
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAntifouling hydrogel film based on a sandwich array for salivary glucose monitoring†
Zifeng Zhang ab,
Shiwen Wangb,
Guanjiang Liub,
Debo Hubc,
Bei Yangbc,
Qing Daiabc and
Qian Dou*b
ab,
Shiwen Wangb,
Guanjiang Liub,
Debo Hubc,
Bei Yangbc,
Qing Daiabc and
Qian Dou*b
aSchool of Materials Science and Engineering, Zhengzhou University, Zhengzhou 450001, China
bDivision of Nanophotonics, CAS Key Laboratory of Standardization and Measurement for Nanotechnology, CAS Center for Excellence in Nanoscience, National Center for Nanoscience and Technology, Beijing 100190, China. E-mail: douq@nanoctr.cn
cCenter of Materials Science and Optoelectronics Engineering, University of Chinese Academy of Sciences, Beijing 100049, China
First published on 12th August 2021
Abstract
A glucose biosensor prepared using interpenetrating polymer network (IPN) hydrogel as a sensing material is the subject of growing interest due to its fast response and high sensitivity. However, the IPN hydrogel circumvents the traditional antifouling strategy, which often requires thick antifouling coating that can result in poor glucose sensitivity owing to its energetic physical barrier (greater than 43 nm); thus a complex, time-consuming and high-cost salivary preprocessing is needed to remove protein contaminants before salivary glucose detection using the IPN hydrogel. This limits its practical application in trace salivary glucose-level monitoring. Herein, a new hydrogel film based on a sandwich array (HFSA) with a weak physical barrier, which exhibits superior antifouling and sensitivity in salivary glucose detection is reported. HFSA relies on the formation of the sandwich structure containing substrate-grafted, surface-grafted zwitterionic polymer brushes (pSBMA) and phenylboronic acid (PBA)-functionalized hydrogel. The synergistic effect originating from pSBMA brushes on the surface of HFSA and inside the HFSA matrix provides a suitable physical barrier (∼28 nm) and a robust hydration layer for HFSA, which can enhance its sensitivity and antifouling. The results show that HFSA reduce the adsorption of nonspecific protein in 10% saliva by nearly 90% and enhanced the glucose sensitivity by 130%, compared to the IPN hydrogel film. These results demonstrate that HFSA exhibits significant potential as an antifouling and sensitive glucose probe for QCM sensors in non-invasive salivary glucose monitoring.
Introduction
Non-invasive glucose monitoring is an important method for the self-management of diabetics.1–4 Recently, body fluids, such as saliva,5,6 sweat,7–11 tear, or urine12–14 have been investigated intensively to achieve blood glucose monitoring. Saliva has become an ideal marker for non-invasive glucose monitoring due to the high correlation between salivary glucose level and blood glucose level, as well as the intrinsic advantages including its safe, convenient and collectible in real-time.15–17 However, there are two crucial issues that must be addressed before detecting salivary glucose. Firstly, the concentration of glucose in the saliva is only 1 to 10% of that in blood, thus needs a highly sensitive glucose biosensor.18 Secondly, the highly nonspecific interaction with the glucose biosensor surface caused by protein in saliva makes the loss of sensitivity and accuracy of the sensor.19–21 Quartz crystal microbalance (QCM) has been developed for salivary glucose monitoring and has become successful for the online detection of many other target molecules, such as protein, bacteria and nucleic acids because of its low cost, the ability of real-time detection and label-free property.22–26 For example, in our recent work, we have reported a glucose-sensitive QCM sensor based on interpenetrating polymer network (IPN) hydrogel film, which has realized the typical salivary glucose monitoring (0–50 mg L−1) in diluted saliva. However, the hydrogel film cannot provide robust resistance to protein contaminants in saliva, and the total content of proteins in saliva is about 71–2232 mg L−1, which is 1.9–4133 fold higher than the typical glucose level (0.54–37.8 mg L−1). Therefore, in order to achieve trace salivary glucose level monitoring, a complex salivary preprocessing (such as PVDF film, solid-phase extraction, 100 °C for 30 min and ion exchange resin) is needed to remove the protein before salivary glucose detection using IPN hydrogel,27 thus limiting its practical application in salivary glucose monitoring.Driven by the increase in problems induced by protein fouling, numerous antifouling materials, such as monolayer protein,28 Nafion,29,30 poly(ethyleneglycol) (PEG),31 poly(sulfobetaine methacrylate) (pSBMA)32 and self-assembled lubricin (LUB)33 have been developed to form a protective antifouling coating to reduce nonspecific protein adsorption.34,35 It has been proposed that the formation of a hydration layer on a non-fouling material prevents protein adsorption by forming a physical barrier. The strength of the physical barrier is determined by many factors but the thickness of the antifouling coating is one of the most important factors.36 In general, the thick antifouling coating can form an energetic physical barrier. Although the energetic physical barrier can effectively reduce nonspecific protein adsorption from complex media such as undiluted blood plasma, the interaction of glucose molecules with glucose-sensitive hydrogel matrix can also be impeded.37–41 This leads to poor glucose sensitivity, which cannot meet the requirements of low salivary glucose levels. But then, if the antifouling coating is too thin, their ability to form a physical barrier will be so greatly impaired that no significant performance of protein resistance can be achieved.
Recently, three-dimensional porous antifouling coating42 has been reported to overcome the drawback of the traditional antifouling coating. In order to enhance the sensitivity, nanomaterials, such as nanoparticles should be added to the porous antifouling coating. However, the porous coating pores are susceptible to plugging by nanoparticles because their diameter is larger than that of the pores in the coating, thus still causing sensitivity loss of the sensor. Therefore, the key challenge for salivary glucose detection is how to provide a suitable physical barrier for glucose biosensors so that they can achieve superior protein resistance and glucose sensitivity.
In the present work, we hypothesize that a weak physical barrier can minimize the glucose sensitivity loss and enhance the protein resistance of the QCM sensor if we simultaneously utilize thin pSBMA brushes on the surface and within the IPN hydrogel to provide a robust hydration layer via synergistic effect. Currently, the synergistic antifouling effect is mainly focused on the simple combination of multiple antifouling components. However, these multiple components do not necessarily have excellent biofouling resistance because of their poor biocompatibility.43 Herein, we designed and synthesized a new hydrogel film based on the sandwich array (HFSA) with substrate-grafted and surface-grafted pSBMA brushes as antifouling components and phenylboronic acid (PBA)-functionalized hydrogel as glucose-sensitive component (Fig. 1a). HFSA can achieve ultra-low protein fouling and high glucose sensitivity, which are mainly attributed to the synergistic effect originating from substrate-grafted and surface-grafted antifouling pSBMA brushes, and the presence of a robust hydration layer and weak physical barrier. The experimental results demonstrated that the surface of HFSA with a 28 nm physical barrier has a low detection limit of 3 mg L−1 and achieves typical salivary glucose level (0–50 mg L−1) monitoring without complex salivary preprocessing compared to IPN hydrogel. Therefore, this study presents a new strategy for improving the antifouling properties and sensitivity of glucose sensors.
