
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
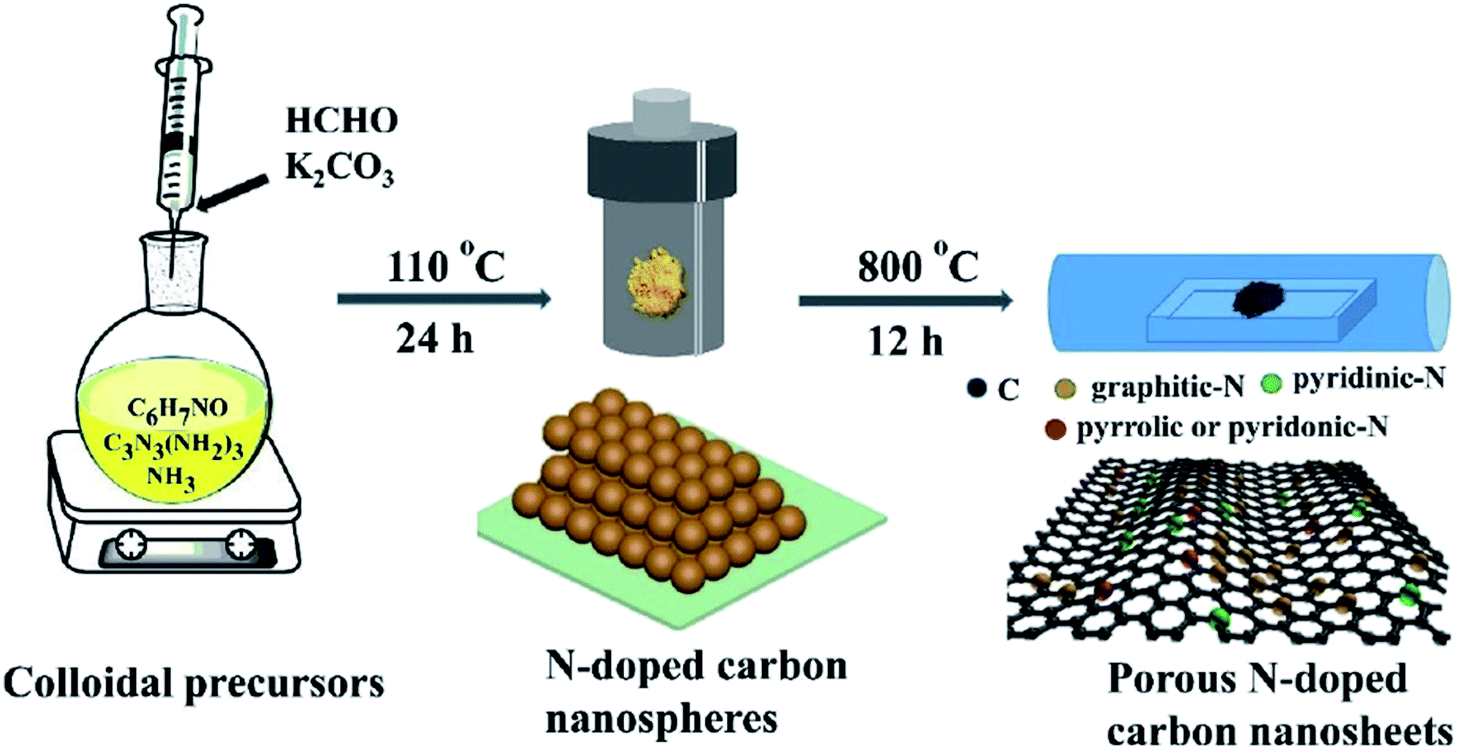
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent advances in metal-free heteroatom-doped carbon heterogonous catalysts
Yalda Rangraz and
Majid M. Heravi *
*
Department of Chemistry, School of Physics and Chemistry, Alzahra University, Vanak, Tehran, Iran. E-mail: m.heravi@alzahra.ac.ir; mmh1331@yahoo.com
First published on 5th July 2021
Abstract
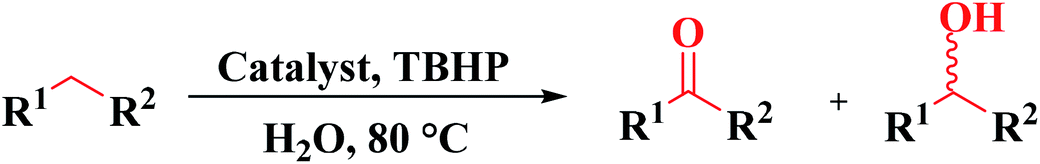
The development of cost-effective, efficient, and novel catalytic systems is always an important topic for heterogeneous catalysis from academia and industrial points of view. Heteroatom-doped carbon materials have gained more and more attention as effective heterogeneous catalysts to replace metal-based catalysts, because of their excellent physicochemical properties, outstanding structure characteristics, environmental compatibility, low cost, inexhaustible resources, and low energy consumption. Doping of heteroatoms can tailor the properties of carbons for different utilizations of interest. In comparison to pure carbon catalysts, these catalysts demonstrate superior catalytic activity in many organic reactions. This review highlights the most recent progress in synthetic strategies to fabricate metal-free heteroatom-doped carbon catalysts including single and multiple heteroatom-doped carbons and the catalytic applications of these fascinating materials in various organic transformations such as oxidation, hydrogenation, hydrochlorination, dehydrogenation, etc.
1. Introduction and scope of the review
Heterogeneous catalytic systems not soluble in reaction mixtures have the benefits of better handling properties and easy separation, from an industrial point of view.1,2With the rapidly growing demand for effective organic reactions, developing novel heterogeneous catalysts is highly needed according to the concept of “Green Sustainable Chemistry”. It has been estimated that almost all large-scale production of organic and inorganic chemicals (commercially produced chemical products), energy conversion, pollution mitigation processes, environmental protection, and crude oil refinery utilize heterogeneous catalysts3–6 and it is no exaggeration to say that heterogeneous catalysts play a key role in creating a more sustainable and cleaner world.7
Metals, especially transition metals, have been extensively applied as homogeneous and heterogeneous catalysts in a series of important organic transformations in the forms of nanoparticles (NPs), clusters, coordination metal complexes, or free ions.8–12
Notwithstanding their high efficiency, these catalysts have negative environmental impacts and suffer from the high price, susceptibility to gas poisoning, poor durability, and low selectivity.13–15 Thus, metal-free catalysts are essential for environmental remediation as well as large-scale commercial applications.16 The replacement of metal heterogeneous catalysts with metal-free catalysts can remarkably address the environmental pollution issues arising from the leaching of metal nanoparticles during usage in the petroleum and pharmaceutical industries.17
In this regard, inexpensive, readily available, environmentally friendly, and stable carbon-based materials (including ordered mesoporous carbon (OMCs), carbon nanotubes (CNTs), graphene oxide (GO), activated carbon (ACs), etc.) have drawn increasing attention in the field of heterogeneous catalysis (Fig. 1).18–29
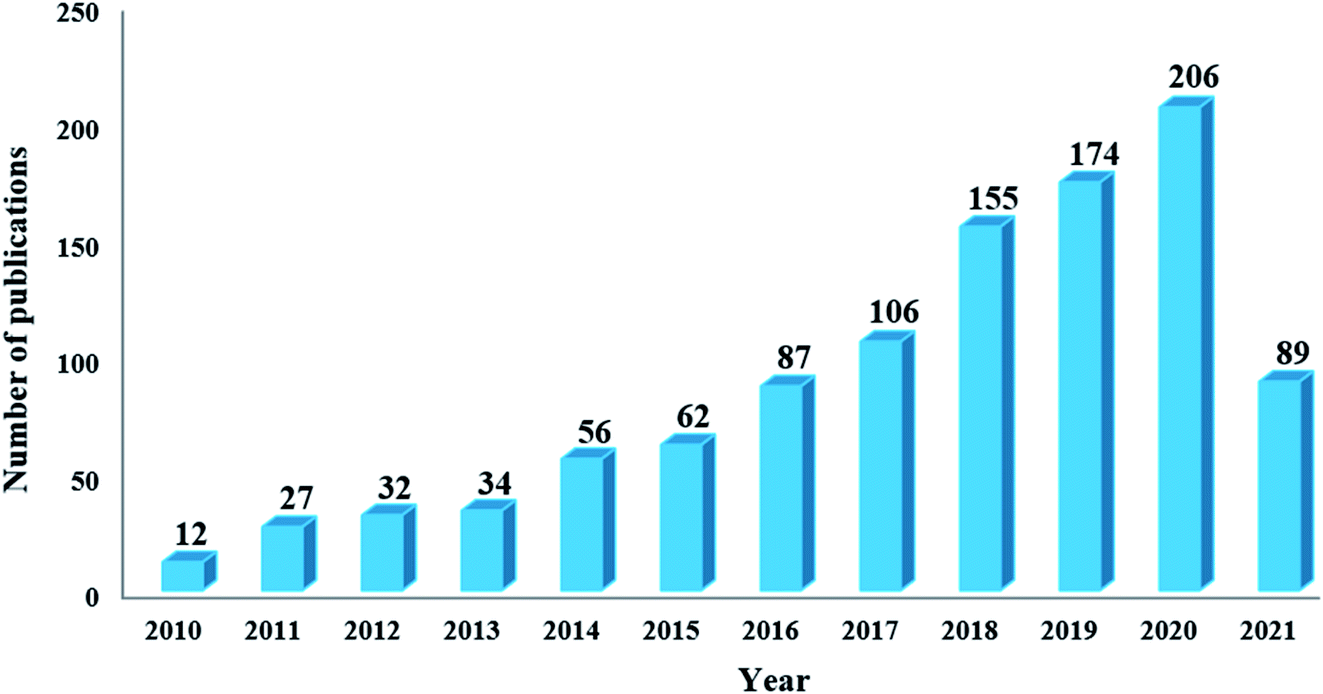 | ||
| Fig. 1 Number of publications obtained from a Scopus search using “carbon-based materials and heterogeneous catalysis” as keywords. | ||
However, pristine carbon materials are inert because uniform charge distribution which is the common property of all carbon materials blocks the electron transfer and decreases the interaction between the carbon materials and the reactants.30–32
Introducing heteroatoms like nitrogen, phosphorus, boron, and sulfur into sp2-hybridized carbon frameworks can cause the charge delocalization and change of the electronic structure into metal-like, which helps to obtain significant catalytic performance in a similar way of metal-based catalyst or can be applied as a superior alternative to the metal catalyst in industrial processes.33–40
In addition, plentiful defects generated by the differences in coordination ability, bond length, and atomic radius of carbon atoms and heteroatoms can serve as actives sites, which are beneficial for in situ charge transfer to the reactants, thus activating the substrates to promote catalytic reactions.41–48
In 1926, Rideal and Wright for the first time reported that the presence of N atoms in carbon framework has a promotional effect on the catalytic performance of charcoal in the oxidation of oxalic acid.49 Even though results were promising, heteroatom-doped carbon catalysts did not attract as much attention as they would attract nowadays.
Recently, these materials have emerged as one of the hot topics in the field of metal-free catalysis for the synthesis of organic compounds. In the past few years, several reviews have been published toward the applications of carbon-based materials especially N-doped carbons in heterogeneous catalysis.7,8,23,50–52 However, a comprehensive review that covers the application of various metal-free heteroatom-doped carbon catalysts (including single-element B-, N-, S-, and P-doped, dual-elements doped, and triple-elements doped carbon catalysts) in the field of organic transformations is highly desirable.
We are interested in novel heterogeneous catalysis, trying to design, prepare, and use them in our contemplated reactions in our laboratory, which delightfully have been mostly successful.53–68
Following, the same interest we have collected and reviewed the most recent activities on the applications of different organic transformations being performed effectively under heterogeneous catalysis.69–75
In this review, we will mainly focus on the latest developments (2015 onwards) of heteroatom-doped carbon materials (single-element B-, N-, S-, and P-doped, dual-elements doped, and triple-elements doped carbons) as promising metal-free heterogeneous catalysts, including the catalysts preparation methods with an emphasis on their application in various organic transformations (oxidation, hydrogenation, hydrochlorination, dehydrogenation, coupling, etc.).
1.1 Heteroatom-doped carbon materials
Heteroatom-doped carbon materials, especially graphene, carbon nanotubes, and new generations of heteroatom-doped porous carbons with various structures and morphologies are widely examined as metal-free catalysts.The incorporation of heteroatoms into the backbone of carbon nanotubes or pores into the walls changes their physicochemical and electronic properties, resulting in an increased ability to catalyze some chemical transformations.103
More importantly, the rich pore structure of porous carbon materials distinguishes them from traditional carbons in their tunable channels and pore size, and large specific surface area.114,115
Based on their pore sizes, porous carbon materials can be classified into three groups: micropores < 2 nm, 2 nm < mesopores < 50 nm, and macropores > 50 nm, according to the recommendation of the International Union of Pure and Applied Chemistry (IUPAC).116,117
For a long time, activated carbons synthesized by chemical or physical activation and pyrolysis of starting materials like wood, coal, and fruit shells, have been the most frequently applied form of porous carbons.7,118,119 However, because of uncertain structures of different precursors, the pore size and surface chemistry have been uncontrollable. In recent years, the designed synthesis of porous carbons through self-assembly of precursors controlled hydrothermal treatment, and carbonization has gained remarkable attention owing to the exact control and simple modification of the final materials.120
In 1999, Ryoo and co-workers121 reported the first example to fabricate highly ordered carbon molecular sieves by utilizing sucrose and MCM-48 as carbon source and template and opened a route to apply porous carbons in many fields.
Porous carbon can also be doped with heteroatoms, similar to graphene, and CNTs. The introduction of foreign atoms is an effective way of modifying the surface properties of porous carbons, at the same time providing materials with appealing features for many more tasks.122
1.2 Synthetic strategies for heteroatom-doped carbon materials
Two strategies are frequently applied to prepare heteroatom-doped carbons:123–126 (1) post-synthetic modification of pre-synthesized carbon materials with heteroatom-containing agents (such as NH3, urea, melamine, H2S, phosphoric acid, sulfur, sodium thiosulfate, polyrhodanine, cysteine, etc.) chemically or thermally, (2) in situ doping (summarized in Scheme 1). In the in situ doping method, both the synthesis of carbon materials and the doping of heteroatom happen at the same time. One of the most utilized in situ doping methods to prepare heteroatom-doped carbons including CNTs, graphene, or microporous carbon materials is the chemical vapor deposition (CVD) due to it is appropriate to obtain well-distributed heteroatoms into the carbon framework. Also, direct carbonization of heteroatom-containing species or a mixture of carbon- and heteroatom-containing precursors that has been widely applied to attain heteroatom-doped porous carbons can be classified into three groups: hard-template method, soft-template method, and template-free (self-template) method.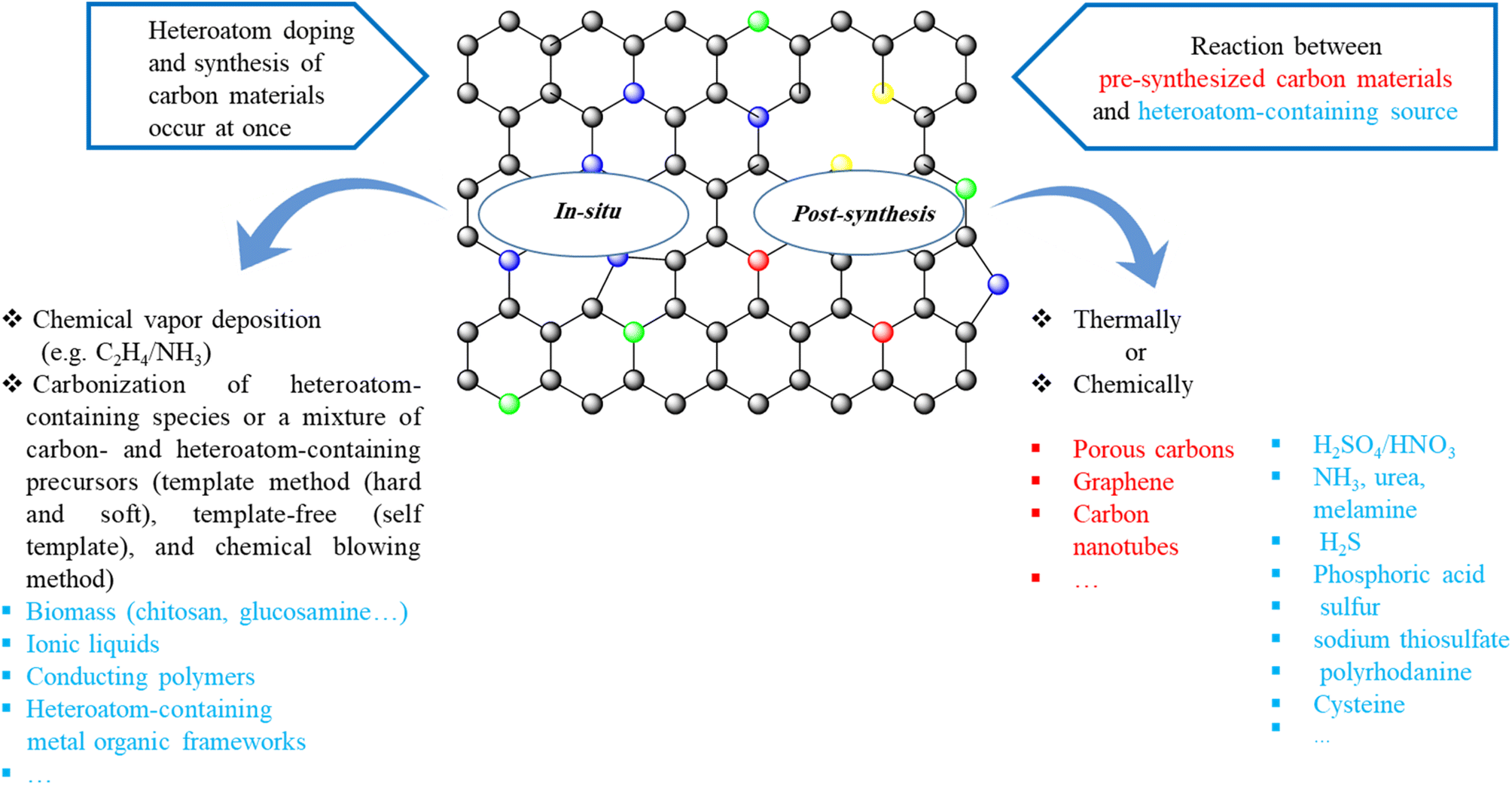 | ||
| Scheme 1 Summary of methods to synthesize heteroatom-doped carbon materials. | ||
The hard-template synthesis strategy includes multiple stages: (a) preparation of rigid template (e.g., SBA-15 mesoporous silica), (b) infiltration of template pores with precursors containing carbon (and heteroatom), (c) carbonization of the precursors and (d) removal of the template.
The hard-template method has important advantages and leads to the synthesis of heteroatom-doped mesoporous carbons with different morphologies and high specific surface areas, but this method is time-consuming, complicated, and inappropriate for mass production.127–129
In addition, the chemicals that are utilized to remove the templates, are hazardous and corrosive (such as basic (NaOH) or acidic (HF) solutions).111
Soft-templating is an attractive alternative method that provides many benefits in terms of time, processing, and large-scale production.130 This method commonly involves the organic–organic co-assembly between organic precursors and amphiphilic molecules like block copolymers and surfactants, and carbonization treatment. The thermally unstable templates are removed during carbonization and generate abundant mesopores in resulting heteroatom-doped carbon structures.131 Notwithstanding the above-mentioned advantages, the soft template method also shows limitations. Most of the soft templates (block copolymers and surfactants) are relatively high-cost, and their recovery is difficult.127
Thus, the development of new template-free methods is highly desired. These methods generally include the direct transformation of molecular precursors into heteroatom-doped porous carbon materials without adding any external template.
In comparison with template methods, template-free synthesis protocols are facile and benign, although the morphology cannot be controlled as elaborately as that of template-synthesized porous carbon and it is difficult to obtain highly porous carbons.132
There exist many heteroatom-containing sources such as biomass, ionic liquids (ILs), polymers, metal–organic framework (MOF), and so on. Besides, among various heteroatom-containing sources, NH3,36,133–135 elemental sulfur/thiophene,136,137 triphenylphosphine,138,139 and diborane,140,141 are the most common candidates for N-, S-, P- and B-doping carbon materials, respectively.
Recently, the chemical blowing strategy, which yields micropores during the carbonization process through gas evolution by decomposing ammonium chloride (NH4Cl) into HCl and NH3, was introduced as a new method to generate highly microporous heteroatom-doped carbons. The possible combination of this approach with thermally decomposable templates, like calcium carbonate (CaCO3) or magnesium acetate (Mg(OAc)2), could increase the chemical blowing effect and further introduce auxiliary mesopores.142
The dissociation temperature of heteroatom-containing species is one of the most important factors in selecting them as a precursor for in situ doping. It is desired that the dissociation temperature of these compounds be close to the growth temperature of the carbon.143 The pyrolysis temperature strongly influences the generation of C–C and C–heteroatom bonds. Generally, a high temperature is favorable for C–C bonds. It is accepted that the post-treatment procedure leads to heteroatom doping only on the surface of carbon materials whereas the direct carbonation method results in the uniform introduction of the heteroatom into the entire carbon framework.18,144
Therefore, the selection of the method will significantly influence the conductivity, acidic or basic properties, oxidation stability, and ultimately the performance of the material in a given utilization.145
2. Recent progress on heteroatom-doped carbon materials as heterogeneous catalysts
2.1 Single heteroatom-doped carbons
Commonly, there are four types of doped nitrogen atoms in a carbon matrix,148,149 including graphitic N, pyridinic N, pyrrolic N, and pyridinic N-oxide (Fig. 2a) that can be identified using X-ray photoelectron spectroscopy (XPS). The N1s peaks at B.E. of 398.3–399.8, 400.1–400.5, 401.0–401.4, and 404.0–405.6 eV are related to nitrogen atoms in the species of pyridinic, pyrrolic, graphitic, and different N-oxide species, respectively, (Fig. 2b).150–152
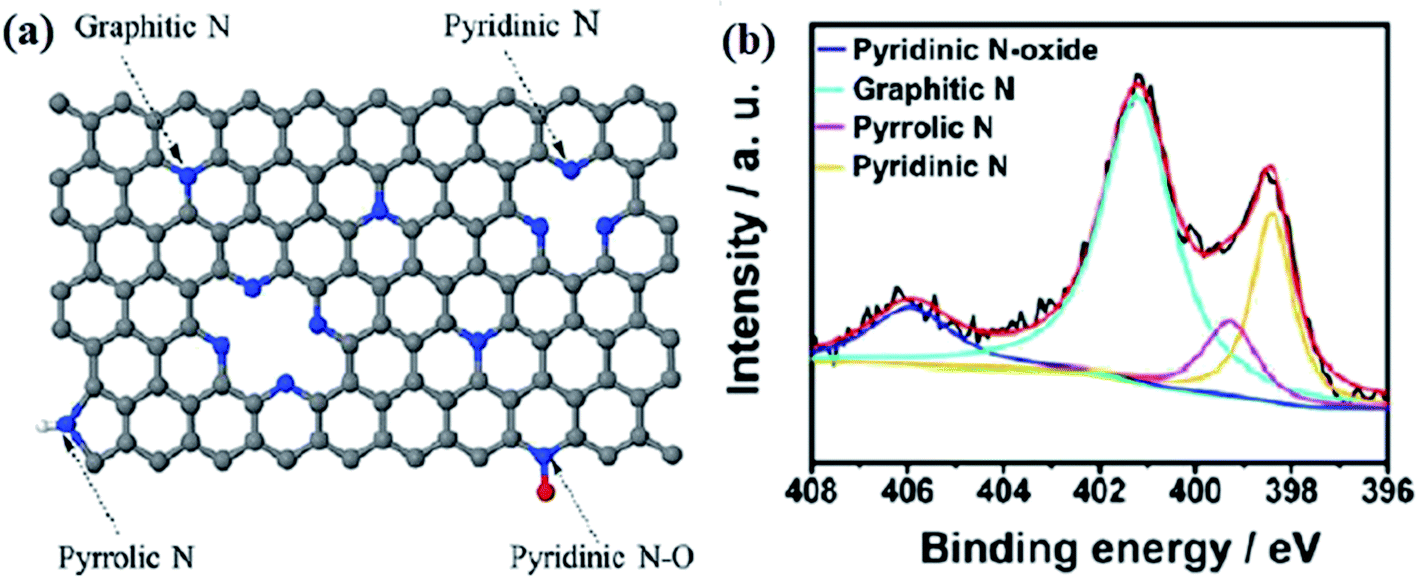 | ||
| Fig. 2 (a) Bonding configurations of nitrogen atoms in carbon networks. Reprinted with permission from ref. 125. Copyright 2017 American Chemical Society and (b) XPS spectra of N atoms in different configurations. Reprinted with permission from ref. 152. Copyright 2014 American Chemical Society. | ||
Graphitic nitrogens, which are also known as quaternary nitrogens, replace carbon atoms situated in the graphitic planes and bond to three C atoms. These N species use four electrons to generate bonds of σ and π, and a remaining electron to occupy the higher-energy π* state, resulting in the electron-donor property of graphitic nitrogen.153,154
Pyridinic nitrogen refers to nitrogen atoms located at the defects or graphitic edges. For the pyridinic N, 2 electrons are applied to generate σ-bonds with C atoms, the other 2 electrons to generate a lone pair, and the fifth electron is in the N π-state.146,155
Pyrrolic nitrogens are incorporated into five-member heterocyclic rings and bond to two carbon atoms. These species are thermally unstable, therefore their concentration in carbon materials is low after high-temperature pyrolysis.156,157
Pyridinic N-oxides refer to nitrogen atoms bonded to one oxygen atom and two carbon atoms.158,159 According to experimental and theoretical investigations, N species of graphitic, pyridinic, and pyrrolic reveal efficient interactions with reactants, and hence, they are responsible for N-doped carbon catalysts.125,160–163
For example, the has been determined as the active site for the cathodic oxygen reduction reaction in acidic conditions.164,165
In another case, the graphitic nitrogen was found to be essential for the reduction of nitroaromatics16 and oxidation of benzylic alcohols.166 Also, it has been displayed that the pyrrolic nitrogen promoted the adsorption of the reactants in the acetylene hydrochlorination reaction.167
Normally, the presence of nitrogen atoms in the carbon skeleton affords basic property, which boosts the interaction between carbon surface and acidic molecules.168–172
More importantly, N doping can greatly influence the charge distribution and spin density of carbon atoms because of the different electronegativity of carbon (2.55) and nitrogen (3.04), leading to the formation of defects in the carbon framework. This in turn can alter the electronic structure of carbon compounds.123,148,173,174
But it has a bigger atomic radius and lower electronegativity (2.19) than nitrogen and carbon atoms. Thus, P doping can produce defect sites and effectively modify the chemical reactivity and electronic properties of carbon materials (pristine carbons). These surface modifications result in an improvement in the performance of catalysts.18,156,182
2.2 Dual- and tri-heteroatom doped carbons
Co-doping by two or three heteroatoms with different electronegativities can display a unique electron distribution and further enhances the catalytic activity of the carbon materials. This intrinsic feature of carbocatalyst can be related to the synergistic effect of incorporated elements.1862.3 Catalytic applications
In recent years, the utilization of metal-free heteroatom-doped carbon catalysts in chemical reactions has shown rapid growth. The main purpose of this review is to highlight the current researches in the field of organic transformations catalyzed by heteroatom-doped carbons reported in the literature.Different applications of these catalysts in the organic reactions are summarized in this section.
NC-950 was found to be highly effective for the selective hydrogenation of nitro compounds in hexane at 90 °C using hydrazine hydrate (N2H4·H2O) as the reductant, affording respective amines in yields ranging from 87.5 to 100% (Scheme 2). The catalytic activity of NC-950 was compatible or even superior to those of the reported metal catalysts.
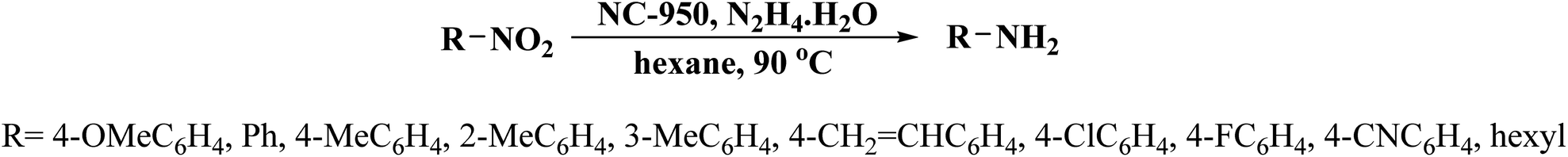 | ||
| Scheme 2 Reduction of nitro compounds using NC-950. | ||
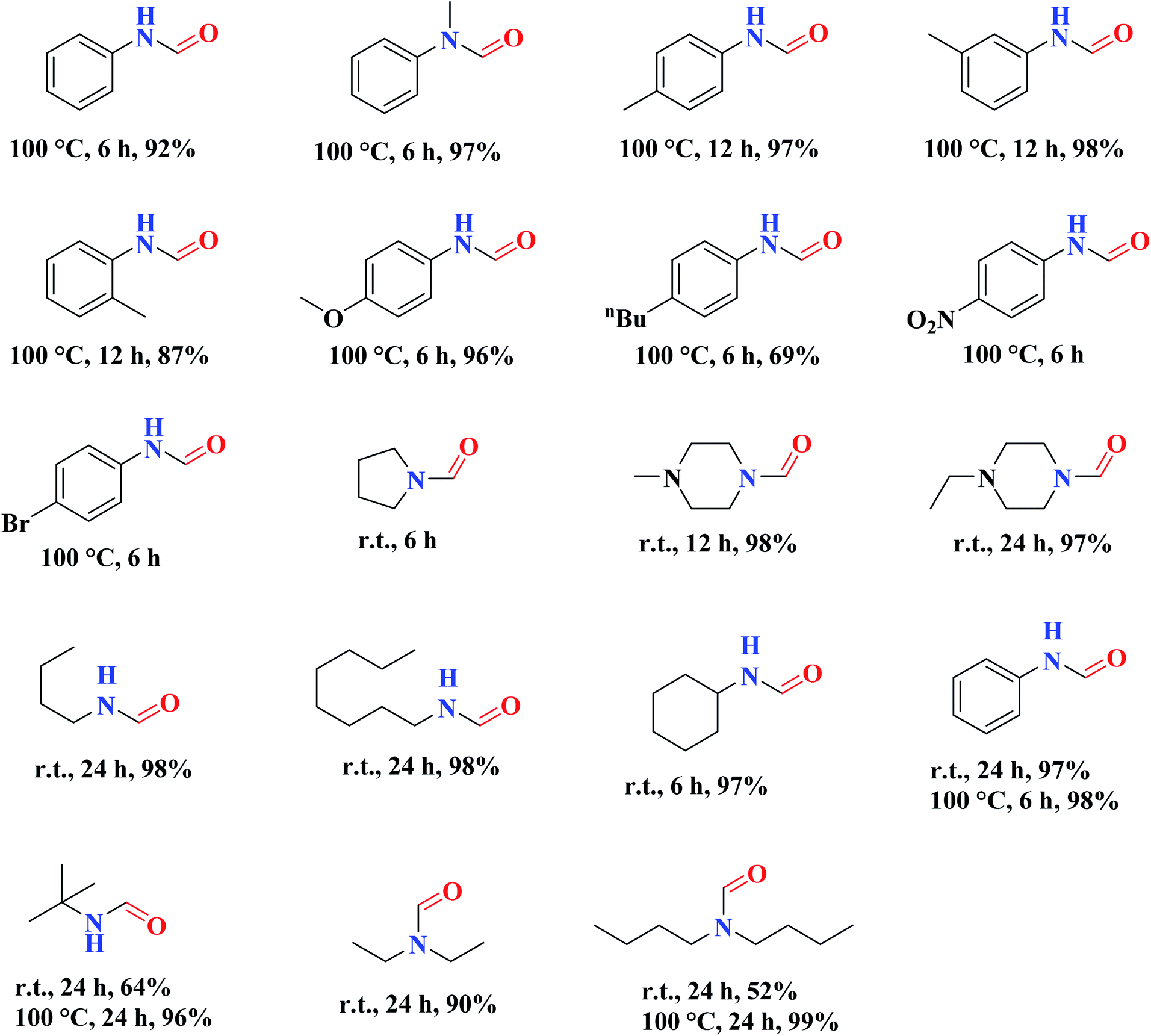
The use of nitrogen-doped graphene nanosheets, NG, as metal-free catalysts in the N-formylation of amines was described by Chen et al.188 NG catalysts were synthesized through direct calcination and activation of a mixture of urea, graphene nanosheets, and potassium carbonate, followed by leaching with HCl solution and washing with deionized H2O to eliminate residual K. A broad range of amines (aliphatic, aromatic and heterocyclic amines) were effectively transformed into the desired products in the attendance of NGU/K-900 (calcination at 900 °C) and CO2/PhSiH3 under mild conditions (Table 1). Also, the catalyst displayed outstanding reusability during 12 cycles with almost unchanged catalytic efficiency.
|
|---|
 |
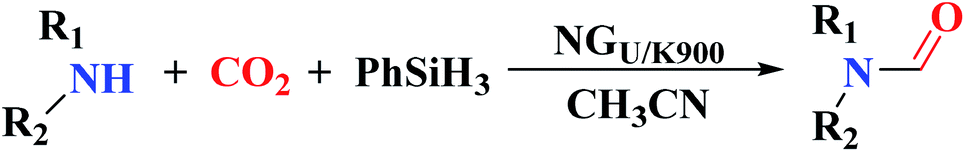
Very recently, another strategy for the preparation of a series of N-doped carbon nanotubes (NCNTs) was reported by Li et al.189 (Fig. 3). The resulting materials were synthesized using the pyrolysis of in situ polymerized pyrrole on the surface of CNTs under a N2 flow at different temperatures.
