
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProtein detection enabled using functionalised silk-binding peptides on a silk-coated optical fibre†
Patrick K. Capon abc,
Aimee J. Horsfallabc,
Jiawen Libcd,
Erik P. Schartnerabc,
Asma Khalidce,
Malcolm S. Purdeyabc,
Robert A. McLaughlinbcd and
Andrew D. Abell*abc
abc,
Aimee J. Horsfallabc,
Jiawen Libcd,
Erik P. Schartnerabc,
Asma Khalidce,
Malcolm S. Purdeyabc,
Robert A. McLaughlinbcd and
Andrew D. Abell*abc
aSchool of Physical Sciences, The University of Adelaide, Adelaide, SA, Australia. E-mail: andrew.abell@adelaide.edu.au
bInstitute for Photonics and Advanced Sensing, The University of Adelaide, Adelaide, SA, Australia
cAustralian Research Council Centre of Excellence for Nanoscale BioPhotonics, Australia
dAdelaide Medical School, Faculty of Health and Medical Sciences, The University of Adelaide, Adelaide, SA, Australia
eDepartment of Physics, School of Science, RMIT University, Melbourne, VIC, Australia
First published on 24th June 2021
Abstract
We present a new coating procedure to prepare optical fibre sensors suitable for use with protein analytes. We demonstrate this through the detection of AlexaFluor-532 tagged streptavidin by its binding to D-biotin that is functionalised onto an optical fibre, via incorporation in a silk fibroin fibre coating. The D-biotin was covalently attached to a silk-binding peptide to provide SBP–biotin, which adheres the D-biotin to the silk-coated fibre tip. These optical fibre probes were prepared by two methods. The first involves dip-coating the fibre tip into a mixture of silk fibroin and SBP–biotin, which distributes the SBP–biotin throughout the silk coating (method A). The second method uses two steps, where the fibre is first dip-coated in silk only, then SBP–biotin added in a second dip-coating step. This isolates SBP–biotin to the outer surface of the silk layer (method B). A series of fluorescence measurements revealed that only the surface bound SBP–biotin detects streptavidin with a detection limit of 15 μg mL−1. The fibre coatings are stable to repeated washing and long-term exposure to water. Formation of silk coatings on fibres using commercial aqueous silk fibroin was found to be inhibited by a lithium concentration of 200 ppm, as determined by atomic absorption spectroscopy. This was reduced to less than 20 ppm by dialysis against water, and was found to successfully form a coating on optical fibres.
Introduction
A wide range of fluorescent molecules are available for the detection of biological analytes, which function by measuring a change in fluorescence output on binding.1–4 However, the associated sensing experiments are often limited to in vitro studies, as minimally invasive delivery of light to an in vivo site is challenging. This is largely due to the low penetration of light through tissue.5 One solution is to use an optical fibre to deliver excitation light, and collect the subsequent fluorescence emission from the location of the fluorophore.6–10 Furthermore, an optical fibre-based probe can be prepared by coating the fibre tip in a matrix, such as a polymer or electrolyte layer, within which a fluorescent sensor molecule is embedded.11–13 Production of such an optical fibre probe provides advantages of small device size,14 precise spatio-temporal control of the fibre tip,10 and delivery of light in difficult to reach locations,15 such as those required for in vivo studies.16,17Optical fibre probes have been developed for a number of applications, including the detection of biological analytes such as metal ions,18–21 small biomolecules,22,23 and pH measurement.24–27 However, there are few reports on the detection of proteins and other large biomolecules using such systems.28,29 This is a significant limitation since biological processes and associated disease states are invariably controlled by these large biomolecules.30,31 Advances are required in optical fibre coating procedures if this unmet need is to be realised.32 Such an advance must address the key problem that the large size of a protein may limit its diffusion into a fibre coating matrix, and hence limit its ability to reach the embedded sensor molecule. This contrasts small analytes that readily diffuse through the matrix. Here we present a new coating procedure to prepare such an optical fibre probe for protein sensing and demonstrate it with the detection of streptavidin using D-biotin adhered to the fibre tip. Specifically, D-biotin is attached to a silk-binding peptide (SBP) by an amide coupling to provide SBP–biotin. This peptide conjugate is then immobilised onto an optical fibre by incorporation into a silk fibroin layer on the fibre tip. Two coating methods were developed to investigate if fibres bearing surface bound SBP–biotin, or those with SBP–biotin incorporated throughout the silk matrix, were able to detect streptavidin in solution. We show that this control over the SBP–biotin location is critical for successful streptavidin detection. Our new fibre coating technology paves the way for development of further fluorescence based optical fibre probes for protein sensing in vivo.