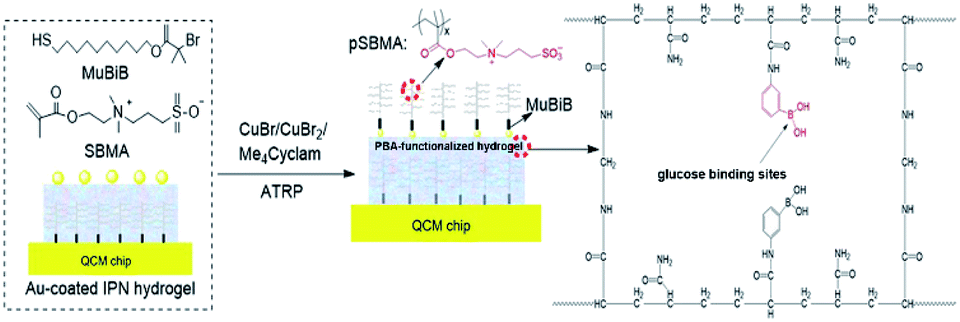 | ||
| Fig. 1 Schematic diagram showing the synthesis procedure for the HFSA. (b) An HFSA-coated quartz chip (i). An SEM image of the cross-sectional HFSA (ii). A schematic diagram illustrating the protein resistance and glucose sensitivity of the HFSA (iii). (c) The traditional antifouling coating modified hydrogel film has poor glucose sensitivity. | ||
Results and discussion
HFSA was prepared to enhance the antifouling and sensitivity of the sensor in this work (Fig. 1). Briefly, Au film was deposited onto the surface of IPN hydrogel by magnetron sputtering to facilitate the modification of the initiator. The attachment of the initiator was achieved by the bifunctional molecules that contained an ATRP initiator at one end (a bromoisobutyrate moiety) and a thiol at the other end, to allow one-step functionalization of the surface with the MUBiB initiator via the formation of an alkanethiol SAM. The MUBiB chains attached to the Au film still possess –C(CH3)2Br groups, which could serve as initiating sites for sequential ATRP for the preparation of pSBMA brushes.45 HFSA was synthesized by grafting the thin pSBMA brushes on the surface of IPN hydrogel via surface-initiated ATRP. The structure of HFSA consists mainly of the substrate and surface-grafted pSBMA brushes as antifouling elements, PBA-functionalized hydrogel as glucose-sensitive elements.An obvious rainbow colour can be seen on the surface of the HFSA-coated quartz chip (Fig. 2a(i)), indicating that the HFSA was uneven. Fig. 2a(ii) shows that the cross-sectional HFSA is compact, which can increase the number of boric acid groups in the hydrogel matrix and enhance glucose sensitivity.46 The schematic diagrams of the protein resistance and glucose sensitivity of HFSA were shown in Fig. 2a(iii). Owing to the synergistic effect of surface and substrate-grafted pSBMA brushes, HFSA can be highly resistant to bacterial adhesion and biofilm formation via a robust hydration layer. Moreover, owing to the synergistic protein-resistive effect, the surfaced-grafted pSBMA brushes have a thin layer, which can form a weak physical barrier. As a consequence, with a weak physical barrier on the surface and a robust hydration layer, HFSA can achieve a high glucose sensitivity.47 However, in contrast, in order to obtain the ultra-low protein fouling, the traditional thick and dense antifouling coating are distributed on the surface of the hydrogel (Fig. 2b). This could result in poor sensitivity owing to the energetic physical barrier on its surface.
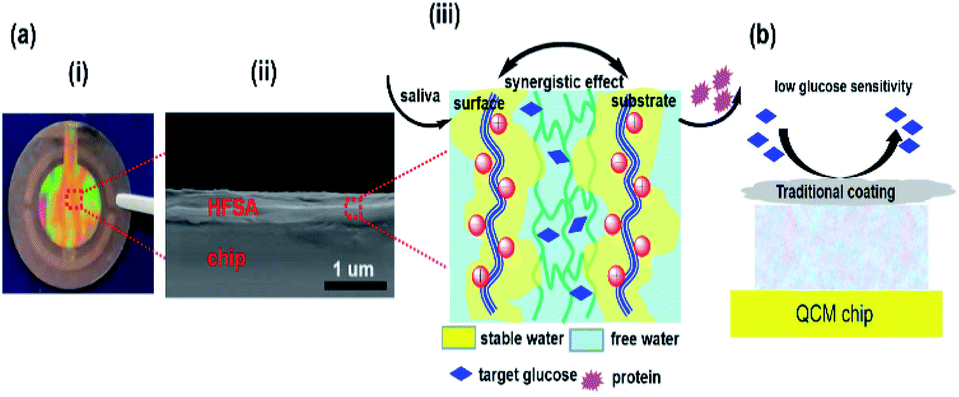 | ||
| Fig. 2 (a) An HFSA-coated quartz chip (i). An SEM image of the cross-sectional HFSA (ii). A schematic diagram illustrating the protein resistance and glucose sensitivity of the HFSA (iii). (b) The traditional antifouling coating modified hydrogel film has poor glucose sensitivity. | ||
The chemical reaction occurring at the surface of IPN hydrogel was characterized by FT-IR (Fig. 3a). The characteristic peaks at 1340 cm−1, 1660 cm−1, and 3310 cm−1 in FTIR of IPN hydrogel are for stretching vibrations of –B(OH)2, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C in the aromatic ring, and O–H, respectively (Fig. 3a(I)). After the introduction of the Au film, the characteristic peaks of IPN hydrogel disappeared (Fig. 3a(II)), which might be attributed to the absorption of infrared light by the Au film. After the introduction of the MUBiB initiator on IPN hydrogel, there was no obvious characteristic peak (Fig. 3a(III)). This is because there was just a very thin layer of initiator immobilized on the surface of IPN hydrogel.48 The bands at 1364 cm−1, 1490 cm−1, and 1750 cm−1 are mainly attributed to the bending vibrations of C–H in CH3, the stretching vibration of C–N in N+(CH3)2, and stretching vibrations of CO in –COOC segments respectively, which demonstrated that the pSBMA brushes were successfully attached to the surface of IPN hydrogel (Fig. 3a(IV)).49 To further confirm the surface properties of HFSA during each step of its synthesis process, XPS was utilized as the method for tracking the surface composition variation of the initiator on IPN hydrogel and HFSA. Table S1† lists the detailed data from XPS scans on different surfaces. According to the data, the characteristic signal of bromine (2.63%, Br3d) appeared with a binding energy of about 69 eV (Fig. 3b), indicating the existence of an initiator on the surface of IPN hydrogel. After the ATRP reactions occurred for 0.5 h, the characteristic signal of bromine disappeared while the characteristic signal of sulfobetaine (S2p at 167 eV, N1S at 402 eV) was obviously observed as compared with the survey scan spectrum of MUBiB initiator modified IPN hydrogel, indicating the pSBMA brushes were successfully grown from the surface of IPN hydrogel. Some researchers50 suggested that the disappearance of the bromine signal might be because the XPS method only measures a depth of ∼10 nm, the chains on the surface are entangled and the living chains-end may not be located directly on the outermost layer. AFM images revealed the morphological features of different surfaces. In Fig. 3c, the IPN hydrogel was a little rough, and the roughness was 24 nm. After the introduction of the Au film, the roughness of IPN hydrogel increased to 50 nm (Fig. 3d). The Au film-coated IPN hydrogel showed a folded appearance. The roughness of MUBiB initiator modified IPN hydrogel decreased to 31 nm as compared with that of Au film-coated IPN hydrogel (Fig. 3e), which might be attributed to a homogeneous and very thin layer of the initiator immobilized on the surface of IPN hydrogel. After incorporating pSBMA brushes onto the surface of IPN hydrogel via surface-initiated ATRP, the surface roughness (Rms = 30 nm) barely increased as compared with that of the initiator-modified IPN hydrogel, indicating the formation of homogeneous pSBMA brushes (Fig. 3f). The homogeneous pSBMA brushes can achieve exceptional resistance to protein adsorption, presumably because these brushes present a high-enough surface density of SBMA moieties at the solid/water interface to prevent the adsorption of proteins.27 These results suggested that HFSA had been successfully prepared.
C in the aromatic ring, and O–H, respectively (Fig. 3a(I)). After the introduction of the Au film, the characteristic peaks of IPN hydrogel disappeared (Fig. 3a(II)), which might be attributed to the absorption of infrared light by the Au film. After the introduction of the MUBiB initiator on IPN hydrogel, there was no obvious characteristic peak (Fig. 3a(III)). This is because there was just a very thin layer of initiator immobilized on the surface of IPN hydrogel.48 The bands at 1364 cm−1, 1490 cm−1, and 1750 cm−1 are mainly attributed to the bending vibrations of C–H in CH3, the stretching vibration of C–N in N+(CH3)2, and stretching vibrations of CO in –COOC segments respectively, which demonstrated that the pSBMA brushes were successfully attached to the surface of IPN hydrogel (Fig. 3a(IV)).49 To further confirm the surface properties of HFSA during each step of its synthesis process, XPS was utilized as the method for tracking the surface composition variation of the initiator on IPN hydrogel and HFSA. Table S1† lists the detailed data from XPS scans on different surfaces. According to the data, the characteristic signal of bromine (2.63%, Br3d) appeared with a binding energy of about 69 eV (Fig. 3b), indicating the existence of an initiator on the surface of IPN hydrogel. After the ATRP reactions occurred for 0.5 h, the characteristic signal of bromine disappeared while the characteristic signal of sulfobetaine (S2p at 167 eV, N1S at 402 eV) was obviously observed as compared with the survey scan spectrum of MUBiB initiator modified IPN hydrogel, indicating the pSBMA brushes were successfully grown from the surface of IPN hydrogel. Some researchers50 suggested that the disappearance of the bromine signal might be because the XPS method only measures a depth of ∼10 nm, the chains on the surface are entangled and the living chains-end may not be located directly on the outermost layer. AFM images revealed the morphological features of different surfaces. In Fig. 3c, the IPN hydrogel was a little rough, and the roughness was 24 nm. After the introduction of the Au film, the roughness of IPN hydrogel increased to 50 nm (Fig. 3d). The Au film-coated IPN hydrogel showed a folded appearance. The roughness of MUBiB initiator modified IPN hydrogel decreased to 31 nm as compared with that of Au film-coated IPN hydrogel (Fig. 3e), which might be attributed to a homogeneous and very thin layer of the initiator immobilized on the surface of IPN hydrogel. After incorporating pSBMA brushes onto the surface of IPN hydrogel via surface-initiated ATRP, the surface roughness (Rms = 30 nm) barely increased as compared with that of the initiator-modified IPN hydrogel, indicating the formation of homogeneous pSBMA brushes (Fig. 3f). The homogeneous pSBMA brushes can achieve exceptional resistance to protein adsorption, presumably because these brushes present a high-enough surface density of SBMA moieties at the solid/water interface to prevent the adsorption of proteins.27 These results suggested that HFSA had been successfully prepared.
