2 could gave good yields. Under the above optimized conditions, compounds 2–11 were then treated with AlCl3 in CHCl3, and corresponding aromatic derivatives 2a, 3a, 5a, 5b, 6a and 6b were obtained from compounds 2–8 in good yields, and the reaction time could be greatly shortened (Table 1).
| Entry | Cassane diterpenoids | Aromatic derivatives | Start time (h) | End time (h) | Isolated yields |
|---|---|---|---|---|---|
| 1 | 1 | 1a | Instantly | Instantly | 90 |
| 2 | 2 | 2a | Instantly | 0.67 | 82 |
| 3 | 3 | 3a | Instantly | 0.08 | 84 |
| 4 | 4 | 2a | Instantly | 0.5 | 85 |
| 5 | 5 | 5a + 5b | 0.05 | 4 | 45:28 |
| 6 | 6 | 6a + 6b | 0.05 | 4 | 47:24 |
| 7 | 7 | 2a | 0.08 | 0.83 | 75 |
| 8 | 8 | 2a | 0.25 | 1 | 72 |
To gain better insight into the mechanism for the AlCl3-promoted aromatization of certain cassane diterpenoid, an aromatization reaction mechanism of 1 was suggested in Scheme 1. It was proposed to start with the coordination of AlCl3 with the oxygen atom of 6-OH to give a, followed by the attack of 7-OH to AlCl3 to yield b and subsequent ligand exchange that generated c with protonation of the C-7 hydroxy group. Next, loss of the C-7 protonated hydroxy group could give d, and subsequent 1,2-hydride shift from C-8 to C-7 would afford the allylic carbocation e, which would be susceptible to a further 1,2-hydride shift from C-9 to C-8 to afford f, making it easy to the formation of g with triene fragment in C ring. Finally, the rearrangement of double bond would provide the aromatic derivative 1a. The above route about AlCl3-promoted formation of triene g is supported by DFT calculations at B3LYP/6-31G (d,p). As shown in Fig. 6, AlCl3 coordinated with hydroxy group at C-6 in 1 to give intermediate a, which is more stable than 1 by 25.3 kcal mol−1, suggesting this complexation process are spontaneous. Then, attack of C-7 hydroxy on AlCl3 took place through transition state TS1 to give intermediate b with an activation free energy of 10.4 kcal mol−1 relative to a. This was followed by the ligand exchange and dehydration via intermediate c and transition state TS2 to generate d. The barrier for this step was calculated to be 16.6 kcal mol−1, which is 1.9 kcal mol−1 lower compared to 1. Subsequently, d was converted to f through twice 1,2-hydride shift via transition states TS3, TS4 and allylic carbocation intermediate e. The free energy barrier for twice hydride shift were calculated to be 11.8 and 11.2 kcal mol−1, respectively. Finally, g equipped with triene segment in C ring was formed through transition state TS5 with a barrier of 3.8 kcal mol−1. The above-mentioned results indicated that the rate-limiting step of the above reaction proceeds through transition state TS2 with a barrier of 16.6 kcal mol−1, and TS2 is more stable than 1 by 1.9 kcal mol−1, so above reaction could happen easily at room temperature.
