
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA new solvate of clonixin and a comparison of the two clonixin solvates†
Yunping Zhoujina,
Mingtao Zhang b,
Sean Parkinc,
Tonglei Lid,
Faquan Yu*a and
Sihui Long*a
b,
Sean Parkinc,
Tonglei Lid,
Faquan Yu*a and
Sihui Long*a
aKey Laboratory for Green Chemical Process of Ministry of Education, Hubei Key Laboratory of Novel Reactor and Green Chemical Technology, Hubei Engineering Research Center for Advanced Fine Chemicals, School of Chemical Engineering and Pharmacy, Wuhan Institute of Technology, 206 1st Rd Optics Valley, East Lake New Technology Development District, Wuhan, Hubei 430205, China. E-mail: fyuwucn@gmail.com; longsihui@yahoo.com; Sihuilong@wit.edu.cn; Tel: +86 27 87194980
bComputational Center for Molecular Science, College of Chemistry, Nankai University, Tianjin, China
cDepartment of Chemistry, University of Kentucky, Lexington, Kentucky 40506, USA
dDepartment of Industrial and Physical Pharmacy, Purdue University, West Lafayette, Indiana 47907, USA
First published on 16th July 2021
Abstract
A new solvate of clonixin (CLX), a dimethylacetamide (DMA) solvate, has been obtained by crystal growth in DMA. The new form was characterized by NMR, single-crystal X-ray diffraction, and PXRD. The crystal structure is stabilized by a strong hydrogen bond between the carboxylic acid OH of CLX and the DMA carbonyl, the strength of which is on par with those of the four solvent-free forms of CLX and the DMF solvate. These previously known forms are based on either the acid–acid homosynthon or the acid–pyridine heterosynthon, depending on the dihedral angle between the two aromatic rings of CLX, or the heterodimer between CLX and DMF. The new solvate loses DMA to convert into form I of CLX, as confirmed by differential scanning calorimetry (DSC) and powder X-ray diffraction (PXRD), similar to how the DMF solvate does. A comparison of the two solvates was carried out and theoretical studies were performed to shed light on the conformational difference between the two CLX molecules in the two solvates and the packing differences between them. The insight gained on this solvatomorphic system could aid the design of new solvates and cocrystals of CLX.
1 Introduction
Many compounds are known to exist in different solid forms, be it polymorphs, solvates (including hydrates), or as cocrystals.1–4 Different solid, particularly crystalline, forms of a given compound can have different properties, which have attracted great interest from researchers.5–8 Cocrystals have been hotly pursued in the past two decades due to controlling and exploiting these ramifications. In pharmaceutical science, cocrystals are of particular significance.9–12Solvates, regarded as one type of cocrystal by some practitioners,13 have been widely studied and have well established applications.14,15 Although solvates may have a tendency to lose their solvent content and some organic solvents present in solvatomorphs of a therapeutically indicated drug substance might increase toxicity risk, Byrn16 has provided rational reasons for their continued study. The solvatomorph could be the penultimate solid form of the drug substance, having been specifically chosen for recovery or purification.17,18 A solvate could induce a preferable morphology compared with its solvent-free counterpart.19 High quality crystals suitable for single-crystal structure determination of the drug substance might only be obtainable as solvates. The desolvated form of a solvate could facilitate its dissolution.20,21 From a business perspective, solvates can be considered as new patentable entities.22,23 For example, Trametinib (Mekinist, a trimetinib tablet) is a MEK inhibitor developed by GlaxoSmithKline (GSK). The formulation is a DMSO solvate and was approved by the US FDA on May 29, 2013. This drug can be used to treat unresectable or metastatic melanoma with BRAF (murine sarcoma virulence tumor-causing homolog B1 gene) V600E or V600K mutation.15
Clonixin [2-(2-methyl-3-chloroanilino)nicotinic acid, a. k. a. CLX] (Scheme 1), is a classic non-steroidal anti-inflammatory drug (NSAID) with important applications ranging from anti-inflammatory and analgesic24,25 to platelet-inhibition26,27 and acute migraine relief.28,29 Due to its pharmaceutical stability and bioavailability issues, currently microemulsions30 and clonixin lysinate are used in clinical formulations.31
 | ||
| Scheme 1 Structure of CLX. | ||
Due to both conformational flexibility and the presence of multiple functional groups, i.e. carboxylic acid and pyridine, CLX is known to exist in at least four solvent-free forms, demonstrating both synthon polymorphism, conformational polymorphism, and tautomeric polymorphism.32
The carboxylic acid and pyridine functional groups on CLX are good hydrogen bond formers. We envisioned it could form hydrogen bonds with other compounds, forming solvates or cocrystals depending on the state (liquid or solid) of the coformer (s) involved. Solvates and cocrystals of pharmaceuticals have been proven to be promising alternative formulation approaches.9,15,33,34
The vision of forming multicomponent crystals of CLX was first realized by the harvest of a DMF solvate of CLX, i.e. CLX·DMF (designated S1).35 In the solvate, a hydrogen bond is formed between the carboxylic OH of CLX and the carbonyl O of DMF, and the two common synthons, i.e. acid–acid homosynthon and acid–pyridine heterosynthon, are not observed. The strength of the new hydrogen bond was found to be on par with that of the acid–acid dimer and the acid–pyridine catemer. Meanwhile, two cocrystals of CLX have also been synthesized with L-proline and piroxicam as coformers.36,37
This phenomenon intrigued us: could this hydrogen bond be robust enough to lead to other solvates with DMF analogs such as dimethylacetamide (DMA), formamide, acetamide, propionamide, butyramide, etc., as they can also provide a hydrogen bond acceptor by virtue of the amide carbonyl? To investigate, we set out to study the crystal growth of CLX in a series of solvents analogous to DMF.