Results and discussion
System design & fibre preparation
The new coating procedure developed here for production of optical fibre probes to sense proteins was demonstrated with a proof-of-concept system that exploits the strong, highly specific, interaction between biotin and streptavidin.33 D-Biotin was covalently attached to the N-terminus of a silk-binding peptide (SBP, sequence SYTFHWHQSWSS)34 by a diisopropylcarbodiimide and Oxyma mediated amide coupling to provide SBP–biotin (Fig. 1). The SBP was selected for use here as we have previously shown that it binds to silk fibroin coatings on optical fibres.35 Optical fibres were coated with silk fibroin and the new peptide SBP–biotin to provide an optical fibre probe for streptavidin, where the streptavidin is detected through collection of the fluorescence signal from an AlexaFluor-532 tag. This fibre coating was carried out by either a one-step or two-step process, referred to as methods A and B (see Fig. 1 and Experimental section in the ESI†). Method A involves submersion of the optical fibre tip in an aqueous mixture of both silk fibroin and SBP–biotin in a single silk-coating step. The silk fibroin was then cured by dipping the functionalised optical fibre tip in methanol.36 This results in distribution of SBP–biotin throughout the silk fibroin coating (Fig. 1, method A). In contrast, method B involves two distinct fibre coating steps. First, the fibre tip was dipped into an aqueous solution of silk fibroin, which results in formation of a silk layer on the fibre tip, that was then cured in methanol.36 The silk-coated fibre was subsequently dipped in a separate aqueous solution of SBP–biotin. This two-step process, with SBP–biotin added last, results in localisation of the SBP–biotin to the exterior of the silk fibroin coating (Fig. 1, method B). The silk fibroin concentration was held constant at 32.5 mg mL−1 for all fibre coating solutions. It was expected that the large size of streptavidin (∼52.8 kDa) would preclude its diffusion through the silk fibroin matrix, and thus not reach the majority of the SBP–biotin sensor component embedded within the silk fibroin through fibre coating method A. However, isolation of SBP–biotin to the exterior of the silk coating, achieved through coating fibres with silk fibroin using method B, was expected to allow the streptavidin to access and bind D-biotin, as diffusion through the silk matrix is not required. Thus, fibre probes bearing surface bound SBP–biotin were expected to result in superior detection of streptavidin compared to those with SBP–biotin distributed throughout the silk layer. Comparison of the two coating methods discussed in this work will therefore inform upon future design of subsequent optical fibre based protein sensors.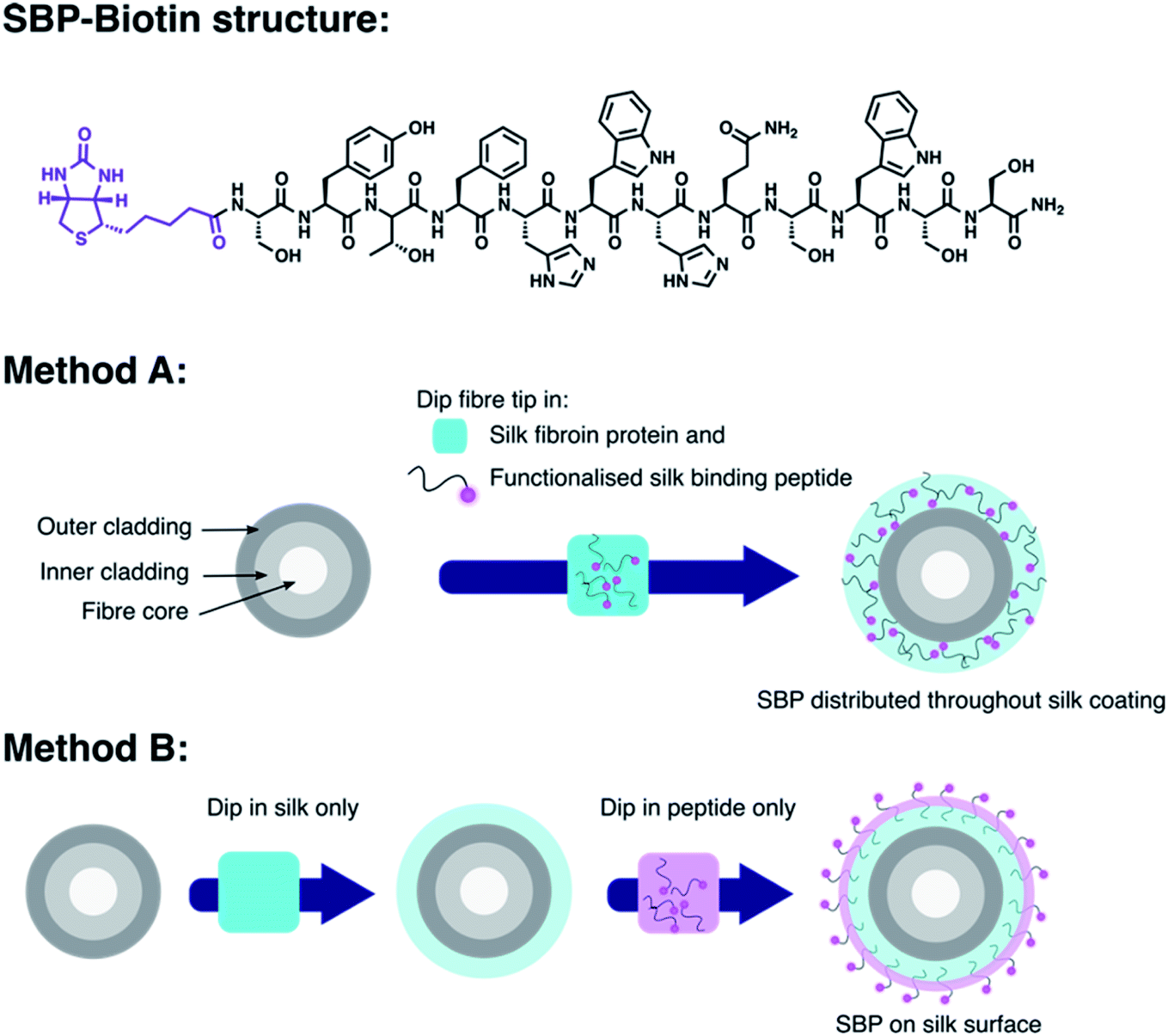 | ||
| Fig. 1 Top: structure of the functionalised silk-binding peptide, SBP–biotin, prepared by N-terminal attachment of D-biotin (purple) to a SBP (black). Middle: fibre coating method A is a single step process, where the optical fibre tip (shown as an end on cross section) is dipped into an aqueous mixture of silk fibroin protein (light blue) and SBP–biotin. The fibre is then removed and dipped in 90% aqueous methanol. This provides a fibre probe with the SBP–biotin distributed throughout the silk fibroin layer. Method B consists of two fibre dip coating steps, where the optical fibre tip is first dipped in a solution of silk fibroin only followed by 90% methanol (shown by the blue coating). The silk coated fibre tip is then dipped in an aqueous solution of the SBP–biotin to provide a fibre probe with the SBP–biotin decorated on the outer surface of the silk fibroin coating (shown by the purple layer). | ||
Silk characterisation
Prior to investigating the detection of streptavidin with SBP–biotin coated fibres, we attempted to use commercial silk fibroin purchased from Merck as a 50 mg mL−1 aqueous solution (Supelco 5154-20 mL) to coat optical fibres. This was used rather than the in-house purified silk fibroin used in previous work,37,38 to allow a direct comparison of the two silk sources and thus improve access to our fibre coating technology for those unable to prepare silk fibroin in-house. We have previously shown the in-house prepared silk fibroin can provide a coating of silk fibroin and SBP–TAMRA (a conjugate of the SBP and 5(6)-carboxyTAMRA, see Experimental section in the ESI†) on optical fibres.35 We therefore attempted to use the commercial silk fibroin in the same manner. Optical fibre tips were dip-coated in a mixture