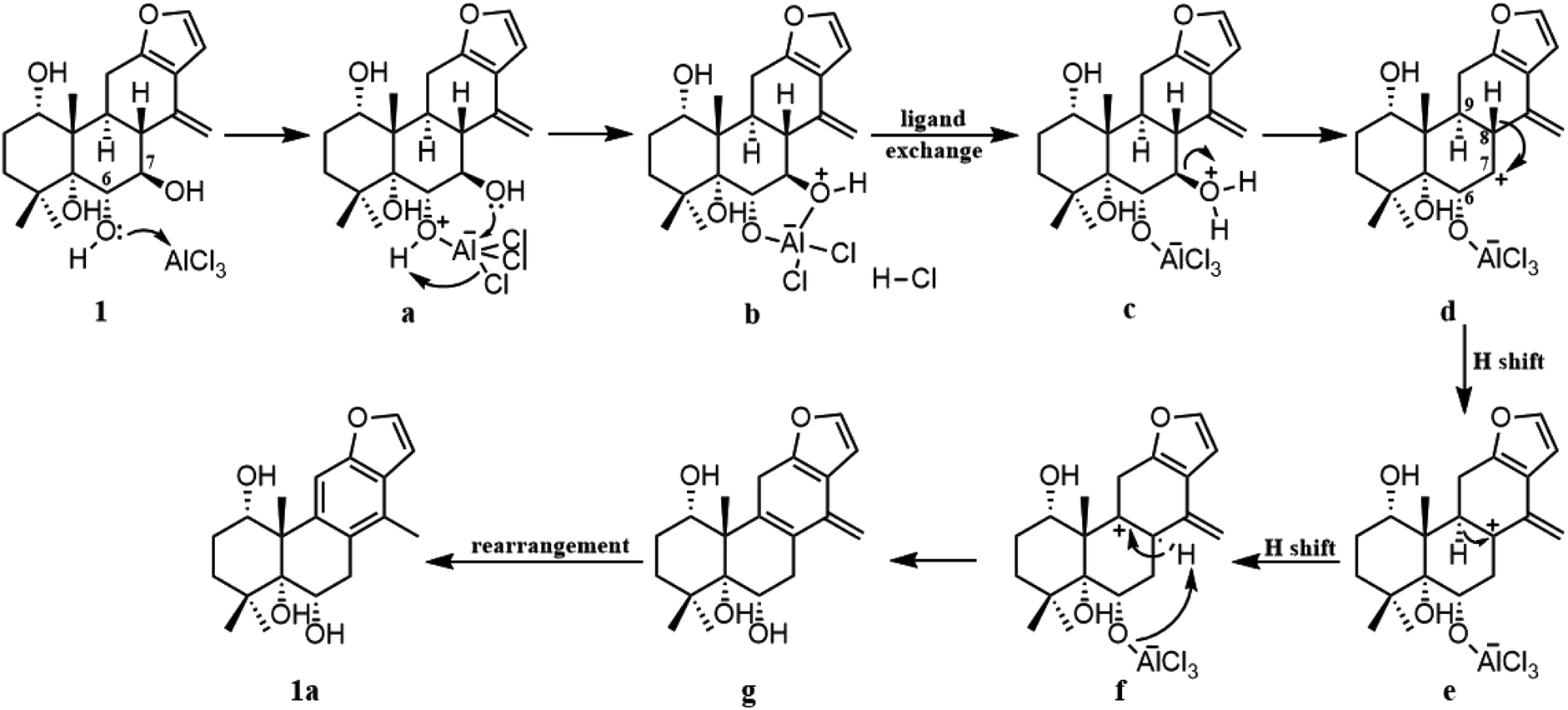 | ||
| Scheme 1 Putative mechanism for the AlCl3-mediated aromatization of 1. | ||
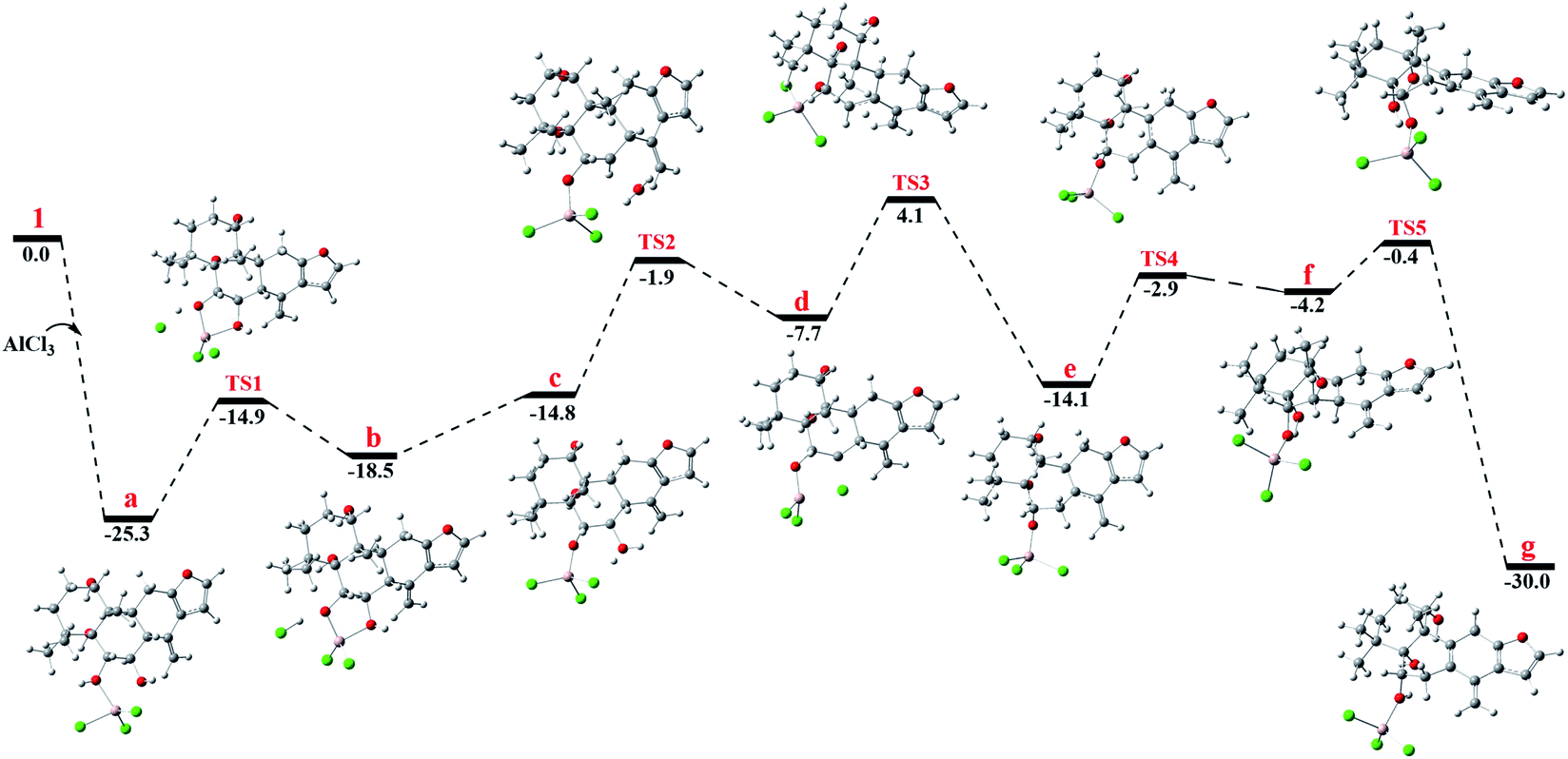 | ||
| Fig. 6 The free energy profile for the aromatization of 1 promoted by AlCl3. | ||
For the mechanism described above, it is reasonable for compounds 1–4, 7 and 8. As for compound 5 or 6, the absence of 7-oxy function led to more difficult aromatization of C ring compared to compounds 1–4, presumably there is no obvious leaving group, which is needed for the formation of carbocation or unsaturated double bond, and longer reaction time also provided evidence to support this. On the other hand, the dehydrogenation of diene in C ring of compound 5 or 6 might be due to air oxidation in the presence of AlCl3, and stabilization of 5 or 6 could be the driving force in losing the hydrogen by air oxidation to form the aromatic derivative 5a or 6a.21 Furthermore, compared with compound 5 or 6, degree of unsaturation and chemical formula of side product 5b or 6b remained unchanged, prompting that acid-induced rearrangement took place, and consequently the molecular was fully delocalized and conjugated. Further studies however are required to verify the mechanism proposed above.
In the photochemical reactions of compounds 2 and 3, besides compounds 2a and 3a, aromatic derivatives 2b and 3b, and oxidative products 2c and 3c also been obtained. We speculated that they may be generated through photo-induced aromatization and oxidation owing to the existence of double bond between C-14 and C-17, and research is continuing to confirm the exact reaction mechanism.
4 Conclusions
In this study, three methods of aromatizing several cassane diterpenoids were described. The aromatization of cassane diterpenoids occurred in CDCl3 reminded us that deuterated solvents display somewhat different chemical nature with their ordinary counterparts and may contribute to the transformation of specific compounds. For natural products researchers, particular attention should be paid to this point. The photochemical reactions of cassane diterpenoids reported in this study also provided us with new prospective of influence of UV light on the natural products. Additionally, compounds 2a, 2c, 3a, 3c, 5a, 5b and 6a have been isolated from plants of Caesalpinia genus, revealing that several biochemical processes may be related to laboratory studies. The chemical transformations reported here would provide information related to the possible biogenesis of these diterpenoids and could be useful for the synthesis of these biologically interesting compounds.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 31670359), Liao Ning Revitalization Talents Program (No. XLYC1905019) and Natural Science Foundation of Liaoning Province (No. 201602691).Notes and references
- W. H. Jing, X. X. Zhang, H. X. Zhou, Y. Wang, M. Q. Yang, L. P. Long and H. Y. Gao, Fitoterapia, 2019, 134, 226–249 CrossRef CAS PubMed.