Here we report the discovery and characterization of a new solvate of CLX, a DMA solvate, and the comparison of the two CLX solvates. The new form was fully characterized by NMR, single-crystal X-ray diffraction, and PXRD, and its phase behavior was investigated by DSC and TGA. Moreover, we also performed theoretical calculations to provide an explanation for the rise of the solvatomorphic system. The insight gained from the experimental and theoretical investigation should provide information pertinent to the design of more solvates and cocrystals of CLX.
2 Experimental section
2.1 Materials
All chemicals were purchased from commercial sources: 2-chloro-nicotinic acid, 3-chloro-2-methylaniline and p-toluenesulfonic acid were from Aladdin Industrial Corporation; pyridine and other solvents were from Sinopharm Chemical Reagent Co., Ltd, and were used as received.2.2 Synthesis
CLX was synthesized according to a literature procedure routinely applied in our laboratory and purified by column chromatography and recrystallization.352.3 Crystallization
Quench cooling was initially employed for the solvate screening, and slow evaporation was performed in order to confirm the result and obtain quality crystals suitable for structure determination by the single-crystal X-ray diffraction.38 All crystals were harvested directly from their mother liquor.2.4 Characterization
1H NMR, DSC, and TGA were employed to confirm the association of solvent with CLX. 1H NMR spectrum was recorded on a Premium Shielded spectrometer (USA) in DMSO-d6. Thermal analyses were performed on SII instruments DSC6220 (Japan). Tzero pans and aluminum hermetic lids were used for measuring, in general, a few milligrams of samples. A heating rate of 10 °C min−1 was employed. Thermogravimetric analysis (TGA) tests were carried out on SDT Q600 of TA Instrument (USA) with a few milligrams of ground sample placed in an open aluminum pan and a heating rate of 10 °C min−1 applied under N2 atmosphere.Crystallographic data for CLX·DMA were collected at 293(2) K on a Rigaku Oxford diffractometer using a CuKα radiation (λ = 1.54184 Å). Cell refinement and data reduction were done using CrysAlisPro. Structure solution and refinement were carried out using the SHELXS and SHELXL programs, respectively.
PXRD data for the solvate and desolvated sample were collected on a Rigaku X-ray diffractometer with CuKα radiation (40 kV, 15 mA, λ = 1.5406 Å) between 5.0–50.0° (2θ) at ambient temperatures.
2.5 Computational details
The molecules were optimized at the B3LYP/6-31G(d) level of theory based on the initial crystal structures, which were confirmed to be stable conformations (except for a dihedral angle restriction of the CLX–DMA single hydrogen-bond structure to maintain the conformation in the crystal, but without other geometrical constraints), and then single-point calculations at the B3LYP/DEF2TZVP level of theory were performed to obtain more accurate energies for hydrogen bonding and π–π interactions. Meanwhile, considering the solvating effect of DMF or DMA, the IEFPCM solvation model was employed. The dispersion energy contributions to lattice energies were calculated using the GD3 program of Grimme.39 All calculations were performed with Gaussian16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 40 and conducted on a Linux cluster. Hirshfeld surface analyses were performed with CrystalExplorer 17.41
40 and conducted on a Linux cluster. Hirshfeld surface analyses were performed with CrystalExplorer 17.41
3 Results and discussion
3.1 Crystal structures