of commercial silk fibroin and SBP–TAMRA according to method A, which should result in distribution of the SBP–TAMRA throughout the silk layer. Fluorescence spectra were collected, however there was no signal observed corresponding to SBP–TAMRA, which occurred consistently over multiple fibres. This suggested that the commercial silk fibroin cannot form a coating on optical fibres. As the final silk fibroin processing step is dialysis from LiBr,37,38 we hypothesised a difference in lithium salt content may affect the production of a silk coating on the fibres. Therefore, atomic absorption spectroscopy (AAS) was conducted on both silk samples to investigate their lithium content (at 50 mg mL−1, see Experimental section in the ESI† and Fig. 2H). The commercial silk fibroin sample was found to contain a 10-fold higher lithium concentration than the in-house silk fibroin sample (200 vs. 20.0 ppm, see Table 1). The commercial sample was then further dialysed against 1 L of water for 48 h with the water changed every ∼12 h, and the resulting dialysed silk solution analysed by AAS to find <1 ppm lithium (Table 1).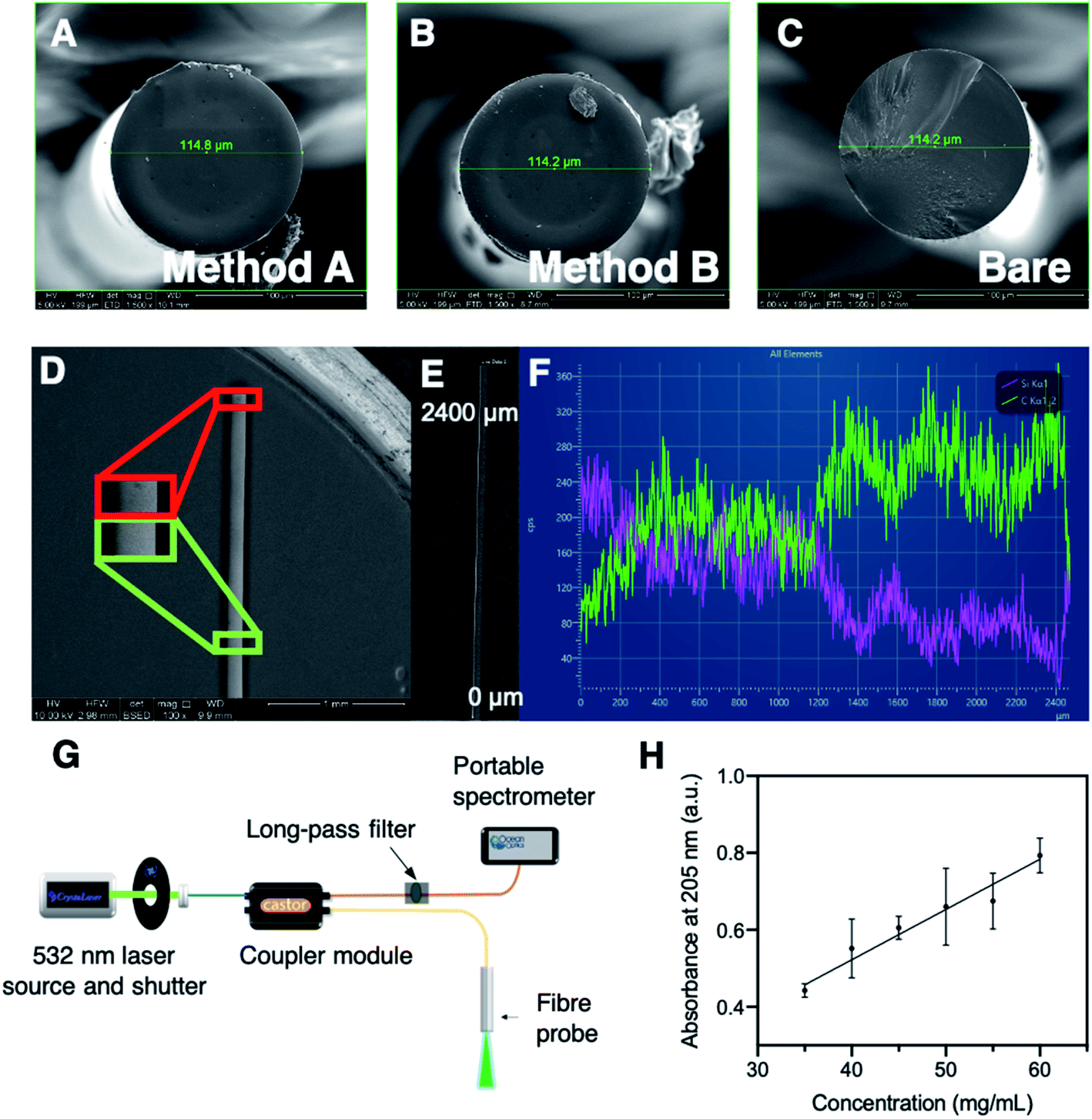 | ||
| Fig. 2 Top: scanning electron microscopy images in secondary electron mode of (A) a fibre coated by method A, (B) a fibre coated by method B, and (C) a bare fibre, which indicate no difference in topology between methods A and B. (D) backscattered electron (BSE) image of an optical fibre coated with silk fibroin by method A showing a minimal difference between the tip of the fibre and a point approximately 2200 mm down the fibre. (E) BSE image indicating the area in which the energy dispersive X-ray (EDX) line scan was conducted. (F) EDX line scan spectra for C Kα emission line (green) and Si Kα emission line (purple). (G) Schematic of the optical system used for fluorescence spectra collection. Blue: single-mode fibre, orange: multimode fibre, yellow: double-clad fibre. (H) Calibration curve for silk fibroin absorbance at 205 nm against concentration (mg mL−1). The equation of best fit was calculated using GraphPad Prism 9 linear regression with a y intercept set to 0, to give absorbance = 0.01306 * concentration (in mg mL−1). Goodness of fit (Sy.x) 0.05892. Data is plotted as mean ± standard deviation of three reads. | ||
| Sample | Approximate lithium concentration (ppm) |
|---|---|
| In-house silk fibroin solution | 20.0 |
| Merck silk fibroin solution | 200 |
| Dialysed Merck silk fibroin solution | <1 |
In order to investigate if the lowered lithium content in the dialysed silk sample affected silk coating production, an optical fibre was dip-coated in a mixture of dialysed commercial silk fibroin and SBP–TAMRA according to method A, then fluorescence spectra collected. A fluorescence signal from SBP–TAMRA was observed, of similar magnitude to that obtained using the in-house silk fibroin. This indicated that the silk coating was successfully formed using the dialysed commercial silk fibroin. Overall, it was found that the as-received commercial silk fibroin cannot form a coating on optical fibres, which is likely due to the observed increase in lithium content. Therefore, we recommend that the lithium content of aqueous silk fibroin solutions is tested, and additional dialysis conducted to reduce the lithium concentration below 20 ppm before attempting to coat optical fibres following our procedures.
Throughout the silk characterisation process, we found that the silk fibroin is stable under refrigeration at ∼4 °C for 3–4 months, after which severe aggregation occurs and results in a thick silk gel that is not easily handled, and cannot be disaggregated. The aggregation process can be slowed by storage at −80 °C, and by centrifugation of the silk fibroin sample when small aggregates are observed in solution. However, removal of these aggregates changes the concentration of the remaining silk fibroin supernatant. Therefore, we investigated whether the UV absorbance of an aqueous silk solution could be used to ensure that all silk solutions were used at the same concentration for improved reproducibility of the fibre coatings. To this end, a calibration curve for silk fibroin concentration was constructed based on absorbance at 205 nm, which predominantly originates from amide bonds (Fig. 2H, see Experimental section in the ESI†).39 The concentration of all silk fibroin solutions used during the course of this work were calculated with reference to this calibration curve to ensure fibre coating results were comparable (see eqn (S1) in the ESI†). Note that these concentrations are relative to the original 65 mg mL−1 sample, as the extinction coefficient of the silk fibroin was not calculated. All silk fibroin solutions used in this work for fibre coating were prepared in-house, at a final concentration of 32.5 mg mL−1 to ensure consistency.