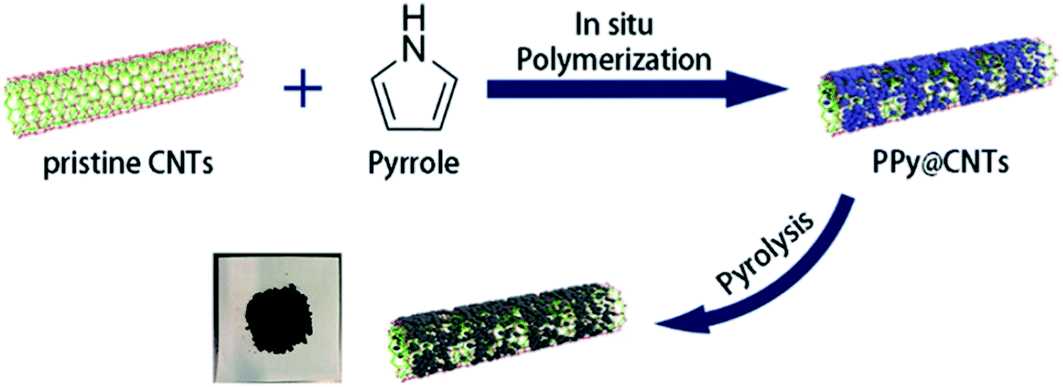 | ||
| Fig. 3 Synthesis of N-doped CNTs. Reprinted with permission from ref. 189. Copyright 2020 American Chemical Society. | ||
To improve the catalytic performance, the outside diameter of CNTs, carbonization temperature, and the concentration of pyrrole were examined. The characterization data exhibited that the optimal NCNTs-800 had a unique structure with similar contents of graphitic and pyrrolic N.
The catalytic efficiency of NCNTs-800 was assessed for the selective reduction of various nitroaromatics, affording excellent selectivity even in the attendance of a fragile iodo group (Scheme 3). In addition, the catalyst was used in seven successive runs with a negligible decrease in activity.
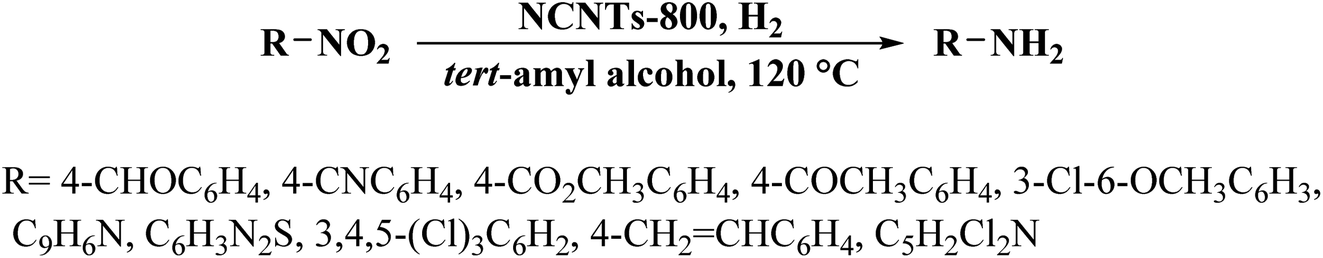 | ||
| Scheme 3 Reduction of different nitroarenes over NCNTs-800. | ||
In 2019, Hao and co-workers190 introduced a series of N-doped carbon nanotubes (CNTs) with diverse nitrogen amounts and species and defects as metal-free catalysts for the hydrogenation of nitrobenzene (Fig. 4). These catalysts were obtained via an in situ preparation and post nitridation treatment. The potential active centers of N-doped CNTs were examined in terms of DFT calculations of H adsorption on CNTs and N species as well as catalytic activity in the reduction of nitrobenzene. N-doped carbon nanotubes synthesized by post nitridation showed good performance, achieving about 60% conversion of nitrobenzene and around 90% selectivity for aniline. Also, the catalyst afforded stable product yield during six runs. Pyrrolic N species in the N-doped CNTs were found to be active sites for hydrogen activation using chemisorption and dissociation of molecules of hydrogen during the reaction.
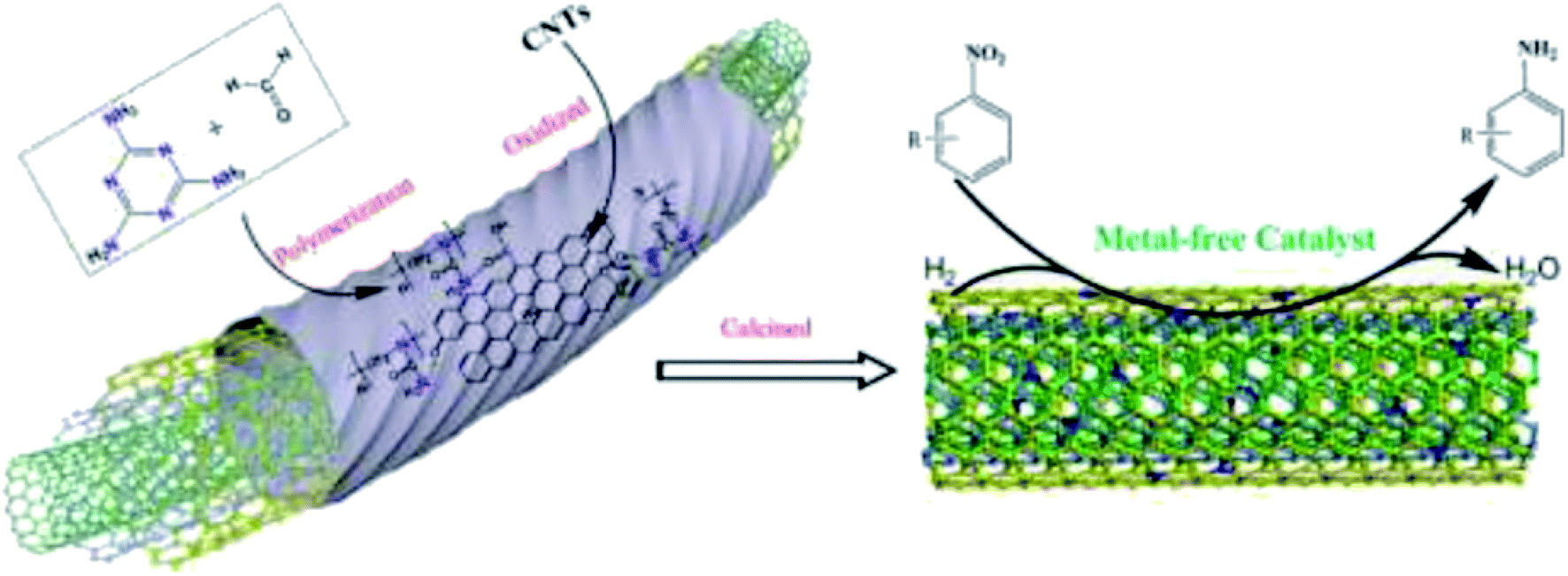 | ||
| Fig. 4 Preparation of N-CNTs and catalytic application in nitrobenzene hydrogenation. Reprinted with permission from ref. 190. Copyright 2020 Elsevier. | ||
Other metal-free N-CNTs were prepared with a pyrolysis process using protoporphyrin IX or melanin as the dopant by Li et al.16 (Fig. 5). Multiwalled carbon nanotubes with diverse diameters were used as the catalyst, and their efficiency was compared with carbon black and graphene. The amount of graphitic N was efficiently controlled by changing the heating rate during the carbonization process. The characterization data showed that the N heteroatoms remarkably destroyed the carbon skeleton of CNTs, resulting in a reduction in the crystallinity. The synthesized N-CNTs were assessed for the reduction of 4-nitrophenol to 4-aminophenol under 120 °C and 2 MPa H2 and N-CNTs with outer diameters of ≤20 nm fabricated using melanin at 800 °C indicated remarkable enhanced catalytic performance. The applicability and chemoselectivity of the optimal catalyst were further explored for a wide variety of substituted nitroarenes including electron-withdrawing and electron-donating and groups and products were afforded in high yield and selectivity (Scheme 4). The authors found a good correlation between the graphitic N amount of N-CNTs and catalytic performance. Moreover, recycling tests displayed that the catalyst was highly stable without the loss of its catalytic activity after six times.
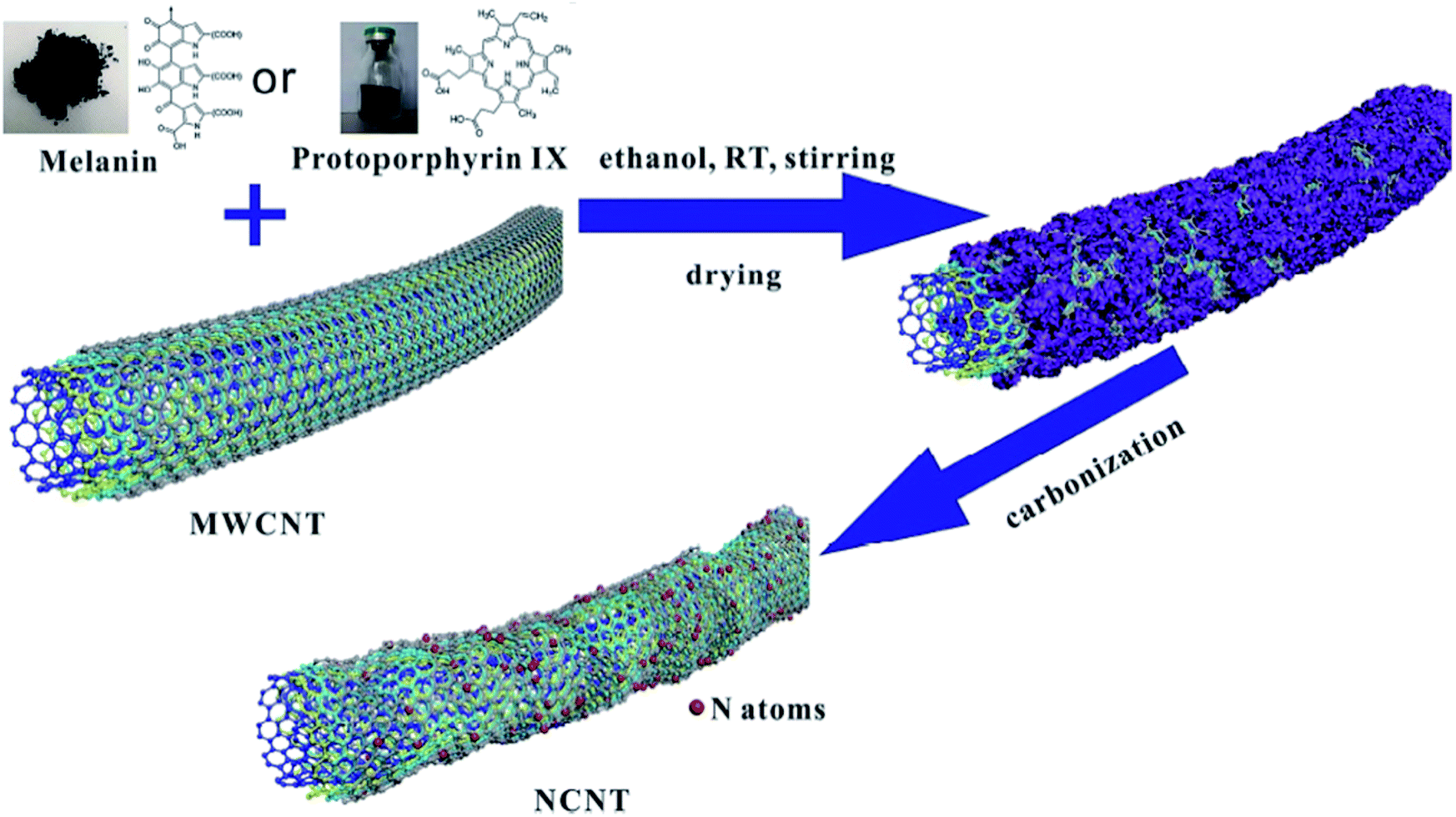 | ||
| Fig. 5 Preparation of NCNTs. Reprinted with permission from ref. 16. Copyright 2019 Elsevier. | ||
 | ||
| Scheme 4 Chemoselective reduction of different nitroarenes with m-NCNTs. | ||
Xi and co-workers191 described a simple one-pot hydrothermal synthesis procedure for the preparation of an effective nitrogen-doped holey graphene (NHG), in which graphene oxide was served as a starting material, NH3 as a nitrogen source, and hydrogen peroxide as etching agent (Fig. 6). During the formation of this carbocatalyst, several reactions could happen. GO nanosheets were self-assembled, reduced, chemically etched, and simultaneously doped with N.
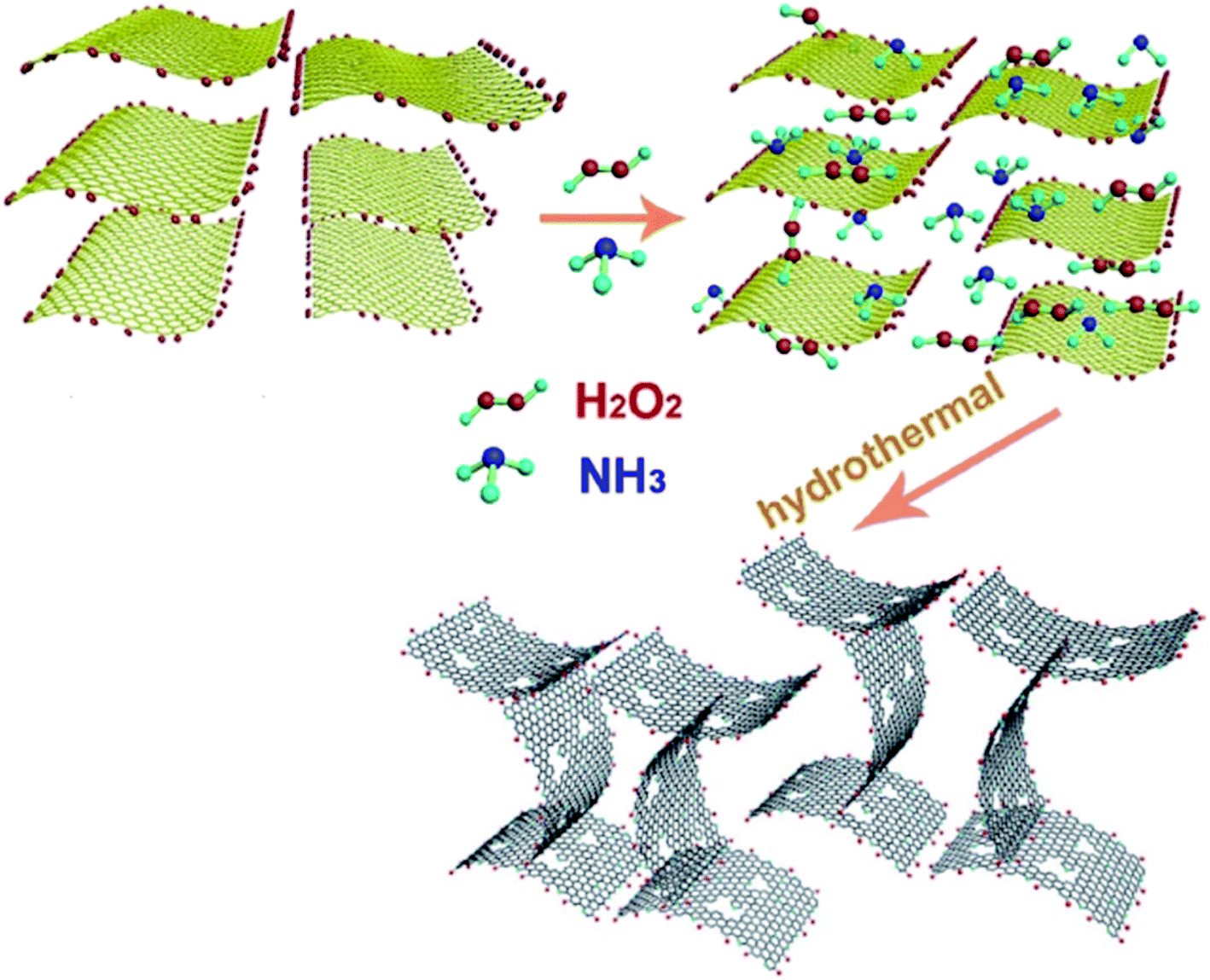 | ||
| Fig. 6 Preparation of NHG. Reprinted with permission from ref. 191. Copyright 2019 Elsevier. | ||
This carbocatalyst with high N loading, unique hierarchical porous structure, large specific surface area, and good hydrophilicity exhibited an excellent catalytic activity for the reduction of five organic dyes (MB, RhB, CR, MO, and 4-NP) in the presence of NaBH4 as a reductant in aqueous solution at ambient temperature. The authors also extended the application of the NHG to the hydrogenation of different substituted nitrobenzenes to study its applicability. These reactions were performed using NaBH4 in H2O/EtOH mixture at ambient temperature and provided the corresponding anilines in superb yields (Scheme 5). In 4-NP reduction, the catalyst delivered a TOF higher than those of the many noble-metal-based catalysts, commercial Pd/C, and other previously reported carbocatalysts.
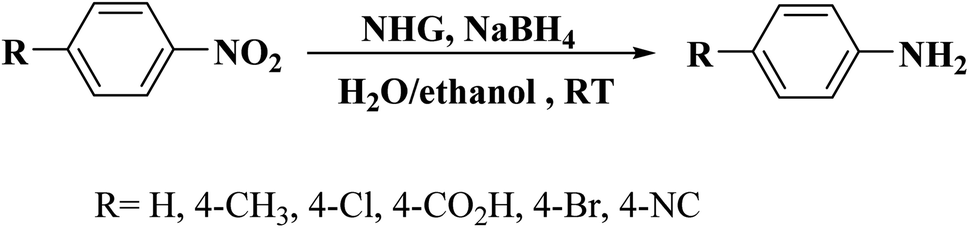 | ||
| Scheme 5 Hydrogenation of nitrobenzenes over NHG. | ||
Yuan et al.192 reported sol–gel routes to synthesize two N-doped porous carbon materials (NC-1 and NC-2) using sucrose and citric acid as carbon precursors and in situ-formed aluminophosphate framework as a template (1 and 2 are the P/Al ratios in the starting CA/AlPO composite). These materials acted as highly effective metal-free catalysts for the hydrogenation of nitrobenzene in an aqueous solution using sulfide and showed much superior catalytic efficiency in comparison with graphite oxide or carbon black catalysts reported in the literature. In addition, NC-2 displayed better removal performance compared with NC-1 material. Also, Both NC-1 and NC-2 were reused for more than five cycles. The presence of numerous nitrogen- and oxygen-containing functional groups on the surface of the N-doped carbon improved their catalytic activity. Furthermore, other features of these catalysts such as tunable surface polarity, enhanced electron transfer ability, and large surface area, were key parameters in increasing their catalytic performance.
In 2017, a well-defined 3-D N-doped graphene foam (3D-NGF) was reported by Liu et al.193 For the preparation of 3D-NGF, the achieved graphene oxide was spread in H2O by sonication and frozen with liquid N. In the following, the resultant material was processed in a vacuum freeze-drying apparatus for 2 days to prepare GO foam. Finally, the GO form was treated with the mixture gas of 50% NH3 and 50% Ar flowing at 750 °C for 2 h. The 3D-NGF demonstrated high catalytic activity and excellent durability toward the reduction of p-nitrophenol to p-aminophenol because of the 3D foam-like structure as well as the synergistic effect between the nitrogen-doping, leading to change in electronic property. Also, the 3D structure can efficiently inhibit the agglomeration of the graphene, and thus provides an effective interface for the hydrogenation to take place.
Dai and colleagues194 developed a hydrothermal strategy for the fabrication of N-doped reduced graphene oxide meshes from pristine GO sheets in the presence of NH4OH as a nitrogen source at elevated temperature (Fig. 7). The N-RGO meshes functioned as an active catalyst to reduce 4-nitrophenol.
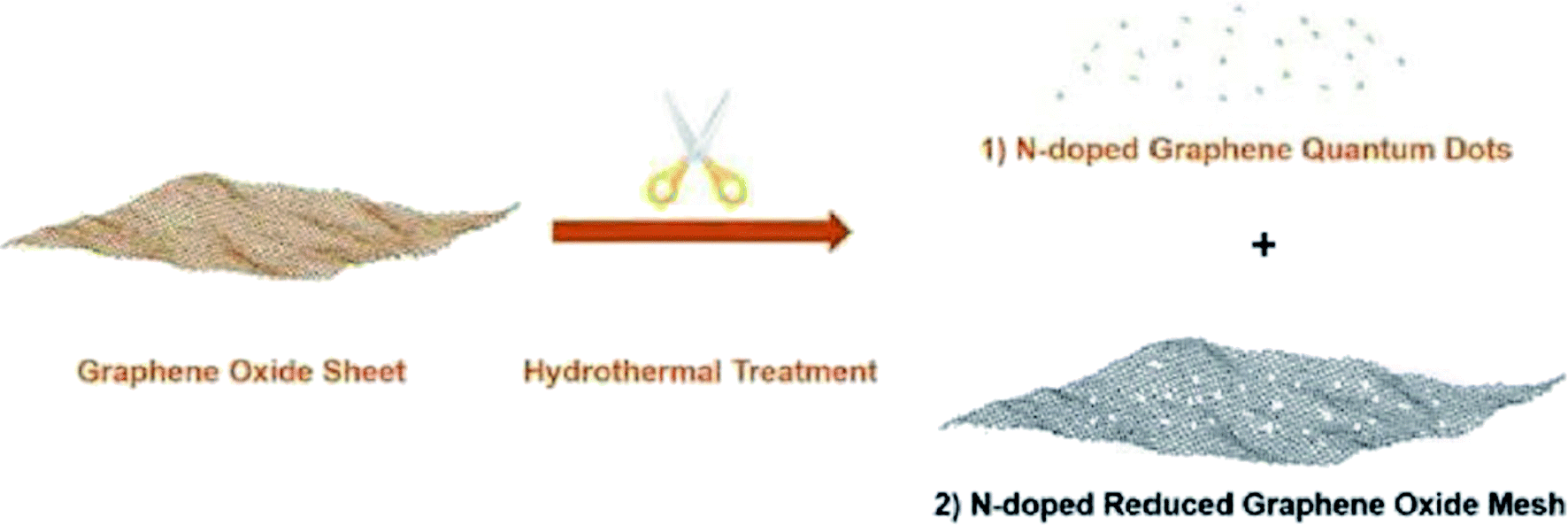 | ||
| Fig. 7 Preparation of N-GQDs and N-RGO mesh. Reprinted with permission from ref. 194. Copyright 2018 John Wiley and Sons. | ||
These N-RGO meshes demonstrated new catalytic behaviors of dramatically increased catalytic efficiency, adjustable catalytic kinetics and reusability, and strikingly reduced activation energy. Such catalytic features were attributed to the synergetic effects between graphitic N atoms and structural defects resulting from the selective etching of nanopores. Also, the N-RGO meshes were capable to conduct reactions in continuous flow.
In 2017, Wang and Chen195 used the N-doped carbon (N-C) catalytic system derived from ZIF-8 as a precursor and sacrificial agent for the reduction of 4-nitrophenol to 4-aminophenol (Fig. 8). Compared to some of the previously metal catalysts, N-C showed good performance, high durability, recyclability, and minimal pollution. The N-C catalysts synthesized through carbonization at various temperatures possessed diverse catalytic performance as well as different N amounts. Among them, N-C800 with the highest pyridine nitrogen amount displayed superior catalytic performance.
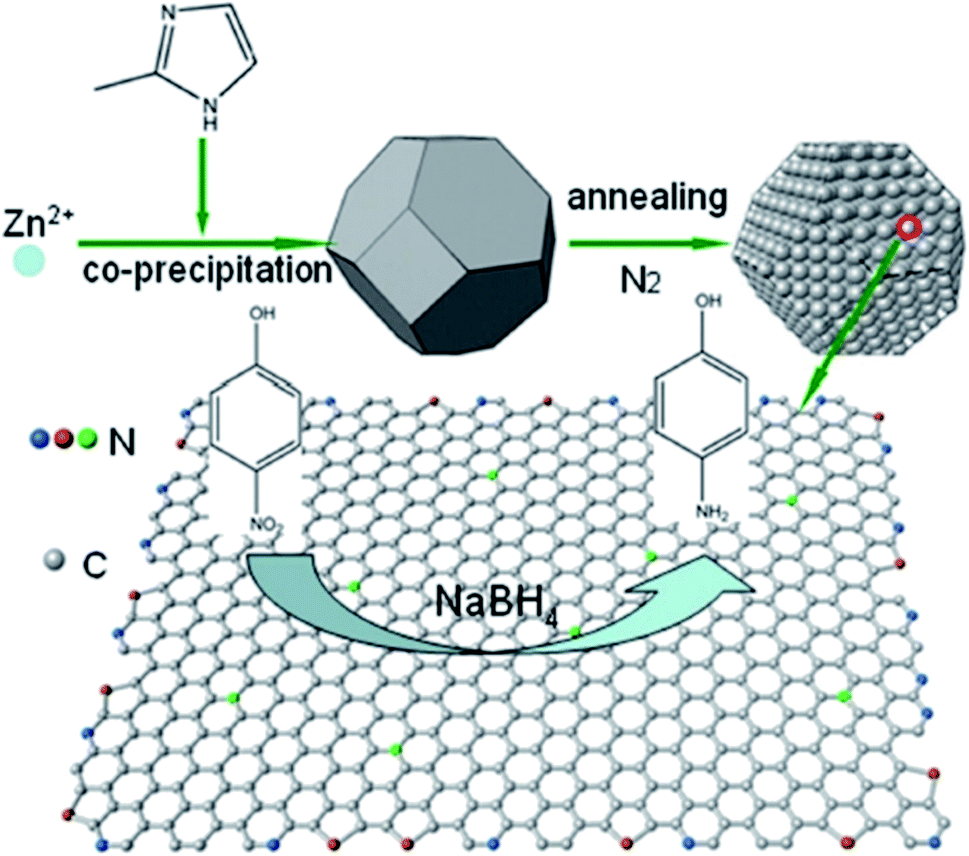 | ||
| Fig. 8 Preparation of N-C for the reduction of 4-nitrophenol. Reprinted with permission from ref. 195. Copyright 2018 John Wiley and Sons. | ||
In 2017, Wang and co-workers196 developed a new protocol to prepare sulfurized graphene (SG) nanomaterials through a ball-milling approach. The activity of SG with a uniform dispersion of S and flake-like morphology was tested in the hydrogenation of p-nitrophenol to p-aminophenol in the attendance of sodium borohydride as the reductant (Scheme 6). The catalytic mechanism of SG was examined by density functional theory (DFT). The effects of the kinetic parameters including reaction temperatures, catalyst amounts, initial reducer concentrations, and various initial p-NP concentrations were investigated. Moreover, the thermodynamic factors such as activation entropy and enthalpy were calculated.
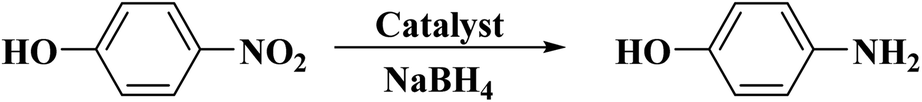 | ||
| Scheme 6 Hydrogenation of p-nitrophenol using SG. | ||
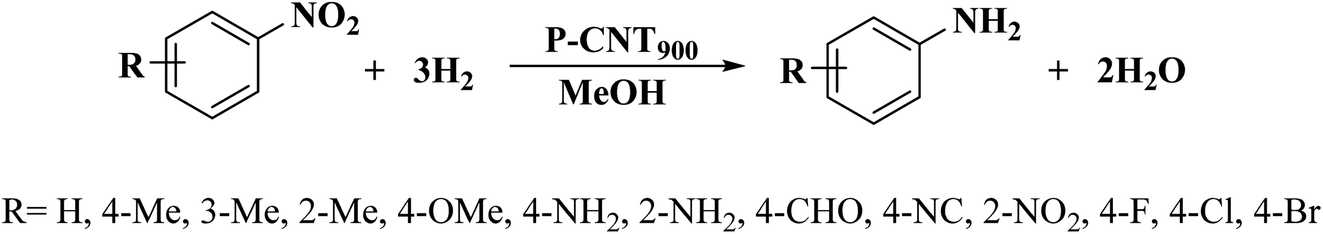
The use of phosphorus-doped carbon nanotubes (P-CNT) as a catalyst in the reduction of nitrobenzene to aniline using H2 as a reducer was described by Chen and co-workers197 in 2019.