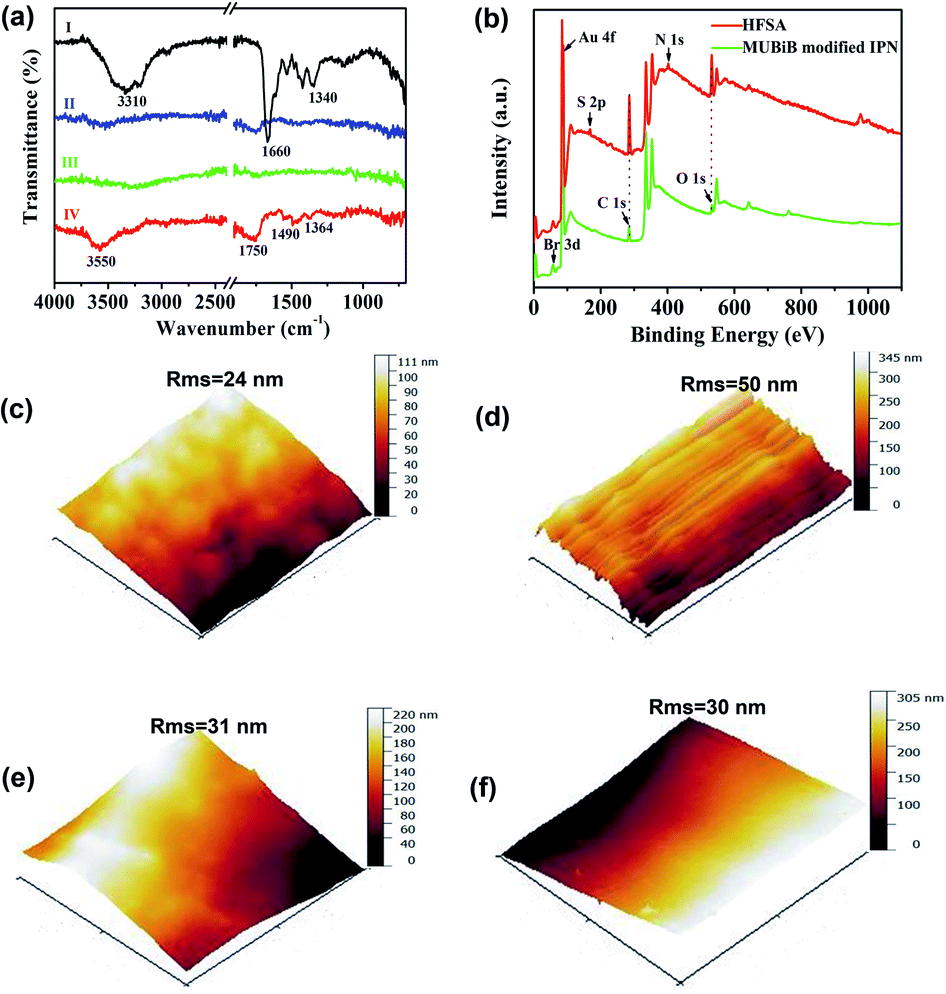 | ||
| Fig. 3 (a) FT-IR spectra of (I) IPN hydrogel. (II) Au film-coated IPN hydrogel. (III) Initiator modified IPN hydrogel. (IV) HFSA. (b) XPS spectra of initiator modified IPN hydrogel and HFSA. AFM images of different surfaces: (c) IPN hydrogel. (d) Au film-coated IPN hydrogel. (e) Initiator modified IPN hydrogel. (f) HFSA. | ||
The common methods for grafting of antifouling materials from polymeric membrane surfaces were through surface-initiated ATRP, in which the first step is conventional to introduce the initiator onto the film surface via chemical methods. However, the modification of the initiator in the film needs harsh reaction conditions51,52 including an ice bath, toxic organic solvents (such as tetrahydrofuran and dichloromethane), and nitrogen protection.
To overcome the above drawbacks, the Au film was deposited onto the surface of IPN hydrogel via magnetron sputtering, in this work, which allowed one-step functionalization of the surface of the IPN-hydrogel film with the sulfhydryl initiator. According to the deposition rate (60 nm min−1) of the Au film, we controlled the thickness of the Au film by adjusting the deposition time. If the deposition time was 3 s, the thickness of the Au film would be expressed as 3 nm. The total amount of proteins in the saliva is about 71–2232 mg L−1.53 To adjust the pH of the sample solution, the sample solutions consisting of pH = 7.5 PBS/saliva mixture (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) were used in the QCM test in this study. As a result, the concentration of protein in saliva could be diluted twice as much. Therefore, 500 mg L−1 Muc., 500 mg L−1 Lys., 500 mg L−1 BSA, and 500 mg L−1 Fib. were used in QCM tests to study the protein resistance of HFSA.
:1 v/v) were used in the QCM test in this study. As a result, the concentration of protein in saliva could be diluted twice as much. Therefore, 500 mg L−1 Muc., 500 mg L−1 Lys., 500 mg L−1 BSA, and 500 mg L−1 Fib. were used in QCM tests to study the protein resistance of HFSA.
As can be seen in Fig. 4a, the protein resistance and glucose sensitivity showed a clear relationship with the thickness of the Au film. An obvious increase in the frequency shifts of Au films at thicknesses of 3 nm and 30 nm, respectively, when the four single protein solutions were pumped into the HFSA-coated QCM sensor. The increase in the frequency shift indicated a mass increase on the surface of the sensor, which was physically correlated with the increased mass of protein vibrating with the sensor.54 This may be attributed to the low surface coverage of pSBMA brushes, thus could reduce the hydration of the brushes and cause protein adsorption.55 A positive frequency shift was observed, and a similar result was also reported by Healy et al.,56 in which they observed a positive frequency shift when 300 mg L−1 Fib. was adsorbed onto the IPN film surface. They described that this is due to changes in viscosity and density of the bulk fluid compared to those of PBS. The effect of polymerization time of ATRP on protein resistance and glucose sensitivity is shown in Fig. 4b. In our recent work, we have demonstrated that the thickness of pSBMA brushes linearly increased with the increase in polymerization time (R2 = 0.994). Therefore, the thickness of surface-grafted pSBMA can be calculated based on the linear equation (y = 0.482x + 13.8).27 The frequency shifts were within 7 Hz and 35 Hz when four single protein solutions and 50 mg L−1 glucose solution were pumped into the HFSA-coated QCM sensor, which demonstrated that excellent protein resistance and glucose sensitivity occurred at a polymerization time of 30 min. According to the linear equation, the thickness of pSBMA brushes is 28 nm. In order to better understand the effect of a physical barrier on sensitivity, the physical barrier is expressed as the thickness of pSBMA brushes in this work. With longer polymerization time (such as 90 min), the glucose sensitivity and protein resistance decreased, which was attributed to the energetic physical barrier of pSBMA brushes and surface defects. Earlier studies55 have shown that the packing density played a key role in protein resistance. The uniform and suitable thickness of pSBMA brushes facilitated the achievement of superior protein resistance owing to an energetic physical barrier. However, very thick pSBMA brushes could lead to a strong dipole interaction between zwitterionic pairs, which would reduce the hydration of pSBMA brushes and cause protein adsorption. In general, a negative frequency shift should be observed when the glucose molecules were adsorbed on the HFSA-coated QCM sensor. The unexpected positive frequency shift may be attributed to the transport of water out of the hydrogel owing to the increasing cross-link density caused by the adsorption of glucose molecules, causing the mass of the sensing layer to decrease.47 Therefore, to obtain the HFSA with high glucose sensitivity and protein resistance, the thickness of Au film was selected as 4 nm and the polymerization time was selected as 30 min in this work. To study the effect of surface morphologies of Au films on glucose sensitivity, SEM images were collected to observe surface morphologies of IPN hydrogel coated with different thicknesses of Au film as shown in Fig. 4c–e. As shown in Fig. 4c, Au films on IPN hydrogel primarily consist of a percolating film with multiple voids at the film thickness of 4 nm, which facilitates the permeability of glucose. While, in comparison, Au films on IPN hydrogel exhibit an almost continuous structure with tiny voids at a thickness of 10 nm (Fig. 4d) and a continuous dense structure at a thickness of 30 nm (Fig. 4e), which resulted in a low permeability of glucose. A similar result was also reported by Yakubovsky et al.57