Fibre characterisation
Scanning electron microscopy (SEM) was conducted in secondary electron (SE) and backscattered electron (BSE) modes, along with energy dispersive spectroscopy (EDS), on optical fibres coated with silk by both methods A and B to determine the surface topology of the fibre tip, as shown in Fig. 2A–C and the Experimental section in the ESI.† The optical fibre tips consisted of a smooth surface with minimal features of note, with no observable difference in the surface topology between methods A and B. One of the method B coated fibres was observed to have a small bubble of silk on the tip (Fig. 2B), however this was not seen for subsequent SE imaging of method B coated fibres. The BSE imaging was also consistent with these observations, where BSE mode provides an image with contrast based on atomic number, with low atomic number elements coloured darker, as they scatter electrons less than higher atomic number elements (Fig. 2D–F). A fibre coated by method A was imaged side-on in BSE mode to ascertain the point on the fibre at which the silk coating ended. Unfortunately, the difference between silk coated and non-coated fibre was difficult to determine by BSE imaging (Fig. 2D). Instead, an EDS line scan was used to approximate the end of the silk coating by collecting emission spectra along the first ∼2400 μm of the fibre with a side on view (Fig. 2E and F). Spectra obtained by EDS contain emission lines characteristic to each element present. Here the Kα emission lines for C (0.27 keV) and Si (1.74 keV) were observed and suggested that the silk coating (indicated by the C signal) was predominantly on the first ∼1200 μm from the fibre tip, while the remainder of the fibre largely exhibited the Si Kα line (as the fibre is made of SiO2). This is consistent with the BSE imaging indicating the silk layer is only present toward the fibre tip. No clear difference between methods A and B was observed in BSE imaging (data not shown). Overall, the SEM observations are consistent with those for our recent report of an optical fibre pH sensor.35Detection of fluorescently tagged streptavidin
The detection of streptavidin with SBP–biotin coated fibres prepared by method A or B was investigated to determine which coating method was more effective for detection of streptavidin in solution (and therefore has the potential for application to other protein sensors in the future). First, all fibres used for sensing were connected to a fibre-coupled laser system prepared in-house and baseline (i.e. uncoated fibre) fluorescence spectra collected (ex. 532 nm, see Experimental section in the ESI† and Fig. 2G for a description of the optical system). Full fluorescence spectra are provided in Fig. S7 in the ESI† for every stage of the following process. Three optical fibres were then coated with SBP–biotin by method A, and three optical fibres were coated with SBP–biotin by method B. Additionally, three control fibres were prepared by coating the fibre with silk fibroin only, as an indirect control to determine if the streptavidin bound non-specifically to the silk fibroin layer in the absence of SBP–biotin (see Experimental section in the ESI†). Each fibre was dipped in a solution of AlexaFluor-532 tagged streptavidin (30 μg mL−1 in 100 mM phosphate buffer at pH 7.2) for 30 seconds. Fluorescence spectra were collected for each fibre and are labelled as ‘pre-wash’ in Fig. 3A. The intensity of all spectra was integrated between 565 and 600 nm to minimise noise (see Fig. 3B for a sample spectrum and Fig. S7 in the ESI† for all full spectra). All fibres were then washed three times by submerging the fibre tip in phosphate buffer (100 mM, pH 7.2) for 30 seconds per wash. Fluorescence spectra were again collected for each fibre and are labelled as ‘washed’ in Fig. 3A. The baseline, pre-wash, and washed spectra were compared for each fibre coating method to determine which set of fibres was successful in detection of the AlexaFluor-532 tagged streptavidin.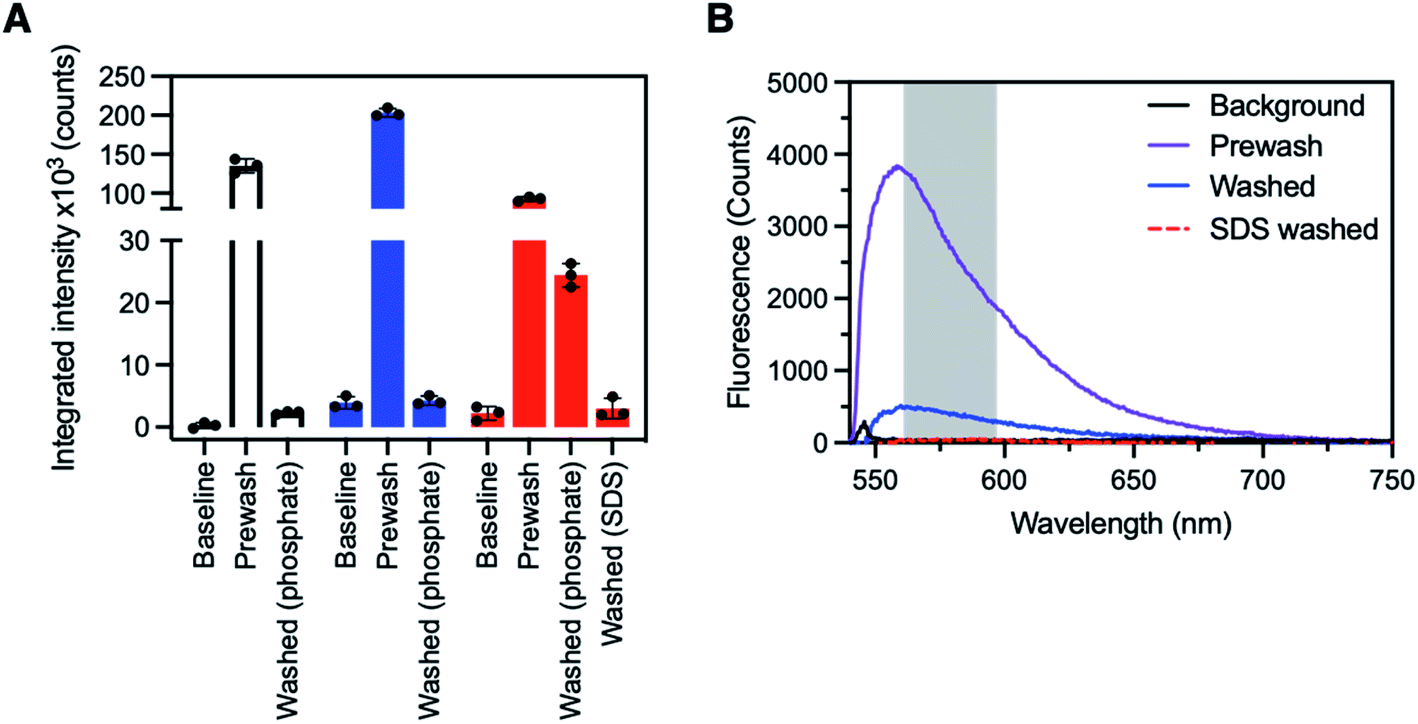 | ||
| Fig. 3 (A) Sensing of AlexaFluor-532 tagged streptavidin with SBP–biotin coated fibre probes. One fibre is shown for each coating type, plotted as the mean ± standard deviation of three individual fluorescence reads. Data for all three fibres for each fibre coating type is available in the ESI.† Left, white; a control fibre coated with silk fibroin only, where all prewash fluorescence was lost upon washing with phosphate buffer (100 mM, pH 7.2). Middle, blue; a fibre coated with SBP–biotin and silk fibroin by method A, where all prewash fluorescence was again lost upon washing with phosphate buffer. Right, red; a fibre coated with SBP–biotin and silk fibroin by method B, where approximately 30% of the prewash signal was retained after washing with phosphate buffer, then all fluorescence removed by further washing of the fibre with aqueous sodium dodecyl sulfate (SDS, 2%). The excitation wavelength for all spectra was 532 nm, with intensity integrated between 565 and 600 nm for each fluorescence spectrum. (B) Sample full spectra for an optical fibre coated with silk fibroin and SBP–biotin by method B. Black: background fluorescence before exposure to AlexaFluor-532 tagged streptavidin. Purple: fluorescence spectrum after dipping in streptavidin solution. Blue: fluorescence spectrum after washing 3 × 30 s in phosphate buffer (100 mM, pH 7.2). Red, dashed: fluorescence spectrum after washing 3 × 30 s in sodium dodecyl sulfate (SDS, 2% aqueous). Excitation wavelength for all spectra was 532 nm. The region marked by the grey box indicates the area where intensity was integrated to provide the data in panel A. | ||
The pre-wash spectra for the control fibres (no SBP–biotin present) had an average integrated intensity of 135![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 counts, indicating that AlexaFluor-532 tagged streptavidin was present on the fibre tips (Fig. 3A, white bars, also see ESI, Fig. S3–S6†). After washing, the fluorescence intensity was reduced to 2400 counts, which indicates that streptavidin was removed. This suggests the streptavidin was non-specifically bound to the silk fibroin layer on the control fibres. The washed fluorescence intensity for the fibres coated with silk fibroin and SBP–biotin by method A was similarly reduced in comparison to the pre-wash intensity (203000 reduced to 4200 counts, Fig. 3A blue bars). The washed intensity was of similar magnitude to the baseline intensity, which indicates that there was no detectable streptavidin remaining on the fibre tip. This shows that when SBP–biotin is embedded within the silk matrix on the fibre tip (method A), it does not selectively bind to fluorescently tagged streptavidin. This observation is consistent with the hypothesis that streptavidin cannot diffuse through the silk fibroin coating to reach the majority of the embedded SBP–biotin. In contrast, the post-wash fluorescence intensity observed for the fibres coated with silk fibroin and SBP–biotin by method B was significantly greater than the baseline intensity (24400 vs. 2200 counts, Fig. 3A red bars and Fig. 3B). As the washing process was shown to remove any non-specifically bound streptavidin on the control and method A coated fibres, the fluorescence signal remaining after washing is attributed to AlexaFluor-532 tagged streptavidin that is bound to SBP–biotin. Hence, streptavidin binds to SBP–biotin that is decorated on the outside of the silk fibroin coating on an optical fibre tip (method B). Finally, we investigated if streptavidin could be removed from these fibres by washing with sodium dodecyl sulfate (SDS), which is known to denature streptavidin.40 The SDS wash was used as a second indirect control to confirm the presence of a specific bond between biotin (in SBP–biotin) and streptavidin. The fibres were washed three times by dipping the fibre tips in 2% v/v aqueous SDS for 30 seconds per wash. Fluorescence spectra were collected, with the resulting integrated intensity found to be 2400 counts (Fig. 3A, ‘washed SDS’ and Fig. 3B). Thus, it is clear that streptavidin is removed from the optical fibre tip by washing the fibre with SDS.