Furthermore, diversely functionalized nitroarenes with broad industrial interests were successfully converted into relevant products with superb yields and selectivities (Scheme 7). P-CNT was synthesized by pyrolysis a mixture of CNT, Ph4PCl, and K2CO3 at 900 °C under N2 atmosphere for 2.0 h. Besides, recycling tests of the P-CNT displayed that the catalyst was highly durable and maintained its activity and selectivity for up to eight runs. In addition to reduction, catalytic transfer hydrogenation of aniline was performed with P-CNT by applying various hydrogen sources such as formic acid/triethyl amine (HCOOH/Et3N), carbon monoxide/water (CO/H2O), and hydrazine hydrate (N2H4·H2O).
 | ||
| Scheme 7 Reduction of nitrobenzenes catalyzed by P-CNT. | ||
In another study, polymerization and carbonization of phytic acid were described by Zou and co-workers198 to fabricate carbon catalyst with an adjustable concentration of P-dopant and lattice defect (Fig. 9). The as-prepared catalyst (PV-900) exhibited excellent performance, outstanding selectivity, and durability in the reduction of nitroarene compounds (Scheme 8), much higher than nickel, metal-oxide, and carbon-based catalysts which were recently reported. DFT calculations indicated that the combination of lattice defect and P-dopant in carbon can change the band structure as a metal-like and cause remarkable electron delocalization, and thus easily activate both nitro group and H2. Additionally, the reduction efficiency was linearly dependent on the defect concentration and P-doping.
 | ||
| Fig. 9 Preparation of PV-carbon catalyst. Reprinted with permission from ref. 198. Copyright 2017 John Wiley and Sons. | ||
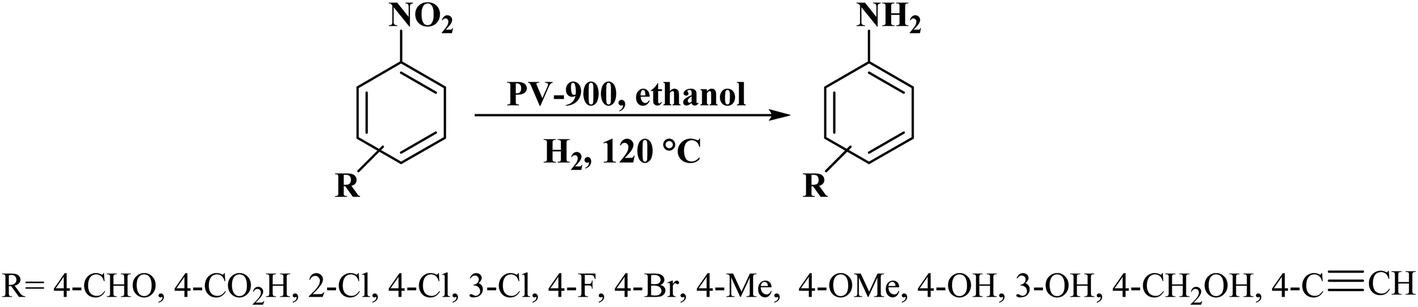 | ||
| Scheme 8 Reduction of nitroarene compounds catalyzed by PV-900. | ||
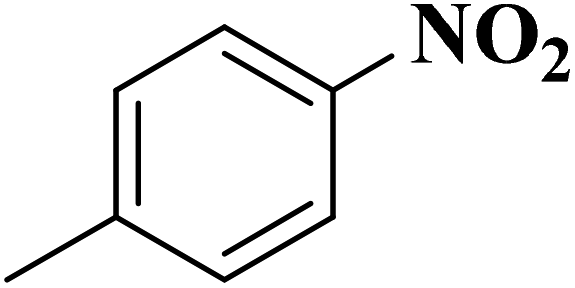
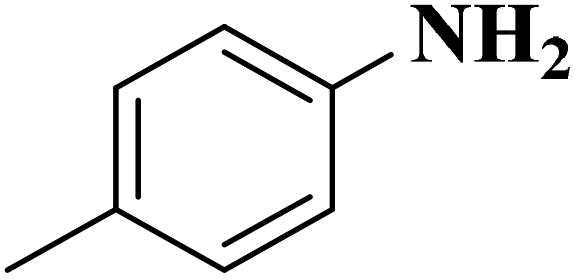
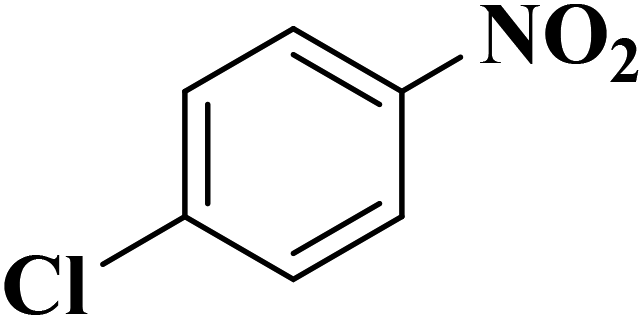
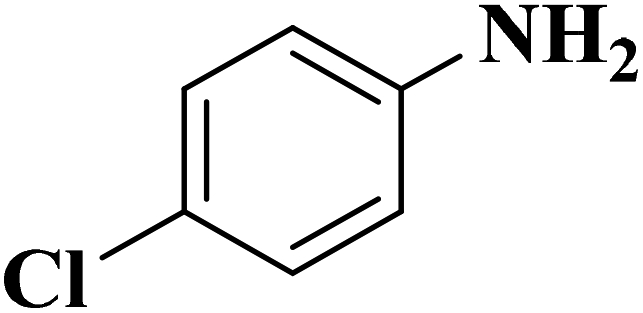
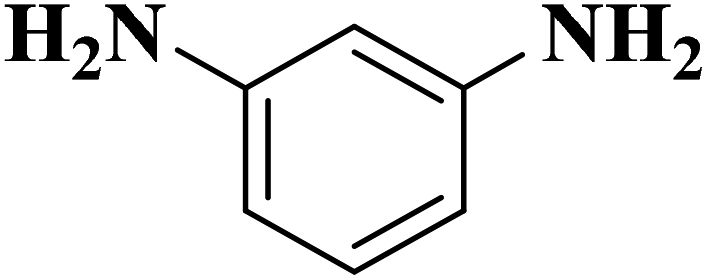
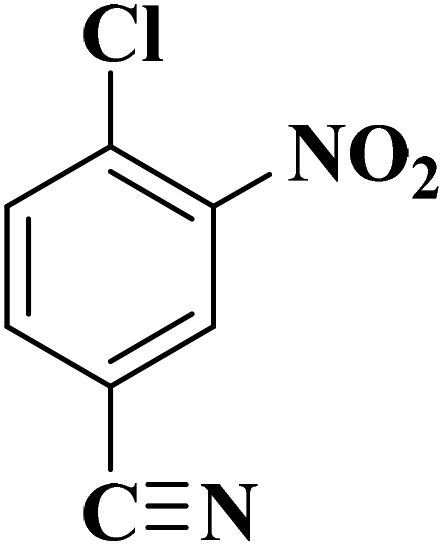
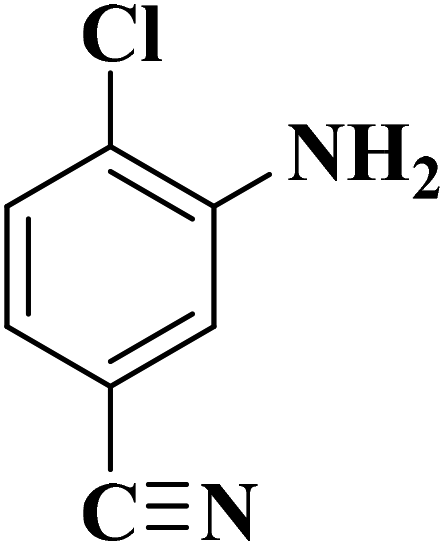
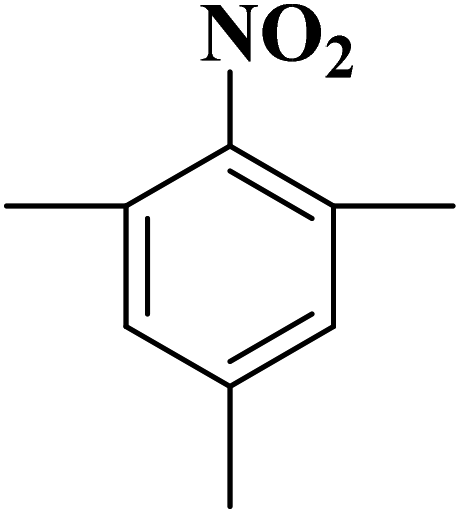
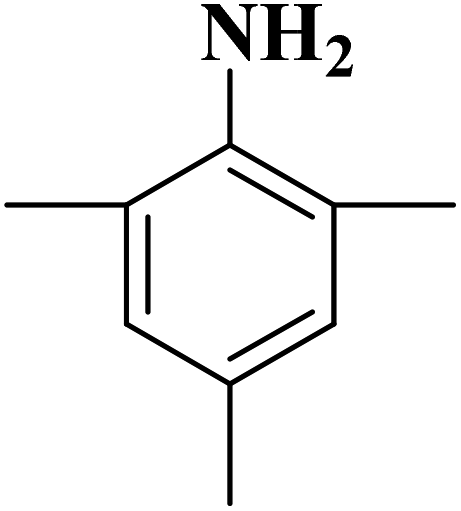
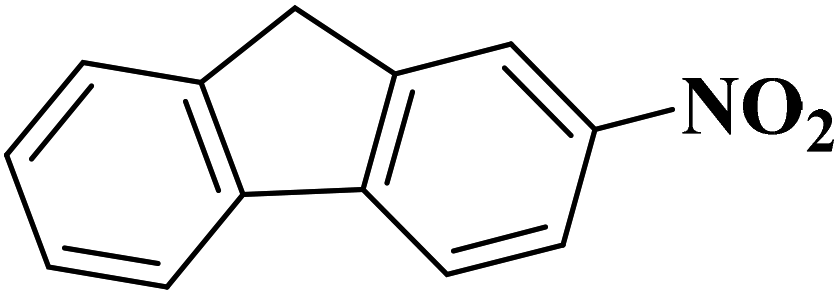
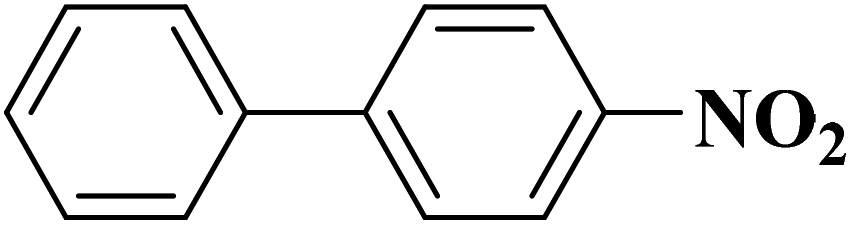
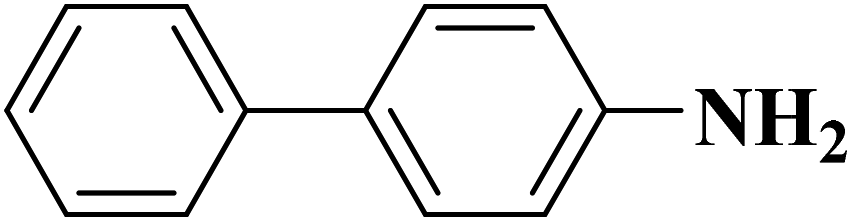
Su et al.199 synthesized two kinds of boron-doped carbon materials (carbon nanotubes (B-CNTs) and onion-like carbon (B-OLC)) through high-temperature carbonization of acid-treated CNTs or nanodiamond and boron acid as B precursor. The catalytic performances of the catalysts were surveyed in the hydrogenation of nitroarenes (Table 2) and compared with pristine CNTs and OLC samples. The results showed that substitutional boron species BC3 played an important role in increasing the catalytic activity and efficient application of hydrazine hydrate.
| Substrate | Product | Catalyst | Con. (%) | Sel. (%) |
|---|---|---|---|---|
 |
 |
B-OLC-2 | >99 | 96.5 |
| B-CNTs-2 | >99 | 97.3 | ||
 |
 |
B-OLC-2 | >99 | 94.5 |
| B-CNTs-2 | >99 | 96.3 | ||
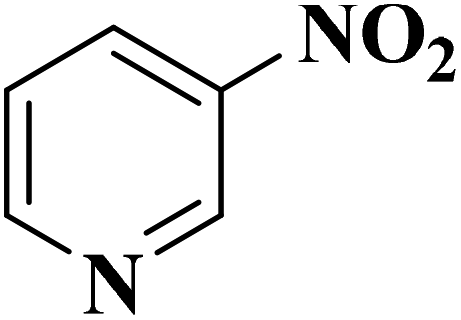 |
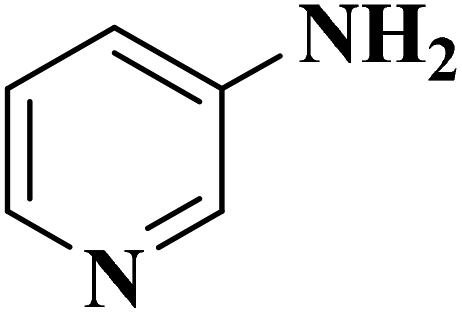 |
B-OLC-2 | >99 | 93.0 |
| B-CNTs-2 | >99 | 94.9 | ||
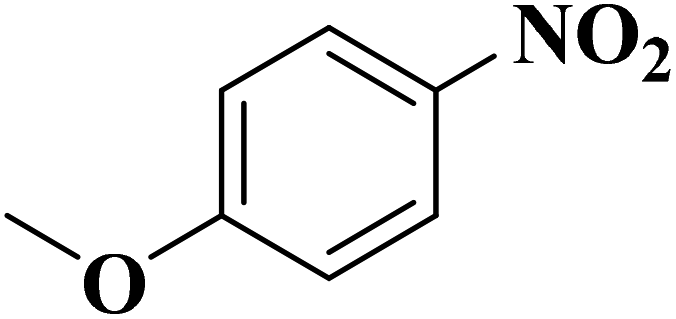 |
 |
B-OLC-2 | >99 | 96.2 |
| B-CNTs-2 | >99 | 97.1 | ||
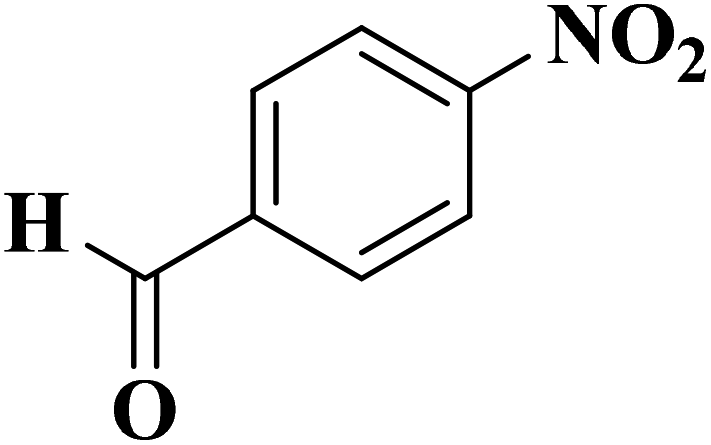 |
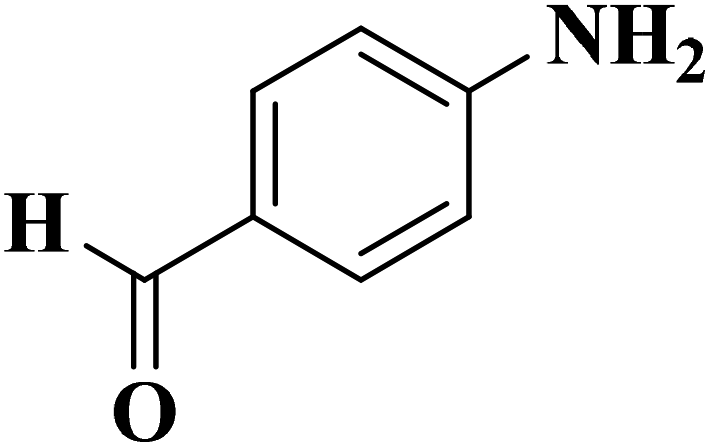 |
B-OLC-2 | >99 | 96.8 |
| B-CNTs-2 | >99 | 97.4 | ||
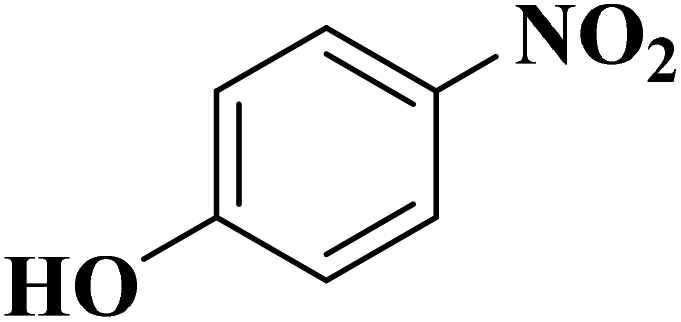 |
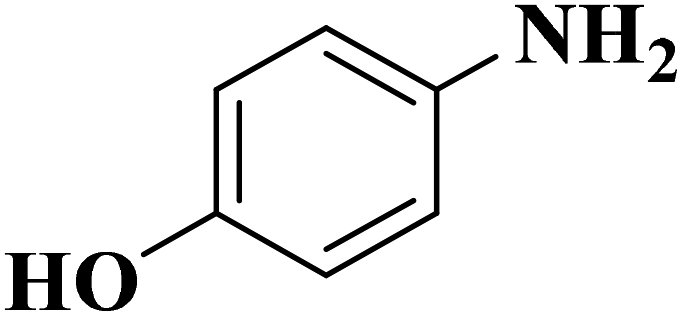 |
B-OLC-2 | >99 | 95.4 |
| B-CNTs-2 | >99 | 93.9 | ||
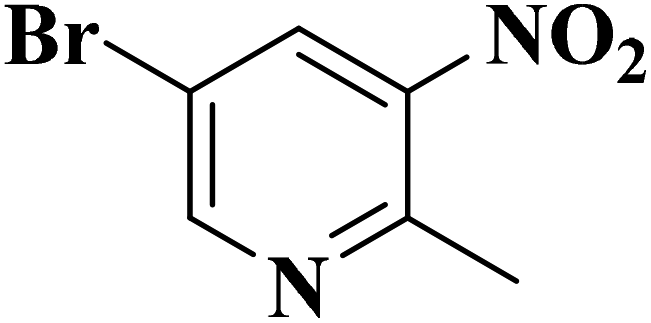 |
 |
B-OLC-2 | >99 | 93.2 |
| B-CNTs-2 | >99 | 92.9 | ||
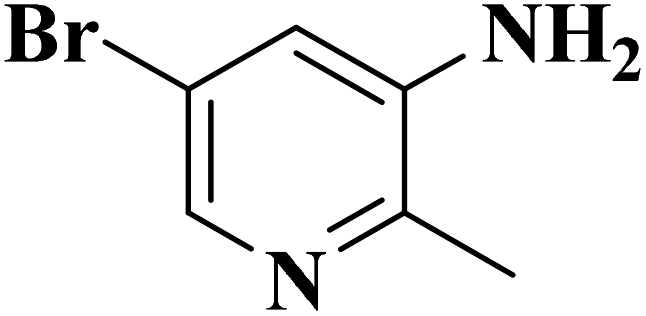
 |
 |
B-OLC-2 | >99 | 8902 |
| B-CNTs-2 | >99 | 90.3 | ||
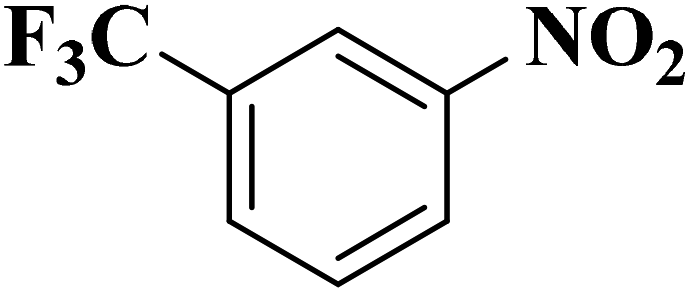 |
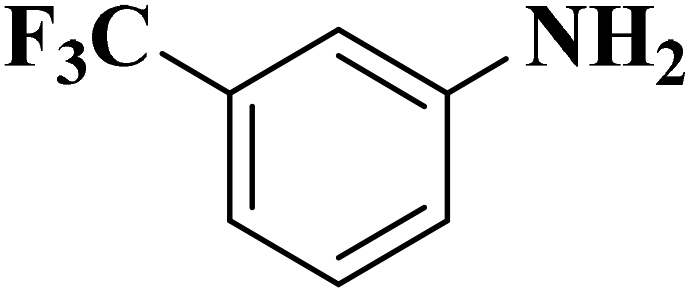 |
B-OLC-2 | >99 | 94.2 |
| B-CNTs-2 | >99 | 93.1 | ||
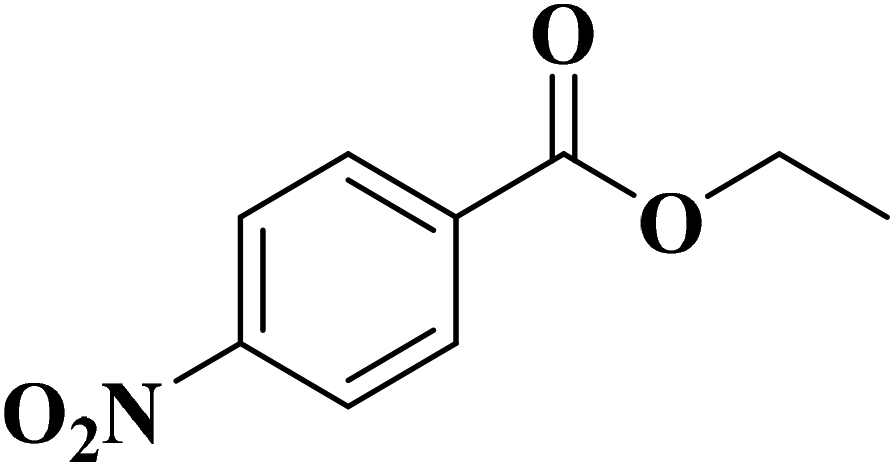 |
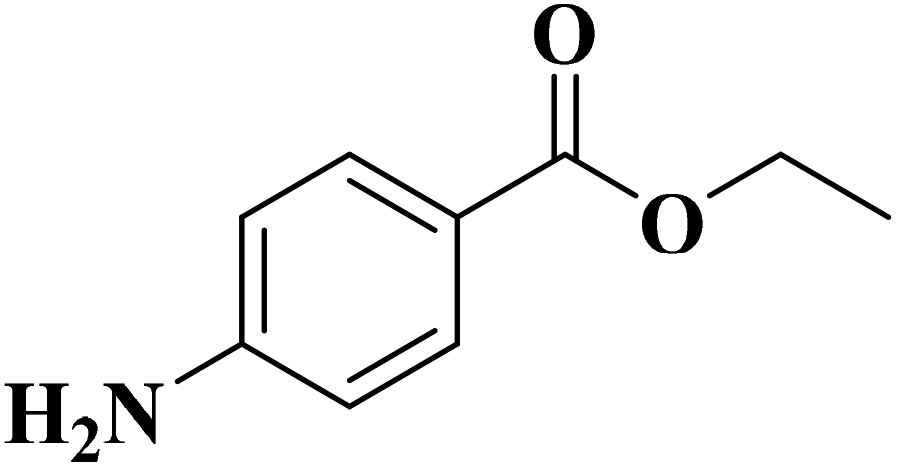 |
B-OLC-2 | >99 | 89.2 |
| B-CNTs-2 | >99 | 90.3 | ||
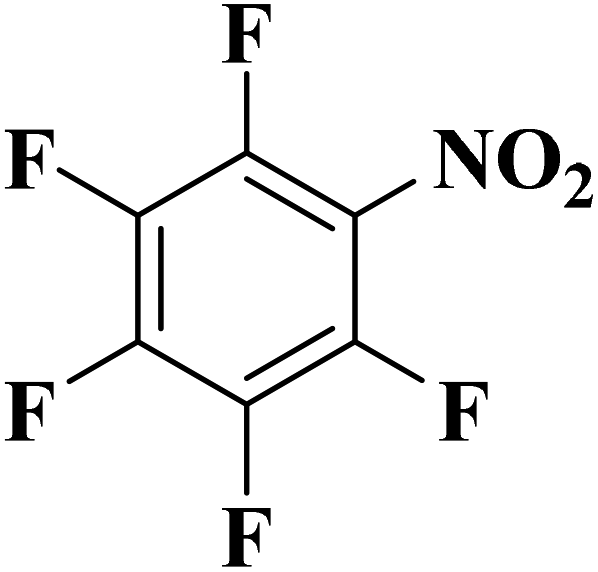 |
 |
B-OLC-2 | >99 | 91.9 |
| B-CNTs-2 | >99 | 90.2 | ||
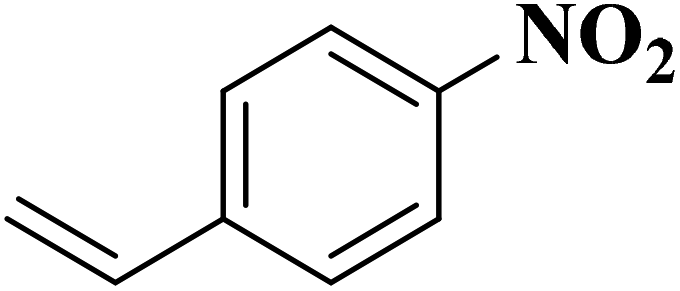 |
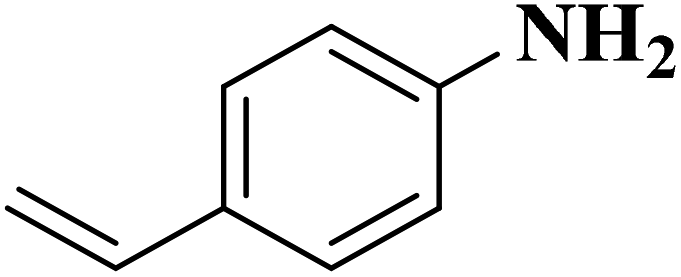 |
B-OLC-2 | >99 | 88.4 |
| B-CNTs-2 | >99 | 90.9 | ||
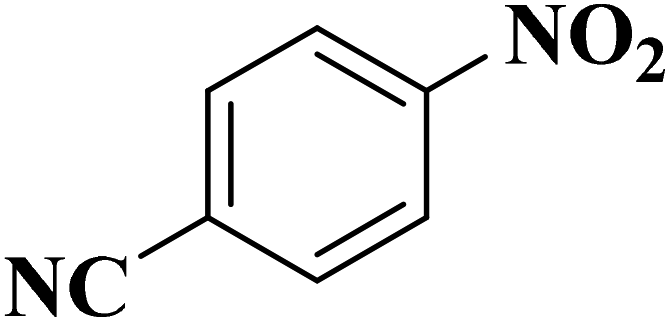 |
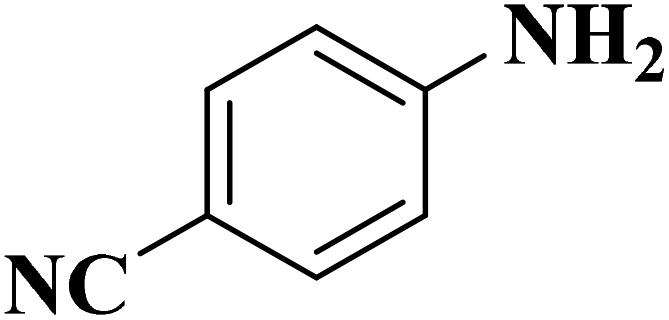 |
B-OLC-2 | >99 | 92.2 |
| B-CNTs-2 | >99 | 91.2 | ||
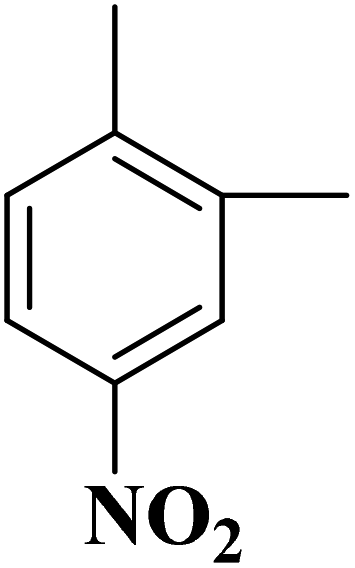 |
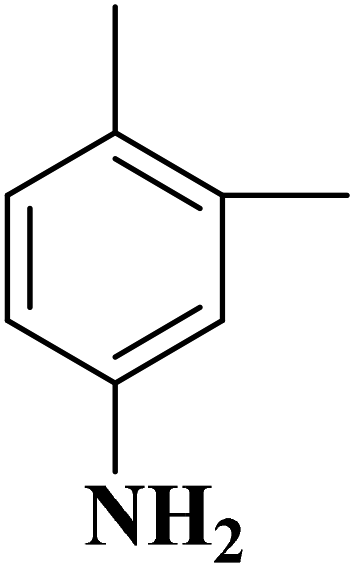 |
B-OLC-2 | >99 | 96.9 |
| B-CNTs-2 | >99 | 97.1 | ||
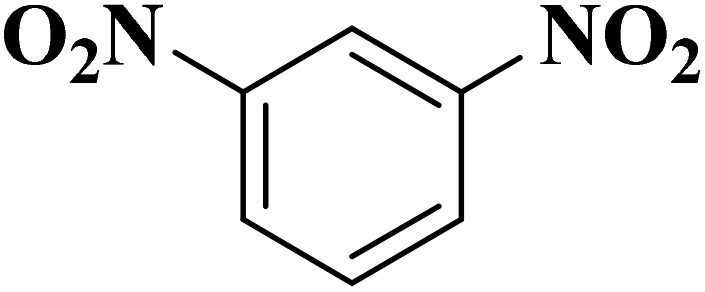 |
 |
B-OLC-2 | >99 | 96.7 |
| B-CNTs-2 | >99 | 97.1 | ||
 |
 |
B-OLC-2 | >99 | 90.2 |
| B-CNTs-2 | >99 | 90.6 | ||
 |
 |
B-OLC-2 | >99 | 96.1 |
| B-CNTs-2 | >99 | 95.3 | ||
 |
 |
B-OLC-2 | >99 | 97.2 |
| B-CNTs-2 | >99 | 97.0 | ||
 |
 |
B-OLC-2 | >99 | 97.9 |
| B-CNTs-2 | >99 | 98.0 |
Song and colleagues200 developed a “solid dual-ions-transformation reaction” strategy to prepare environmentally friendly S,N co-doped hollow carbon nanotubes (Fig. 10). The procedure used for the fabrication of S,N co-doped hollow carbon nanotubes involved three steps: the preparation of CdS nanowires as both source of sulfur and hard template; in situ self-polymerization of dopamine on the surface of CdS (PDA acted as the sources of N and C as well as the reductant); and carbonization. Sulfur and nitrogen amount in the final materials could be simply adjusted by altering the carbonization temperature. The catalytic activity of S,N co-doped carbon nanotubes was evaluated in the hydrogenation of 4-aminophenol at ambient temperature using NaBH4.
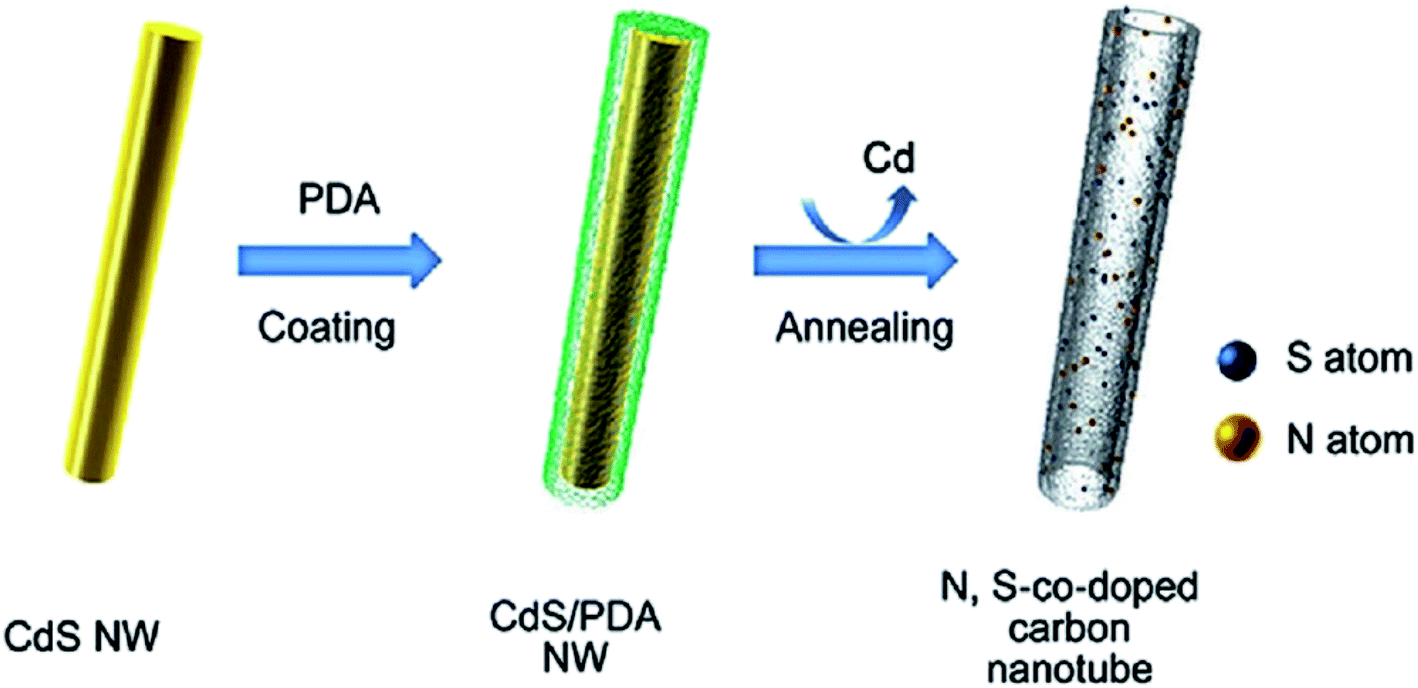 | ||
| Fig. 10 Preparation of S,N co-doped hollow carbon nanotubes. Reprinted with permission from ref. 200 Copyright 2016 John Wiley and Sons. | ||
In 2020, a convenient two-steps method to fabricate polymer-derived N,S co-doped carbons (PDNSC-X) was offered by Long et al.201 The starting material was prepared from polymerization of trithiocyanuric acid, glyoxal, and melamine. Then, the pyrolysis of synthesized polymer containing numerous S and N active species as a precursor at four different temperatures (600, 700, 800, and 900 °C) under N2 atmosphere afforded PDNSC-X materials (Fig. 11). PDNSC-800 displayed outstanding catalytic performance and high selectivity toward the hydrogenation of a broad variety of functionalized nitrobenzenes (Scheme 9) as well as the selective oxidation of ethylbenzene duo to the abundant N- and S-containing active sites, a hierarchically porous structure, synergistic effects of a high surface area, and defect formation. In addition, the PDNSC-800 catalyst exhibited high durability and reusability in both reactions even after eight runs. Also, this study clarified the relationship between the different doped atoms in N,S co-doped catalysts and superb selectivity in non-metallic catalytic synthesis.