- S. Mahdjour, M. Harche-Haid, A. Haidour, R. Chahboun and E. Alvarez-Manzaneda, Org. Lett., 2016, 18, 5864–5967 CrossRef PubMed.
- M. Chatzopoulou, A. Antoniou, E. N. Pitsinos, M. Bantzi, S. D. Koulocheri, S. A. Haroutounian and A. Giannis, Org. Lett., 2014, 16, 3344–3347 CrossRef CAS PubMed.
- P. Gutierrez, J. Altarejos, P. J. Linares-Palomino, R. Chahboun and E. Alvarez-Manzaneda, Org. Chem. Front., 2018, 5, 2537–2541 RSC.
- H. Zentar, F. Arias, A. Haidour, R. Alvarez-Manzaneda, R. Chahboun and E. Alvarez-Manzaneda, Org. Lett., 2018, 20, 7007–7010 CrossRef CAS PubMed.
- E. N. Pitsinos, I. Mavridis, E. Tzouma and V. P. Vidali, Eur. J. Org. Chem., 2020, 4730–4742 CrossRef CAS.
- R. W. Jiang, S. C. Ma, P. P. H. But and T. C. W. Mak, J. Nat. Prod., 2001, 64, 1266–1272 CrossRef CAS PubMed.
- K. Pudhom, D. Sommit, N. Suwankitti and A. Petsom, J. Nat. Prod., 2007, 70, 1542–1544 CrossRef CAS PubMed.
- A. Balmain, J. D. Connolly, M. Ferrari, E. L. Ghisalberti, U. M. Pagnoni and F. Pelizzoni, J. Chem. Soc., 1970, 19, 1244–1245 Search PubMed.
- S. K. Kalauni, S. Awale, Y. Tezuka, A. H. Banskota, T. Z. Linn, P. B. S. Asih, D. Syafruddin and S. Kadota, Biol. Pharm. Bull., 2006, 29, 1050–1052 CrossRef CAS PubMed.
- L. Wu, J. Luo, Y. M. Zhang, X. B. Wang, L. Yang and L. Y. Kong, Fitoterapia, 2014, 93, 201–208 CrossRef CAS PubMed.
- T. Z. Linn, S. Awale, Y. Tezuka, A. H. Banskota, S. K. Kalauni, F. Attamimi, J. Y. Ueda, P. B. S. Asih, D. Syafruddin, K. Tanaka and S. Kadota, J. Nat. Prod., 2005, 68, 706–710 CrossRef CAS PubMed.
- I. Kitagawa, P. Simanjuntak, T. Mahmud, M. Kobayashi, S. Fujii, T. Uji and H. Shibuya, Chem. Pharm. Bull., 1996, 44, 1157–1161 CrossRef CAS.
- K. O. Pascoe, B. A. Burke and W. R. Chan, J. Nat. Prod., 1986, 49, 913–915 CrossRef CAS.
- S. K. Kalauni, S. Awale, Y. Tezuka, A. H. Banskota, T. Z. Linn and S. Kadota, J. Nat. Prod., 2004, 67, 1859–1863 CrossRef CAS PubMed.
- J. L. Zhang, Z. H. Chen, J. Xu, J. Li, Y. F. Tan, J. H. Zhou, W. C. Ye, H. Y. Tian and R. W. Jiang, RSC Adv., 2015, 5, 76567–76574 RSC.
- S. R. Peter, W. F. Tinto, S. Mclean, W. F. Reynolds and M. Yu, J. Nat. Prod., 1997, 60, 1219–1221 CrossRef CAS.
- F. M. Salih, A. E. Pillay and A. Al-Hamdi, J. Photochem. Photobiol., B, 2003, 70, 91–97 CrossRef CAS.
- P. Zhao, H. Q. Chen, H. Wang, J. Xu, M. C. Wang, Y. L. Wang, D. Q. Jin and Y. Q. Guo, Phytochem. Lett., 2013, 6, 606–609 CrossRef CAS.
- G. X. Ma, J. Q. Yuan, H. F. Wu, L. Cao, X. P. Zhang, L. J. Xu, H. Wei, L. Z. Wu, Q. X. Zheng, L. Y. Li, L. J. Zhang, J. S. Yang and X. D. Xu, J. Nat. Prod., 2013, 76, 1025–1031 CrossRef CAS.
- T. Narender, S. Sarkar, K. Rajendar and S. Tiwari, Org. Lett., 2011, 13, 6140–6143 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra03636j |
| This journal is © The Royal Society of Chemistry 2021 |