A new solvate (CLX·DMA, designated S2) was obtained for CLX (Fig. 1). S2 crystallized as colorless needles from DMA. It is monoclinic, space group P21/c (Z = 4). Crystallographic data for the new form and the DMF solvate are listed in Table 1, and the complete CIF file is provided in the ESI.† There is one formula unit in the asymmetric unit of S2. The crystallographically independent molecule CLX has a near planar conformation with a dihedral angle between the two aromatic rings of 2.60(7)°, similar to that of form IV of solvent-free CLX. In contrast, the same molecule in CLX·DMF has a highly twisted conformation with the corresponding dihedral angle of 66.51(5)°, similar to that of form I of CLX (68.22(5)°). A superposition of the two molecules is provided in Fig. 2. | ||
| Fig. 1 Crystals and structure of two CLX solvates.35 Scale bar 0.2 mm. | ||
| S1 | S2 | |
|---|---|---|
| Formula | C16H18ClN3O3 | C17H20ClN3O3 |
| Formula weight | 335.78 | 349.81 |
| Crystal size (mm) | 0.30 × 0.20 × 0.10 | 1.00 × 0.20 × 0.10 |
| Crystal system | Triclinic | Monoclinic |
| Space group | P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/c |
| a/Å | 8.4310(2) | 15.4376(4) |
| b/Å | 9.1450(2) | 5.92226(13) |
| c/Å | 11.1420(2) | 19.0732(6) |
| α/° | 103.6360(8) | 90 |
| β/° | 93.1390(8) | 100.548(3) |
| γ/° | 107.8450(9) | 90 |
| Z, Z′ | 2, 1 | 4, 1 |
| V/Å3 | 787.16(3) | 1714.31(8) |
| Dcal/g cm−3 | 1.417 | 1.355 |
| T/K | 90.0(2) | 293(2) |
| Abs coeff. (mm−1) | 0.262 | 2.150 |
| F(000) | 352 | 736 |
| Range (deg) | 1.90–27.49 | 4.717–66.856 |
| Limiting indices | −10 ≤ h ≤ 10 | −18 ≤ h ≤ 17 |
| −11 ≤ k ≤ 11 | −6 ≤ k ≤ 7 | |
| −14 ≤ l ≤ 14 | −22 ≤ l ≤ 21 | |
| Completeness to 2θ | 99.9% | 99.5% |
| Unique reflections | 2845 | 9510 |
| R1[I > 2σ(I)] | 0.0402 | 0.0405 |
| wR2 (all data) | 0.1039 | 0.1155 |
| CCDC refcode/accession code | FITBIW | 2079841 |
 | ||
| Fig. 2 Superposition of the two molecules in the asymmetric unit of CLX·DMF and CLX·DMA. | ||
Although the flat molecule of CLX prefers to form an acid–acid homodimer in its solvent free forms, due to the presence of DMA, neither the acid–acid homosynthon nor the acid–pyridine heterosynthon is observed in the new solvated crystal structure. Instead, the hydrogen bond is between the carboxylic OH of CLX and the carbonyl O of DMA (Fig. 3), similar to that of CLX·DMF. This hydrogen bond should be stronger than or at least on par with the other two types considering the shorter bond distance of 1.74 Å and the near linear alignment of the three atoms (173.73° for bond angle), which is close to those of CLX·DMF. The intramolecular hydrogen bond has parameters of 1.910 Å and 145.37°. Similar to DMF, DMA provides a strong hydrogen-bond acceptor for interaction with CLX.
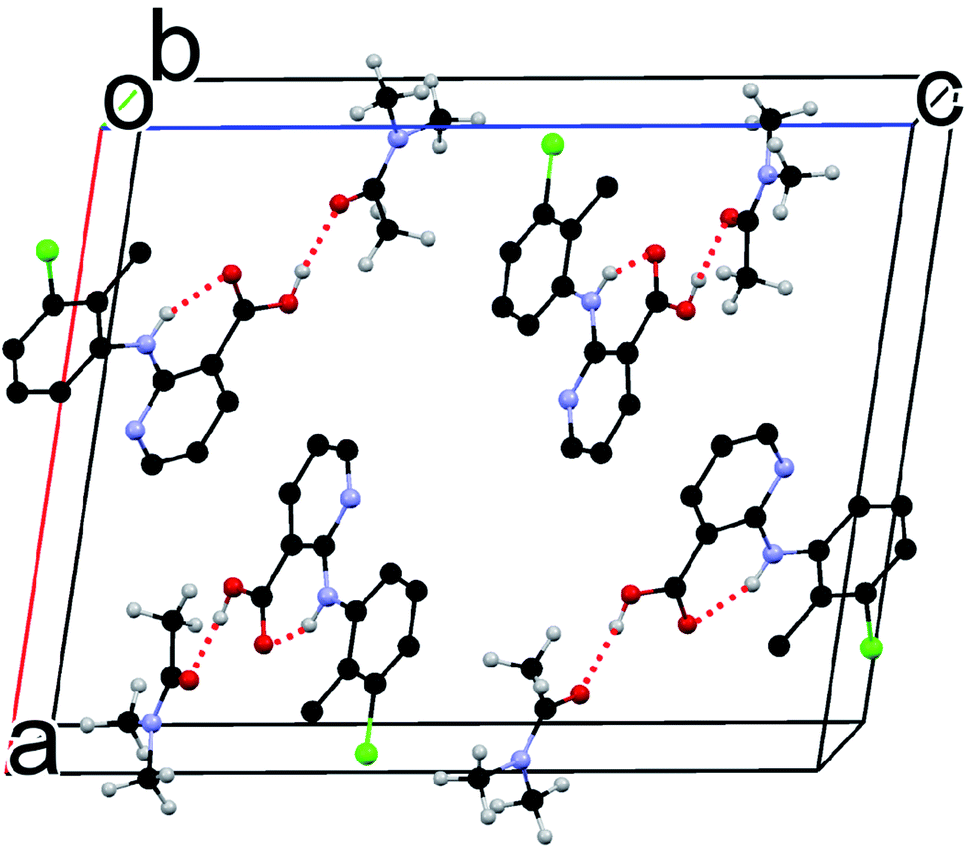 | ||
| Fig. 3 Crystal packing of S2 (for clarity, hydrogens not involved in H-bonding are omitted). | ||
3.2 Thermal properties
DSC was conducted to investigate the thermal properties of S2. The DSC thermogram of the solvate is shown in Fig. 4. It shows two main thermal events: the first with an onset temperature of 87.3 °C and heat of fusion of 59.8 J g−1 corresponds to the loss of the solvent DMA and transformation into form I of CLX, while the second with an onset temperature of 240.1 °C and heat of fusion 42.1 J g−1 matches the melting of form I. The unevenness of the DSC curve could be due to the accumulation of DMA in the DSC pan since it is a solvent with a high boiling point. The desolvation was also confirmed by the TGA study, as suggested by a weight loss of ∼22.5% (theoretical value is 24.9%), suggesting an essentially 1:1 ratio of CLX and DMA (Fig. 5). In addition, the 1:1 ratio of DMA and CLX is also verified by a 1H NMR study as indicated by the 1:1:1:1 ratio of the integers of the CH3 (chemical shift 2.32 ppm) from CLX and the three CH3 groups (chemical shifts 2.94 ppm, 2.78 ppm and 1.95 ppm) from DMA (ESI†).