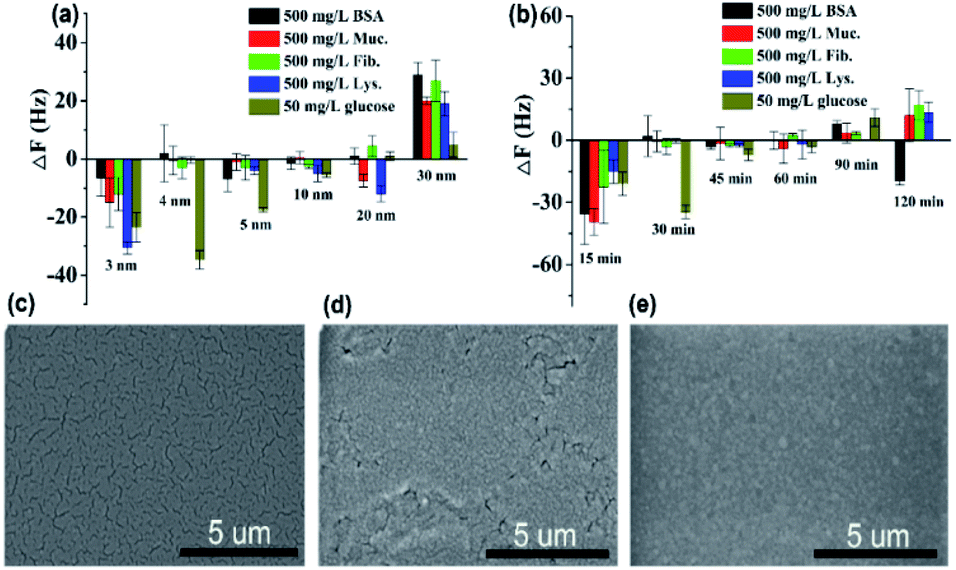 | ||
| Fig. 4 (a) Effect of thickness of Au film on protein resistance and glucose sensitivity of HFSA. Flow rate: 960 μL min−1; incubation time: 5 min. (b) Effect of polymerization time of ATRP on protein resistance and glucose sensitivity of HFSA. Flow rate: 960 μL min−1; incubation time: 5 min. Error bars indicate the s.d. of three replicating measurements. SEM images of the surface morphologies of IPN hydrogel coated with different thickness of Au film. (c) The thickness of Au film: 4 nm. (d) The thickness of the Au film: 10 nm. (e) The thickness of the Au film: 30 nm. | ||
Fig. 5a shows that the QCM sensor based on the HFSA reveals little fluctuation in frequency shifts (1.1 Hz, the fundamental frequency of this system is 5 × 106 Hz, only 0.22 millionth of the fundamental frequency) over 240 min in pH = 7.5 PBS solution. This demonstrates that the HFSA-coated QCM sensor has good stability. The thickness of the film is an important parameter that dictates the performance of the HFSA. The different thicknesses of HFSA was obtained by controlling the thickness of IPN hydrogel. The different thickness of IPN hydrogel was obtained at different spinning speed. We performed an experiment to study the effect of HFSA with different thicknesses on protein resistance and glucose sensitivity. The film thicknesses were 220, 252, 300, 380 and 650 nm (Fig. S1†). From our experimental results, we find that, the thickness of the HFSA hydrogel has no conspicuous effect on protein resistance. This is mainly attributed to the strong hydration capacity of pSBMA brushes via ionic solvation.32 It was observed that the frequency shift in 50 mg L−1 glucose solution is increased as the film thickness is raised from 220 nm to 380 nm (Fig. S2†). This is likely because the thicker the HFSA, the more glucose molecules it can bind.58 Nonetheless, HFSA with a thickness of 650 nm had poor glucose sensitivity. A similar result was also observed by Dou et al.46 In their study, they demonstrated that the hydrogel film with a thickness of 600 nm had poor glucose sensitivity due to its poor viscoelasticity. Therefore, the thickness of HFSA was selected as 380 nm in this work. In order to avoid the error of thickness of HFSA using AFM measurement, SEM is further used to measure the thickness of HFSA. As shown in Fig. S3,† the thickness of HFSA obtained by SEM is 370 nm, which is similar to that (380 nm) obtained by AFM. The density of glucose in the saliva is only 1 to 10% of that in blood, and the typical salivary glucose level in humans is between 0.54 mg L−1 and 37.8 mg L−1.18 Therefore, the glucose concentration range of 0.0 to 50 mg L−1 was selected to study the glucose sensitivity in this work. The shift in the resonance frequency was plotted as a function of glucose concentration under different pH conditions (Fig. 5b). ΔF became more negative with increasing pH under all three investigated pH conditions. Phenylboronic acid exists in an aqueous solution in two forms: a negatively charged dissociated state and an uncharged non-dissociated state. A dissociation equilibrium exists between these two states (Fig. S4†). Non-dissociated phenylboronic acid is a flat triangle and forms an unstable complex with glucose, while dissociated phenylboronic acid has a tetrahedral structure and can form cyclic lactones with glucose molecules via the reversible interaction of diol-containing glucose molecules and the hydroxyl group of dissociated phenylboronic acid.59–61 The magnitude of the sensitivity of the sensor is larger at high pH (7.5) than at low pH (6.8), presumably due to the increased population of the dissociated phenylboronic acid form, which has a higher glucose affinity.62 The sensor shows good linearity from 0.0 to 50 mg L−1 (Fig. 5c), adequately encompassing the range of glucose concentration in saliva under physiological conditions. The linear correlation coefficients were 0.912 (pH = 6.8), 0.979 (pH = 7.3) and 0.965 (pH = 7.5). Fig. 5d shows the glucose response of the QCM sensor based on HFSA and IPN at pH = 7.5. It should be noted that the thickness of HFSA (380 nm) and IPN (440 nm)44 were selected as the optimized thickness toward glucose sensitivity. As shown in Fig. 4d, when the glucose concentration was increased to 50 mg L−1, the total frequency of HFSA and IPN was shifted by 35 Hz and 27 Hz, respectively. Obviously, HFSA exhibited ∼130% increase in glucose sensitivity compared with IPN. This is mainly attributed to HFSA possessing a weak physical barrier and robust hydrated layer via synergistic effect originating from substrate-grafted and surface-grafted pSBMA brushes. The effect of the hydrated layer on glucose sensitivity has also been demonstrated by our recent work.47 Subsequently, in order to obtain the low detection limit (LOD) of the HFSA-coated sensor, we gradually increased the glucose concentration. When the glucose concentration was increased to 3 mg L−1, ΔF had a relatively obvious increase (Fig. 5e). So the LOD of the sensor was 3 mg L−1, which is sufficient for applications in typical salivary glucose monitoring. In general, to confirm the repeatability of the sensor, three cycles of testings are needed.63 Therefore, five cycles of testing were selected in this study. In the present study, repeatability testing was performed by alternatively pumping PBS solution (pH = 7.5, glucose-free) and glucose solution (10, mg L−1) into the flow cell. As illustrated by Fig. 5f, HFSA still has a