000 counts, indicating that AlexaFluor-532 tagged streptavidin was present on the fibre tips (Fig. 3A, white bars, also see ESI, Fig. S3–S6†). After washing, the fluorescence intensity was reduced to 2400 counts, which indicates that streptavidin was removed. This suggests the streptavidin was non-specifically bound to the silk fibroin layer on the control fibres. The washed fluorescence intensity for the fibres coated with silk fibroin and SBP–biotin by method A was similarly reduced in comparison to the pre-wash intensity (203000 reduced to 4200 counts, Fig. 3A blue bars). The washed intensity was of similar magnitude to the baseline intensity, which indicates that there was no detectable streptavidin remaining on the fibre tip. This shows that when SBP–biotin is embedded within the silk matrix on the fibre tip (method A), it does not selectively bind to fluorescently tagged streptavidin. This observation is consistent with the hypothesis that streptavidin cannot diffuse through the silk fibroin coating to reach the majority of the embedded SBP–biotin. In contrast, the post-wash fluorescence intensity observed for the fibres coated with silk fibroin and SBP–biotin by method B was significantly greater than the baseline intensity (24400 vs. 2200 counts, Fig. 3A red bars and Fig. 3B). As the washing process was shown to remove any non-specifically bound streptavidin on the control and method A coated fibres, the fluorescence signal remaining after washing is attributed to AlexaFluor-532 tagged streptavidin that is bound to SBP–biotin. Hence, streptavidin binds to SBP–biotin that is decorated on the outside of the silk fibroin coating on an optical fibre tip (method B). Finally, we investigated if streptavidin could be removed from these fibres by washing with sodium dodecyl sulfate (SDS), which is known to denature streptavidin.40 The SDS wash was used as a second indirect control to confirm the presence of a specific bond between biotin (in SBP–biotin) and streptavidin. The fibres were washed three times by dipping the fibre tips in 2% v/v aqueous SDS for 30 seconds per wash. Fluorescence spectra were collected, with the resulting integrated intensity found to be 2400 counts (Fig. 3A, ‘washed SDS’ and Fig. 3B). Thus, it is clear that streptavidin is removed from the optical fibre tip by washing the fibre with SDS.
In summary, optical fibre probes with SBP–biotin isolated to the exterior of the silk fibroin layer on the fibre tip were able to detect AlexaFluor-532 tagged streptavidin (method B). However, fibres with SBP–biotin distributed throughout the silk fibroin layer were not able to detect streptavidin (method A). This is consistent with the hypothesis that streptavidin cannot diffuse through the silk fibroin matrix to reach the embedded SBP–biotin sensor component. The preparation of SBP–biotin and silk fibroin coated fibres by method B is thus optimum for detection of streptavidin. Our SBP–biotin and streptavidin model system demonstrates that coating method B is amenable to sensing large analytes such as proteins, while coating method A is not suitable for proteins of a comparable size to (or larger than) streptavidin.
Limit of detection for streptavidin with SBP–biotin coated fibres
An approximate concentration limit for detection of AlexaFluor-532 tagged streptavidin using SBP–biotin coated fibre probes was determined. Three fibre probes prepared by method B were dipped into a solution of AlexaFluor-532 tagged streptavidin at 7.8 μg mL−1 (100 mM phosphate buffer, pH 7.2). The fibres were washed once by dipping for 30 seconds in phosphate buffer (100 mM, pH 7.2) to remove any non-specifically bound streptavidin, then fluorescence spectra were collected and the integrated intensity plotted (Fig. 4, blue bars). Some variability in the integrated intensity was apparent between fibre probes, where the AlexaFluor-532 fluorescence intensity (blue) was well separated from the baseline intensity (white) for fibres #1 and #3, but fibre #2 was within error of the baseline intensity (Fig. 4). Therefore, fibre #2 was not able to distinguish between the streptavidin solution (7.8 μg mL−1) and the baseline fluorescence of the fibre.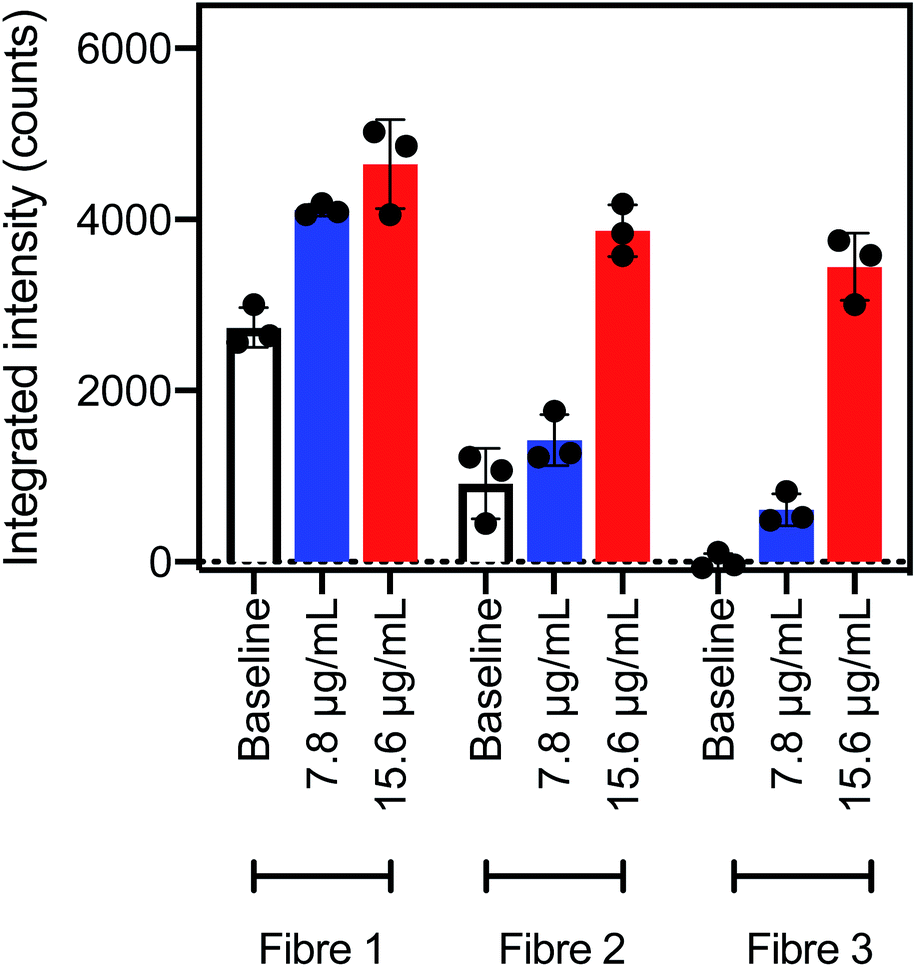 | ||
| Fig. 4 Integrated intensity between 565 and 600 nm for fibre probes coated with silk fibroin and SBP–biotin by method B. White, the baseline signal obtained before exposure to streptavidin; blue, signal after dipping in AlexaFluor-532 tagged streptavidin at 7.8 μg mL−1; and red, signal after dipping in AlexaFluor-532 tagged streptavidin at 15 μg mL−1. The excitation wavelength was 532 nm for all spectra, data is plotted as mean ± standard deviation of three reads. | ||
The same three fibre probes coated with SBP–biotin by method B were next washed three times in 2% aqueous SDS to remove all bound streptavidin, as shown by the earlier results in the streptavidin detection experiments. These fibres were then dipped in a second solution of AlexaFluor-532 tagged streptavidin at a higher concentration of 15 μg mL−1 for 30 seconds, then washed once with phosphate buffer. Fluorescence spectra were collected and the resultant integrated intensity (Fig. 4, red bars) was found to be clearly distinguished from the baseline signal (Fig. 4, white bars) for all three fibres. Therefore, all three fibres can detect streptavidin at a concentration of 15 μg mL−1.