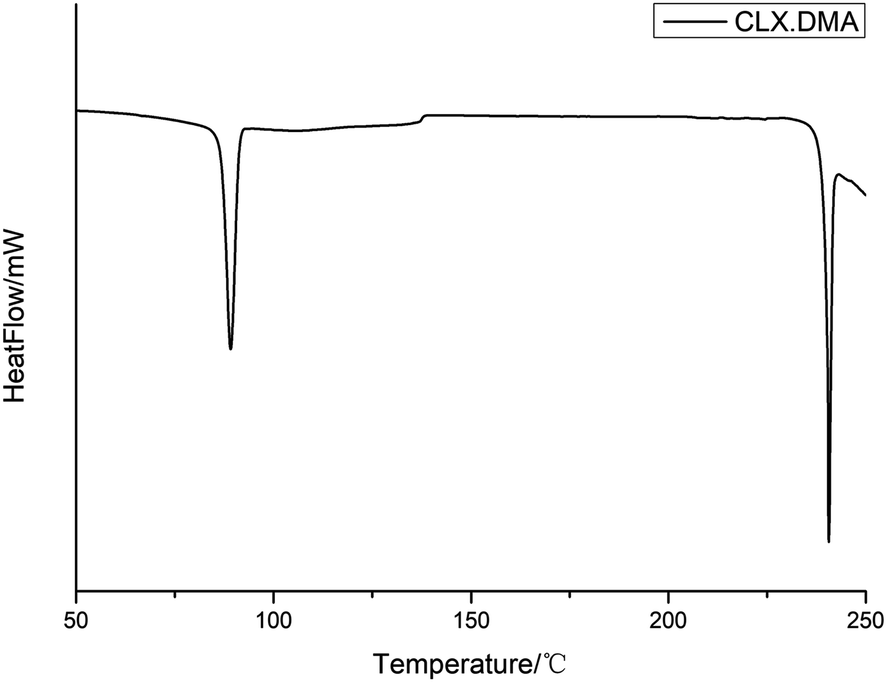 | ||
| Fig. 4 DSC thermogram of CLX·DMA. | ||
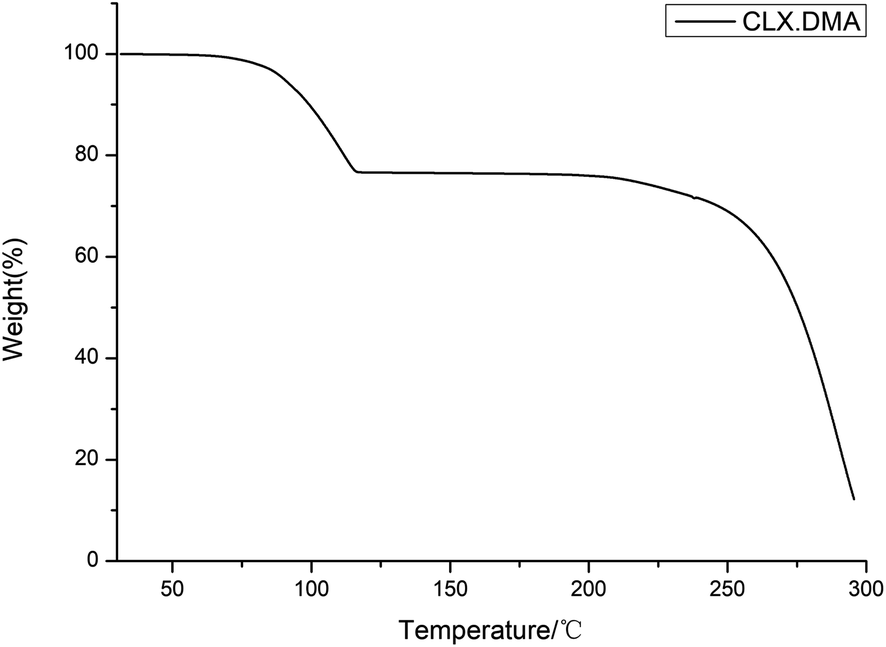 | ||
| Fig. 5 TGA thermogram of CLX·DMA. | ||
Fig. 6 shows the PXRD pattern of the solvate collected at room temperature, along with PXRD patterns calculated from the single-crystal structure determined at 293 K, as well as the PXRD patterns of CLX·DMA after desolvation and form I of CLX, which confirm the phase transition from the solvate to form I of CLX after thermal treatment. As can be seen, when the solvate was heated to 90 °C and kept at that temperature for two hours, the resulting colorless sample showed a PXRD pattern matching that of form I (Fig. 6).