high sensitivity to glucose under various glucose concentrations after five cycles. Moreover, as listed in Table S2,† the relative standard deviations (%RSD) of the frequency response for the HFSA and IPN hydrogel at glucose concentrations of 10 mg L−1 are 4.3%, and 2.5% (ref. 27) (n = 5), respectively, which further demonstrated that HFSA has good repeatability. The response to interferences such as fructose was not tested in this study, and this is because there are almost no other saccharides except for glucose in saliva.53 Moreover, a recent study by Dou et al.,64 which investigated the influence of possible competitive binding of interference (0.1 mM) on glucose detection, demonstrated that the PBA-functionalized hydrogel film-coated QCM sensor can effectively detect glucose despite the presence of other saccharides, such as fructose, maltose, and lactose. To study the protein resistance of different film-coated QCM sensors, adsorption from a single protein solution of BSA, Muc., Fib., and Lys. on the IPN hydrogel, pSBMA coating-modified PBA-functionalized hydrogel, and HFSA was measured by the QCM sensor (Fig. 6a). There were obvious changes in frequency when the Lys. solution was pumped into the IPN hydrogel-coated QCM sensor, which demonstrated that the IPN hydrogel had high fouling from Lys. solution. The detailed surface composition of IPN hydrogel is shown in Table S3,† indicating that there was a little antifouling material (0.162% S2p) on the surface of the IPN hydrogel. In contrast, there was no obviously measurable change in frequency when a single protein solution was pumped into HFSA at a polymerization time of 30 min (28 nm physical barrier on its surface), which demonstrated that HFSA had excellent protein resistance owing to the robust hydration layers. However, high fouling from a single protein solution was observed on the pSBMA coating modified PBA-functionalized hydrogel at a similar physical barrier on its surface (Fig. S5†). Although the pSBMA coating modified PBA-functionalized hydrogel exhibited excellent protein resistance at a polymerization time of 60 min (43 nm physical barrier on its surface), it has poor glucose sensitivity.27
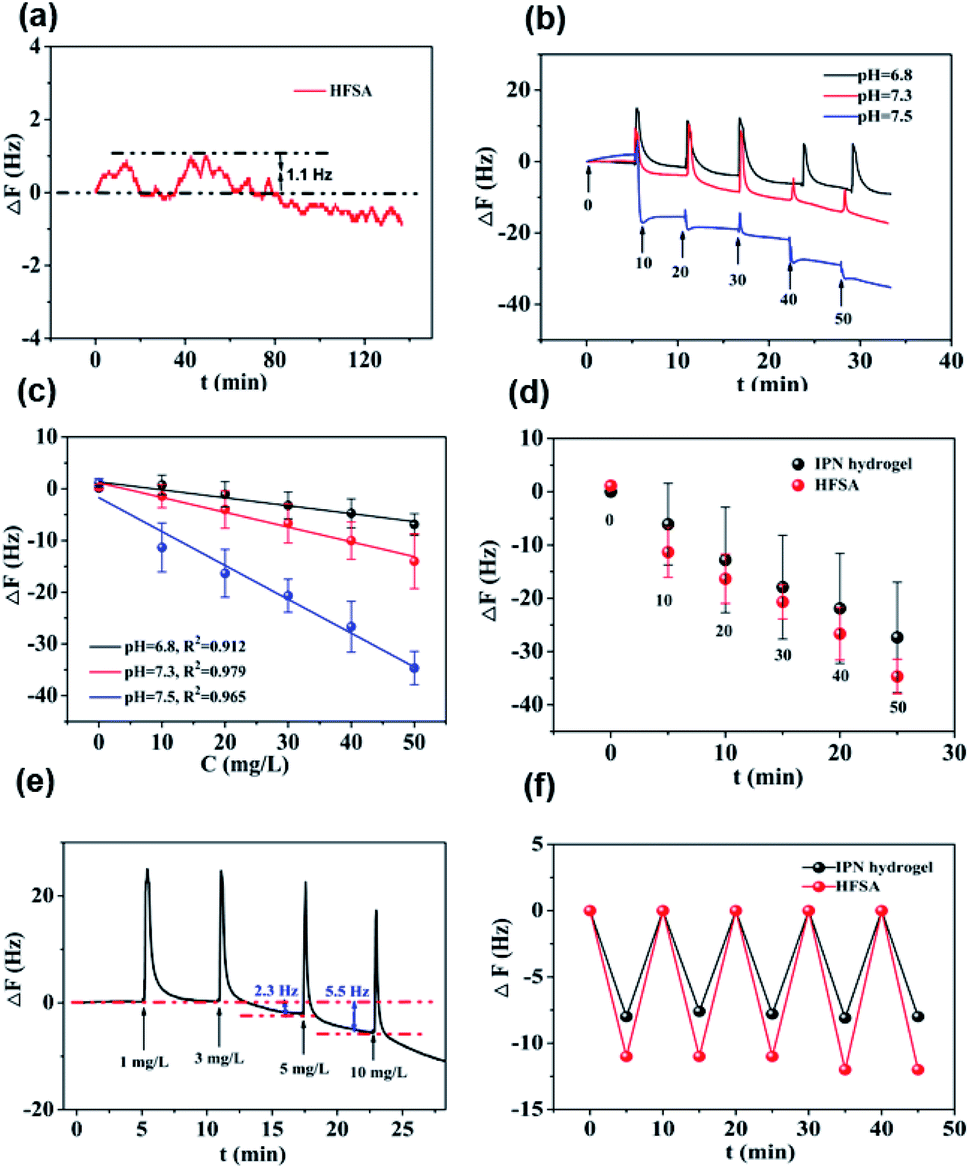 | ||
| Fig. 5 The stability, sensitivity, LOD and repeatability of the QCM sensor based on HFSA to identify glucose. (a) The sensor has good stability and the ΔF was only ∼1.1 Hz within almost 2 h. (b) The ΔF became more negative with the increase in pH. The numbers in (b) represent the glucose level. (c) Favorable linear relationship between ΔF and different glucose concentrations from 0 to 50 mg L−1 at different pH values. Error bars indicate the s.d. of three replicating measurements. (d) The QCM sensor based on HFSA has better glucose sensitivity than the QCM sensor based on IPN. Error bars indicate the s.d. of three replicating measurements; the numbers in (d) represent the glucose level. (e) The low detection limit of the QCM sensor based on HFSA was 5 mg L−1. (f) The QCM sensor based on HFSA and IPN hydrogel has a good repeatability, respectively. Experimental conditions: flow rate: 960 μL min−1; incubation time: 5 min. | ||
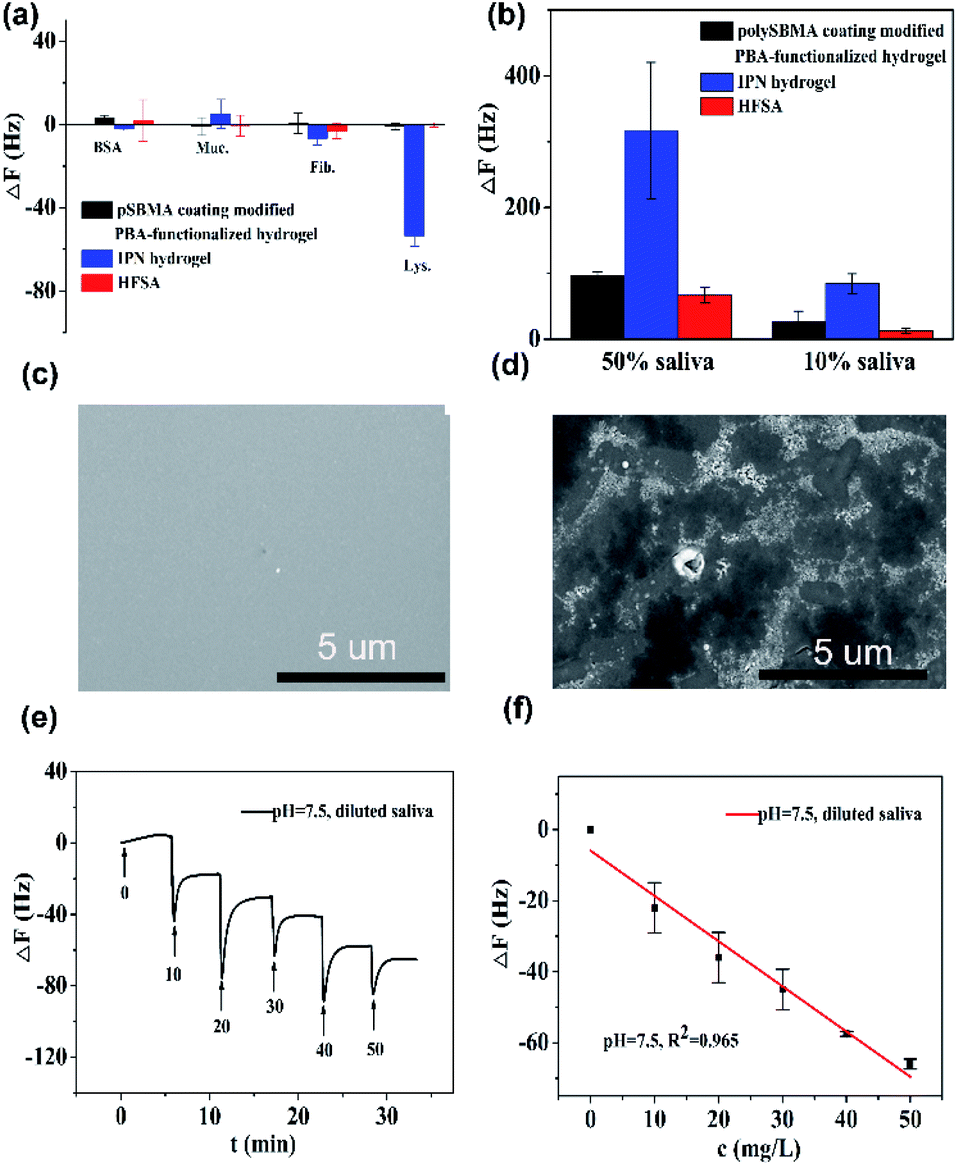 | ||
| Fig. 6 (a) Adsorption of 500 mg L−1 BSA, 500 mg L−1 Muc., 500 mg L−1 Fib., and 500 mg L−1 Lys. on IPN hydrogel, pSBMA coating modified PBA-functionalized hydrogel and HFSA at pH = 7.5, respectively. Flow rate: 960 μL min−1; incubation time: 5 min. (b) Adsorption of 50% and 10% saliva on IPN hydrogel, pSBMA-coating modified PBA-functionalized hydrogel, and HFSA. Flow rate: 960 μL min−1; incubation time: 5 min. SEM images of (c) HFSA (d) IPN hydrogel after 48 h incubation in the human saliva. (e) The detection of glucose in diluted saliva by HFSA-coated QCM sensor. Flow rate: 960 μL min−1; incubation time: 5 min. (f) Relationship between frequency shift and glucose concentration. Flow rate: 960 μL min−1; incubation time: 5 min. | ||
Saliva samples were provided through the project “early identification, early diagnosis and cutting point of diabetes risk factors” (2016YFC1305700). All participants were between 18–82 years old and they provided written informed consent. The donors fasted for 8 h prior to collecting samples. Participation in these studies was voluntary, and the medical ethics committee of Jiangsu Provincial Center for Disease Control and Prevention (JSJK2017-B003-02). China had approved the study protocols. To reduce the impact of differences in individual saliva content and acquire enough saliva samples, we mixed the saliva samples that originated from multiple people. Before the measurement, ion chromatography was used to detect the concentration of glucose in the mixed saliva. Only the glucose concentration of the mixed saliva is ≤1 mg L−1 that can be applied for glucose detection. In order to evaluate the protein resistance of HFSA in the real sample, diluted human saliva samples were pumped into flow cells mounted with an HFSA-coated QCM quartz chip. As shown in Fig. 6b, The HFSA-coated QCM sensor has better antifouling performance than IPN hydrogel-coated sensor in diluted saliva. The frequency shift (8 Hz) of HAS was 10.4-fold lower than that (83 Hz) of IPN hydrogel in 10% saliva, indicating that the HFSA reduces 90% nonspecific protein adsorption than the IPN hydrogel. To further verify the protein resistance of different films, SEM images of HFSA and IPN hydrogel film after incubating in human saliva for 48 h were collected and are shown in Fig. 6c and d compared with the IPN hydrogel film, there were almost no obvious biological molecules adhering on the surface of HFSA, showing that the HFSA has