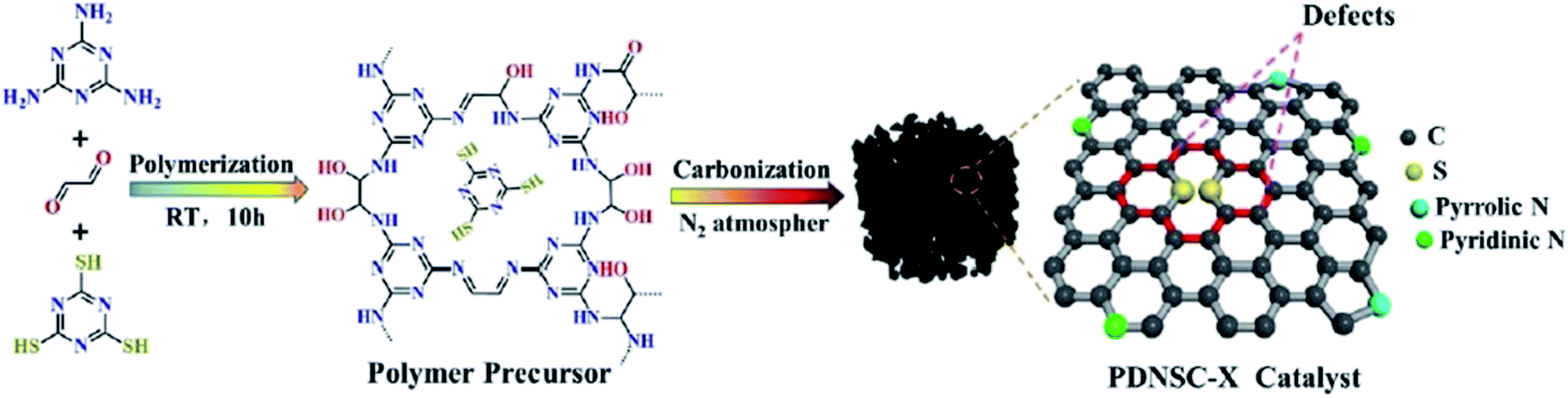 | ||
| Fig. 11 Preparation of PDNSC-X catalyst. Reprinted with permission from ref. 201. Copyright 2020 Royal Society of Chemistry. | ||
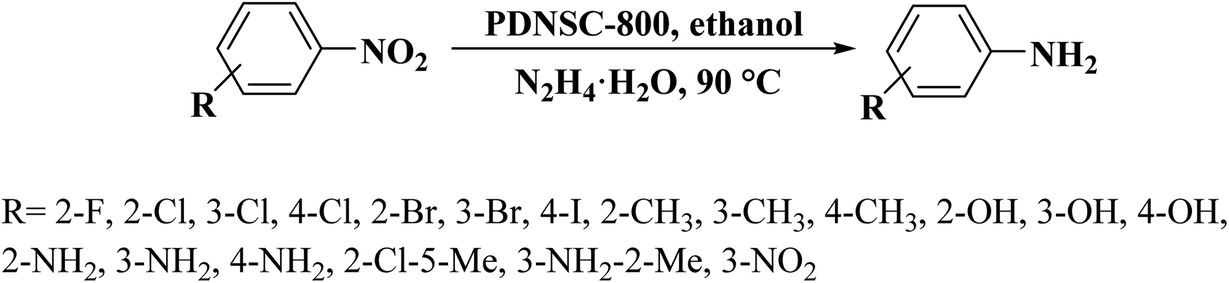 | ||
| Scheme 9 Hydrogenation of nitrobenzenes using PDNSC-800. | ||
In 2018, Li et al.202 employed ball-milling and thermal methods for the preparation of B and N co-doped graphene-like carbon (BNG) materials (Fig. 12). To prepare BNG catalysts, initially, the edge-selectively functionalized graphene nanoplatelets (EFGnPs) were synthesized using ball milling of the pure graphite, and then the obtained EFGnPs and boric acid were pyrolyzed in various temperatures. The catalytic performance of these materials was tested in the reduction of 4-chloronitrobenzene to 4-chloroaniline and among them, BNG-800 was the best catalyst (was much more active than other catalysts). Authors found that the type of B species possessed a great influence on the reaction and the BNG bearing the most B/N showed the best catalytic efficiency, demonstrating that the doped B/N in BNG was one of the important active centers in promoting this reaction.
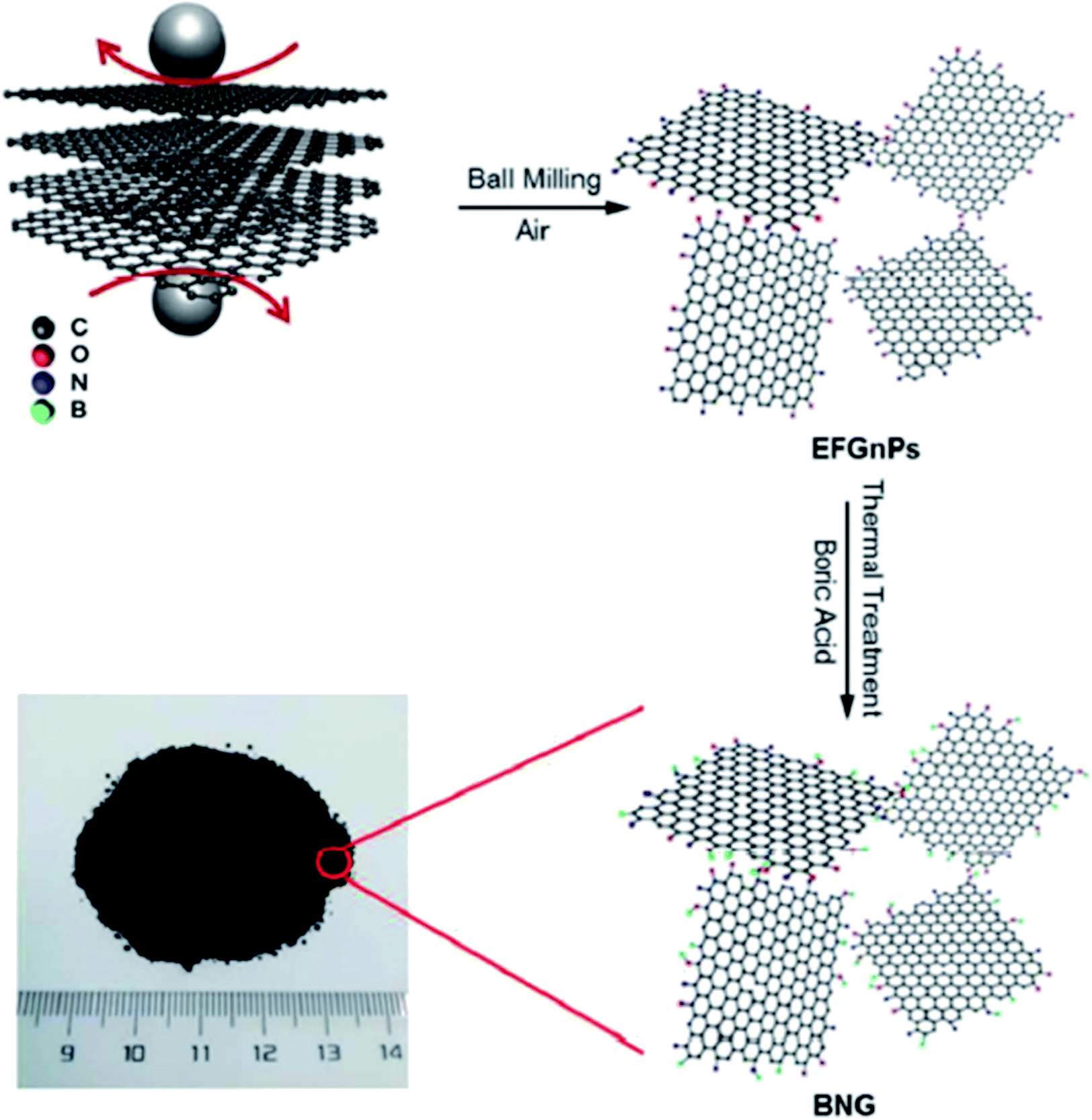 | ||
| Fig. 12 Preparation of BNG. Reprinted from ref. 202. Royal Society of Chemistry. | ||
The scope of the reaction was explored for a broad range of functionalized nitroarenes; high levels of yield were observed for the corresponding anilines under the optimized reaction conditions (Scheme 10).
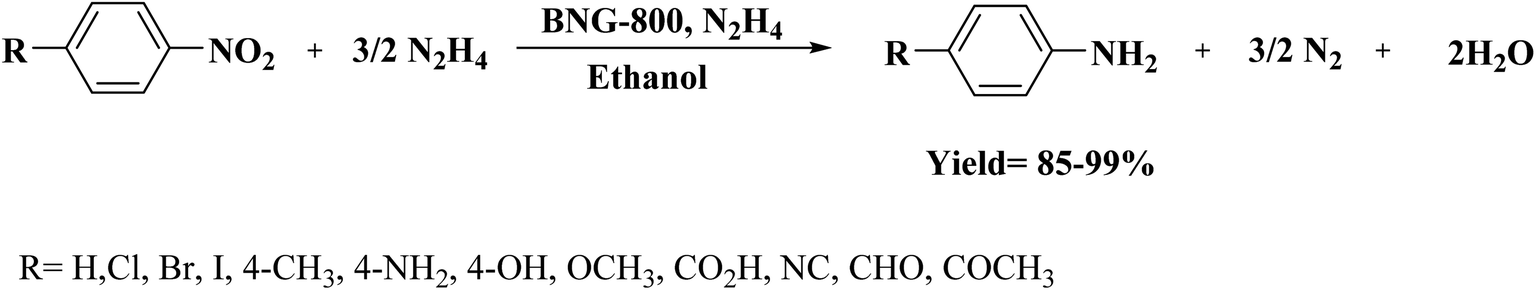 | ||
| Scheme 10 Hydrogenation of nitroarenes catalyzed by BNG-800. | ||
Also, the BNG-800 could be easily and effectively reused for six successive cycles without the formation of dehalogenation products for halogen-substituted nitroarenes.
In 2019, Chung and co-workers203 reported the synthesis of three-dimensional boron and nitrogen heteroatom-doped porous carbons (3DBNPCs) through carbonization of the mixture of nitrogen-doped porous carbons (NPC) (derived from ZIF-8) and boric acid at different temperatures (Fig. 13). As fabricated B-NPC-1200 exhibited superior catalytic prowess in the hydrogenation of p-NP (Scheme 11) with a high reaction rate constant and low activation energy. The doping of boron atoms into nitrogen-doped 3D porous carbon considerably improved the electrical conductivity and the catalytic activity of B-NPC-1200. Also, the synergistic effects between oxygen, boron, and nitrogen atoms could create the strongly active sites that were responsible for the lowering of the activation energy in the structure of the catalyst.
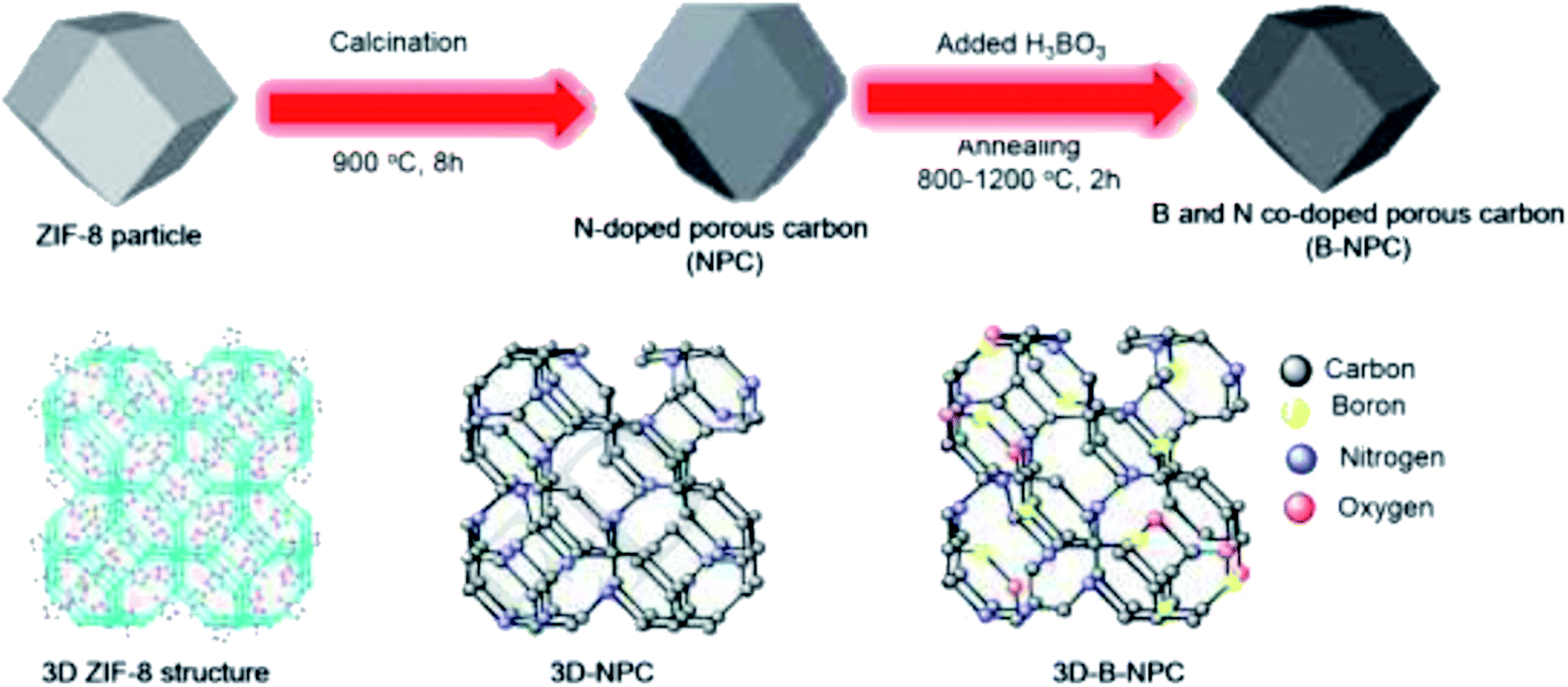 | ||
| Fig. 13 Preparation of B-NPC. Reprinted with permission from ref. 203. Copyright 2019 Elsevier. | ||
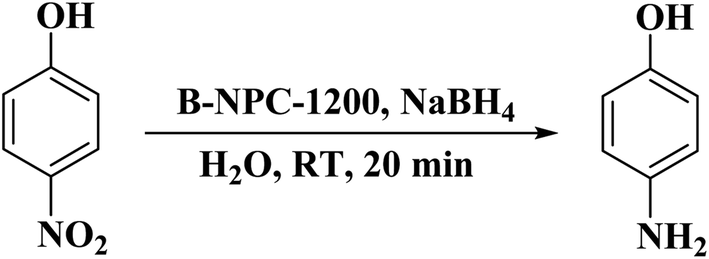 | ||
| Scheme 11 Reduction of p-NP catalyzed by B-NPC-1200. | ||
The B-NPC-1200 was further applied for the reduction of various nitro aromatics. Besides, the catalyst could be reused in five successive cycles without an obvious decrease in its activity. The authors suggested that the content and type of boron precursor can influence the structure of catalyst including properties and morphologies.
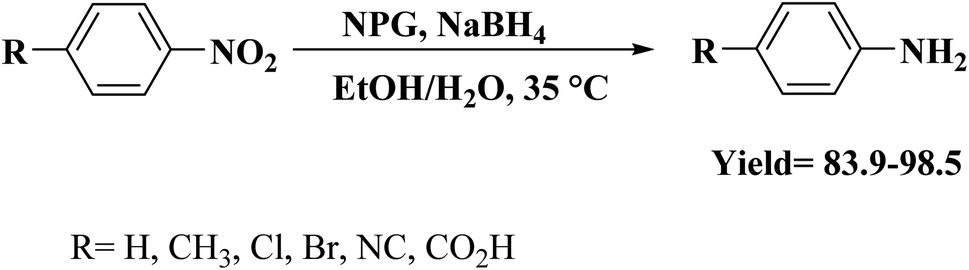
In 2018, Xi and coworkers204 described an effective and facile route to prepare N,P-dual-doped graphene (NPG) carbocatalyst. The synthesis process consisted of oxidation of graphite powders, microwave exfoliation of graphite oxide, and finally, annealing of microwave-exfoliated graphite (MEG) and hexachlorocyclotriphosphazene (HCCP) mixture under an inert atmosphere at 700 °C (Fig. 14). HCCP functioned as both P and N sources. NPG showed excellent efficiency, selectivity, durability, and good reusability toward hydrogenation of 4-NP in aqueous media.
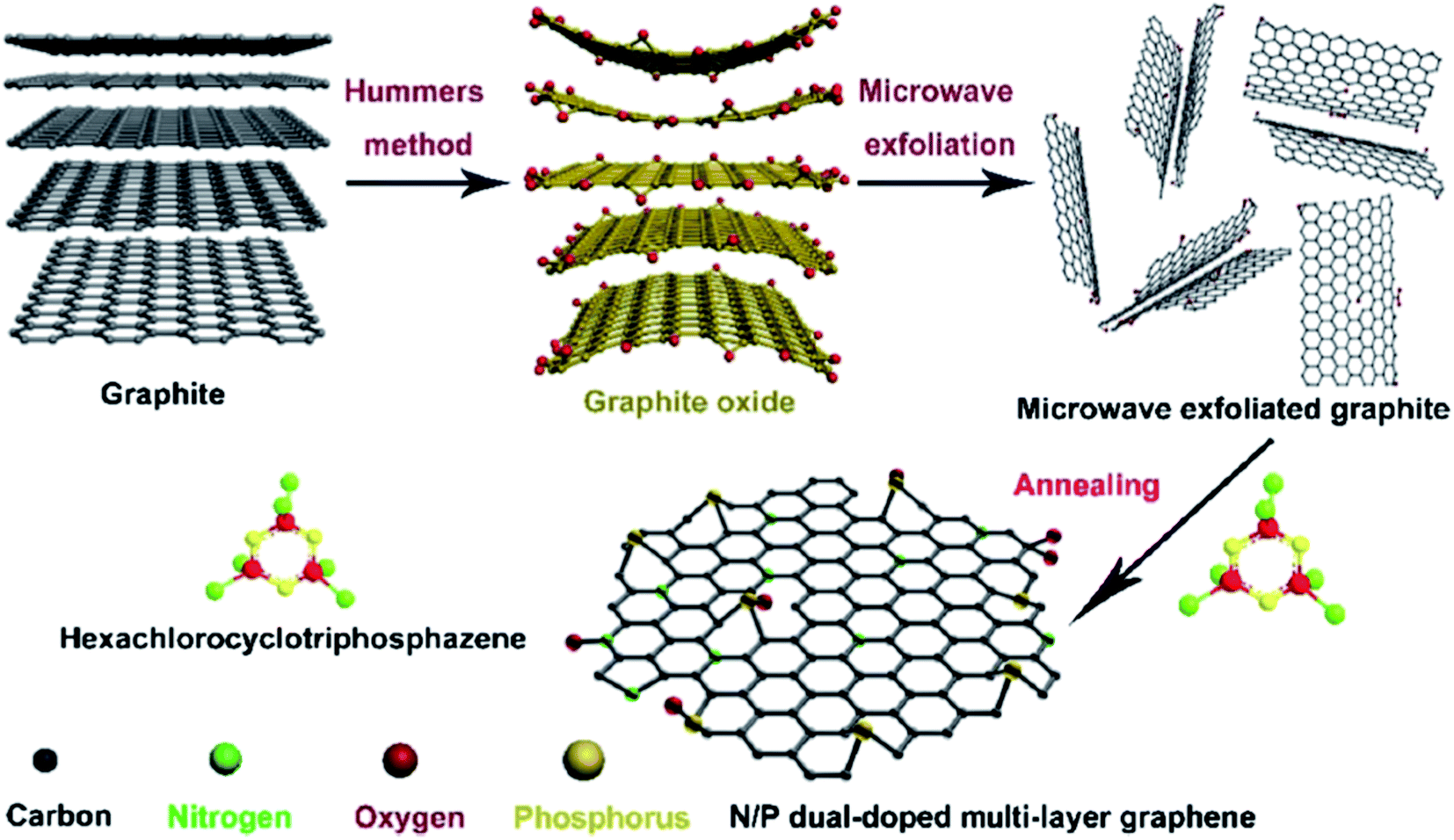 | ||
| Fig. 14 Preparation of NPG. Reprinted with permission from ref. 204. Copyright 2018 Elsevier. | ||
The catalytic performance was higher compared with other carbocatalysts based on graphene and even similar to that of commercial Pd/C (5%) and many previously reported noble metal-based catalysts.
Furthermore, the application of NPG catalyst could be extended to the hydrogenation of other substituted nitroarenes (Scheme 12).
 | ||
| Scheme 12 Hydrogenation of nitroarenes catalyzed by NPG. | ||
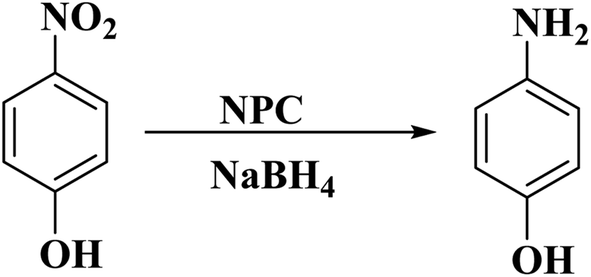
Recently, Pu and co-workers186 reported the synthesis of a metal-free catalyst containing nitrogen and phosphorus co-doped carbon (NPC) material (Fig. 15). The abundant and benign α-cellulose crystallite served as a carbon source, while ammonium phosphate supplied the nitrogen and phosphorus components. The sample was fabricated through the mixing of precursors, freeze-drying followed by pyrolyzing under nitrogen at 800 °C. The catalytic efficiency of the heterogeneous cellulose-derived N/P-doped carbon material with flake-like morphology was assessed for the reduction of p-NP (Scheme 13), affording high TOF of 2 × 10−5 with an apparent kinetic rate constant of 0.0394 min−1, activation energy of 21.55 kJ mol−1, and excellent yield because of the synergistic effect of phosphorus and nitrogen dopants.
 | ||
| Fig. 15 Preparation of NPC. Reprinted with permission from ref. 186. Copyright 2020 Elsevier. | ||
 | ||
| Scheme 13 Reduction of p-NP in the presence of NPC. | ||
Structural engineering of carbon-based materials is an innovative method to adjust their chemical and physical features, which are directly attributed to their catalytic behaviors.
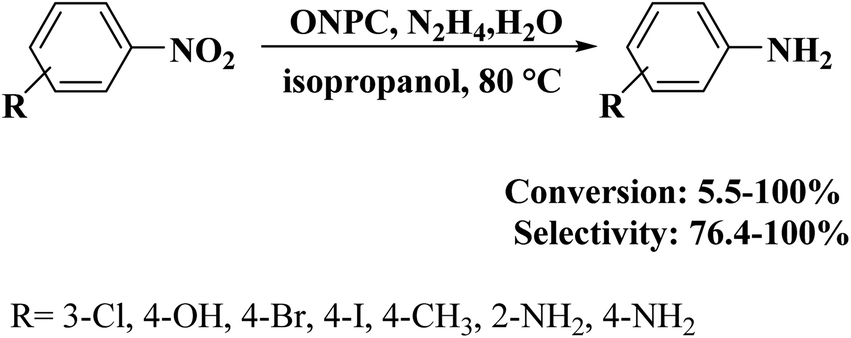
An environmentally benign and inexpensive procedure was disclosed to obtain oxygen and nitrogen co-doped porous carbon (ONPC) using coal tar- and residual oil-derived asphaltene as starting material, by Shen and colleagues205 in 2018. Firstly, the synthesized asphaltene as parent carbon was oxidized by HNO3 to dope O groups and next activated using potassium hydroxide combined with urea to introduce N species. The resulting ONPC was able to catalyze the hydrogenation of nitroarenes to corresponding anilines in the presence of hydrazine hydrate (Scheme 14). This catalyst indicated a significantly better catalytic activity relative to carbon black, activated carbon (AC), and un-doped porous carbon (PC), owing to co-doping of two different atoms (O and N), developed pore structure, and huge surface area. Experimental studies using model catalysts confirmed that C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups were more favorable oxygen active species for the reduction of nitrobenzene. When N atoms were introduced into PC, the generated nitrogen species synergistically cooperated with oxygen groups, resulting in high catalytic activity.
O groups were more favorable oxygen active species for the reduction of nitrobenzene. When N atoms were introduced into PC, the generated nitrogen species synergistically cooperated with oxygen groups, resulting in high catalytic activity.
 | ||
| Scheme 14 Hydrogenation of nitroarenes catalyzed by ONPC. | ||
In a very recent study, Zhang et al.206 developed N-doped activated carbon-based catalysts for acetylene hydrochlorination through a melamine/formaldehyde/polyvinyl alcohol (PVA) coating pyrolysis approach (Fig. 16) and investigated the effects of the synthesis parameters, such as the calcination temperature of the precursors, mass ratio of AC to melamine, the content of PVA, and the molar ratio of formaldehyde to melamine. The results of various analyses demonstrated that N species of graphitic-N and pyridinic-N and defect sites were the main reasons for high catalytic efficiency with a conversion of acetylene up to 75% and gas hourly space velocity of 50 h−1 under 220 °C in 120 h. Also, the numerous mesopores of catalyst retarded the process of deactivation and accelerated the reactants transfer.
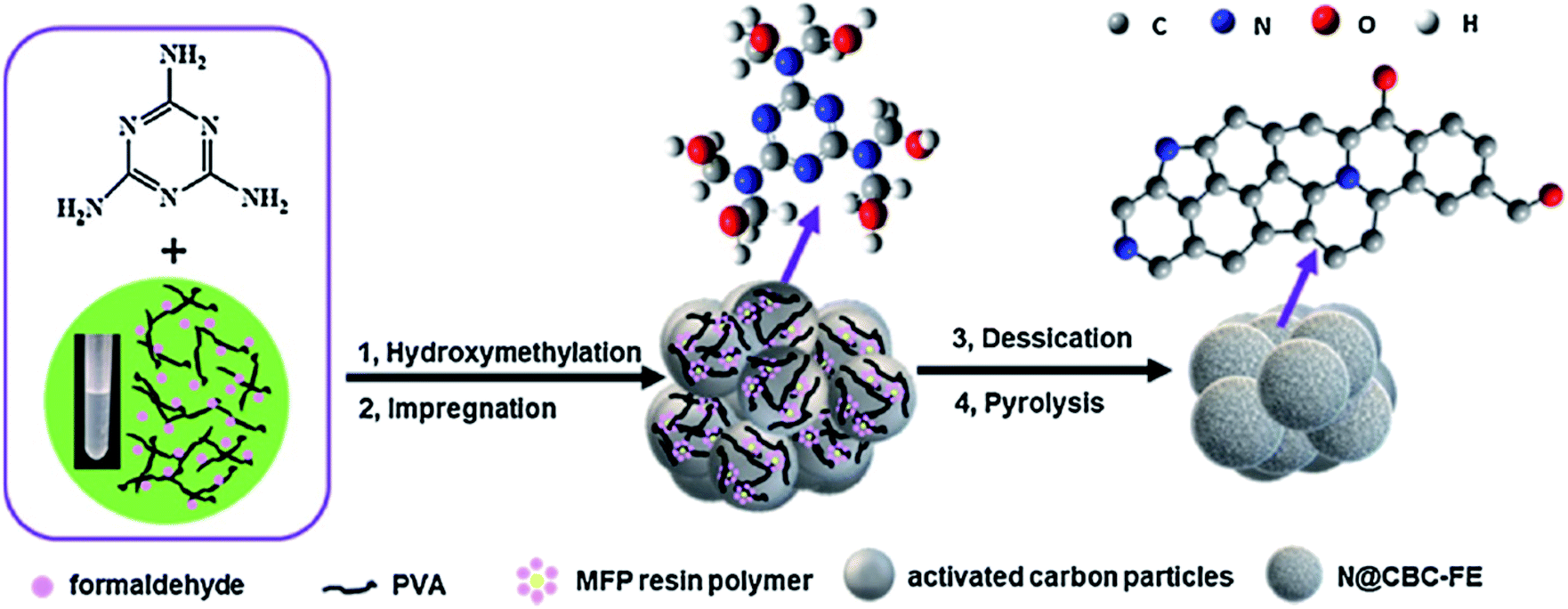 | ||
| Fig. 16 Preparation of N@CBC-FE catalysts. Reprinted with permission from ref. 206. Copyright 2021 Elsevier. | ||
Recently, a simple method for the preparation of a series of carbon materials with rich C defects and high nitrogen content was developed by Zhu and colleagues207 (Fig. 17). These materials were synthesized through polymerization of diaminopyridine by ammonium persulfate and followed by pyrolysis in different temperatures and were employed as a catalyst for hydrochlorination of acetylene. NC-800 displayed outstanding activity and durability with acetylene conversion of 98%, C2H2 GHSV = 30 h−1 under 220 °C. The exceptional catalytic performance of NC-800 was related to strong C2H2 and HCl adsorption, a high defect amount, a large specific surface area and a good level of active pyridine and pyrrole N contents.