 | ||
| Fig. 6 Experimental PXRD patterns of CLX·DMA before and after desolvation and form I of CLX and calculated PXRD patterns of CLX·DMA and form I of CLX. | ||
3.3 Computational analyses
DMA differs from DMF by the addition of a methyl group. Since both compounds act as a hydrogen-bond acceptor due to the presence of the amide carbonyl, one might expect the two solvates to be similar in the solid state. Yet, they are dramatically different, as evidenced by the conformational difference between the CLX molecules as well as the overall packing in each solvate.In CLX·DMF, CLX takes on a twisted conformation, and it forms a heterodimer with DMF. The dimer could be viewed as being sustained by a strong O–H⋯O hydrogen bond with parameters of 1.771 Å and 178.22° and a weak sp2C–H⋯O hydrogen bond with parameters of 2.274 Å and 136.25° (Fig. 7a). In addition, π–π interaction between the pyridine ring and another DMF molecule underneath it is observed (Fig. 8b).
 | ||
| Fig. 7 (a) The heterodimer based on R22(7) hydrogen bond motif of S1; (b) the heterodimer based on D2 motif of S2; (c) the hypothetical heterodimer based on R22(8) motif of S2. | ||
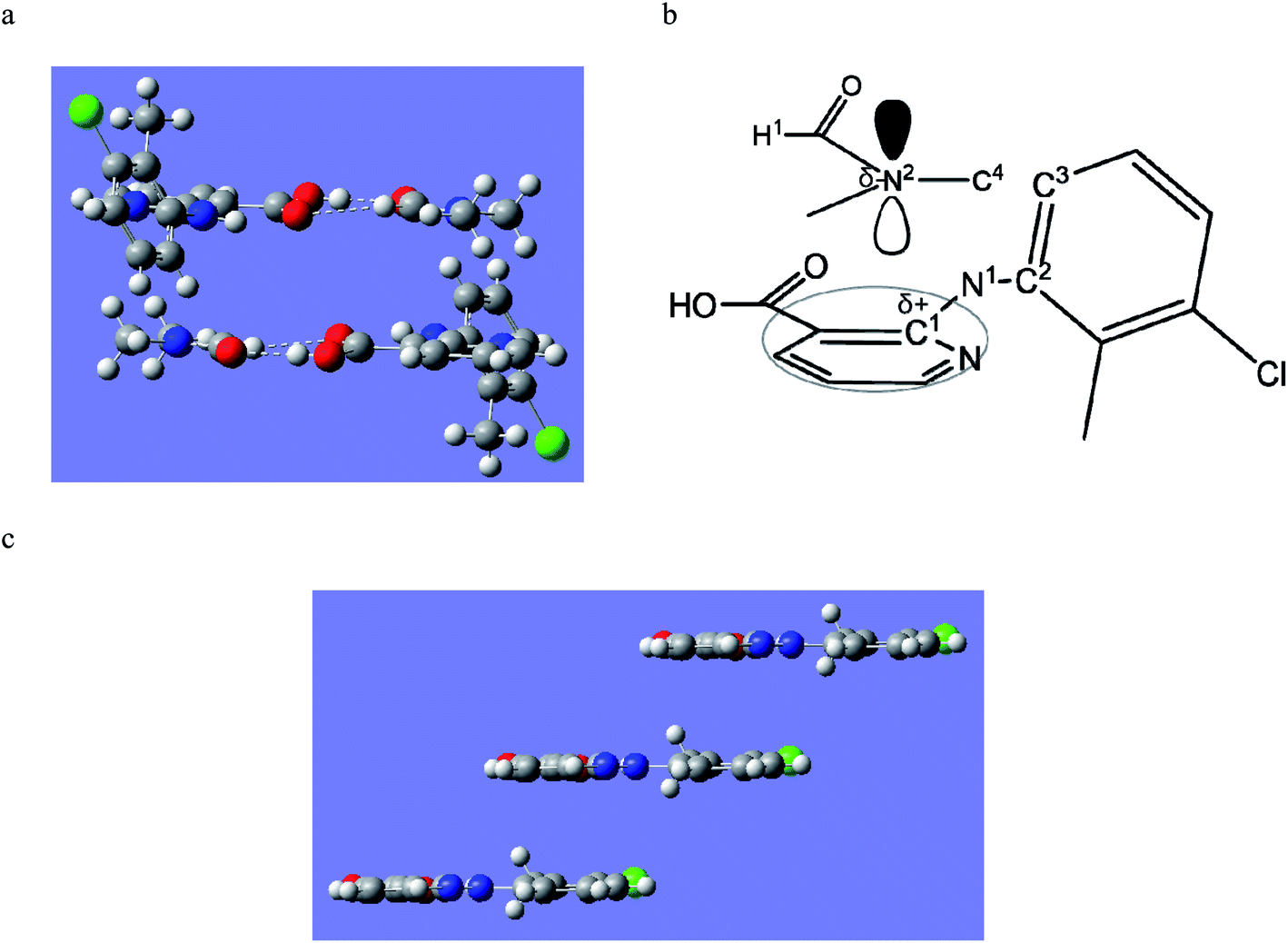 | ||
| Fig. 8 (a) The π–π interaction between CLX and DMF in S1; and (b) the bonds involved in the π–π interaction; (c) the π–π stacking of CLX in S2. | ||
In DMA, as aforementioned, the CLX molecule is in a planar conformation, and it also forms a dimer with DMA (Fig. 7b). Yet, the dimer is based only on the carboxylic O–H⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C hydrogen bond, as the H covalently bonded to sp2C in DMF is replaced with sp3CH3, and no π–π interaction between CLX and DMA is observed. Why would not CLX·DMA pack in a similar fashion to CLX·DMF?