In summary, optical fibre probes coated with SBP–biotin with method B were able to detect streptavidin at 15 μg mL−1. Fibres #1 and #3 also detected streptavidin at 7.8 μg mL−1, however fibre #2 could not distinguish between a 7.8 μg mL−1 streptavidin sample and a baseline measurement. The detection limit of AlexaFluor-532 tagged streptavidin using optical fibre probes coated with SBP–biotin by method B was thus determined to be 15 μg mL−1 in accordance with the definition by Armbruster and Pry of “the lowest analyte concentration likely to be reliably distinguished from the blank and at which detection is feasible”.41 Recently reported surface plasmon resonance based optical fibre probes for streptavidin42 and C-reactive protein43 have a comparable detection limit of 0.1 to 20 μg mL−1, suggesting that silk fibroin and SBP coatings prepared by method B are a viable option for the production of optical fibre probes for protein sensing.
Retention of method B coatings on fibres
The stability of coatings prepared by method B to repeated submersion in water was investigated to simulate repeated usage of the optical fibre probes. A model SBP was prepared where D-biotin was replaced by the fluorophore 5(6)-carboxyTAMRA to provide the functionalised SBP, SBP–TAMRA (see Experimental section in the ESI†).35 This allowed the stability of the coating to be monitored by collection of the TAMRA fluorescence signal, where a loss of fluorescence implies a loss of SBP–TAMRA from the fibre tip. SBP–TAMRA directly replaced SBP–biotin in the procedure for coating method B, which resulted in isolation of SBP–TAMRA to the outer surface of the silk layer on the optical fibre. Fluorescence spectra were collected using the described optical system (Fig. 2G). When fibres coating with SBP–TAMRA with method B were washed in water with a 3 × 30 s protocol, 90% of the fluorescence signal was retained in comparison to control fibres, while soaking the fibres in water for 18 h resulted in 68% fluorescence retention compared to control fibres. See the ESI† for a full description of this washing and calculation of fluorescence retention process, along with Fig. S8.† Overall, the stability of silk and SBP–TAMRA coatings prepared by method B on optical fibres was in agreement with the observed stability of fibres coated with SBP–TAMRA by method A (see ESI, Fig. S8–S11†).35Conclusion
We have presented a new approach to prepare optical fibre probes for protein sensing, and have detected streptavidin using fibre probes coated with SBP–biotin as a model system. However, the SBP–biotin sensor component can be substituted for other sensors, through covalent attachment of a sensor to the N-terminus of the SBP. This will enable development of optical fibre probes for other protein analytes using our silk coating technology. The fibre coating must be prepared following method B, developed here to isolate the sensor component to the external surface of the silk layer, as we have shown that only these fibres are amenable to protein detection.The streptavidin detection model system was enabled by attachment of D-biotin to the fibre tip. First, D-biotin was attached to the N-terminus of a silk-binding peptide (SBP) by an amide coupling reaction to provide SBP–biotin. The SBP was selected as it is known to bind to a silk fibroin coating on optical fibres, which therefore ensures that SBP–biotin is adhered to the silk coated fibre tip. We developed two methods to prepare these optical fibre probes in order to control the location of SBP–biotin: either distributed throughout the silk coating (method A), or isolated to the outer surface of the silk layer (method B). These methods were shown to form a silk coating that is stable to repeated or long-term exposure to water. The detection of streptavidin with optical fibre probes prepared by each method was compared in order to investigate the diffusion of streptavidin through the silk matrix. It was found that only fibres bearing SBP–biotin on the outer surface of the silk layer were able to detect streptavidin (method B), with the experimental limit of detection found to be 15 μg mL−1. This comparison demonstrates that the precise location of the sensor component (SBP–biotin in our model), with respect to the fibre coating, is a critical design consideration for preparation of optical fibre probes for protein sensing.
Finally, the coating of optical fibres with in-house prepared or commercial silk fibroin was also compared to allow wider access to our fibre coating technology. We determined that the commercial silk fibroin did not successfully form a coating on optical fibres, however this was rectified by dialysis against water. Analysis by atomic absorption spectroscopy suggested that a low lithium content (≤20 ppm) in the silk solution was an important factor in forming a silk fibroin coating on optical fibres. Therefore, our fibre coating technology requires that the lithium content of all aqueous silk fibroin solutions is checked prior to coating optical fibres, and dialysis be performed to reduce the lithium content below 20 ppm.