excellent resistance to biofouling. This is in agreement with the unobvious frequency shift of the hydrogel in diluted saliva. In order to quantify glucose in real samples, diluted human saliva samples (1:9 in PBS) were used as carrier solutions into flow cells mounted with the HFSA. Fig. 6e shows the detection of glucose in 10% diluted saliva by HFSA-coated QCM sensor. As the glucose concentrations gradually increased, the frequency shift became more negative. However, the detection of glucose in 10% diluted saliva by the IPN hydrogel had a larger frequency shift (138 Hz) than that (65 Hz) of HFSA due to nonspecific protein adsorption (Fig. S6a†). This can make it difficult to recognize small frequency changes generated by the binding of glucose molecules with their receptors from the total frequency response.65 Moreover, the IPN hydrogel-coated QCM sensor had poor linearity with R2 = 0.884 (Fig. S6b†) owing to the nonspecific protein adsorption. Unlike the IPN hydrogel-coated QCM sensor, the response of glucose in 10% diluted saliva by HFSA had good linearity with R2 = 0.965 (Fig. 6f). The HFSA-coated QCM sensor could detect the typical saliva glucose level (0–50 mg L−1) in diluted saliva without complex salivary preprocessing. Moreover, the limit of detection is 3 mg L−1, which is comparable or lower than those for most similar technology (Table S4†). These results demonstrate that HFSA can be used as a candidate for sensing glucose in saliva.
Experimental section
Materials
Fibrinogen (Fib.), fraction I from bovine plasma, lysozyme (Lys.), and mucin (Muc.) from bovine submaxillary gland were purchased from Shanghai Yuanye Biotechnology Co. Ltd. Bovine serum albumin (BSA), copper(I) bromide (99%), copper(II) bromide (99%) and Me4Cyclam (98%) were obtained from Sigma-Aldrich. N-(3-Sulfopropyl)-N-(methacryloxyethyl)-N,N-dimethylammoniumbetaine (SBMA, 99%), 2-hydroxy-2-methylpropiophenone (HMPP, 99%) were purchased from J&k. ω-Mercaptoundecyl bromoisobutyrate (MUBiB, ≥95%) was obtained from Shanghai D&B Biological Science Technology Co. Ltd. N,N-Methylenebisacrylamide (Bis, 98%) was purchased from Sinopharm Chemical Reagent Co. Ltd. 3-Acrylamidophenylboronic acid (PBA, 98%) was obtained from Ark Pharm. Acrylamide (AM, 98.5%) was purchased from Xilong Chemical Industry. Sulfuric acid (H2SO4, AR), hydrogen peroxide (H2O2, 30% aqueous solution), ethanol (C2H5OH, AR), dimethyl sulfoxide (DMSO, AR), glucose (C6H1206, AR), sodium phosphate dibasic dodecahydrate (Na2HPO4, AR), and potassium dihydrogen phosphate (KH2PO4, AR) were purchased from Beijing Chemical Factory. Water used in the experiments was purified using a Millipore water purification system (UPR-II-10TNZP, Sichuan Youpu Ultrapure Technology Co. Ltd.). The saliva was collected from volunteers.Preparation of Au film-coated IPN hydrogel
The IPN hydrogel film was prepared based on our recent work.27 First, Au crystals were sonicated in a piranha solution (98% H2SO4:30% H2O2 = 7:3) for 10 min to eliminate organic substances and were thereafter rinsed with distilled water and dried under N2 stream. The cleaned Au crystal was then immersed in a 1 × 10−3 M ω-mercaptoundecyl bromoisobutyrate initiator solution by forming a self-assembled monolayer (SAM) at room temperature for 24 h. Before polymerization, Au crystals were coated with a SAM, rinsed with pure ethanol, and then dried under an N2 stream. Second, ten milliliters of a mixture containing ethanol and distilled water (1:1; v/v) was degassed using three freeze–pump–thaw cycles. After that, it was transferred under N2 atmosphere to a Schlenk tube containing CuBr (19.1 mg, 133 μM), CuBr2 (5.9 mg, 26.5 μM), and Me4Cyclam (40.0 mg, 160 μM). In a separate Schlenk tube, the catalyst solution (blue solution) was mixed with a monomer SBMA (1500 mg, 5.4 mmol). The polymerization solution was then transferred to a reactor containing Au crystals coated with a SAM. The polymerization reaction was carried out at 30 °C under N2 atmosphere, and the samples were withdrawn at different times to obtain polySBMA brushes with varying lengths. Au crystals were coated with polySBMA brushes were washed with ethanol, followed by water; and were then stored in phosphate buffer saline (PBS). Third, a pre-polymer solution consisting of 25% PBA, 2% BIS, 71% AM, and 2% HMPP (by mass) in DMSO solvent was prepared. After that, 25 μL of the prepared pre-polymer solution was deposited onto the upper electrode of polySBMA brush-coated Au crystal for 30 min, and then spun at a speed of 3500 rpm for 1 min. The coated Au crystals were subsequently irradiated with ultraviolet light (λ = 365 nm) under N2 atmosphere for 60 min for UV curing. Finally, the obtained IPN hydrogel film-coated Au crystals were repeatedly rinsed with ethanol, followed by distilled water. As shown in Fig. S7,† the MUBiB initiator were bifunctional molecules that contained a bromoisobutyrate moiety at one end and a thiol (–SH) at the other end. In order to allow one-step functionalization of the IPN surface with the MUBiB initiator via the formation of Au–s bond, the Au film was deposited onto the surface of IPN hydrogel by magnetron sputtering in a dc magnetic of 5 × 10−3 T. Details of the sputtering system have been described elsewhere.44 Briefly, no intentional heating of the IPN hydrogel film was done during the sputtering deposition of the Au film. The sputtering deposition was carried out at the gas pressure range of 1.0 × 10−2 to 6.0 × 10−2 Torr in pure argon gas with a power of 32–85 W. The deposition rate of the Au film was 60 nm min−1 in our experiment. Au films with different thicknesses were obtained by adjusting the time of magnetron sputtering.