C hydrogen bond, as the H covalently bonded to sp2C in DMF is replaced with sp3CH3, and no π–π interaction between CLX and DMA is observed. Why would not CLX·DMA pack in a similar fashion to CLX·DMF?
First we wanted to see if conformational variation of the two CLX molecules in the two solvates would lead to a significant difference in conformational energy. It turned out that the twisted and planar conformations have similar conformational energies as suggested by calculations based on different levels of theory, as either ΔE or ΔG is less than 1 kcal mol−1. Thus, conformational energy is not an issue in the molecular packing. Then, what if the planar CLX in S2 forms a hypothetical dimer (Fig. 7c) with DMA in a similar fashion as that of CLX and DMF? The hydrogen bonding energy of the hypothetical dimer of CLX and DMA is calculated to be −11.0 kcal mol−1, which is comparable to that of CLX and DMF dimer (−11.1 kcal mol−1). But if CLX·DMA packs similarly to CLX·DMF, the distance between the carbonyl C of CLX and that of DMA would increase from 3.686 Å to 4.256 Å, due to the size of CH3 in contrast to H, which would cause a close contact between the methyl (C4 in Fig. 8b) on N and the benzene ring (C3 in Fig. 8b). If the same antiparallel π–π interaction in CLX·DMF existed between CLX and DMA, the significant steric hindrance between C4 and C3 would induce a dihedral rotation (C1–N1–C2–C3 in Fig. 8b) which would result in the transformation of CLX from a twisted conformation to planar. In the new dimer in CLX·DMA, the hydrogen bond has strength of −10.4 kcal mol−1, which rivals that of both CLX–DMF dimer and the hypothetical one. Furthermore, the new planar CLX·DMA synthon induces a new packing mode (as shown in Fig. 8c), and either the pyridine ring or the benzene ring can stack with its neighboring synthons, with a stabilization energy of −14.8 kcal mol−1 (−7.4 kcal mol−1 for each side), which is on par with the π–π stacking between CLX and DMF (−14.4 kcal mol−1) in CLX·DMF. Thus, the new packing mode in CLX·DMA is likely optimal and as stable as that of CLX·DMF.
3.4 Hirshfeld analysis
Hirshfeld analysis was also performed for the CLX molecule in S2 and the results are shown in Fig. 9 and 10. It is evident that there are dominant interactions represented by the bright red spots in Fig. 9, which are caused by the hydrogen-bond interaction between the H atom of the carboxylic acid and the O atom of DMA. The hydrogen-bond interaction can also be expressed as a sharp spike in the lower-left corner of the fingerprint plot (Fig. 10). Other close contacts were also apparent in their contributions to the Hirshfeld surface area. H–π interactions can be seen as C⋯H contacts covering 18.9% of the Hirshfeld surface. H⋯Cl contacts can also be found with a proportion of 11.0%. π–π stacking interactions were represented by C⋯C contacts with a proportion of 6.8% and by C⋯N contacts with a proportion of 1.4%. Hirshfeld surface analysis provides a more complete description of the interactions of molecules in the crystal. Taken together, the crystal structures are stabilized by a variety of interactions. | ||