Author contributions
Patrick Capon – conceptualised the research, conducted all experiments, wrote and edited the manuscript, Aimee Horsfall – performed peptide design and assisted with synthesis & purification, edited the manuscript, Jiawen Li – designed and built the custom laser system, prepared optical fibres, edited the manuscript, Erik Schartner – designed and built the custom laser system, wrote custom LabView software, edited the manuscript, Asma Khalid – prepared the in-house silk fibroin, Malcolm Purdey – conceptualised the research, supervised the research, edited the manuscript, Robert McLaughlin – conceptualised the fibre experiments, provided funding and equipment for laser systems, supervised the research, edited the manuscript, Andrew Abell – conceptualised the research, provided funding and reagents for peptide synthesis, supervised the research, edited the manuscript.Conflicts of interest
RAM is a co-founder and a Director of Miniprobes Pty Ltd, a company that develops novel optical imaging systems. Miniprobes Pty Ltd did not contribute to this study.Acknowledgements
The authors acknowledge support from the ARC Centre of Excellence for Nanoscale Biophotonics (CE140100003). PKC would like to thank Matthew Bull for running the atomic absorption spectroscopy experiments, Adelaide Microscopy and Ken Neubauer for assistance with SEM and EDS analysis, and acknowledges PhD scholarship support from the MF and MH Joyner Scholarship in Science and the Norman and Patricia Polglase Supplementary Scholarship. AJH acknowledges PhD scholarship support from a Research Training Program Stipend (RTPS). JL is supported by a Postdoctoral Fellowship (102093) from the National Heart Foundation and an NHMRC Ideas grant (2001646). EPS is supported by an ARC Linkage grant (LP 150100657). AK acknowledges an RMIT Vice Chancellor's Research Fellowship for supporting silk fibroin research. RAM is supported by an NHMRC Development grant (APP1178912) and an NHMRC Ideas grant (2002254). This work was performed in part at the Optofab node of the Australian National Fabrication Facility utilising Commonwealth and SA State Government funding.References
- J. Chan, S. C. Dodani and C. J. Chang, Reaction-Based Small-Molecule Fluorescent Probes for Chemoselective Bioimaging, Nat. Chem., 2012, 4(12), 973–984, DOI:10.1038/nchem.1500.
- A. Fernández and M. Vendrell, Smart Fluorescent Probes for Imaging Macrophage Activity, Chem. Soc. Rev., 2016, 45(5), 1182–1196, 10.1039/C5CS00567A.
- H.-W. Liu, L. Chen, C. Xu, Z. Li, H. Zhang, X.-B. Zhang and W. Tan, Recent Progresses in Small-Molecule Enzymatic Fluorescent Probes for Cancer Imaging, Chem. Soc. Rev., 2018, 47(18), 7140–7180, 10.1039/C7CS00862G.
- A. Kaur and E. J. New, Bioinspired Small-Molecule Tools for the Imaging of Redox Biology, Acc. Chem. Res., 2019, 52(3), 623–632, DOI:10.1021/acs.accounts.8b00368.
- S. L. Jacques, Optical Properties of Biological Tissues: A Review, Phys. Med. Biol., 2013, 58(11), R37–R61, DOI:10.1088/0031-9155/58/11/R37.
- C.-S. Chu, Y.-L. Lo and T.-W. Sung, Review on Recent Developments of Fluorescent Oxygen and Carbon Dioxide Optical Fiber Sensors, Photonic Sens., 2011, 1(3), 234–250, DOI:10.1007/s13320-011-0025-4.
- O. Stevens, I. E. I. Petterson, J. C. C. Day and N. Stone, Developing Fibre Optic Raman Probes for Applications in Clinical Spectroscopy, Chem. Soc. Rev., 2016, 45(7), 1919–1934, 10.1039/C5CS00850F.
- S. Addanki, I. S. Amiri and P. Yupapin, Review of Optical Fibers-Introduction and Applications in Fiber Lasers, Results Phys., 2018, 10, 743–750, DOI:10.1016/j.rinp.2018.07.028.
- A. Canales, S. Park, A. Kilias and P. Anikeeva, Multifunctional Fibers as Tools for Neuroscience and Neuroengineering, Acc. Chem. Res., 2018, 51(4), 829–838, DOI:10.1021/acs.accounts.7b00558.
- J. Li, E. Schartner, S. Musolino, B. C. Quirk, R. W. Kirk, H. Ebendorff-Heidepriem and R. A. McLaughlin, Miniaturized Single-Fiber-Based Needle Probe for Combined Imaging and Sensing in Deep Tissue, Opt. Lett., 2018, 43(8), 1682, DOI:10.1364/OL.43.001682.
- A. Song, S. Parus and R. Kopelman, High-Performance Fiber-Optic pH Microsensors for Practical Physiological Measurements Using a Dual-Emission Sensitive Dye, Anal. Chem., 1997, 69(5), 863–867, DOI:10.1021/ac960917+.
- P. K. Sarkar, A. Halder, A. Adhikari, N. Polley, S. Darbar, P. Lemmens and S. K. Pal, DNA-Based Fiber Optic Sensor for Direct in Vivo Measurement of Oxidative Stress, Sens. Actuators, B, 2018, 255, 2194–2202, DOI:10.1016/j.snb.2017.09.029.
- A. François, H. T. C. Foo and T. M. Monro, Polyelectrolyte Multilayers for Surface Functionalization: Advantages and Challenges, in Advanced Photonics, OSA, Barcelona, 2014, p. JTu4C.1, DOI:10.1364/BGPP.2014.JTu4C.1.
- R. Kopelman and S. Dourado, Is Smaller Better?–Scaling of Characteristics with Size of Fiber-Optic Chemical and Biochemical Sensors, in Chemical, Biochemical, and Environmental Fiber Sensors VIII, International Society for Optics and Photonics, 1996, vol. 2836, pp. 2–11, DOI:10.1117/12.260577.
- B. Rente, M. Fabian, Y. Chen, L. Vorreiter, H. Bustamante, T. Sun and K. T. V. Grattan, In-Sewer Field-Evaluation of an Optical Fibre-Based Condition Monitoring System, IEEE Sens. J., 2020, 20(6), 2976–2981, DOI:10.1109/JSEN.2019.2956826.
- S. Musolino, E. P. Schartner, G. Tsiminis, A. Salem, T. M. Monro and M. R. Hutchinson, Portable Optical Fiber Probe for in vivo Brain Temperature Measurements, Biomed. Opt. Express, 2016, 7(8), 3069–3077, DOI:10.1364/BOE.7.003069.
- J. Li, H. Ebendorff-Heidepriem, B. C. Gibson, A. D. Greentree, M. R. Hutchinson, P. Jia, R. Kostecki, G. Liu, A. Orth, M. Ploschner, E. P. Schartner, S. C. Warren-Smith, K. Zhang, G. Tsiminis and E. M. Goldys, Perspective: Biomedical Sensing and Imaging with Optical Fibers—Innovation through Convergence of Science Disciplines, APL Photonics, 2018, 3(10), 100902, DOI:10.1063/1.5040861.
- T. H. Nguyen, T. Sun and K. T. V. Grattan, A Turn-On Fluorescence-Based Fibre Optic Sensor for the Detection of Mercury, Sensors, 2019, 19(9), 2142, DOI:10.3390/s19092142.
- A. Bachhuka, S. Heng, K. Vasilev, R. Kostecki, A. Abell and H. Ebendorff-Heidepriem, Surface Functionalization of Exposed Core Glass Optical Fiber for Metal Ion Sensing, Sensors, 2019, 19(8), 1829, DOI:10.3390/s19081829.
- S. C. Warren-Smith, S. Heng, H. Ebendorff-Heidepriem, A. D. Abell and T. M. Monro, Fluorescence-Based Aluminum Ion Sensing Using a Surface-Functionalized Microstructured Optical Fiber, Langmuir, 2011, 27(9), 5680–5685, DOI:10.1021/la2002496.