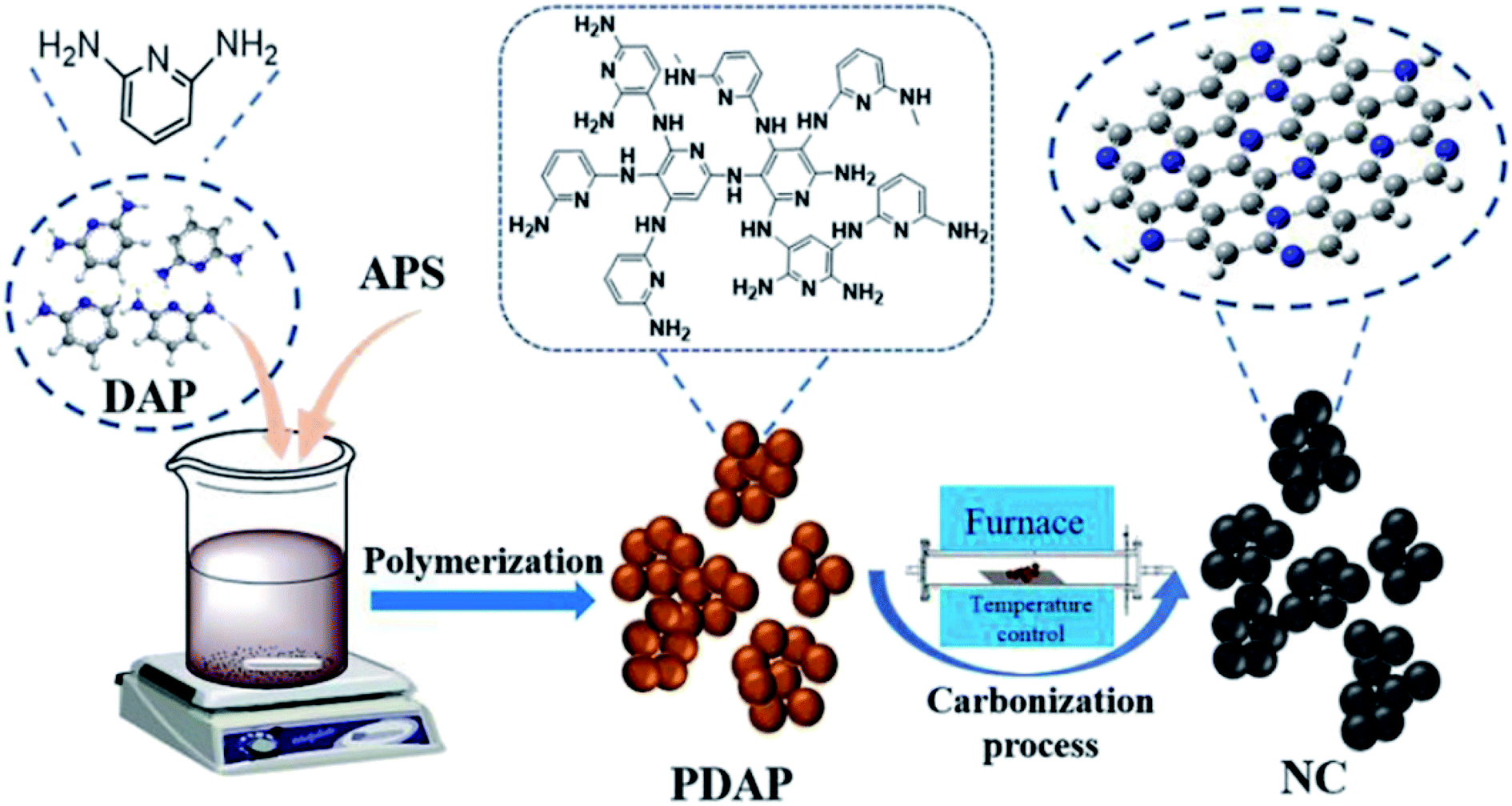 | ||
| Fig. 17 Preparation of NC. Reprinted with permission from ref. 207. Copyright 2021 Elsevier. | ||
Li and co-workers208 fabricated a series of nitrogen-doped carbon materials derived from ZIF-8 by adding melamine to tune the N content. The catalytic application of these samples was investigated in acetylene hydrochlorination and the optimized Z4M1 with the appropriate pore structure and the largest specific surface areas exhibited the highest acetylene conversion under 180 °C and, C2H2 GHSV of 50 h−1, indicating a promising non-metallic catalyst for this reaction. According to TG analysis, the authors suggested that coke deposition was a reason for the deactivation of the catalysts, and the coke deposition gently enhanced with the increasing content of N.
Li and co-workers209 utilized MOF-derived N-doped carbon composites as efficient catalysts in the acetylene hydrochlorination and the optimal 17% ZIF-8/SAC afforded VCM in good yield and high selectivity. These catalysts were prepared through in situ preparation of ZIF-8 in the pores of spherical activated carbon (SAC) (with different contents), followed by calcination at 950 °C to tune the morphology and catalytic performance (Fig. 18). Bamboo-shaped CNTs with a mean diameter of 40–80 nm appeared on the modified SAC surface could modify the porosity of composites, preventing the generation of coke deposition and well-dispersed nanoparticles with a mean size around 10.50 nm could simply form more active centers and efficiency increase the catalytic performance.
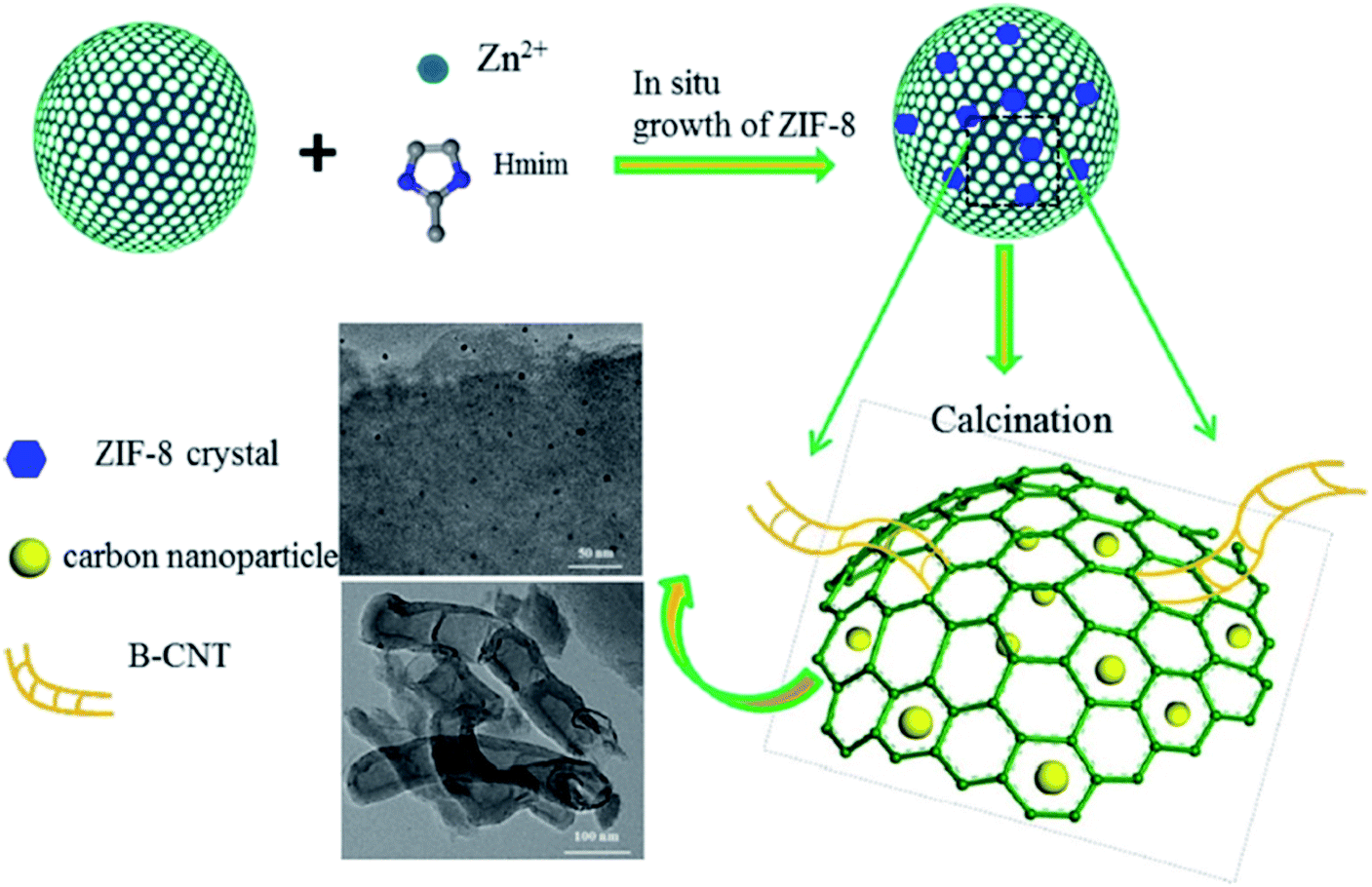 | ||
| Fig. 18 Preparation of ZIF-8/SAC composite. Reprinted with permission from ref. 209. Copyright 2018 Springer Nature. | ||
Also, results of TGA and TPD confirmed that the optimal 17% ZIF-8/SAC could increase the adsorption of the reactants as well as prevent the product adsorption, thereby leading to the reduction in the amount of coking deposition.
In 2017, Wang et al. utilized the ZIF-8-derived N-doped carbon catalyst calcined at 1000 °C for the acetylene hydrochlorination reaction.210 Catalyst showed excellent activity and outstanding durability (92% acetylene conversion was observed with a slight decrease during the 200 h test under atmospheric pressure at 220 °C). Theoretical calculations and experimental studies indicated that neighboring C atoms of pyridinic N atoms were active centers and the main reason for the deactivation of catalyst was coke deposition covering pyridinic N.
Recently, Qi' group211 proposed several sulfur-doped spherical activated carbon (SAC) materials with selecting phenyl disulfide as the S source. The performance evaluation of these catalysts was performed in the acetylene hydrochlorination reaction and 9% S/B-SAC displayed preferable catalytic efficiency, in comparison to the blank carrier under 180 °C and GHSV (C2H2) of 90 h−1.
The characterization data indicated that the presence of S dopants increased the capability of reactants' adsorption and prevented the generation of coke deposition. In addition, the DFT study demonstrated that the incorporation of S atoms altered charge density created more active centers on a carrier, and accelerated the electron transfer between reactants and carrier surface.
In 2015, Zhu and colleagues212 designed a metal-free catalyst for the hydrochlorination of acetylene.
Boron and nitrogen dual doped on graphene oxide (B, N-G) catalyst was obtained by calcination of pre-made B-G intermediate under 20% NH3/Ar at 900 °C. This catalyst displayed acetylene conversion considerably superior compared to N- or B-doped graphene and a little lower than that of Hg and Au catalysts.
TPD experiments and DFT calculations indicated that the synthetic effect of N and B codoping could facilitate the adsorption of HCl, which is the rate-determining step in this reaction.
In 2019, Zhao and his research team213 described the use of N,P-codoped carbon-based materials toward hydrochlorination of acetylene. For the preparation of these catalysts, modified ionic liquids (ILs) such as 1-ethyl-3-methylimidazolium dicyanamide [EMim]+N(CN)2−] and 1-ethylsulfonate-3-methylimidazolium dihydrogen phosphate [ESO3HMim+H2PO4−] were employed as nitrogen and phosphorus sources, respectively (Fig. 19).
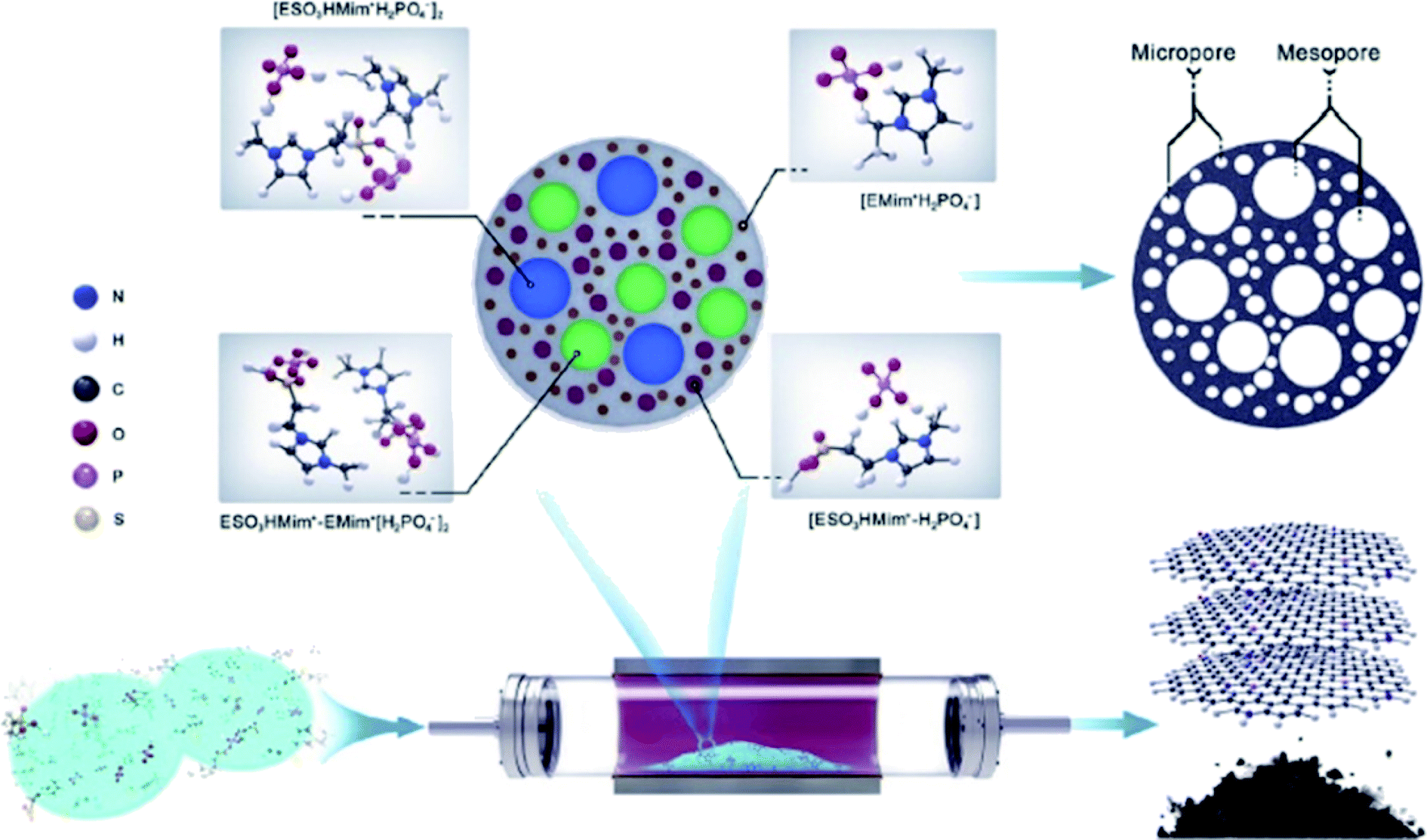 | ||
| Fig. 19 Preparation of NP-C600. Reprinted with permission from ref. 213. Copyright 2019 Elsevier. | ||
The utilization of task-specific ILs can facilitate the uniform introduction of heteroatoms over the entire carbon-based samples.
The outstanding and catalytically beneficial chemical and physical properties can be created in the fabricated carbon-based materials because of the formation of defects as well as uneven charge distribution caused by the presence of various heteroatoms.
The catalytic efficiency of these new N,P-codoped carbon-based catalysts was higher compared to the HgCl2 catalysts and comparable to that of Au-based catalysts.
DFT calculations and experimental results confirmed that P atoms bonded with N in the pyridine structure were responsible for the preferable catalytic activity of the NP-C600 catalyst.
In 2017, Zhu et al.214 used (NH4)2S2O8 (APS) and p-phenyldiamine (pPD) as the sulfur and nitrogen sources to construct several S and N dual-doped carbon materials with various molar ratios of the pPD/APS to catalyze acetylene hydrochlorination. The authors concluded that the pyrrolic nitrogen amount in the catalysts changes with the addition of various S amounts, leading to differences in the catalytic performance. The co-doping of S with N in the catalysts provided the synergistic effect for acetylene hydrochlorination. Also, TPD experiments and DFT calculations revealed that the doping of the S and N-doped carbon catalysts enhanced the adsorption ability of C2H2.
In 2017, a hard templating approach was utilized by the research group of Li215 for the preparation of a sulfur and nitrogen dual-doped mesoporous carbon using sucrose as the precursor of carbon and thiourea as precursors of sulfur and nitrogen in the presence of SBA-15 as the template (Fig. 20). Acetylene hydrochlorination reaction was selected to survey the catalytic efficiency of the resulting material. The authors found that doping of sulfur could effectively improve the pyridinic N content and hence showed increased acetylene conversion relative to the nitrogen-doped carbon catalyst. Based on the C2H2 TPD results and high-resolution XPS data, carbon atoms bonded to pyridinic nitrogen were active centers of catalyst for the reaction.
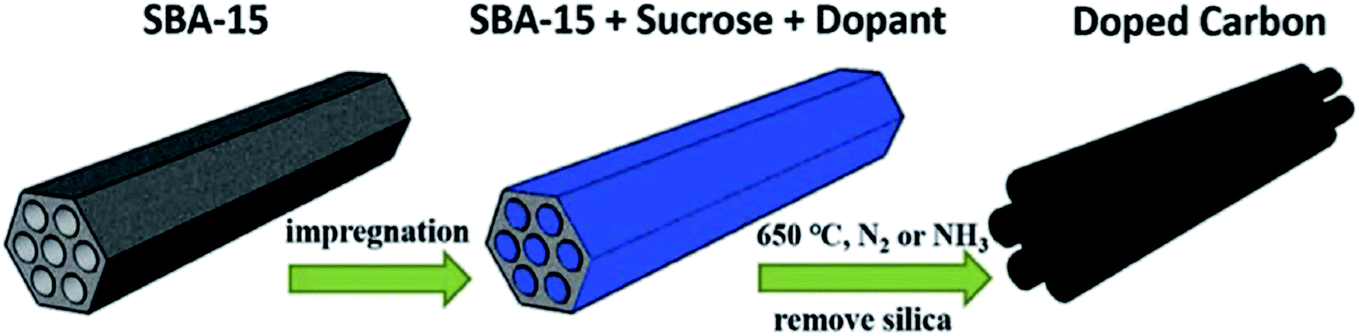 | ||
| Fig. 20 Preparation of S and N co-doped mesoporous carbon. Reprinted with permission from ref. 215. Copyright 2018 Elsevier. | ||
In addition, high nitrogen level, mesoporous structure, and large specific surface area derived from SBA-15 resulted in superb catalytic durability and preferable catalytic activity of the co-doped catalyst processed in NH3.
When NG and NP were quite close, a synergistic effect was displayed between them, which was unbeneficial for the catalytic performance because of the very high energy barriers.
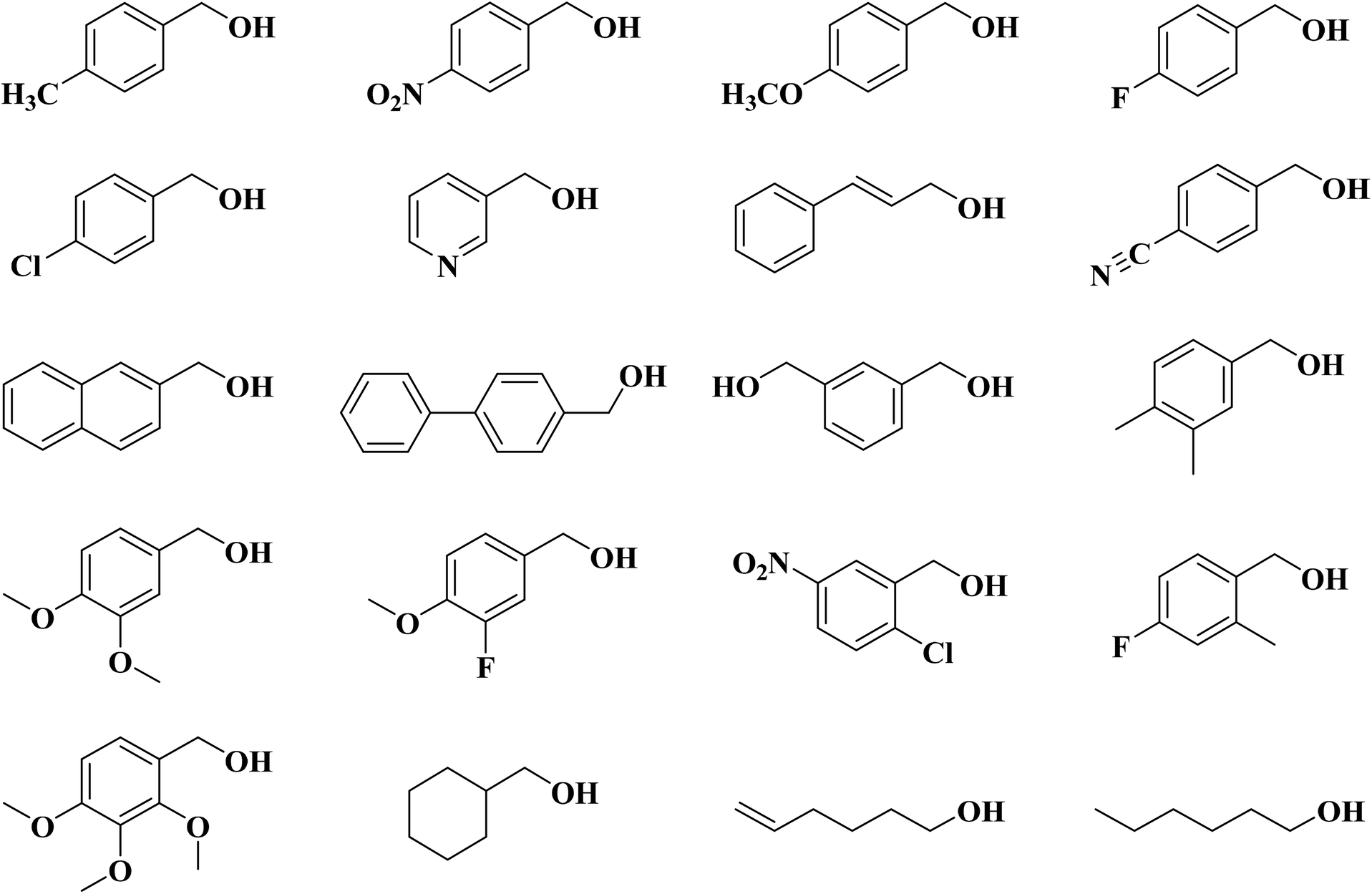
Lin and colleagues217 reported an effective and simple CVD-like method to synthesize four N-doped nanocarbon (NDN) catalysts (including nitrogen-doped onion-like carbon (NOLC), nitrogen-doped oxidized bucky nanodiamond (NOBND), nitrogen-doped CNTs (NCNTs), and nitrogen-doped bucky nanodiamond (NBND)) with enriched pyridinic nitrogen configuration and without any graphitic N species (Fig. 21). Both the ammonia gas and pyrimidine vapor were employed as the N precursors at a relatively low calcination temperature. NBND showed high catalytic prowess towards the oxidation of phenols (Table 3) and complicated alcohols (Table 4) with H2O2, O2, or TBHP as oxidants. The authors also indicated the activation mechanisms and potential roles of N species using aromatic molecules with single N species as probe catalysts. They found that pyridinic nitrogen species played a key role in catalytic reactions.
 | ||
| Fig. 21 Preparation of nitrogen-doped nanocarbon (NDN) materials. Reprinted with permission from ref. 217. Copyright 2019 American Chemical Society. | ||
|
|---|
 |

|
|---|
 |

Wang et al.218 reported the preparation of a series of nitrogen-doped graphene materials and their catalytic performance as metal-free catalysts for oxidation of organic pollutants (p-hydroxyl benzoic acid (PHBA), furfuryl alcohol (FFA), and phenol) through activation of peroxymonosulfate (PMS). N-doped graphenes were fabricated by pyrolyzing MIL-100 (Fe) in the presence of three various nitrogen precursors (urea, melamine, and dicyandiamide) and subsequent acid digestion to eliminate Fe. The specific surface area of catalysts and N doping were important factors affecting the activity of pollutants removal. The mechanism was studied by both quenching experiments (EtOH and NaN3 as the radical scavengers) and electron paramagnetic resonance (EPR, 2,2,6,6-tetramethyl-4 piperidinol, and 5,5-dimethyl-1-pyrroline N-oxide as the trapping agents). Also, furfuryl alcohol and benzoic acid were used as probing reagents for singlet oxygen and hydroxyl/sulfate radicals, respectively. Overcomes affirmed that singlet oxygen was formed and influenced the degradation of the pollutant on N-doped graphene instead of hydroxyl/sulfate radicals regardless of N precursors.
By employing fructose, glucose, and 5-hydroxymethylfurfural (5-HMF) as inexpensive and widely available precursors, the Wen group219 prepared the three kinds of N-doped graphene-like carbon (NG) catalysts via a pyrolysis process. Among them, when 5-HMF was utilized, a thin-layered structure with a large lateral dimension was obtained. Also, the NG derived from 5-HMF demonstrated better efficiency in epoxidation reactions than those of the conventional carbon catalysts and its performance was even comparable to that of a cobalt catalyst.
EPR, TEM, and XPS analyses displayed that the increased catalytic performance arose from the activation ability both for O2 and alkene, which could be ascribed to the graphitic N species and graphitic layered structure.
Sun and co-workers220 utilized a simple route for the preparation of nitrogen-doped reduced graphene oxide (N-rGO) through simultaneous reduction and introduction of nitrogen on GO using ammonium nitrate as N precursor at various temperatures from 300 to 400 °C. The performances of these metal-free N-rGO materials as green catalysts were studied in the aqueous oxidation of phenol solutions by catalytic activation of peroxymonosulfate (PMS).
Characterization data showed that the chemical compositions and crystal/micro-structures of samples are dependent on the thermal annealing temperature. Authors concluded that the improvement of phenol degradation using N-rGO at calcination temperatures between 300 and 325 °C was due to the N heteroatom, whereas these improved performances for N-rGO at calcination temperatures between 350 and 400 °C were due to both relatively high specific surface area and N heteroatoms.
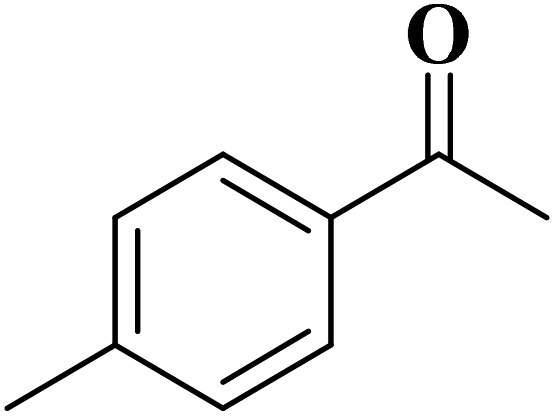
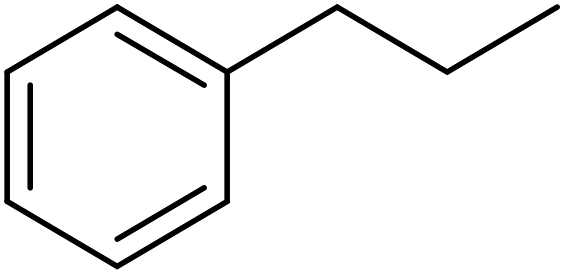
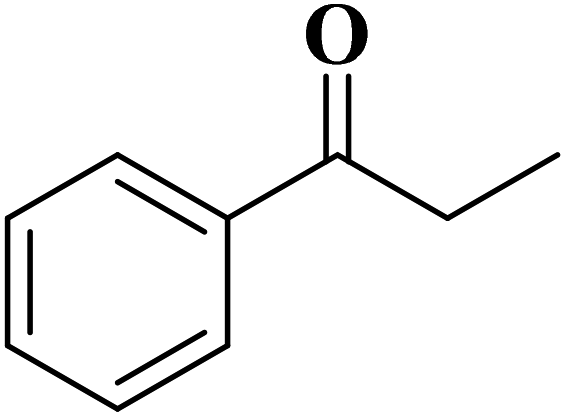
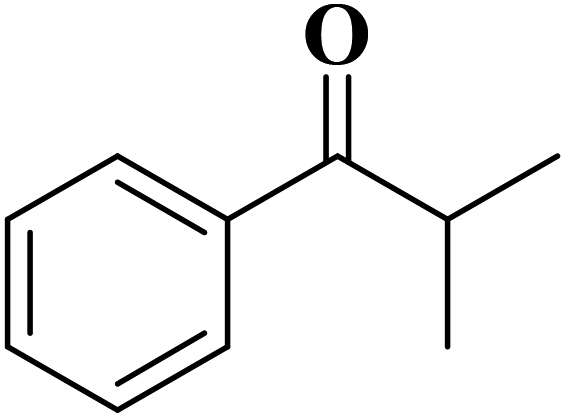
Using melamine and biomass (such as lignin, glucose, and cellulose) as the nitrogen and carbon sources, and the mixture of KCl/ZnCl2 as the solvent and porogen, Liu group221 fabricated a series of mesoporous nitrogen-doped carbons (NDCs) with specific surface area up to 1800 m2 g−1 and N content up to 11.9% based on a “Salt Templating” synthesis method. The M-G-1.5-800 (G= glucose, 1.5 = mass ratio of melamine to glucose, and 800 = calcination temperature) with nitrogen loading of 11.4% indicated high activity toward alkanes oxidation in the presence of tert-butyl hydroperoxide (TBHP) as the oxidant in the aqueous phase (Table 5). In addition, NDCs were applied as ideal supports to stabilize noble metal nanoparticles (e.g., Rh, Ru, Pt, and Pd).
| Substrate | Product | Conv./% | Yield/% |
|---|---|---|---|
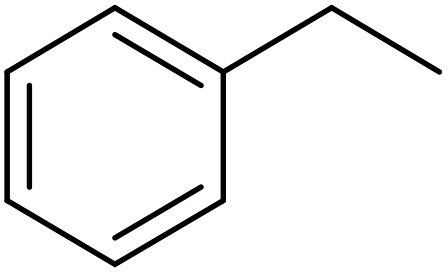 |
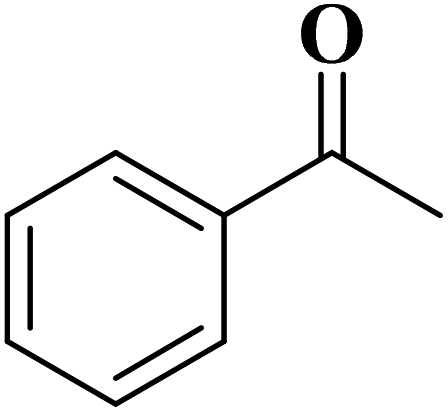 |
93.8 | 91.2 |
 |
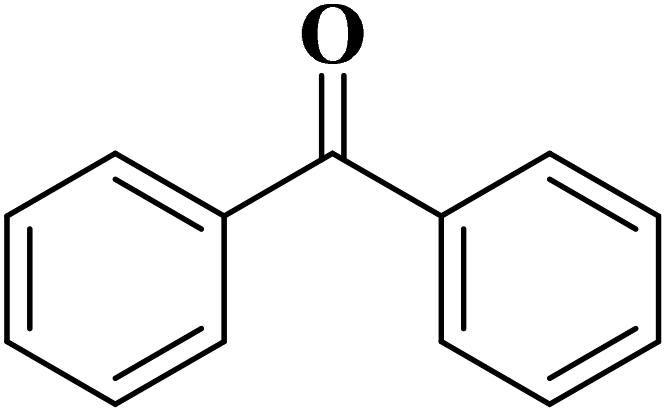 |
97.6 | >99 |
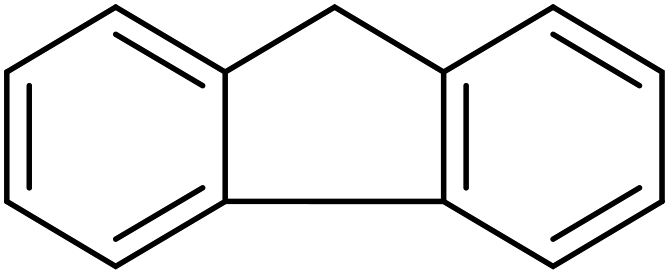 |
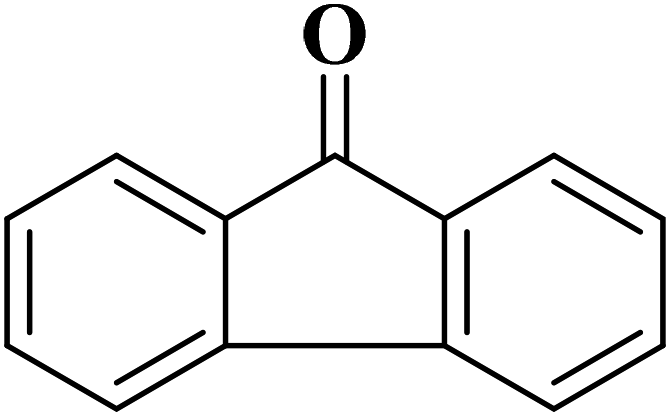 |
>99 | >99 |
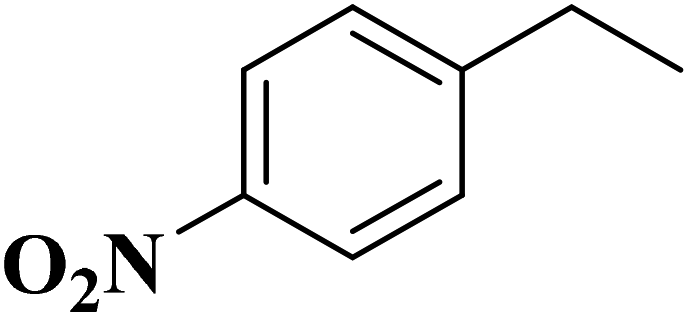 |
 |
98.7 | >99 |
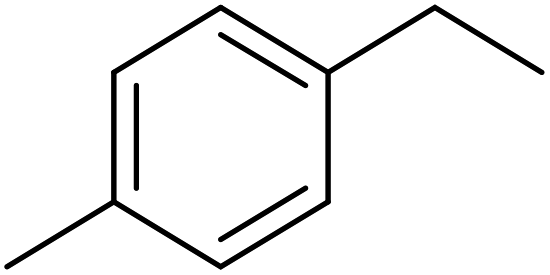 |
 |
89.5 | 60.0 |
 |
 |
85.4 | 82.6 |
 |
 |
73.9 | 51.8 |
 |
 |
51.4 | 48.7 |
 |
 |
— | 26.3 |
 |
 |
12.3 | 9.8 |
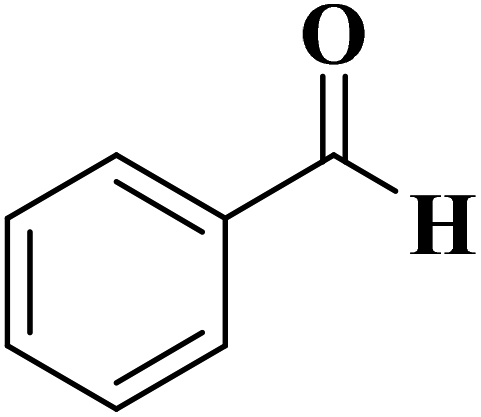
A facile two-step synthetic method including pyrolysis-etching of ZIF-67 was reported by Li and Wang222 for the preparation of highly graphitized N-doped mesoporous carbon materials (Fig. 22). High contents of sp2-bonded carbons, large pore volumes, and specific surface areas, and abundant mesopores were realized after chemical etching of the in situ generated Co nanoparticles. Also, the sizes of mesopores could be controlled through the adjustment of pyrolysis temperature. These N-doped carbons as metal-free catalysts exhibited effective catalytic activities and strong durability in several oxidation reactions like the oxidative coupling of amines (Scheme 15), aerobic oxidation of cyclohexane (Scheme 16) as well as toluene (Scheme 17).