| Fig. 9 Hirshfeld surface of CLX in S2. | ||
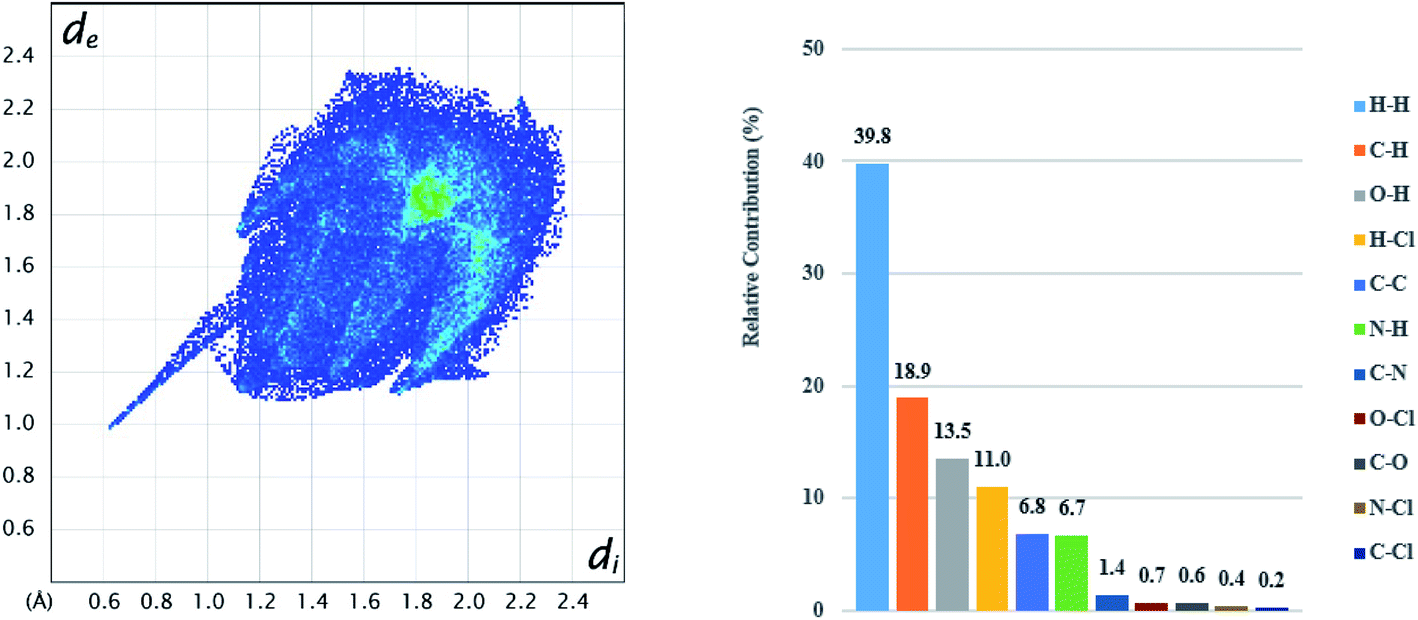 | ||
| Fig. 10 2D fingerprint plots (left) for CLX in CLX·DMA and relative contributions to the Hirshfeld surface (right) by the various intermolecular contacts in the crystal. | ||
4 Conclusions
A new solvate of CLX has been obtained by crystal growth in DMA. The new form was fully characterized by 1H NMR, SCXRD, and PXRD. The crystal structure of the new form is sustained on the hydrogen bond between the carboxylic acid OH of CLX and the DMA carbonyl O, which is similar to CLX·DMF, yet different from the acid–acid homosynthon or the acid–pyridine heterosynthon in the four known solvent-free forms of CLX. Thermal studies of CLX·DMA by DSC and TGA revealed that desolvation led to form I of CLX, similar to the desolvation of CLX·DMF. The dramatic difference in the conformation of the two host molecules in the two solvates and the packing patterns was explained by the analysis of intermolecular interactions in the solid state. Hirshfeld analysis further delineates the contribution of individual interactions to the overall stability of the new solvate. Although the obtained solvates may not serve as new formulation possibilities for CLX, as the solvents are not generally considered safe, the information gained in this study can be applied to potential cocrystal formation, which may provide alternative/superior formulation for the classic NSAID CLX. Using compounds with similar structural characteristics as DMF, i.e., good hydrogen bond acceptors as coformers, there is a good reason to believe cocrystals involving CLX could be discovered, such follow-up work is underway.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
SL thanks Natural Science Foundation of Hubei Province for financial support (2014CFB787). TL is grateful to NSF for supporting the work (DMR1006364).References
- T. L. Threlfall, Analyst, 1995, 120, 2435–2460 RSC.
- J. Bernstein, Polymorphism in Molecular Crystals 2e. International Union of Crystal, 2020 Search PubMed.
- H. G. Brittain, Polymorphism in pharmaceutical solids. CRC Press, 2018 Search PubMed.
- R. Hilfiker, Polymorphism in the pharmaceutical industry. Wiley Online Library, 2006 Search PubMed.
- C. Sun and D. J. Grant, Pharm. Res., 2001, 18, 274–280 CrossRef CAS PubMed.
- M. Pannipara and A. Al-Sehemi, Research Square, 2021 Search PubMed.
- A. Sokal, E. Pindelska, L. Szeleszczuk and W. Kolodziejski, Int. J. Pharm., 2017, 522, 80–89 CrossRef CAS PubMed.
- P. K. Mondal, M. Kiran, U. Ramamurty and D. Chopra, Chem.–Eur. J., 2017, 23, 1023–1027 CrossRef CAS PubMed.
- N. Shan and M. J. Zaworotko, Drug Discov. Today, 2008, 13, 440–446 CrossRef CAS PubMed.
- S. L. Childs, L. J. Chyall, J. T. Dunlap, V. N. Smolenskaya, B. C. Stahly and G. P. Stahly, J. Am. Chem. Soc., 2004, 126, 13335–13342 CrossRef CAS PubMed.
- N. K. Duggirala, M. L. Perry, Ö. Almarsson and M. J. Zaworotko, Chem. Commun., 2016, 52, 640–655 RSC.