Preparation of thiol-coated surface of IPN hydrogels
The Au-coated IPN hydrogels were immersed in a 1 × 10−3 M solution of MUBiB initiator at room temperature for 24 h. Before polymerization, the IPN hydrogel coated with a self-assembled monolayer (SAM) formed by the MUBiB initiator was rinsed with pure ethanol and dried with a stream of nitrogen.Preparation of HFSA via the surface-initiated ATRP
Ten milliliters of ethanol/distilled water mixture (1:1 v/v) was degassed using three freeze–pump–thaw cycles. Afterward, it was transferred under N2 atmosphere to a Schlenk tube containing CuBr (19.1 mg, 133 μM), CuBr2 (5.9 mg, 26.5 μM), and Me4Cyclam (40.0 mg, 160 μM). The blue solution of the catalyst was injected into another Schlenk tube containing the monomer, SBMA (150 mg, 0.54 mmol). Then the polymerization solution was transferred to the reactor containing the IPN hydrogel film coated with a SAM. The reaction was carried out at 30 °C under N2 atmosphere and samples were taken at different polymerization times to obtain varying lengths of pSBMA brushes. The obtained HFSA was washed with ethanol and water and stored in PBS. The traditional pSBMA brushes coating modified PBA-functionalized hydrogel was also prepared according to the above procedure.
Measurement of glucose sensitivity and protein adsorption
Fig. S8† shows the experimental setup, which includes crystal oscillator electronics, frequency counter circuit, flow cell, peristaltic pump, and so on. HFSA was deposited onto the quartz chip and then the quartz-hybrid hydrogel complex resonator was installed in the flow cell of the QCM 200 system (fundamental frequency of 5 MHz). The frequency of the crystal was monitored in real-time using the QCM data acquisition software. After the frequency was stabilized (frequency shift ≤ ±1.1 Hz), the glucose detection capacity of the HFSA was evaluated. Solutions (1 mL) of increasing glucose concentrations (from 0.0 to 50 mg L−1 with PBS) were pumped into the flow cell every 5 min with a flow rate of 960 μL min−1, and glucose levels were converted to the frequency shift (ΔF) by the electrical system. Finally, the frequency shift (ΔF) was recorded for each glucose solution or protein solution. Several cycles of these experiments were conducted at different pH from 6.8 to 7.5. Similarly, protein adsorption onto HFSA was measured according to the above procedure.Characterization
The surface morphology and thickness of HFSA formed on the quartz chips were measured by atomic force microscopy (AFM) in contact mode using a scattering SNOM (Neaspec GmbH). The cross-section of hydrogel was characterized by SEM (Hitachi S-4800). Attenuated total reflection Fourier transform infrared spectra (ATR-FTIR) for membrane surfaces were obtained using a Fourier-transform infrared spectrometer (Nicolet 560). X-ray photoelectron spectroscopy (XPS, ESCALAB250Xi) is a quantitative technique that reveals the elemental compositions of the surface of a material. Nitrogen (N), sulfur (S), bromine (Br) and boron (B) contents were measured by XPS. The photoelectron take-off angle was 15°. HFSA and pSBMA coating modified PBA-functionalized hydrogel film were placed in saliva for 48 h at room temperature. Then quartz chips were rinsed with water and dried with a stream of nitrogen. Finally, the samples were examined by a scanning electron microscope (Hitachi S-4800) after coating with gold.Conclusions
We have successfully prepared a new hydrogel film with excellent protein resistance and glucose sensitivity. HFSA was found to possess better glucose sensitivity than the IPN hydrogel film, which was attributed to a weak physical barrier and a robust hydrated layer formed by ionic solvation. Moreover, HFSA possessed excellent resistance to biofouling from 10% saliva with a reduction in the adsorption of 90% when compared to that of IPN hydrogel. This is attributed to the synergistic antifouling effect of pSBMA brushes. More importantly, HFSA can achieve typical salivary glucose (0–50 mg L−1) detection without complex salivary preprocessing. The enzyme-based electrochemical glucose meters that are already in the market suffer from stability problems caused by temperature, pH, humidity and toxic chemicals. Therefore, the advantages of glucose monitoring by using a QCM sensor are HFSA with antifouling, glucose sensitivity and simple storage conditions over other detection methods like electrochemical sensors. These results indicated that the HFSA-coated QCM sensor has a great potential for sensitive and convenient detection of glucose in real-world applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key Research and Development Program of China (2016YFA0201600), the National Natural Science Foundation of China (grant number 51925203; U2032206), and Science and Technology Service Network Project (STS Program) of the Chinese Academy of Sciences (KFJ-STS-ZDTP-063).Notes and references
- S. Wild, C. Roglic, A. Green, R. Sicree and H. King, Diabetes care, 2004, 27, 1047–1053 CrossRef PubMed.
- D. C. Klonoff, L. Blonde, G. Cembrowski, A. R. Chacra, G. Charpentier, S. Colagiuri, G. Dailey, R. A. Gabbay, L. Heinemann, D. Kerr, A. Nicolucci, W. Polonsky, O. Schnell, R. Vigersky and J. F. Yale, J. Diabetes Sci. Technol., 2011, 5, 1529–1548 CrossRef PubMed.
- I. Mamkin, S. Ten, S. Bhandari and N. Ramchandani, J. Diabetes Sci. Technol., 2008, 2, 882–889 CrossRef PubMed.
- M. C. Torjman, N. Dalal and M. E. Goldberg, J. Diabetes Sci. Technol., 2008, 2, 178–181 CrossRef.
- W. Zhang, Y. Du and M. L. Wang, Sens. Biosens. Res., 2015, 4, 23–29 Search PubMed.
- H. Lee, T. K. Choi, Y. B. Lee, H. R. Cho, R. Ghaffari, L. Wang, H. J. Choi, T. D. Chung, N. Lu, T. Hyeon, S. H. Choi and D. H. Kim, Nat. Nanotech., 2016, 11, 566–572 CrossRef CAS PubMed.
- J. Heikenfeld, Electroanalysis, 2016, 28, 1242–1249 CrossRef CAS.
- W. Gao, S. Emaninejad, D. H. Lien, G. A. Brooks, R. W. Davis and A. Javery, Nature, 2016, 529, 509–514 CrossRef CAS.
- A. Koh, D. Kang, Y. Xue, S. Lee, J. Kim, Y. Huang and J. A. Rogers, Sci. Transl. Med., 2016, 8, 366ra165 CrossRef.
- H. Lee, C. Song, Y. S. Hong, M. S. Kim, H. R. Cho, T. Kang, K. Shin, S. H. Choi, T. Hyeon and D. H. Kim, Sci. Adv., 2017, 3, e1601314 CrossRef PubMed.
- Y. J. Hong, H. Lee, J. Kim, M. Lee, H. J. Choi, T. Hyeon and D. H. Kim, Multifunctional Wearable System that Integrates Sweat-Based Sensing and Vital-Sign Monitoring to Estimate Pre-/Post-Exercise Glucose Levels, Adv. Funct. Mater., 2018, 28, 1805754–1805766 CrossRef.
- Q. Y. Yan, B. Peng, G. Su, B. E. Cohan, T. C. Major and M. E. Meyerhoff, Anal. Chem., 2011, 83, 8341–8346 CrossRef CAS.
- H. Yao, A. J. Shum, M. Cowan, I. Lahdesmaki and B. A. Parviz, Biosens. Bioelectron., 2011, 26, 3290–3296 CrossRef CAS PubMed.
- J. H. Park, J. H. Kim, S. Y. Kim, W. H. Cheong, J. Jang, Y. G. Park, F. Bien and J. U. Park, Sci. Adv., 2018, 4, eaap9841 CrossRef PubMed.
- C. Z. Liao, C. H. Mak, M. Zhang, L. W. Chan and F. Yan, Adv. Mater., 2015, 27, 676–681 CrossRef CAS PubMed.
- T. Pfaffe, J. C. White, P. Beyerlein, K. Kostner and C. Punyadeera, Clin. Chem., 2011, 57, 675–687 CrossRef CAS PubMed.
- S. Chiappin, G. Antonelli, R. Gatti and E. F. De Palo, Clin. Chim. Acta, 2007, 383, 30–40 CrossRef CAS PubMed.
- Y. H. Chen, S. Y. Lu, S. S. Zhang, Y. Li, Z. Qu, Y. Chen, B. W. Lu, X. Y. Wang and X. Feng, Sci. Adv., 2017, 3, e1701629 CrossRef PubMed.
- S. Y. Jiang and Z. Q. Cao, Adv. Mater., 2010, 22, 920–932 CrossRef CAS PubMed.
- G. R. Hendrickson, M. H. Smith, A. B. South and L. A. Lyon, Adv. Funct. Mater., 2010, 20, 1697–1712 CrossRef CAS.
- A. Hucknall, S. Rangarajan and A. Chilkoti, Adv. Mater., 2009, 21, 2441–2446 CrossRef CAS.
- C. Li, X. Chen, F. Y. Zhang, X. X. He, G. Z. Fang, J. F. Liu and S. Wang, Anal. Chem., 2017, 89, 10431–10438 CrossRef CAS PubMed.