- S. Heng, M.-C. Nguyen, R. Kostecki, T. M. Monro and A. D. Abell, Nanoliter-Scale, Regenerable Ion Sensor: Sensing with a Surface Functionalized Microstructured Optical Fibre, RSC Adv., 2013, 3(22), 8308–8317, 10.1039/C3RA40321A.
- K. S. Bronk, K. L. Michael, P. Pantano and D. R. Walt, Combined Imaging and Chemical Sensing Using a Single Optical Imaging Fiber, Anal. Chem., 1995, 67(17), 2750–2757, DOI:10.1021/ac00113a005.
- J. P. Issberner, C. L. Schauer, B. A. Trimmer and D. R. Walt, Combined Imaging and Chemical Sensing of L-Glutamate Release from the Foregut Plexus of the Lepidopteran, Manduca sexta, J. Neurosci. Methods, 2002, 120(1), 1–10, DOI:10.1016/S0165-0270(02)00165-6.
- K. L. Michael and D. R. Walt, Combined Imaging and Chemical Sensing of Fertilization-Induced Acid Release from Single Sea Urchin Eggs, Anal. Biochem., 1999, 273(2), 168–178, DOI:10.1006/abio.1999.4173.
- M. Purdey, J. Thompson, T. Monro, A. Abell and E. Schartner, A Dual Sensor for pH and Hydrogen Peroxide Using Polymer-Coated Optical Fibre Tips, Sensors, 2015, 15(12), 31904–31913, DOI:10.3390/s151229893.
- E. P. Schartner, M. R. Henderson, M. Purdey, D. Dhatrak, T. M. Monro, P. G. Gill and D. F. Callen, Cancer Detection in Human Tissue Samples Using a Fiber-Tip pH Probe, Cancer Res., 2016, 76(23), 6795–6801, DOI:10.1158/0008-5472.CAN-16-1285.
- H. J. McLennan, A. Saini, G. M. Sylvia, E. P. Schartner, K. R. Dunning, M. S. Purdey, T. M. Monro, A. D. Abell and J. G. Thompson, A Biophotonic Approach to Measure pH in Small Volumes in Vitro: Quantifiable Differences in Metabolic Flux around the Cumulus-Oocyte-Complex (COC), J. Biophotonics, 2020, 13(3), e201960038, DOI:10.1002/jbio.201960038.
- E. Benito-Peña, M. G. Valdés, B. Glahn-Martínez and M. C. Moreno-Bondi, Fluorescence Based Fiber Optic and Planar Waveguide Biosensors. A Review, Anal. Chim. Acta, 2016, 943, 17–40, DOI:10.1016/j.aca.2016.08.049.
- S. E. Mowbray and A. M. Amiri, A Brief Overview of Medical Fiber Optic Biosensors and Techniques in the Modification for Enhanced Sensing Ability, Diagnostics, 2019, 9(1), 23, DOI:10.3390/diagnostics9010023.
- H. Goehler, M. Lalowski, U. Stelzl, S. Waelter, M. Stroedicke, U. Worm, A. Droege, K. S. Lindenberg, M. Knoblich, C. Haenig, M. Herbst, J. Suopanki, E. Scherzinger, C. Abraham, B. Bauer, R. Hasenbank, A. Fritzsche, A. H. Ludewig, K. Buessow, S. H. Coleman, C.-A. Gutekunst, B. G. Landwehrmeyer, H. Lehrach and E. E. Wanker, A Protein Interaction Network Links GIT1, an Enhancer of Huntingtin Aggregation, to Huntington's Disease, Mol. Cell, 2004, 15(6), 853–865, DOI:10.1016/j.molcel.2004.09.016.
- M. W. Gonzalez and M. G. Kann, Chapter 4: Protein Interactions and Disease, PLoS Comput. Biol., 2012, 8(12), e1002819, DOI:10.1371/journal.pcbi.1002819.
- B. Leca-Bouvier and L. J. Blum, Biosensors for Protein Detection: A Review, Anal. Lett., 2005, 38(10), 1491–1517, DOI:10.1081/AL-200065780.
- N. M. Green, Adv. Protein Chem., 1975, 29, 85–133, DOI:10.1016/S0065-3233(08)60411-8.
- Y. Nomura, V. Sharma, A. Yamamura and Y. Yokobayashi, Selection of Silk-Binding Peptides by Phage Display, Biotechnol. Lett., 2011, 33(5), 1069–1073, DOI:10.1007/s10529-011-0519-6.
- P. K. Capon, J. Li, A. J. Horsfall, S. Yagoub, E. P. Schartner, A. Khalid, R. W. Kirk, M. S. Purdey, K. R. Dunning, R. A. McLaughlin and A. D. Abell, A Silk-Based Functionalization Architecture for Single Fiber Imaging and Sensing, Adv. Funct. Mater., 2021, 2010713, DOI:10.1002/adfm.202010713.
- D. Huemmerich, U. Slotta and T. Scheibel, Processing and Modification of Films Made from Recombinant Spider Silk Proteins, Appl. Phys. A, 2006, 82(2), 219–222, DOI:10.1007/s00339-005-3428-5.
- A. Khalid, R. Lodin, P. Domachuk, H. Tao, J. E. Moreau, D. L. Kaplan, F. G. Omenetto, B. C. Gibson and S. Tomljenovic-Hanic, Synthesis and Characterization of Biocompatible Nanodiamond-Silk Hybrid Material, Biomed. Opt. Express, 2014, 5(2), 596–608, DOI:10.1364/BOE.5.000596.
- A. Khalid, L. Peng, A. Arman, S. C. Warren-Smith, E. P. Schartner, G. M. Sylvia, M. R. Hutchinson, H. Ebendorff-Heidepriem, R. A. McLaughlin, B. C. Gibson and J. Li, Silk: A Bio-derived Coating for Optical Fiber Sensing Applications, Sens. Actuators, B, 2020, 311, 127864, DOI:10.1016/j.snb.2020.127864.
- N. J. Anthis and G. M. Clore, Sequence-Specific Determination of Protein and Peptide Concentrations by Absorbance at 205 nm, Protein Sci., 2013, 22(6), 851–858, DOI:10.1002/pro.2253.
- J. S. Cheah and S. Yamada, A Simple Elution Strategy for Biotinylated Proteins Bound to Streptavidin Conjugated Beads Using Excess Biotin and Heat, Biochem. Biophys. Res. Commun., 2017, 493(4), 1522–1527, DOI:10.1016/j.bbrc.2017.09.168.
- D. A. Armbruster and T. Pry, Limit of Blank, Limit of Detection and Limit of Quantitation, Clin. Biochem. Rev., 2008, 29(suppl. 1), S49–S52 Search PubMed.
- A. Urrutia, K. Bojan, L. Marques, K. Mullaney, J. Goicoechea, S. James, M. Clark, R. Tatam and S. Korposh, Novel Highly Sensitive Protein Sensors Based on Tapered Optical Fibres Modified with Au-Based Nanocoatings, https://www.hindawi.com/journals/js/2016/8129387/, accessed Jan 4, 2021, DOI:10.1155/2016/8129387.
- W. Wang, Z. Mai, Y. Chen, J. Wang, L. Li, Q. Su, X. Li and X. Hong, A Label-Free Fiber Optic SPR Biosensor for Specific Detection of C-Reactive Protein, Sci. Rep., 2017, 7(1), 16904, DOI:10.1038/s41598-017-17276-3.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra03584c |
| This journal is © The Royal Society of Chemistry 2021 |