- S. Golob, M. Perry, M. Lusi, M. R. Chierotti, I. Grabnar, L. Lassiani, D. Voinovich and M. J. Zaworotko, J. Pharm. Sci., 2016, 105, 3626–3633 CrossRef CAS PubMed.
- A. D. Bond, CrystEngComm, 2007, 9, 833–834 RSC.
- U. J. Griesser, The importance of solvates. John Wiley & Sons, Ltd, 2006 Search PubMed.
- I. Lugowska, H. Koseła-Paterczyk, K. Kozak and P. Rutkowski, OncoTargets Ther., 2015, 8, 2251–2259 CAS.
- S. Byrn, R. Pfeiffer and J. Stowell, Solid-state chemistry of drugs, SSCI, West Lafayette. 2nd edn, 1999 Search PubMed.
- L. Maini, D. Braga, F. Farinella, E. Melotto, M. Verzini, R. Brescello, I. Michieletto and I. Munari, Cryst. Growth Des., 2018, 18, 3774–3780 CrossRef CAS.
- A. Joseph, J. S. R. Alves, C. E. Bernardes, M. F. M. Piedade and M. E. M. da Piedade, CrystEngComm, 2019, 21, 2220–2233 RSC.
- D. Ahuja, P. Bannigan and Å. C. Rasmuson, CrystEngComm, 2017, 19, 6481–6488 RSC.
- Z. Zheng, B. Hou, X. Cheng, W. Liu, X. Huang, Y. Bao, T. Wang, Z. Wang and H. Hao, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2020, 76, 343–352 CrossRef CAS PubMed.
- M. Yan, Y. Liu, J. Xu, L. Yang, L. Zhang, F. Nie and S. Huang, Cryst. Growth Des., 2020, 20, 5387–5394 CrossRef CAS.
- A. Hotter, J. Wieser and A. Pichler, WO2009062964 A1, 2009.
- D. Datta, S. Lahiri, P. Koilkonda, J. Sharma and M. Achar, WO2009128089 A2, 2009.
- J. Finch and T. Dekornfeld, J. Clin. Pharm., 1971, 11, 371–377 CAS.
- H. Paredes, R. Solis, M. Rocha, A. Montecinos and H. Ramos, Curr. Ther. Res., 1986, 40, 86–91 Search PubMed.
- B. Katchen, S. Buxbaum and J. Ning, J. Pharmacol. Exp. Ther., 1973, 187, 152–157 CAS.
- Y. S. Arkel, J. J. Schrogie and R. Williams, J. Clin. Pharm., 1976, 16, 30–33 CrossRef CAS PubMed.
- E. E. Morse, Ann. Clin. Lab. Sci., 1977, 7, 68–72 CAS.
- A. V. Krymchantowski, H. Carneiro, J. Barbosa and C. Jevoux, Arq. Neuro-Psiquiatr., 2008, 66, 216–220 CrossRef PubMed.
- J. M. Lee, K. M. Park, S. J. Lim, M. K. Lee and C. K. Kim, J. Pharm. Pharmacol., 2002, 54, 43–49 CrossRef CAS PubMed.
- Y. I. Hwang, C. J. Kim, G. S. Lee, S. B. Lee and Y. Oh, invention patent, KR20030042935 A, 2003.
- M. Takasuka, H. Nakai and M. Shiro, J. Chem. Soc., Perkin Trans. 2, 1982, 1061–1067 RSC.
- A. V. Trask, W. S. Motherwell and W. Jones, Cryst. Growth Des., 2005, 5, 1013–1021 CrossRef CAS.
- D. P. McNamara, S. L. Childs, J. Giordano, A. Iarriccio, J. Cassidy, M. S. Shet, R. Mannion, E. O'Donnell and A. Park, Pharma Res., 2006, 23, 1888–1897 CrossRef CAS PubMed.
- S. Long, T. Mao, P. Chen, M. Liu, S. Parkin, M. Zhang, T. Li, P. Zhou and F. Yu, ChemistrySelect, 2017, 2, 4942–4950 CrossRef CAS.
- D. Li, M. Kong, J. Li, Z. Deng and H. Zhang, CrystEngComm, 2018, 20, 5112–5118 RSC.
- D. Li, J. Li, Z. Deng and H. Zhang, CrystEngComm, 2019, 21, 4145–4149 RSC.
- M. Liu, G. Shen, Z. Yuan, S. Parkin, F. Yu, M. Zhang, S. Long and T. Li, Cryst. Growth Des., 2018, 18, 7006–7014 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- M. Frisch, G. Trucks, H. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, G. Scalmani, V. Barone, G. Petersson and H. Nakatsuji, Gaussian 16, Gaussian Inc., Wallingford, CT, 2016 Search PubMed.
- M. Turner, J. McKinnon, S. Wolff, D. Grimwood, P. Spackman, D. Jayatilaka and M. Spackman, PhD thesis, University of Western Australia 2018.
Footnote |
| † Electronic supplementary information (ESI) available: Crystal growth results in a series of DMF analogs, the 1H NMR spectrum of CLX·DMA, and crystal structure of the CLX·DMA in the form of crystallographic information file (CIF) are provided. CCDC 2079841. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra03623h |
| This journal is © The Royal Society of Chemistry 2021 |