 | ||
| Fig. 22 Synthesis of N-doped carbons. Reprinted with permission from ref. 222. Copyright 2016 Royal Society of Chemistry. | ||
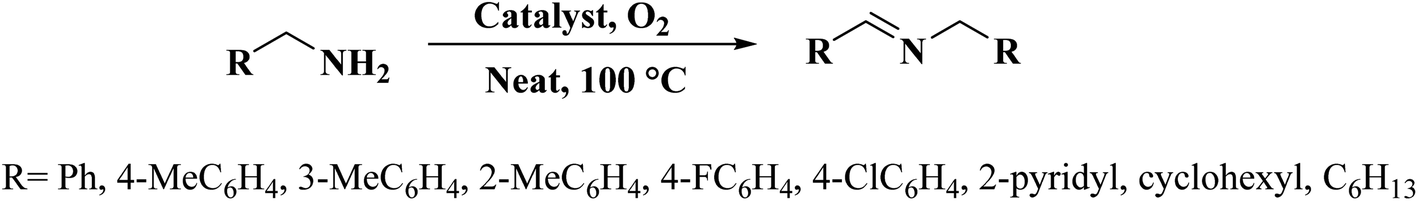 | ||
| Scheme 15 Oxidative coupling of amines. | ||
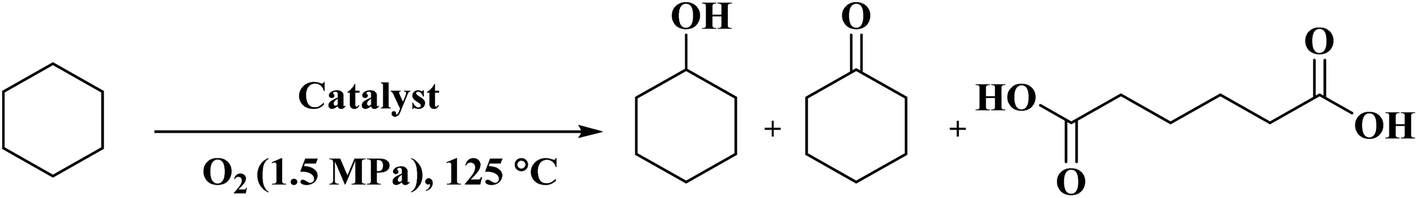 | ||
| Scheme 16 Oxidation of cyclohexane using carbon materials. | ||
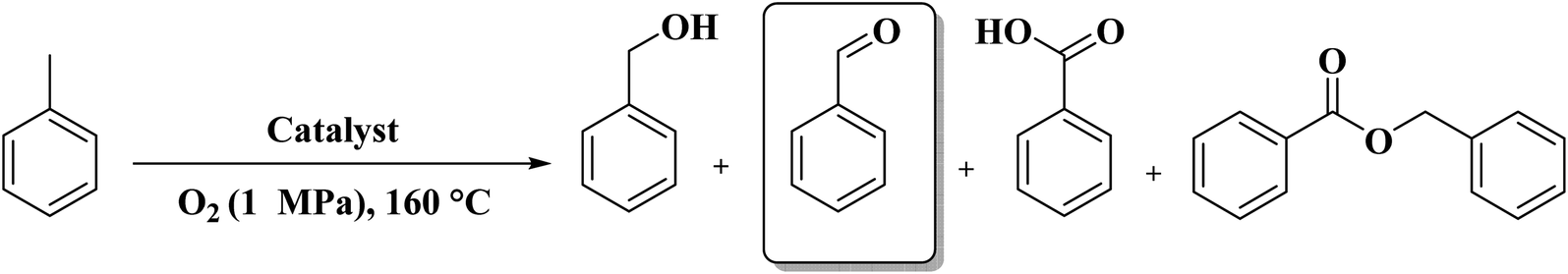 | ||
| Scheme 17 Aerobic toluene oxidation using carbon materials. | ||
Characterization results suggested that the uniform distribution of doped graphitic-type N and the available mesopores created by etching were responsible for the outstanding efficiency of these catalysts.
In 2017, Rizescu and co-workers223 reported two different types of N-containing graphenes. One of the types was prepared through simultaneous reduction of graphene oxide (GO) as starting material and amination with ammonia at three various concentrations. The second type was fabricated by pyrolysis of chitosan at 900 °C under an inert atmosphere (Fig. 23). The performance of these two N-doped Gs was tested for the selective wet oxidation of glucose to succinic acid. These protocols afforded good selectivities and excellent glucose conversions under O2 pressure at 160 °C.
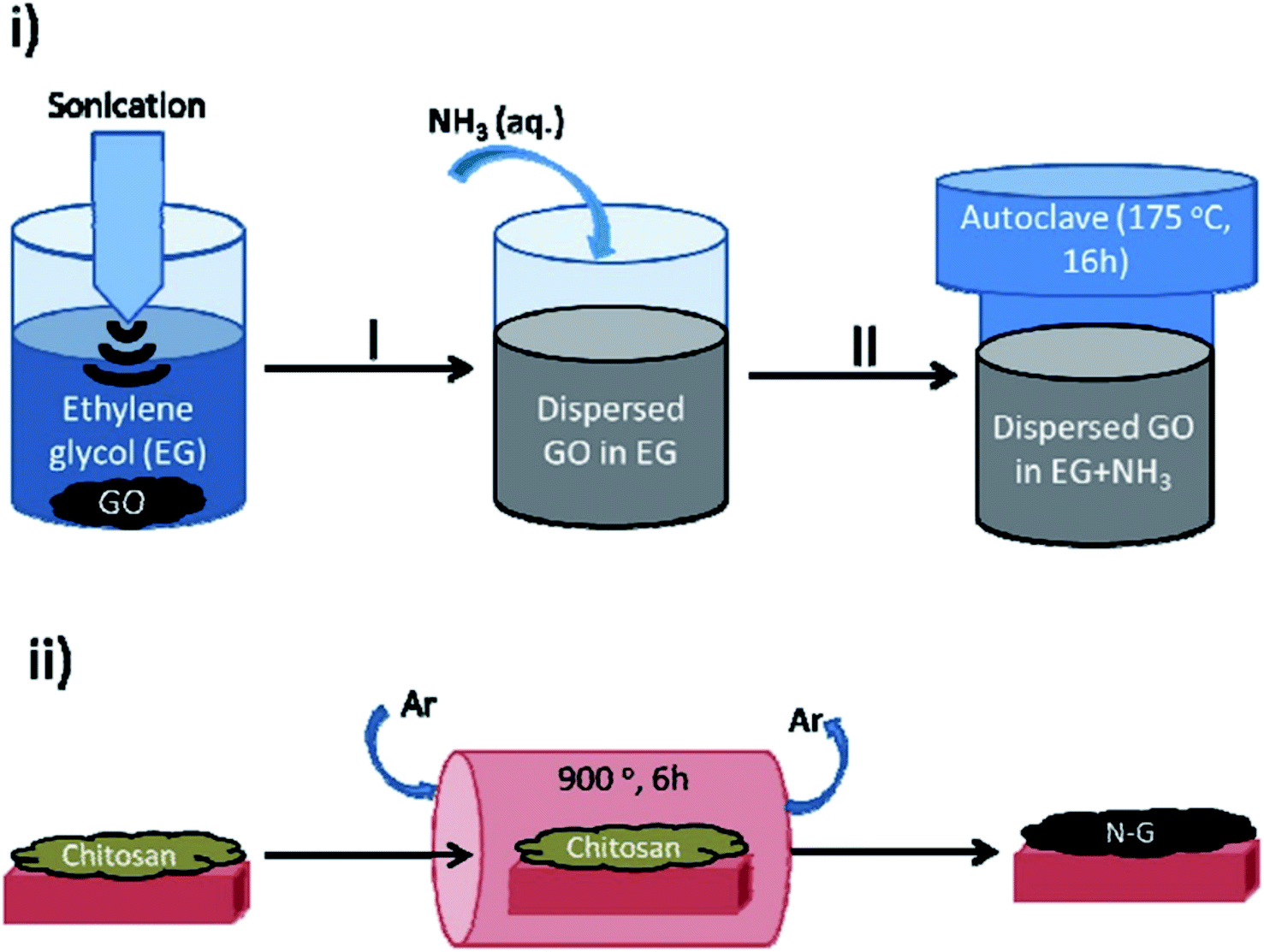 | ||
| Fig. 23 Preparation of (i) NH2-rGO(x) and (ii) N-G samples. Reprinted with permission from ref. 223. Copyright 2017 Royal Society of Chemistry. | ||
Graphenic-type N atoms on graphene were responsible for these performances. Notably, catalysts could be reused for four runs without any decline in selectivity and conversion of the process.
In 2019, Cao and his research team224 demonstrated the preparation of the N-doped carbon nanofibers (N-CNF) via one-step carbonization of urea and bacterial cellulose as low-cost raw materials (Fig. 24).
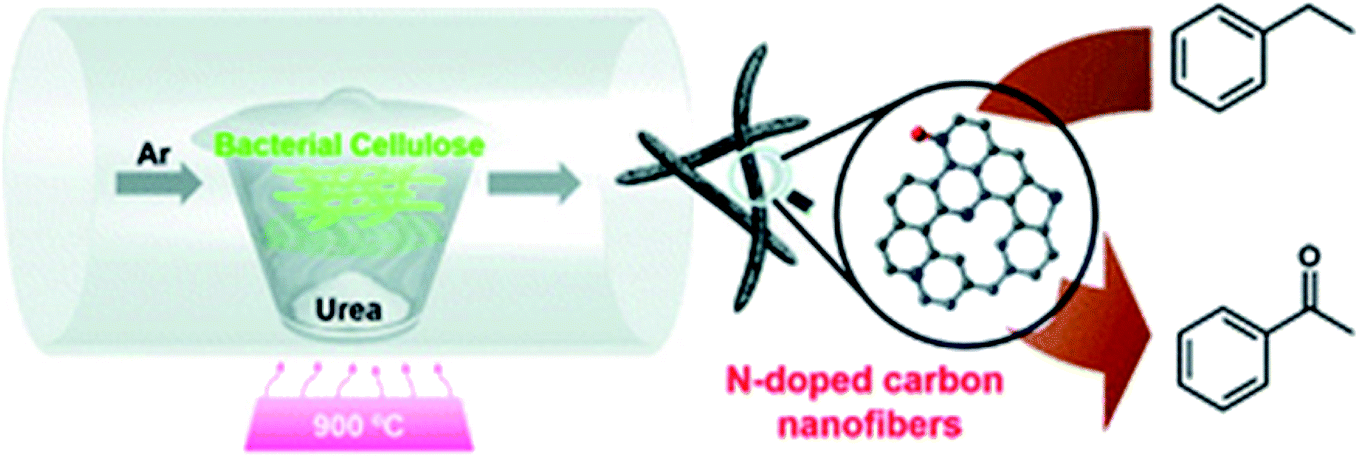 | ||
| Fig. 24 Preparation of NCNF. Reprinted with permission from ref. 224. Copyright 2019 Royal Society of Chemistry. | ||
The obtained N-doped carbon nanofibers indicated excellent selectivity, activity, and good reusability for oxidation of arylalkanes in aqueous solution in the presence of tert-butyl hydroperoxide (TBHP) (Table 6), because of the great nitrogen content, large specific surface area, as well as interconnected nanofibrous structure. Moreover, the catalytic performance of N-CNF was well maintained even when O2 was used as an oxidant.
|
|||
|---|---|---|---|
| Substrate | Product | Con. (%) | Sel. (%) |
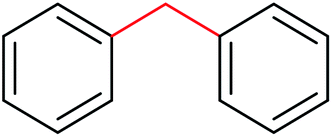
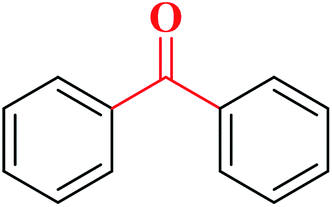
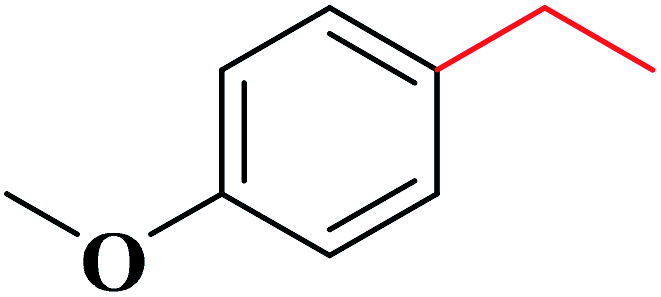 |
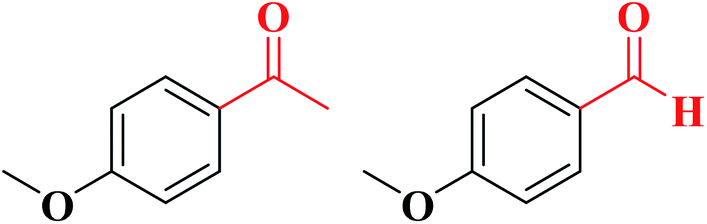 |
78 | 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 :1 |
 |
 |
96 | 99 |
 |
 |
97 | 99 |
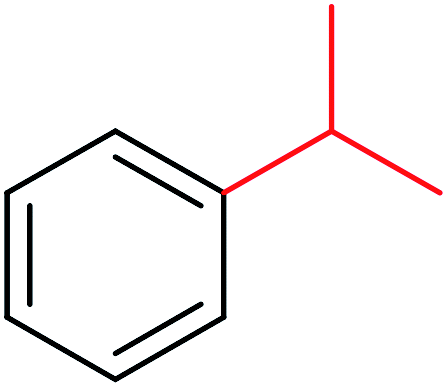 |
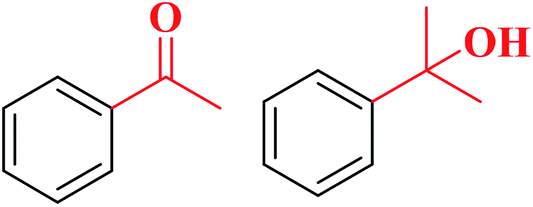 |
99 | 1:2.85 |
 |
 |
99 | 99 |
 |
 |
99 | 99 |
 |
 |
96 | 20 |
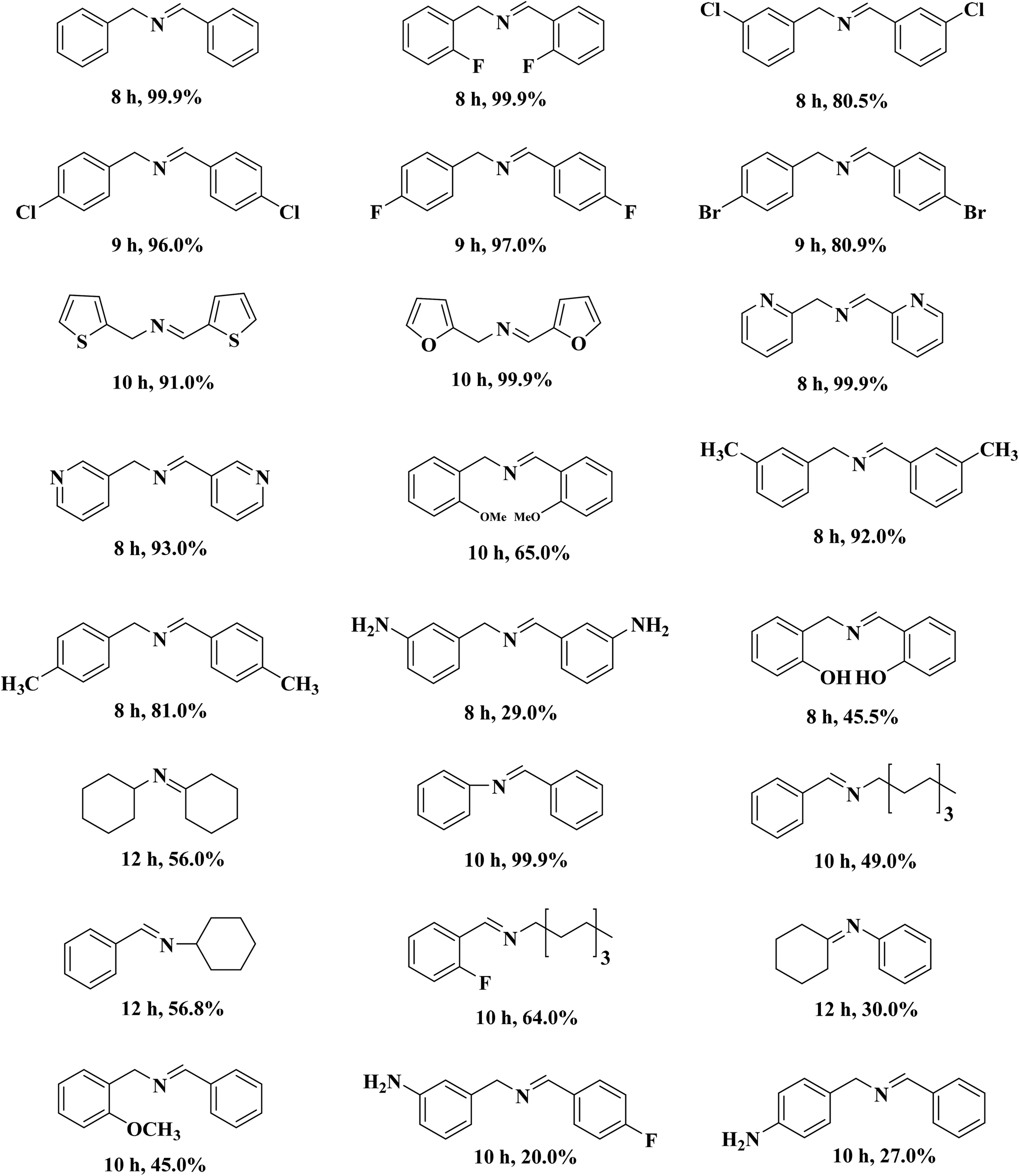
A porous multifunctional and environmentally friendly N-doped carbon catalyst for the oxidation of amines and transfer hydrogenation of nitriles was developed by Li et al.225 (Fig. 25) and diverse substrates were efficiently transformed to the relevant products under the mild conditions using 15 mg NC-800 (Table 7) due to the outstanding surface area and abundant active sites. In particular, this catalyst retained the initial catalytic efficiency at least seven times, which is comparable with metal catalysts. According to experimental and characterization data, the authors concluded that graphite-N atoms are the main active site in NC-800.
 | ||
| Fig. 25 Preparation of NC-800. Reprinted with permission from ref. 225. Copyright 2019 Royal Society of Chemistry. | ||
|
|---|
 |
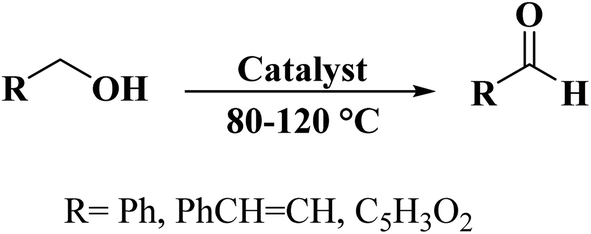
Via treatment of activated carbon (AC) with hydrogen peroxide and ammonia, several N-doped carbons were synthesized by Arai et al.226 The nature and content of species of nitrogen were investigated by XPS analysis to determine the genesis of active species. Graphitic-N species were remarkable to form active sites on the surface of activated carbon.
These catalysts were found to be efficient for the oxidation of alcohols like 5-(hydroxymethyl)-2 furaldehyde, cinnamyl alcohol, and benzyl alcohol (Scheme 18) and, in some cases, even more, selective than those of conventional carbon-supported Ru and Pt catalysts.
 | ||
| Scheme 18 Aerobic oxidation of alcohols using the nitrogen-doped AC catalysts. | ||
An attractive catalytic application of nitrogen- and oxygen-doped activated carbon was also disclosed by the same research team.227 N-doped AC could catalyze the hydrogenation of nitrobenzene using hydrazine but not phenylacetylene. Phenylacetylene hydrogenation could take place when nitrobenzene existed in the reaction mixture. The authors believed that synergistic effects between nitrogen and/or oxygen species of surface and nitrobenzene could accelerate the adsorption of phenylacetylene and thus allowed its hydrogenation.
A hard templating method was applied by Gao et al.228 for the preparation of nitrogen-doped nanocarbons with a mesoporous structure and a large surface area using polypyridyl ligand 4,5-diazafluorene-9-one azine (DAA) as carbon and nitrogen precursor in the presence of SiO2. The achieved meso-N/C-900 indicated good efficiency, selectivity, and reusability toward the aerobic oxidative preparation of nitriles from various alcohols by aqueous ammonia (Scheme 19) due to the efficient pyridinic/pyrrolic-N doping as well as the available mesopores.
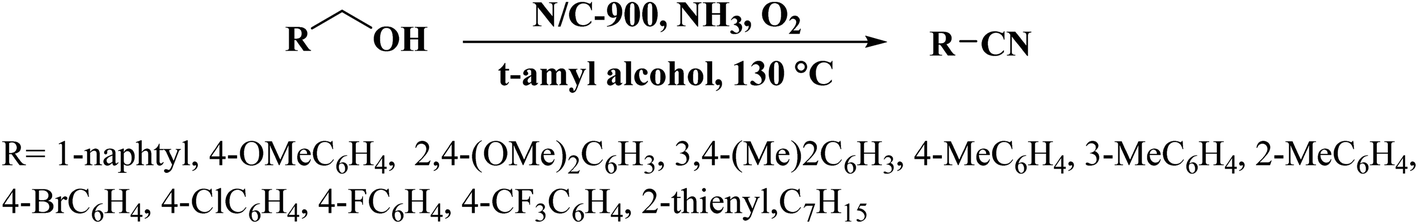 | ||
| Scheme 19 Synthesis of nitriles in the presence of meso-N/C-900. | ||
In 2019, Wang and co-workers229 reported the fabrication of an N-doped and nanofiber-based porous carbon foam through a simple and effective self-foaming method, followed by the carbonization process (Fig. 26). In this method, formaldehyde and resorcinol were utilized as carbon sources and CO2-rich ethanolamine (EAC) served as the polymerization catalyst, N source, and foaming agent.
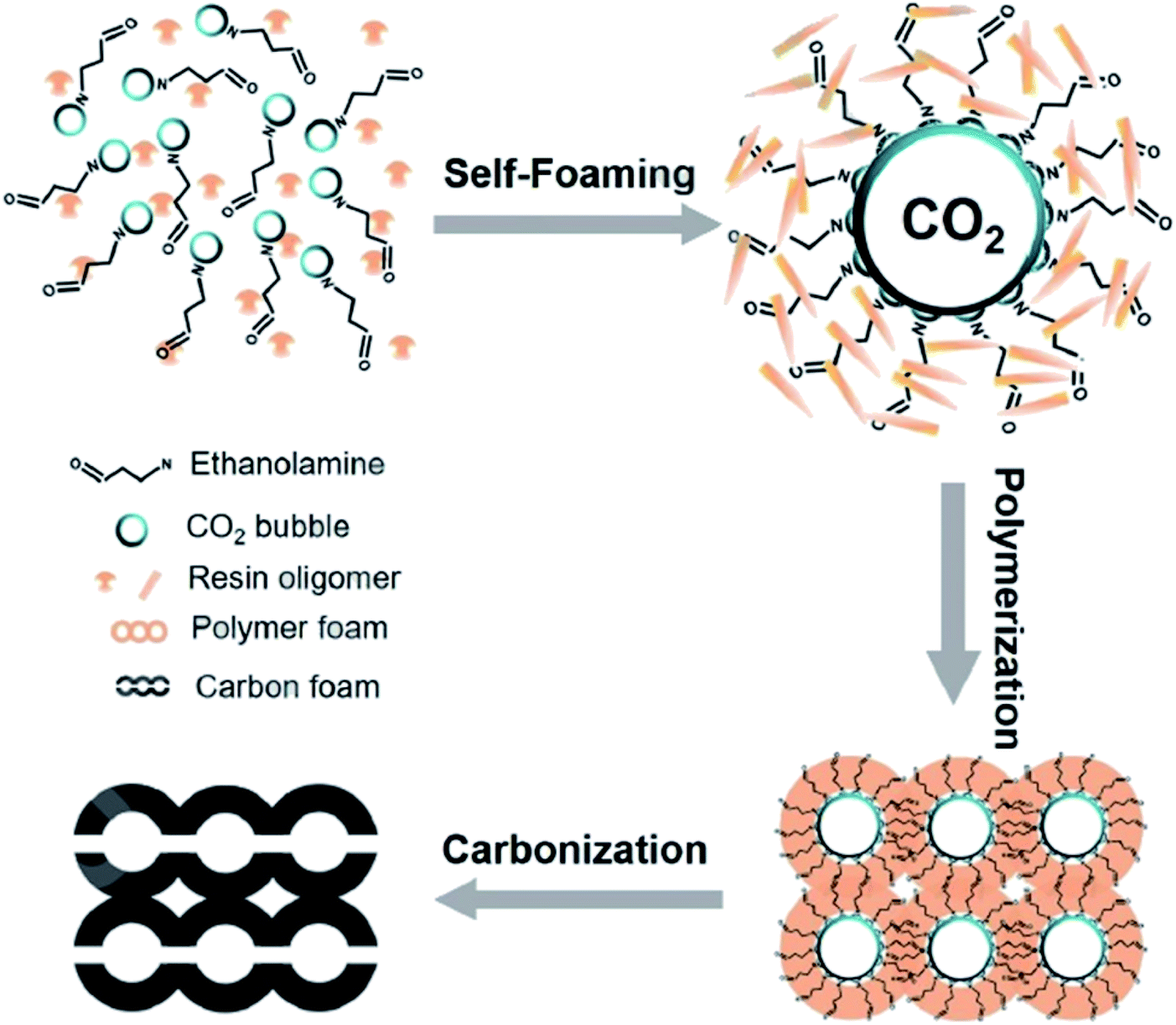 | ||
| Fig. 26 Preparation of foam-like carbon monoliths. Reprinted with permission from ref. 229. Copyright 2020 Elsevier. | ||
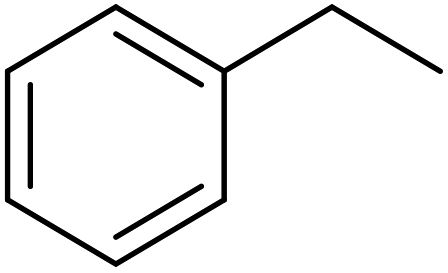
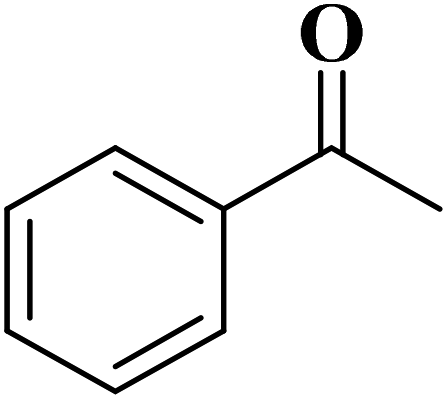
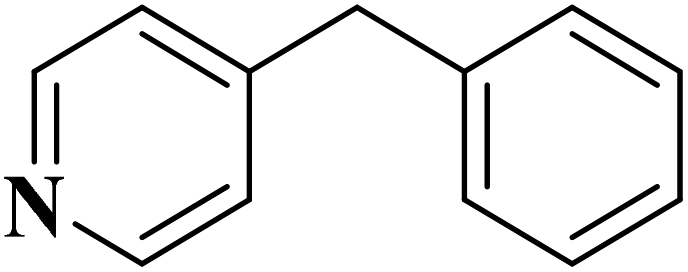
EAC played a critical role in initiating polymerization on the interfaces of bubbles as well as in the release. The hierarchical porous structures provided accessible active sites with high surface area sites while doping with nitrogen changed the electronic properties of the carbon. The N-doped carbon foam was able to catalyze the selective oxidation of C–H in ethylbenzene and benzyl pyridines under atmospheric pressure in water in the presence of tert-butyl hydroperoxide (TBHP) as an oxidant (Table 8). Also, the synthesized carbon foam exhibited good catalytic activity in the scale-up test.