- C. Y. Yao, T. Y. Zhu, Y. Z. Qi, Y. H. Zhao, H. Xia and W. L. Fu, Sensors, 2010, 10, 5859–5871 CrossRef CAS PubMed.
- Y. Tsuge, Y. K. Moriyama, Y. Tokura and S. M. Shiratori, Anal. Chem., 2016, 88, 10744–10750 CrossRef CAS PubMed.
- D. D. Erbahar, I. Gurol, F. Zelder and M. Harbeck, Sens. Actuators. B, 2015, 207, 297–302 CrossRef CAS.
- M. Lazerges, H. B. Perrot, N. Rabehagasoa and C. T. Compere, Biosensors, 2012, 2, 245–254 CrossRef CAS PubMed.
- Z. Z. Zhang, Q. Dou, S. W. Wang, D. B. Hu, B. Yang, Z. P. Zhao, H. L. Liu and Q. Dai, Nanoscale, 2020, 12, 22787–22797 RSC.
- M. M. Picher, S. Kupcu, C. J. Huang, J. Dostalek, D. Pum, U. B. Sleytr and P. Ertl, Lap Chip, 2013, 13, 1780–1789 RSC.
- F. Moussy, D. J. Harrison and R. V. Rajotte, Int. J. Artif. Organs, 1994, 17, 88–94 CrossRef CAS PubMed.
- F. Moussy, S. Jakeway, D. J. Harrison and R. V. Rajotte, Anal. Chem., 1994, 66, 3882–3888 CrossRef CAS PubMed.
- I. Banerjee, R. C. Pangule and R. S. Kane, Adv. Mater., 2011, 23, 690–718 CrossRef CAS PubMed.
- J. M. Wang, H. Sun, J. J. Li, D. Y. Dong, Y. B. Zhang and F. L. Yao, Carbohydr. Polym., 2015, 117, 384–391 CrossRef CAS.
- M. J. Russo, A. F. Quigley, R. M. I. Kapsa, S. E. Moulton, R. Guijt, S. M. Silva and G. W. Greene, Chem. Electro. Chem., 2020, 7, 2851–2861 CAS.
- M. Russo, M. Han, P. Desroches and C. S. Manasa, ACS sensors, 2021, 6, 1482–1507 CrossRef CAS PubMed.
- C. Jiang, G. X. Wang, R. Hein and N. Liu, Chem. Rev., 2020, 120, 3852–3889 CrossRef CAS PubMed.
- S. Herrwerth, W. Eck, S. Reinhardt and M. Grunze, J. Am. Chem. Soc., 2003, 125, 9359–9366 CrossRef CAS PubMed.
- L. Mi, M. M. Giarmarco, Q. Shao and S. Jiang, Biomaterials, 2012, 33, 2001–2006 CrossRef CAS PubMed.
- Y. Chang, Y. J. Shi, C. J. Lai, H. H. Kung and S. Jiang, Adv. Funct. Mater., 2013, 23, 1100–1110 CrossRef CAS.
- S. Campuzano, M. Pedrero, P. Yanezsedeno and J. M. Pingarron, Int. J. Mol. Sci., 2019, 20, 423–442 CrossRef PubMed.
- S. Chen, L. Li, C. Zhao and J. Zhang, Polymer, 2010, 51, 5283–5293 CrossRef CAS.
- I. Banerjee, R. C. Pangule and R. S. Kane, Adv. Mater., 2011, 23, 690–718 CrossRef CAS PubMed.
- J. S. Rio, O. Y. Henry, P. Jolly and D. E. Ingber, Nat. Nanotechnol., 2019, 14, 1143–1149 CrossRef PubMed.
- Y. Y. He, X. Y. Wan, K. C. Xiao, W. W. Lin, J. H. Li, Z. Li, F. Luo, H. Tan, J. S. Li and Q. Fu, Biomater. Sci., 2019, 7, 5369–5382 RSC.
- T. Minami, H. Sato, H. Nanto and S. Takata, JPM. J. Appl. Phys., 1985, 24, 781–784 CrossRef CAS.
- A. Hucknall, D. H. Kim, S. Rangarajan, R. T. Hill, W. M. Reichert and A. Chilkoti, Adv. Mater., 2009, 21, 1968–1971 CrossRef CAS PubMed.
- Q. Dou, Z. F. Zhang, Y. X. Wang, S. W. Wang, D. B. Hu, Z. P. Zhao, H. L. Liu and Q. Dai, ACS Appl. Mater. Interfaces, 2020, 12, 34190–34197 CrossRef CAS PubMed.
- Z. Z. Zhang, Q. Dou, S. W. Wang, D. B. Hu, X. D. Guo, B. X. Liao, Z. P. Zhao, H. L. Liu and Q. Dai, J. Mater. Chem. C, 2020, 8, 9655–9662 RSC.
- P. S. Liu, Q. Chen, S. S. Wu, J. Shen and S. C. Lin, J. Member. Sci., 2010, 350, 387–394 CrossRef CAS.
- L. Ye, Y. B. Zhang, Q. S. Wang, X. Zhou, B. G. Yang, F. Ji, D. Y. Dong, L. Gao, Y. L. Cui and F. L. Yao, ACS Appl. Mater. Interfaces, 2016, 8, 15710–15723 CrossRef CAS PubMed.
- A. Carlmark and E. E. Malmstrom, Biomacromolecules, 2003, 4, 1740–1745 CrossRef CAS PubMed.
- Y. H. Zhu, X. W. Xu, N. D. Brault, A. J. Keefe, X. Han, Y. Deng, J. Q. Xu, Q. M. Yu and S. Y. Jiang, Anal. Chem., 2014, 86, 2871–2875 CrossRef CAS PubMed.
- Y. C. Hu, B. Liang, L. Fang, G. G. Ma, G. Yang, Q. Zhu, S. F. Chen and X. S. Ye, Langmuir, 2016, 32, 11763–11770 CrossRef CAS PubMed.
- K. Nqamchuea, K. B. Chaisiwamonqkhol, C. B. Mcauley and R. G. Compton, Analyst, 2018, 143, 81–99 RSC.
- S. H. Baxamusa and K. K. Gleason, Adv. Funct. Mater., 2009, 19, 3489–3496 CrossRef CAS.
- C. Huang, Y. T. Li, J. B. Krause, N. D. Brault and S. Y. Jiang, Macromol. Rapid Commun., 2012, 33, 1003–1007 CrossRef CAS PubMed.
- E. F. Irwin, J. E. Ho, S. R. Kane and K. E. Healy, Langmuir, 2005, 21, 5529–5536 CrossRef CAS PubMed.
- D. I. Yakubovsky, Y. V. Stebunov, R. V. Kirtaev, G. A. Ermolaev, M. S. Mironov, S. M. Novikov, A. V. Arsenin and V. S. Volkov, Adv. Mater. Interfaces, 2019, 6, 1900196–1900202 CrossRef.
- Z. X. Zhang, Q. Dou, H. K. Gao, B. Bai, Y. M. Zhang, D. B. Hu, A. K. Yetisen, H. Butt, X. X. Yang, C. J. Li and Q. Dai, Adv. Healthcare Mater., 2018, 7, 1700873–1700880 CrossRef PubMed.
- R. J. Ma and L. Q. Shi, Polym. Chem., 2014, 5, 1503–1518 RSC.
- D. Shen, H. J. Yu, L. Wang, A. Khan, F. Haq, X. Chen, Q. Huang and L. S. Teng, J. Controlled Release, 2020, 321, 236–258 CrossRef CAS PubMed.
- A. M. Horgan, A. J. Marshall, S. J. Kew, K. E. S. Dean, C. D. Creasey and S. Kabilan, Biosens. Bioelectron., 2006, 21, 1838–1845 CrossRef CAS PubMed.
- S. A. Asher, V. L. Alexeev, A. V. Goponenko, A. C. Sharma, I. K. Ledenv, C. S. Wilcox and D. J. Finegold, J. Am. Chem. Soc., 2003, 125, 3322–3329 CrossRef CAS PubMed.
- X. Chen, J. Chen, F. B. Wang, X. Xiang, M. Luo, X. H. Ji and Z. K. He, Biosens. Bioelectron., 2012, 35, 363–368 CrossRef CAS PubMed.
- Q. Dou, D. B. Hu, H. K. Gao, Y. M. Zhang, A. K. Yetisen, H. Butt, J. Wang, G. J. Nie and Q. Dai, RSC Adv., 2017, 7, 41384–41390 RSC.
- Q. Dou, S. W. Wang, Z. F. Zhang, Y. X. Wang, Z. P. Zhao, H. J. Guo, H. L. Liu and Q. Dai, Nanoscale, 2020, 12, 19317–19324 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra03517g |
| This journal is © The Royal Society of Chemistry 2021 |