| Substrate | Product | Conversion (%) | Selectivity (%) |
|---|---|---|---|
| a TBHP as oxidant, H2O as solvent at 80 °C. | |||
 |
 |
93 | 91 |
 |
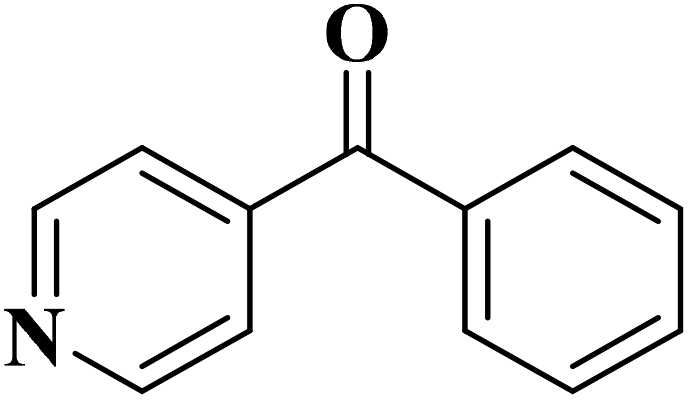 |
84 | >99 |
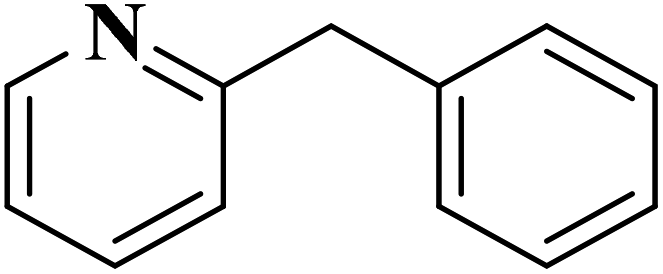 |
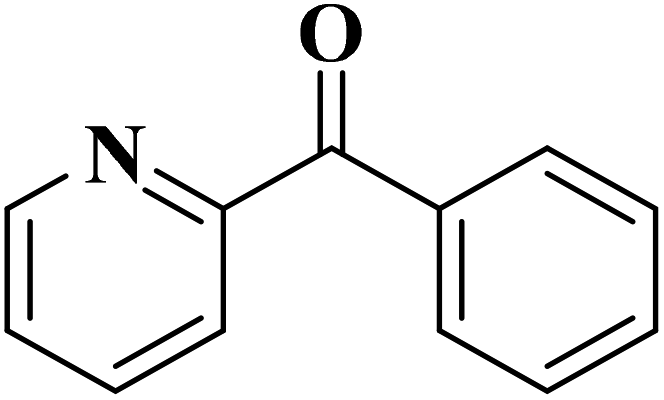 |
64 | >99 |
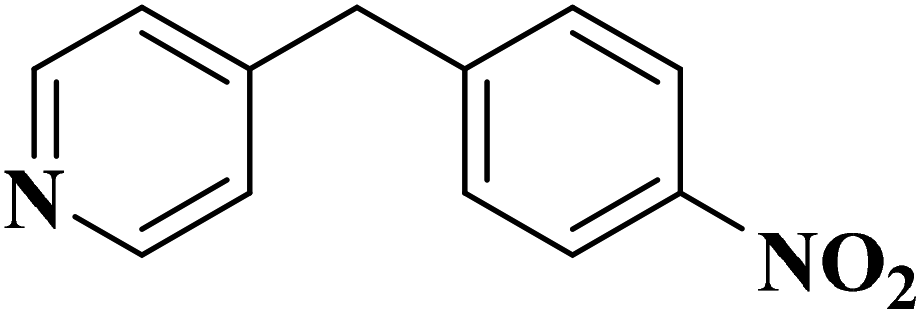 |
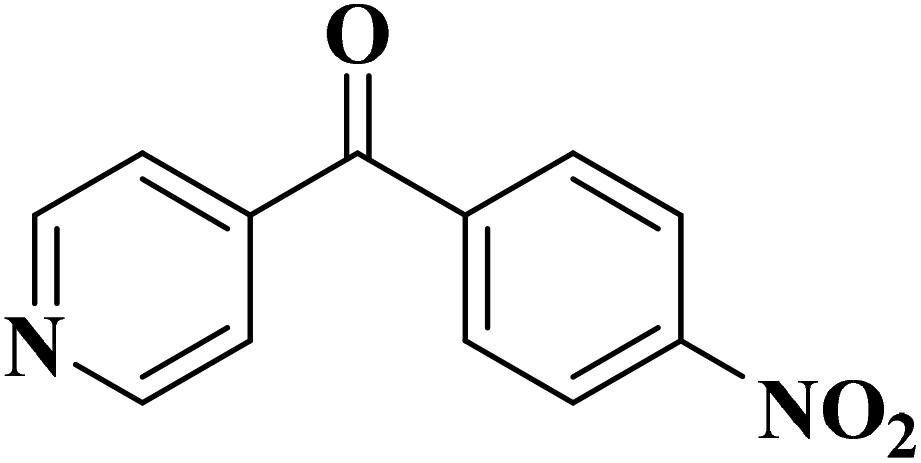 |
>99 | >99 |
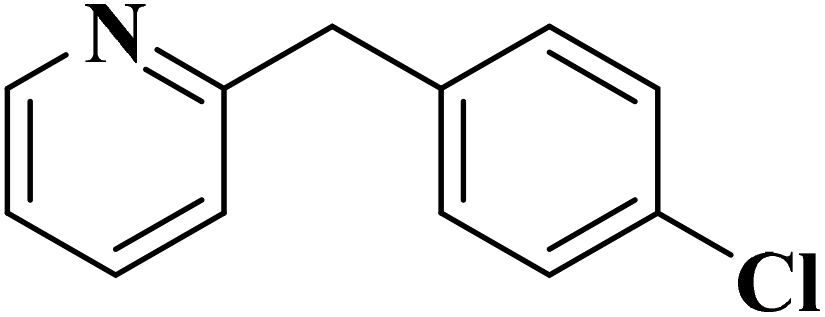 |
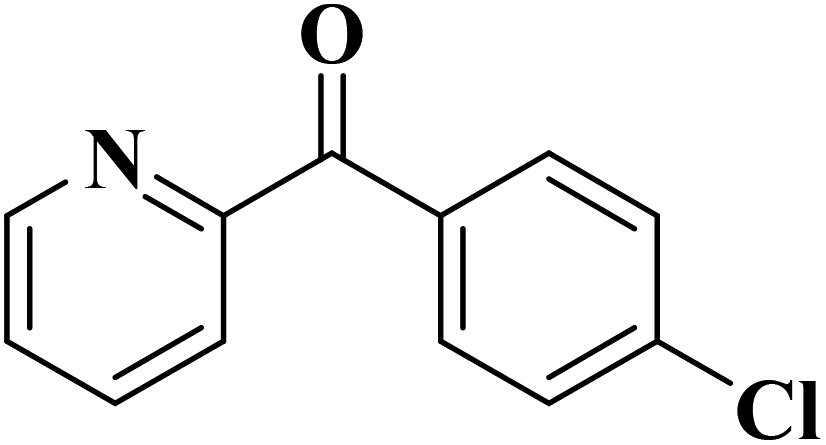 |
24 | >99 |
The use of N-doped porous carbons, as catalysts in the selective aerobic oxidation of 5-hydroxymethylfurfural (HMF), was described by Wang and co-workers230 in 2019. To prepare these materials, chitosan as a natural N-containing macromolecule was pyrolyzed in the presence of K2CO3 as an activator at temperatures ranging from 600 to 900 °C. The N-doped porous carbon at 700 °C exhibited the best catalytic efficiency, affording 2,5-diformylfuran (DFF) in excellent yield and selectivity under 2.0 MPa O2 at 120 °C, for 7.5 h (Scheme 20). Based on the XPS data, graphitic N atoms on the catalyst's surface were essential for the activation of O2 to generate radicals of oxygen that simplified the oxidative dehydrogenation of HMF.
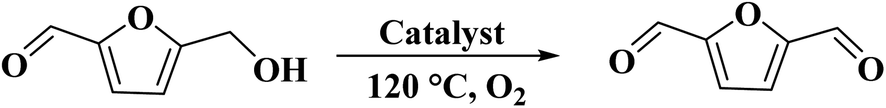 | ||
| Scheme 20 Aerobic oxidation of HMF using N-doped porous carbon-700. | ||
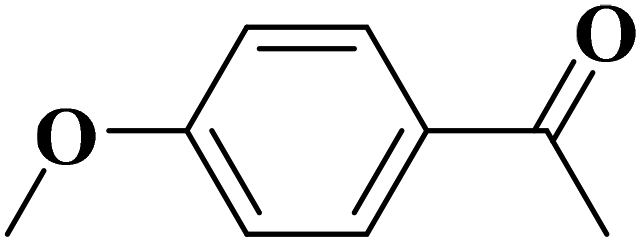
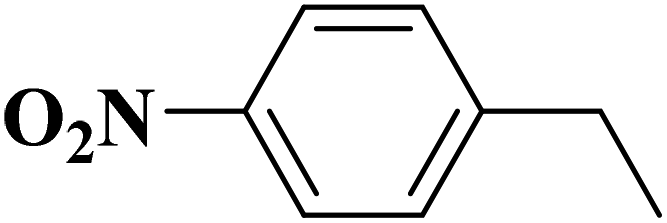
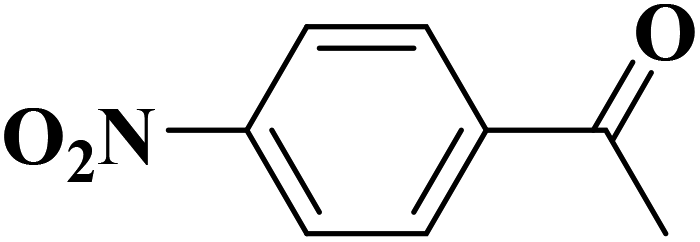
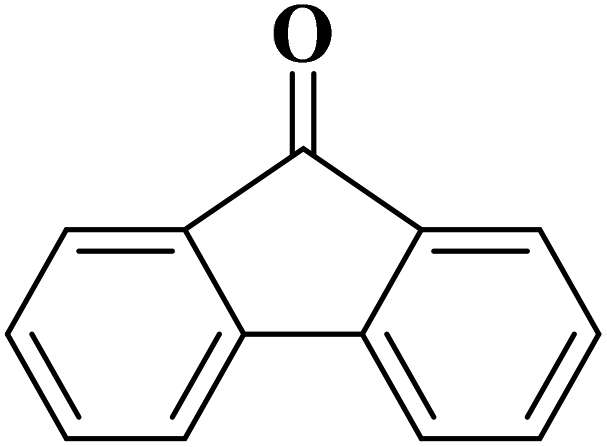
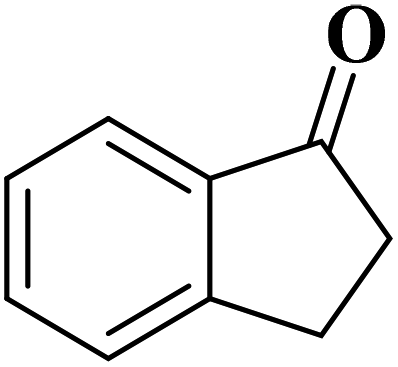
Sun and co-workers231 synthesized two nitrogen-doped porous carbon materials through direct carbonization of the petioles and blades of apricot leaves at 800 °C for 2 h under N2 flow without adding nitrogen precursors (denoted as ALPC and ALBC) (Fig. 27). Characterization results indicated that as-obtained compounds were similar in graphitization degree and element composition, but differ significantly in pore volume and surface area. Such differences can arise from the different contents of proteins, vascular bundles, and inorganic salts in blades and petioles. The catalytic behavior of ALPC and ALBC was evaluated in the oxidation of ethylbenzene and ALPC showed higher catalytic activity owing to the high surface area, and high mean pore size, as well as doped N species. Additionally, the catalytic performance of ALPC was also investigated in the oxidation of different aromatic alkanes and products obtained with high selectivities and diverse yields (Table 9). Furthermore, the ALPC could be used for 5 cycles with a negligible reduction in efficiency presumably owing to the loss of catalyst during the recycling process. The authors noted that the carbon catalysts derived from the blades and petioles of parasol tree leaves and poplar leaves showed the same difference in reaction. These results can provide guidance for preparing carbon materials from leaves.
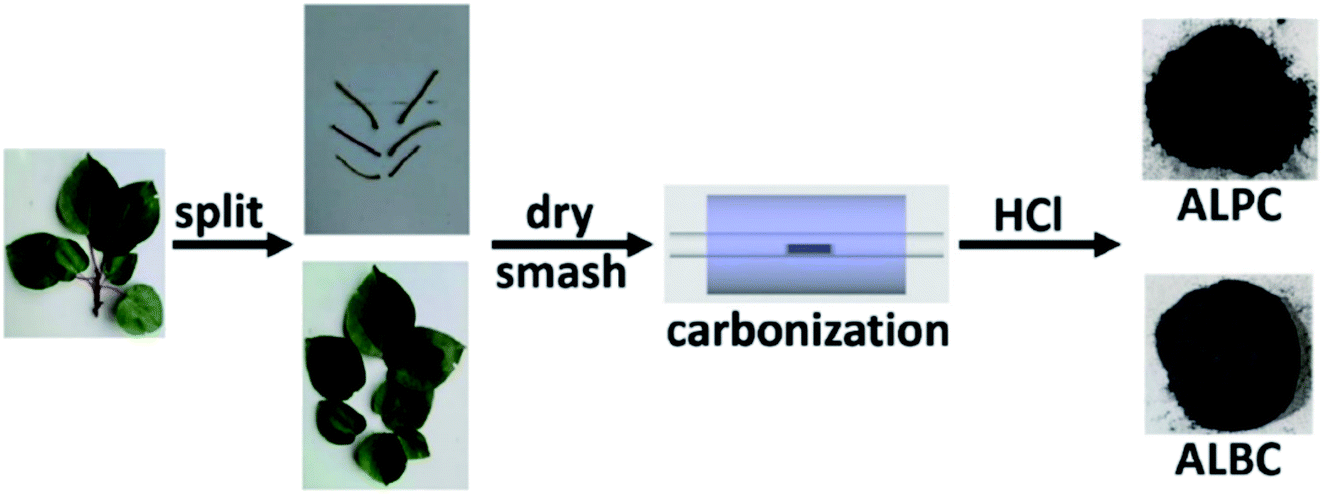 | ||
| Fig. 27 Preparation of ALPC and ALBC. Reprinted with permission from ref. 231. Copyright 2020 Springer Nature. | ||
| Substrate | Product | Con. (%) | Sel. (%) |
|---|---|---|---|
| a ALPC as a catalyst, TBHP as an oxidant at 80 °C. | |||
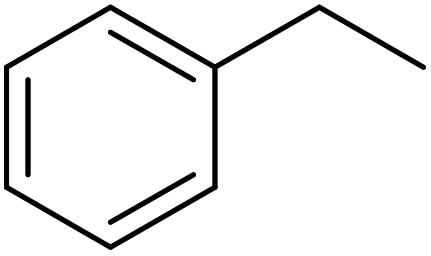 |
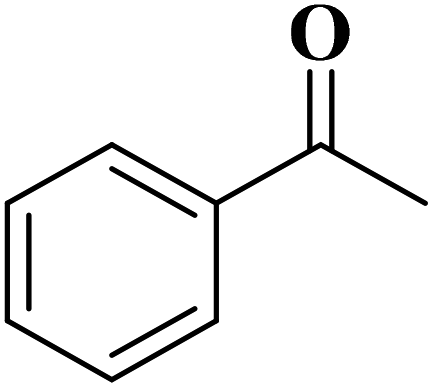 |
93 | 99 |
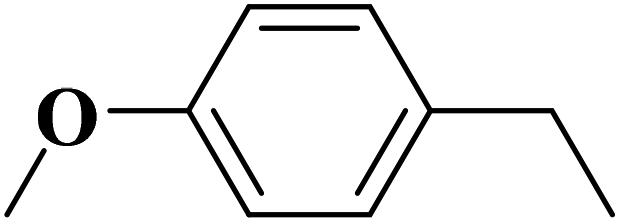 |
 |
15 | 94 |
 |
 |
54 | 98 |
 |
 |
25 | 98 |
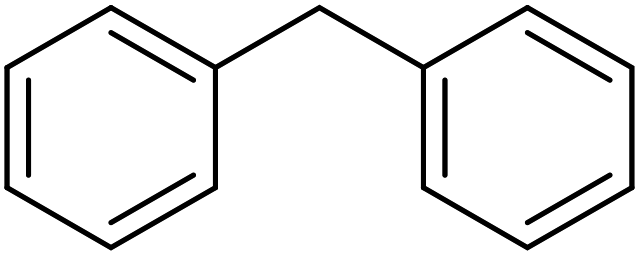 |
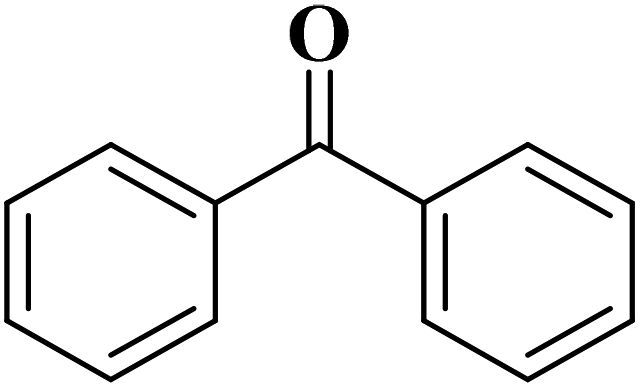 |
65 | 98 |
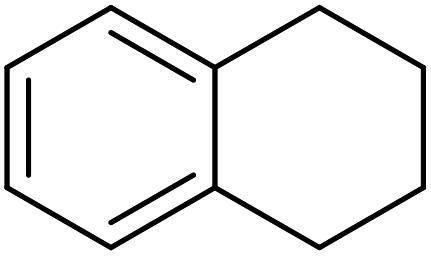 |
 |
83 | 93 |
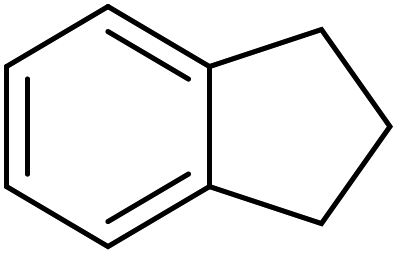 |
 |
81 | 89 |
Verma et al.232 reported an effective, sustainable, and inexpensive chitosan-derived porous carbon nitride (PCNx) catalyst originated from marine waste for selective oxidation of 5-hydroxymethyl-furfural (HMF). The reaction was carried out at 70 °C under ambient air pressure and 2,5-furandicarboxylic acid (FDCA) was attained in high yield (Scheme 21). The graphitic N in the catalyst activated the oxygen and played a main role in the hydrogenation. Additionally, the PCNx showed high reusability, and no remarkable decrease of efficiency was observed during the five consecutive runs.
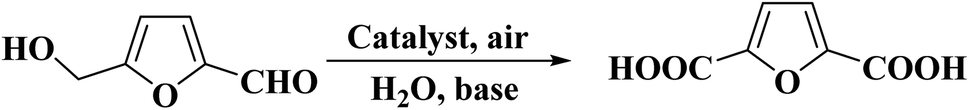 | ||
| Scheme 21 Oxidation of HMF catalyzed by PCNx. | ||
Chitosan as a renewable, abundant, and inexpensive precursor was applied by Zhang and colleagues233 for the manufacturing of nitrogen-doped carbon materials. Also, to increase the nitrogen loading, urea as a commercially available, nitrogen-enriched, and cheap co-precursor was added. The catalytic performance of these materials was largely dependent on the surface area and the type of N species. The NC-950 indicated the best catalytic efficiency for the nitric acid-mediated oxidation of 5-hydroxymethylfurfural (HMF) into 2,5-diformylfuran (DFF) at 100 °C in the presence of f molecular oxygen as the terminal oxidant (Scheme 22). The control tests demonstrated that firstly, HNO3 accelerated reaction, and O2 was used as a secondary oxidant for recovery of HNO3. Additionally, the designed material retained its activity and selectivity for six consecutive recycles.
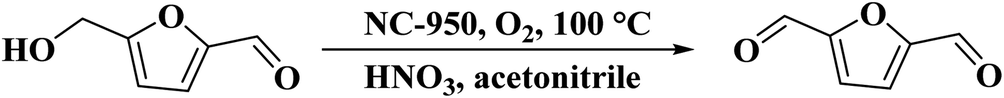 | ||
| Scheme 22 Oxidation of HMF over NC-950. | ||
In 2016, Sun and co-workers89 used a facile pyrolysis method to fabricate high-quality N-doped graphene (NG) nanomaterials using urea as a nitrogen precursor, glucose as a carbon precursor, and ferric chloride as both a catalyst and a template. A relatively high nitrogen doping level and a low oxygen content were obtained at a moderate temperature. The achieved NG effectively catalyzed phenol oxidation by activation of peroxymonosulfate (PMS). Electron paramagnetic resonance (EPR) spectrums showed that both ·OH and SO4−˙ were formed during oxidation processes and played key roles in the oxidation of phenol solutions.
Hou and his research team234 introduced several nitrogen-doped graphenes with different types and amounts of nitrogen through thermal treatment of GO under ammonia flow and their applications were demonstrated in aerobic oxidation of 5-hydroxymethyl-furfural (HMF) using 2,2,6,6-tetramethylpiperidin-oxyl (TEMPO) as co-catalyst. An excellent HMF conversion and very good selectivity of 2,5-diformylfuran (DFF) were provided at 100 °C under air pressure for 6 h (Scheme 23). Based on XPS results and other control tests, the authors found that the graphitic N atoms activated molecular oxygen. They also concluded that the synergistic effect of NG, TEMPO, and molecular oxygen was the reason for the improved catalytic activity. In addition, the apparent activation energy of N-doped graphene for oxidation of HMF was much lower than those of conventional metal catalysts supported on active carbon (Au/C, Pd/C, Pt/C, and Ru/C).
 | ||
| Scheme 23 Oxidation of HMF catalyzed by NG. | ||
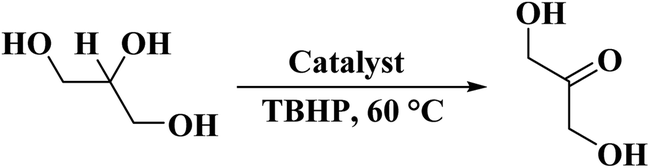
In 2017, Villa et al.235 reported the first metal-free catalyst for the oxidation of glycerol to dihydroxyacetone (DHA) in the presence of TBHP as an oxidant (Scheme 24). Two nitrogen-rich carbon nanotubes were synthesized through catalytic CVD technique at 700 and 800 °C by Fe–Mo–Al catalyst prepared using hydrothermal method and imidazole as nitrogen/carbon feedstocks. Pyridinic nitrogen groups doped in the carbon framework were identified as active centers for the reaction that are probably transformed into pyridine oxime groups generated by edge oxidation of N-doped active carbon by TBHP. The catalyst was found to be recyclable for eight successive runs and provided an environmentally benign method for the transformation of glycerol to DHA.
 | ||
| Scheme 24 Oxidation of glycerol catalyzed by N-CNTs. | ||
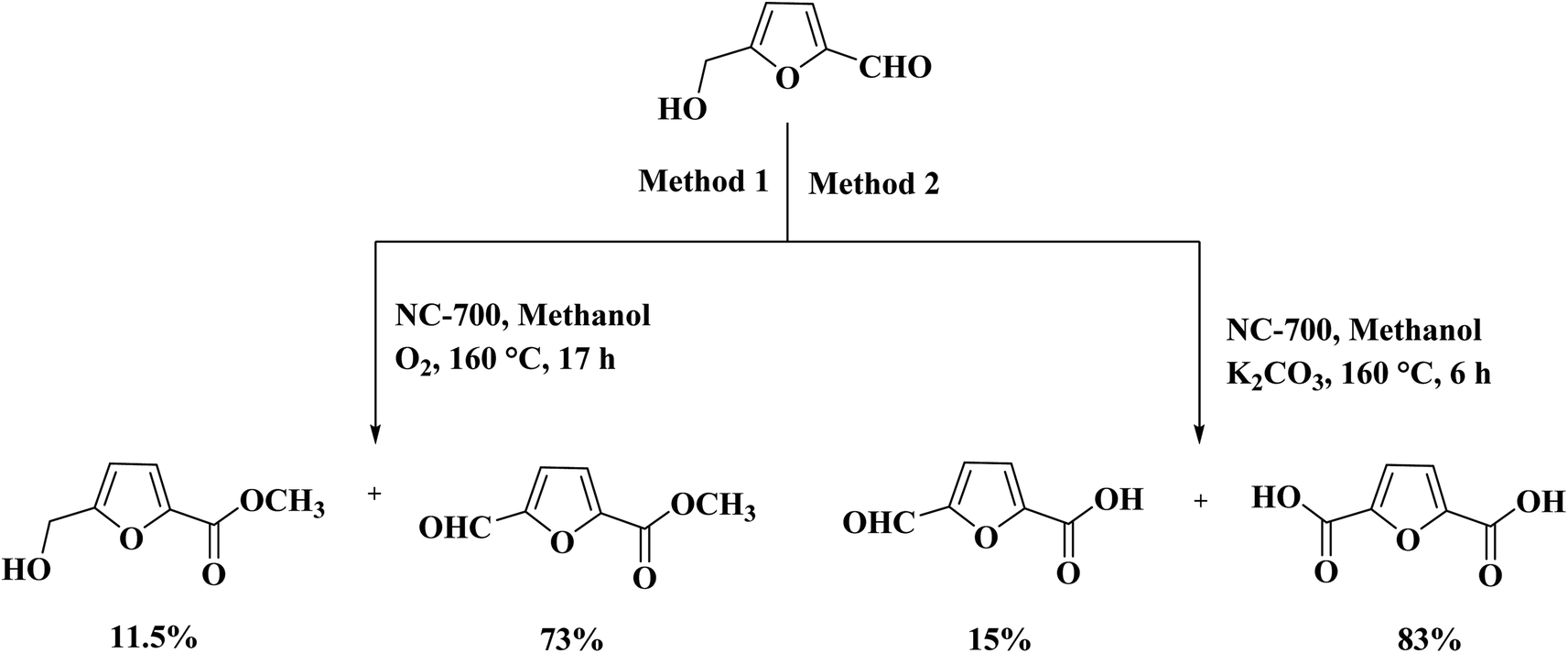
Quite recently, Xu et al.236 presented the synthesis of cheap N-doped carbon (NC) materials through pyrolysis of a mixture of bamboo sawdust, melamine, and K2CO3. The performance of these materials was studied in the selective oxidation of crude 5-hydroxymethyl-furfural (5-HMF) achieved from HFCS-90 into furan-2,5-dicarboxylic acid (FDCA). According to the characterization data and activity tests, the rate of generation of product was greatly dependent on the lattice defects in the carbon framework, the graphitic N content, and the pyrolysis temperature of the catalyst. The NC-700 catalyst afforded full HMF conversion and 83% FDCA yield under the optimized reaction conditions (2 MPa O2, 160 °C, and 6 h) (Scheme 25).
 | ||
| Scheme 25 Oxidation of 5-HMF using NC-700. | ||
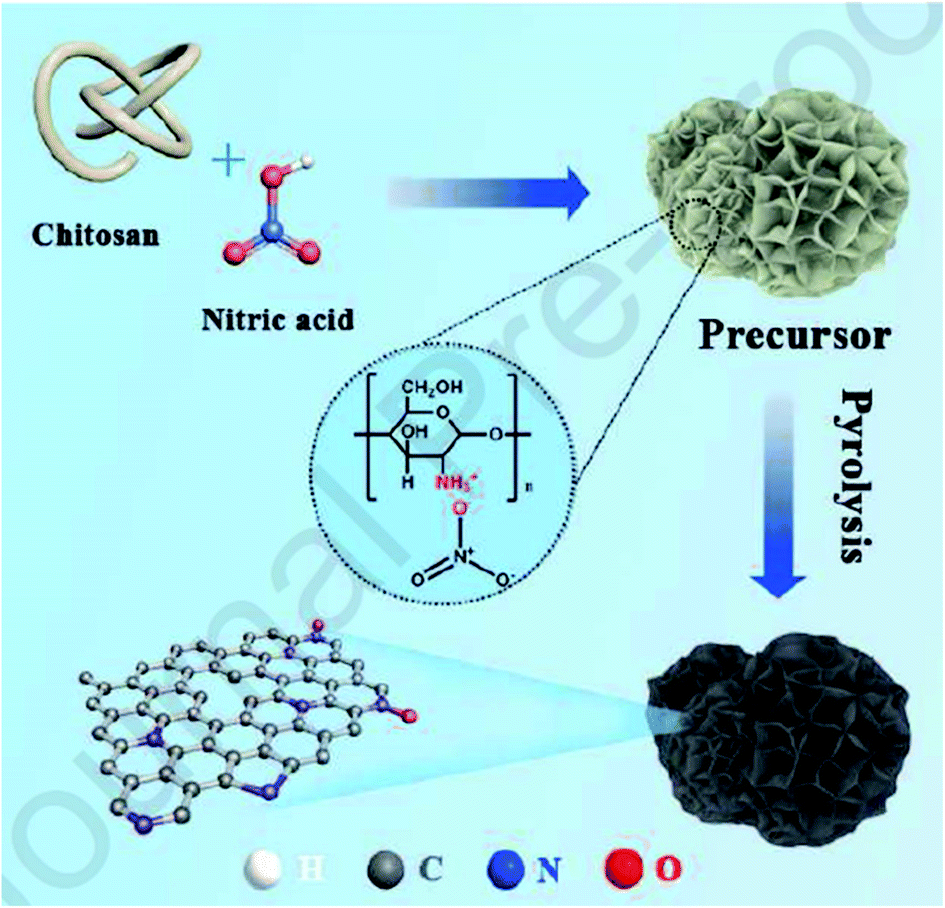
Utilizing chitosan as both C and N sources and HNO3 as a solvent, Liu et al.237 successfully fabricated N-doped defect-rich carbon materials by carbonization at various temperatures (700, 800, and 900 °C) (Fig. 28). The NC-800-5 (5: the ratio of HNO3 to chitosan) was an effective heterogeneous catalyst for the oxidation of D-xylose in alkaline aqueous media, under 1 MPa O2 pressure, at 100 °C, and D-xylonic acid could be furnished in moderate yield (57.4%) for 30 min (Scheme 26). DFT calculation revealed that graphitic N species act as active centers and contribute to the interaction between D-xylose molecular and hydroxyl ion, leading to the generation of germinal diols ion. Subsequently, the cleavage of the C–H bond produces the carboxylic group. Additionally, the catalyst showed good durability and reusability after activation treatment.
 | ||
| Fig. 28 Preparation of NC catalysts. Reprinted ref. 237. Elsevier. | ||
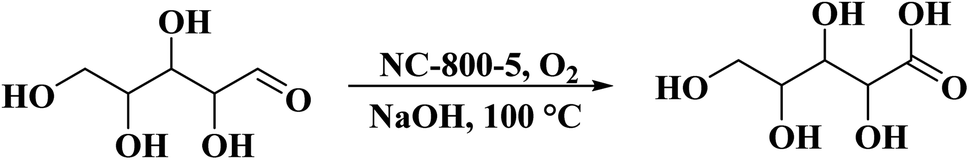 | ||
| Scheme 26 Oxidation of D-xylose in the presence of NC-800-5. | ||
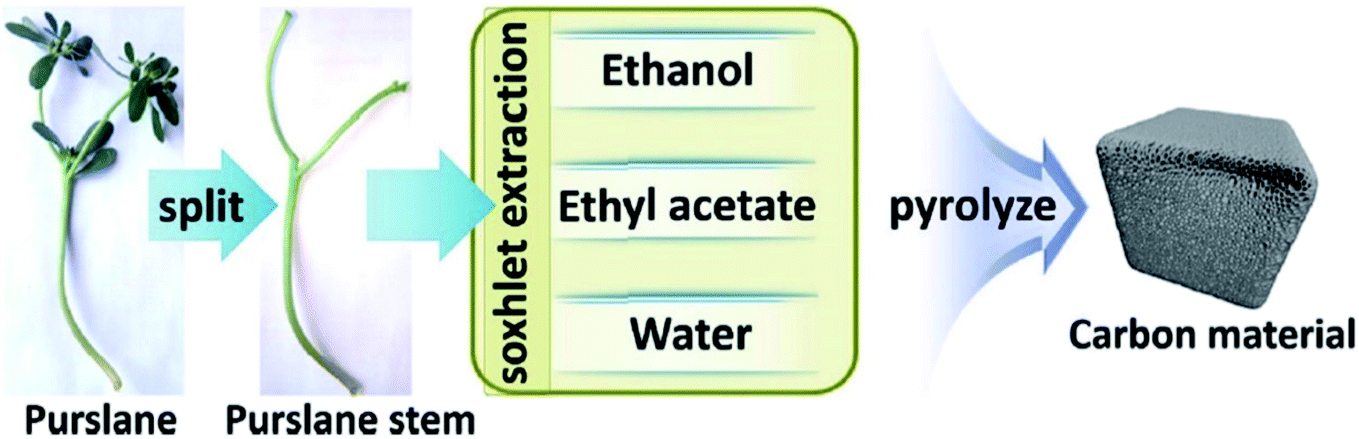
Very recently, Sun and co-workers238 selected purslane stem as a precursor to fabricate nitrogen-doped foam-like carbon catalysts by a simple two-steps method of Soxhlet extraction and pyrolysis (Fig. 29). For the preparation of these materials, firstly, the dry samples were treated with water, ethyl acetate, and ethanol by Soxhlet extraction, and then pyrolyzed under flowing nitrogen flow at 700 °C. The polarity of the solvents used in Soxhlet extraction possess is different, hence they extracted various components from the purslane stem, which possessed considerable influences on the structure and catalytic efficiency of afforded carbon samples. The catalytic ability of these carbon materials was tested in the oxidative coupling of benzylamine and the carbon material derived from the purslane stem pretreated with ethanol (SEC) was found to be the most active catalyst among others because of the many available N-containing active sites, large pore size, and high surface area. Furthermore, diverse amines were transformed into imines using SEC as catalyst under mild conditions (Scheme 27). Also, the SEC showed good durability and could be reused for at least six cycles without a remarkable decrease in activity. In addition, the catalysts synthesized from apricot leaf and celery stem pretreated with ethanol in Soxhlet extraction displayed superior catalytic performance compared with the samples pretreated with water and ethyl acetate.
 | ||
| Fig. 29 Preparation of nitrogen-doped foam-like carbon catalysts. Reprinted with permission from ref. 238. Copyright 2021 Springer Nature. | ||
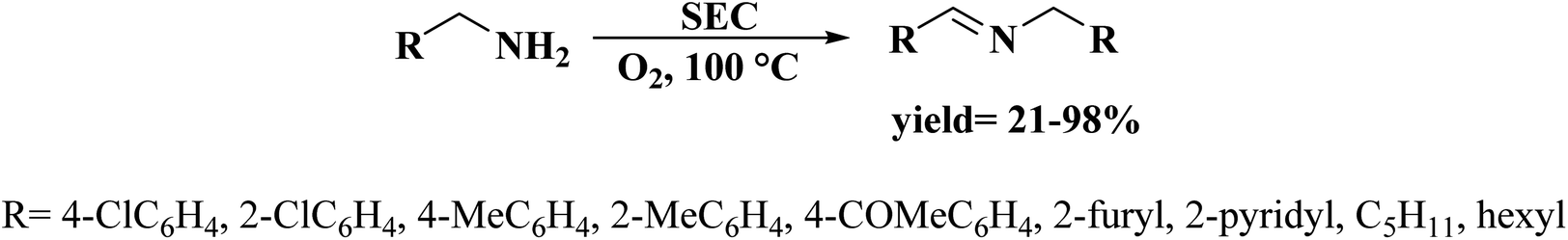 | ||
| Scheme 27 Oxidative coupling of amines catalyzed by SEC. | ||
Dhakshinamoorthy and his research team92 synthesized S-doped graphene [(S)G] from λ-carrageenan as a natural polysaccharide containing sulfate groups through carbonization at 1000 °C, followed by exfoliation (Fig. 30). The utilization of carrageenans with lower sulfate amounts and increasing carbonization temperature to 1200 °C led to the failure of S introduction. XPS results confirmed the attendance of two kinds of S species, alike to sulfide and sulfoxide. (S)G catalyzed the aerobic oxidation of styrenes to their desired benzaldehydes along with the formation of a small amount of styrene oxide (Scheme 28). Unlike (S)G, reduced graphene oxide showed no efficiency under identical reaction conditions. The catalyst was used for two runs without a remarkable decrease in selectivity and conversion.
 | ||
| Fig. 30 Preparation of (S)G. Reprinted with permission from ref. 92. Copyright 2015 Elsevier. | ||
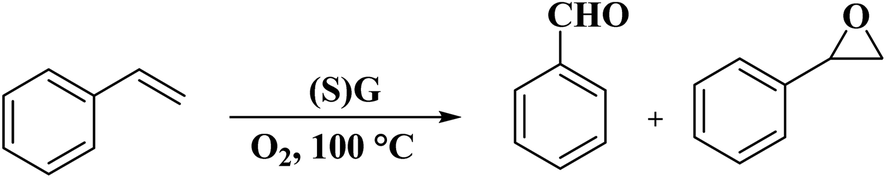 | ||
| Scheme 28 Aerobic oxidation of styrene catalyzed by (S)G. | ||
Chemical activation of polythiophene with potassium hydroxide at various carbonization temperatures was reported by Zeng and co-workers239 for the synthesis of in situ S-doped activated carbons with large specific surface areas and developed total pore volumes.
The ACS-800 demonstrated outstanding catalytic performance for oxidation of 4-chlorophenol (4CP) in 1 h with an apparent rate constant much superior compared to AC-800.
The authors pointed out that the removal of acidic functional groups (like carboxyl groups and sulphone groups) and the incorporation of S at an elevated temperature significantly increased the catalytic efficiency of ACS-800. Results showed that carbonization temperature was effective on the textural features, also could improve the surface chemistry of the materials, thereby enhancing the catalytic performances of carbons.
In comparison tests, ACS-800 indicated higher efficiency in activation of persulfate than those of the conventional catalytic systems (multi-walled carbon nanotube, reduced graphene oxide, zero-valent iron (ZVI), Fe3O4, and Co3O4). In addition, various peroxides and different aqueous organics were utilized to further assess the catalytic performance of ACS-800. Overcomes presented that ACS-800 activated different peroxides and effectively degraded various types of organic pollutants.
In 2019, Wang and co-workers240 used sulfurized graphene (SG) as an efficient catalyst for the oxidation of benzyl alcohol in the presence of H2O2 in a liquid phase reaction system (Scheme 29). SG nanosheets were prepared from the mixture of commercial graphite and sulfur powder through a modified ball-milling strategy (Fig. 31). DFT calculations indicated that the incorporation of the S atoms into the C framework can facilitate excellent catalytic performance by promoting the decomposition of hydrogen peroxide molecules into OH radicals. Moreover, a high-gravity RPB reactor was utilized and the benzyl alcohol conversion was enhanced. Both experimental and CFD simulations displayed that the high gravity level can afford faster surface renewal rate and more turbulent kinetic energy, leading to more effective collision chances between the catalyst and the reactants, finally resulting in better catalytic activity.
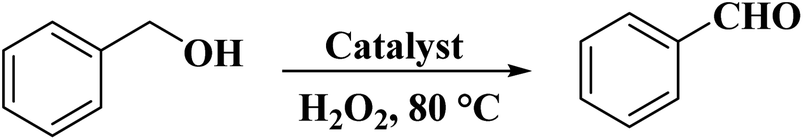 | ||
| Scheme 29 Oxidation of benzylic alcohol catalyzed by SG. | ||
 | ||
| Fig. 31 Preparation of SG. Reprinted with permission from ref. 240. Copyright 2019 Royal Society of Chemistry. | ||
In 2018, Chen and co-workers241 presented the catalytic activities of a series of P-doped carbons (MCel-PCs) in selective oxidation of benzyl alcohol at the atmospheric air as the inexpensive oxidant in the attendance of H2O as the green solvent (Fig. 32). The catalysts were prepared through pyrolyzing microcrystalline cellulose (MCel) activated by the H3PO4 at 800 °C for 2 h under flowing nitrogen. The maximum benzaldehyde yield (99.7%) and turnover frequency value were observed with MCel-PC-4(800) (4: weight ratio of MCel to H3PO4), while the un-doped catalyst only gave a 9.4% yield. According to the experiments and DFT calculations, it was confirmed that the C3PO species were the most catalytic active centers.
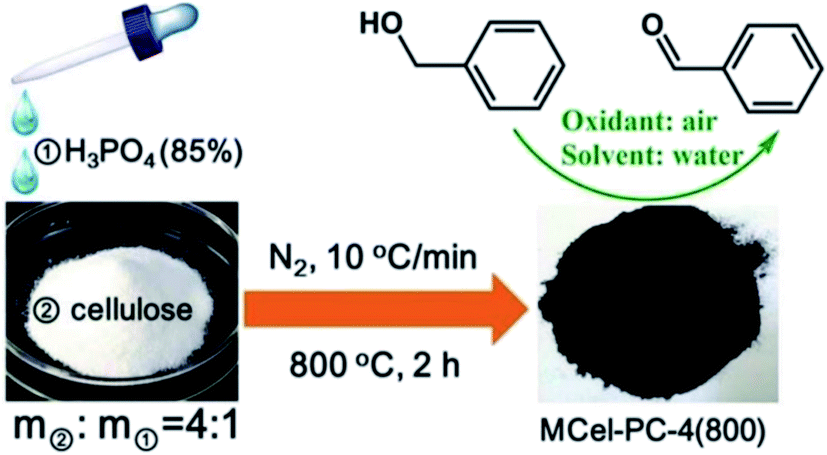 | ||
| Fig. 32 Preparation of MCel-PC4(800) and catalytic application in oxidation of benzyl alcohol. Reprinted with permission from ref. 241. Copyright 2018 Royal Society of Chemistry. | ||
In 2019, Dong et al.242 reported the design of phosphorus-doped carbon materials by applying easily accessible biomass soluble starch and H3PO4 through a two-steps approach including physical mixing and carbonization (Fig. 33). The obtained PC-700 with a highly porous structure and extremely high surface area efficiently behaved in the aerobic oxidation of benzyl alcohol with a superior TOF value compared to other heteroatom-doped carbons which were previously reported (Scheme 30). Also, this catalyst was applicable for the oxidation of different substrates, such as aliphatic, heterocyclic, alicyclic, and aromatic alcohols. The experimental results and characterization data revealed that the P–O–C species and the defects arising from P–O species doping on catalyst were the active centers for reaction. Besides, the recycling tests indicated that the PC-700 retained its initial catalytic activity for eight consecutive recycles.
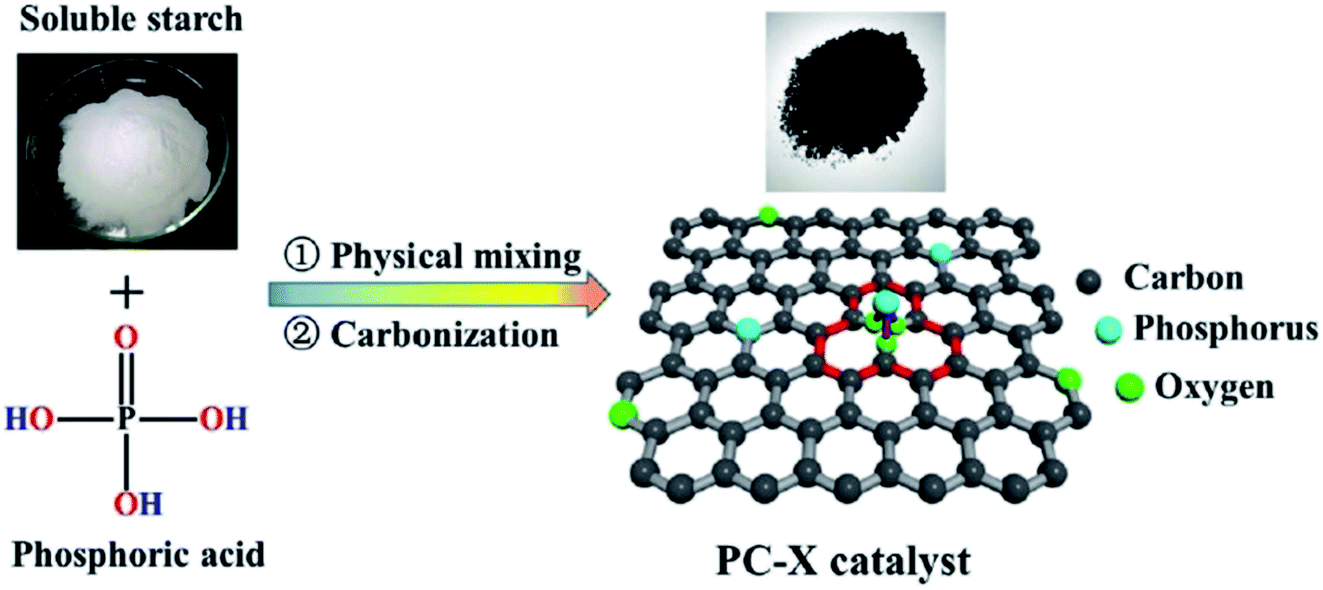 | ||
| Fig. 33 Preparation of PC-X catalyst. Reprinted with permission from ref. 242. Copyright 2019 Royal Society of Chemistry. | ||
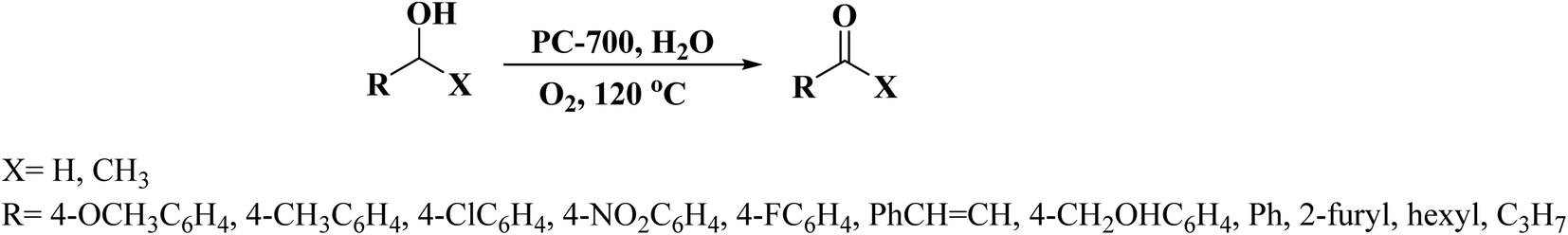 | ||
| Scheme 30 Oxidation of benzyl alcohols over PC-700. | ||
In 2017, the Wang group243 reported the synthesis of phosphorus-doped nanomesh graphene (PG) through a thermal annealing approach (Fig. 34). MgO was used as a template and triphenylphosphine (TPP) served as both carbon and phosphorus source. The as-prepared PG with lamellar hexagonal structure and large specific surface area was applied for aerobic oxidative coupling of amines (Scheme 31) which are key intermediates for the preparation of agricultural chemicals, pharmaceuticals, and fine chemicals. The reactions were performed under neat and mild conditions using O2 as the oxidant. The DFT calculation revealed that phosphorus doping into the graphene matrix exhibited better catalytic performance compared with nitrogen-doped graphene and graphene, because of the longest elongation of O–O bonds and lowest adsorption energy of benzylamine. Additionally, the PG could be easily and effectively reused for six successive cycles.
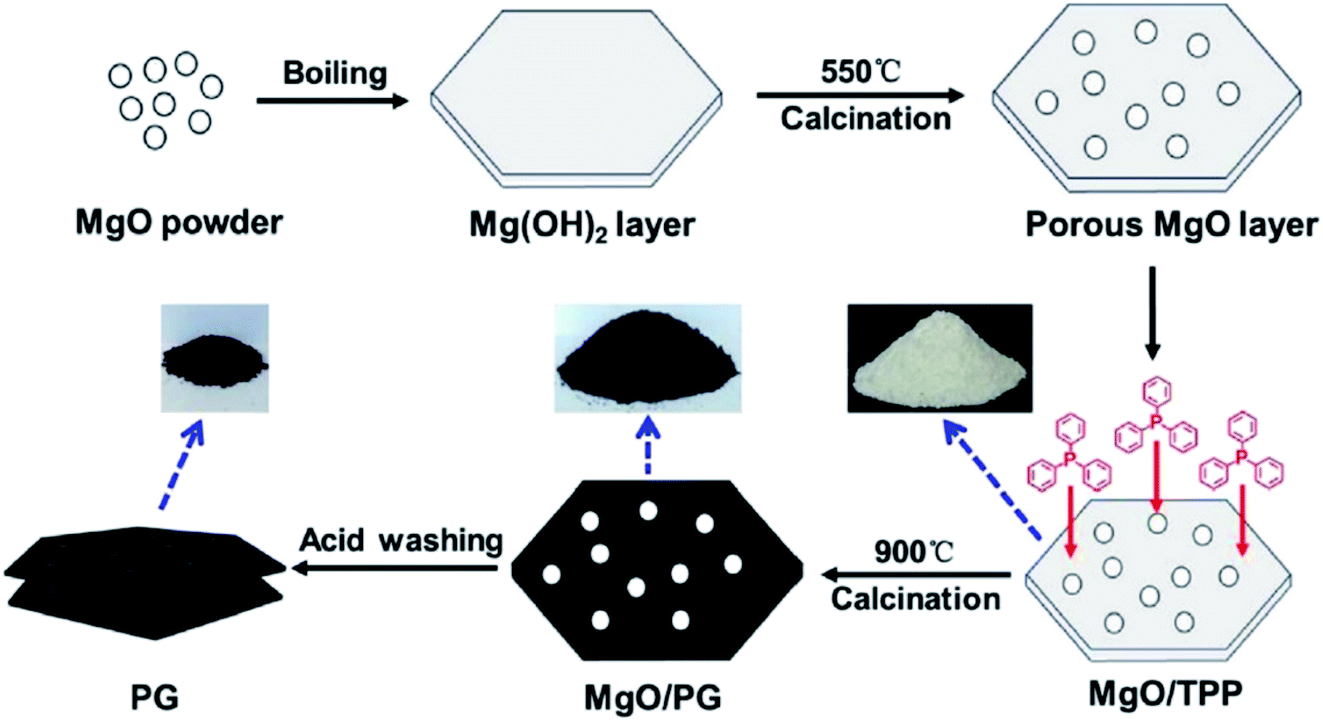 | ||
| Fig. 34 Preparation of PG. Reprinted with permission from ref. 243. Copyright 2017 Elsevier. | ||
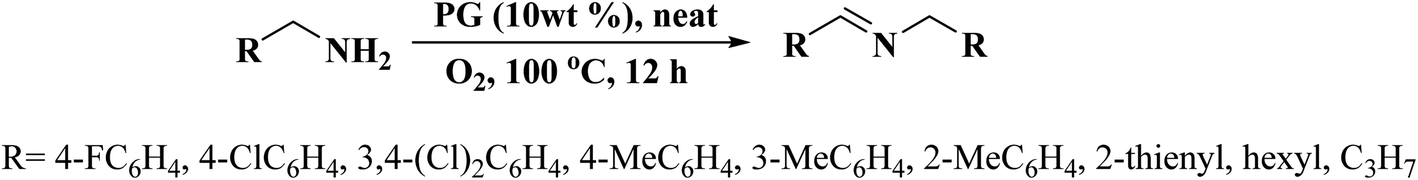 | ||
| Scheme 31 Aerobic oxidative coupling of amines in the presence of PG. | ||
In 2016, Szostak and his research team244 introduced P-dope porous carbon materials by a very facile, rapid, and scalable method (Fig. 35). In this study, phytic acid, an abundant biomass molecule, was selected as starting material. These materials successfully catalyzed aerobic oxidation of both primary and secondary benzyl alcohols to the relevant aldehydes or ketones (Scheme 32). Compared to the N-doped carbon materials, the P-doped carbon materials with higher work functions and lower activation energy showed superior efficiency in catalytic aerobic oxidation. Also, the selectivity trend for electron-deficient and electron-rich substrates was different from other heteroatom doped carbon catalysts.
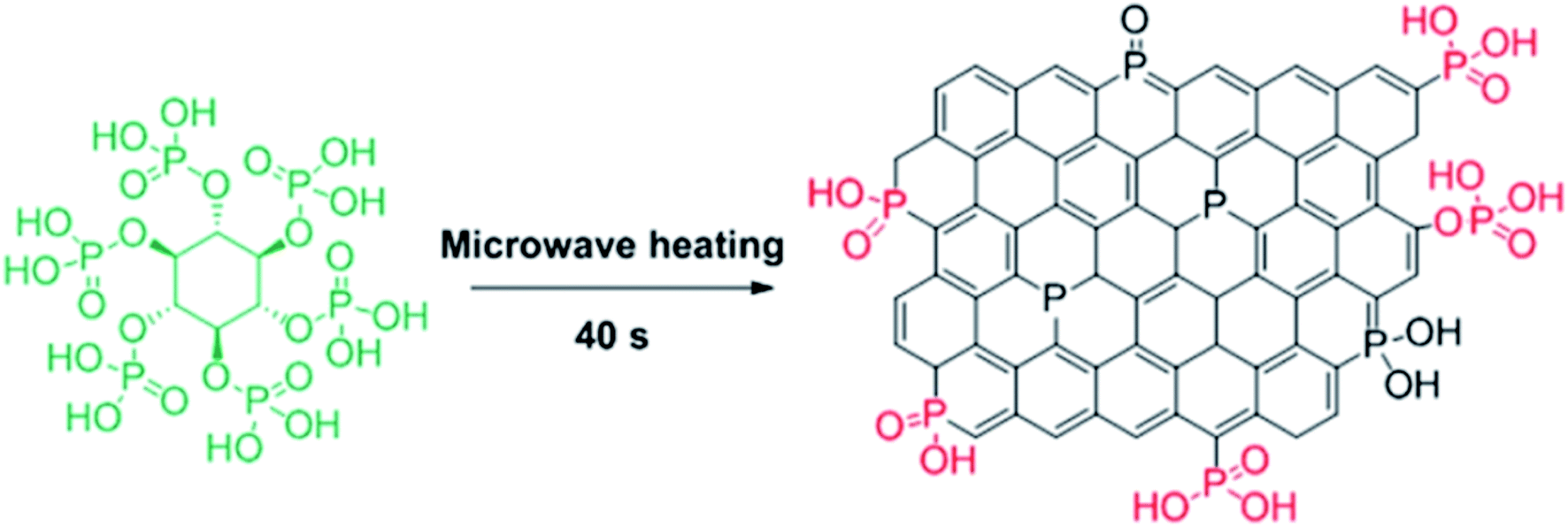 | ||
| Fig. 35 Preparation of PGc. Reprinted with permission from ref. 244. Copyright 2016 American Chemical Society. | ||
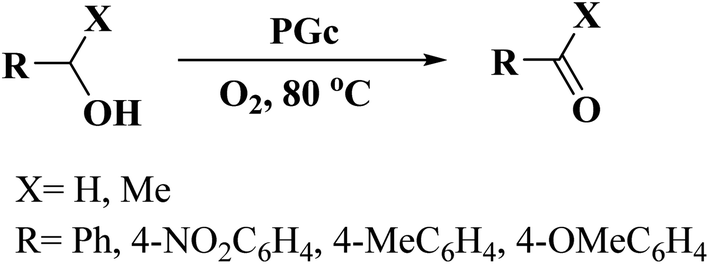 | ||
| Scheme 32 Aerobic oxidation of benzyl alcohols catalyzed by PGc. | ||
An unexpected and unique catalytic mechanism was displayed, which was different from both N-doped graphene and GO afforded using high-temperature nitrification. The outstanding catalytic approach endowed the P-doped materials with not only high catalytic activity but also reusability.
In another study in 2018, Liu and co-workers245 reported that boron-doped mesoporous carbon (B-C) can be used as a high-performance heterogeneous catalyst for the oxidative coupling of amines and the relevant imines were furnished in good to high yields and selectivity (Scheme 33). This heteroatom-doped carbon was synthesized by mixing earth-abundant biomass cellulose and H3BO3, followed by annealing in an inert atmosphere at elevated temperature (Fig. 36). Characterization data, including X-ray absorption near edge structure and X-ray photoelectron spectroscopy, displayed that B heteroatoms mainly existed in the form of CBO2 and acted as catalytic active centers.
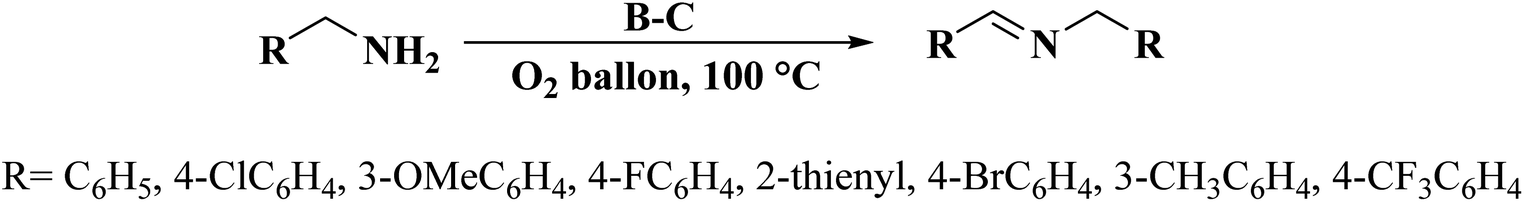 | ||
| Scheme 33 Oxidative coupling of amines in the presence of B-C. | ||
 | ||
| Fig. 36 Preparation of B-doped C. Reprinted with permission from ref. 245. Copyright 2018 American Chemical Society. | ||
Also, the proposed catalytic mechanism according to DFT calculation demonstrated that the synergistic effect of borane and oxygen in CBO2 accelerated the adsorption and dehydrogenation of benzylamine.
In 2018, the application of boron-doped graphene samples (BGs) as catalysts for the gas-phase oxidation of benzyl alcohol to benzaldehyde was reported by Li and co-workers.246 These materials with adjustable B amount of 0–2.90 at% were prepared by tuning the mass ratio of GO/boric acid. XPS analysis demonstrated that the graphitic sp2 boron species (BC3) could considerably increase the content of active sites (CO), as a result enhancing the catalytic activities. The benzyl alcohol conversion using BGs improved 2.35 times and the benzaldehyde selectivity reached 99.2%, compared to the un-doped graphene (G).
Sun and co-workers87 presented a simple one-pot method for the introduction of nitrogen and sulfur heteroatoms into graphene sheets. Ammonium nitrate and diphenyl disulfide served as N and S sources. The S and N co-doped graphene (SNG) sample was utilized as a metal-free catalyst for catalytic oxidation of phenol by activation of peroxymonosulfate (PMS).
SNG exhibited a much higher apparent reaction rate constant than those of GO, rGO, S-rGO, and N-rGO. Kinetic studies demonstrated that reaction temperature, initial phenol concentration, PMS dosage, and catalyst loading significantly influenced the phenol removal efficiency.
According to both theoretical and experimental investigations, the synergistic effect of nitrogen and sulfur co-doping improved the activation of PMS in comparison with the pure and S- (or N-) sole-doped graphene.
Also, classical quenching tests and EPR spectra indicated that both sulfate and hydroxyl radicals were formed and played key roles in phenol catalytic oxidation.
Besides, the theoretical calculations displayed that incorporation of S into N-doped graphene could remarkably change the electrostatic potential of graphene and surface charge distribution. Based on experimental observations, S acted as an efficient co-dopant to further increase the catalytic performance of N-doped graphene in phenol oxidative decomposition with radicals, compared to B, P, and I.
Bordoloi et al.247 prepared nitrogen and boron co-doped hierarchical porous carbon (BxCN) by means of a nanocasting route by utilizing ethylene diamine and CCl4 as the precursor of nitrogen and carbon, dimethylaminoborane as a new boron precursor in the presence of SBA-15 as the hard template (Fig. 37). The newly synthesized material possessed high pore volume, versatile pore diameter, and large specific surface area. The surface area and pore diameter of BxCN could be adjusted by tuning the concentration of the B source. XPS and solid-state MAS NMR results suggested the generation of N-B-C species in the catalyst structure. Moreover, DFT calculation showed the prevailing presence of monosubstituted isomer of b1 type in BxCN.
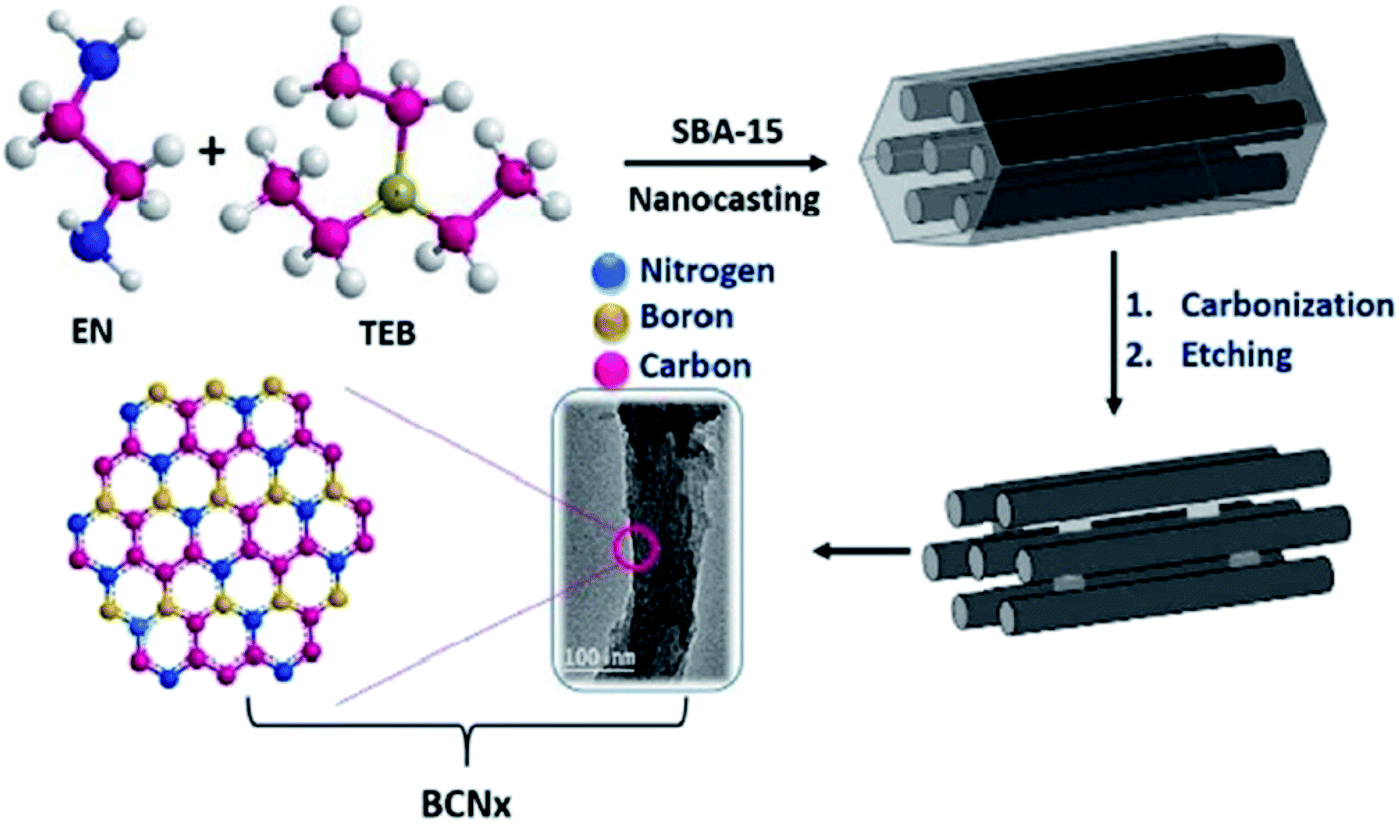 | ||
| Fig. 37 Preparation of BxCN. Reprinted with permission from ref. 247. Copyright 2016 Royal Society of Chemistry. | ||
BxCN was highly effective, selective, and stable for oxidative dehydrogenation of propane at low temperature, providing propylene in 84.6% yield without the attendance of any metal.
Lowering the HOMO energy and enhance in its absolute electronegativity are the main reasons for the improvement of the oxidation capability of the BxCN material.
In a recent study, Luo et al.248 reported a functionalization strategy to incorporate the oxygen atoms into the framework of N-doped carbon nanotubes (fabricated via a chemical vapor deposition technique) through the H2O-assisted ozone (O3/H2O) treatment with NCNT (Fig. 38). These groups showed the specific catalytic property. The as-prepared oNCNT-3 catalyst (treated at vapor generator temperature at 40 °C for 2 h) was found to be effective and selective for oxidative coupling of benzylamine to imine at ambient pressure under solvent-free conditions (Scheme 34). The nitrogen atom adjacent to the CO group modified the reactivity of carbonyl towards the activation of C–H as well as activated O2 molecule for bifunctionality. The benzylamine productivity was highest compared to other carbon-based catalysts which were reported.
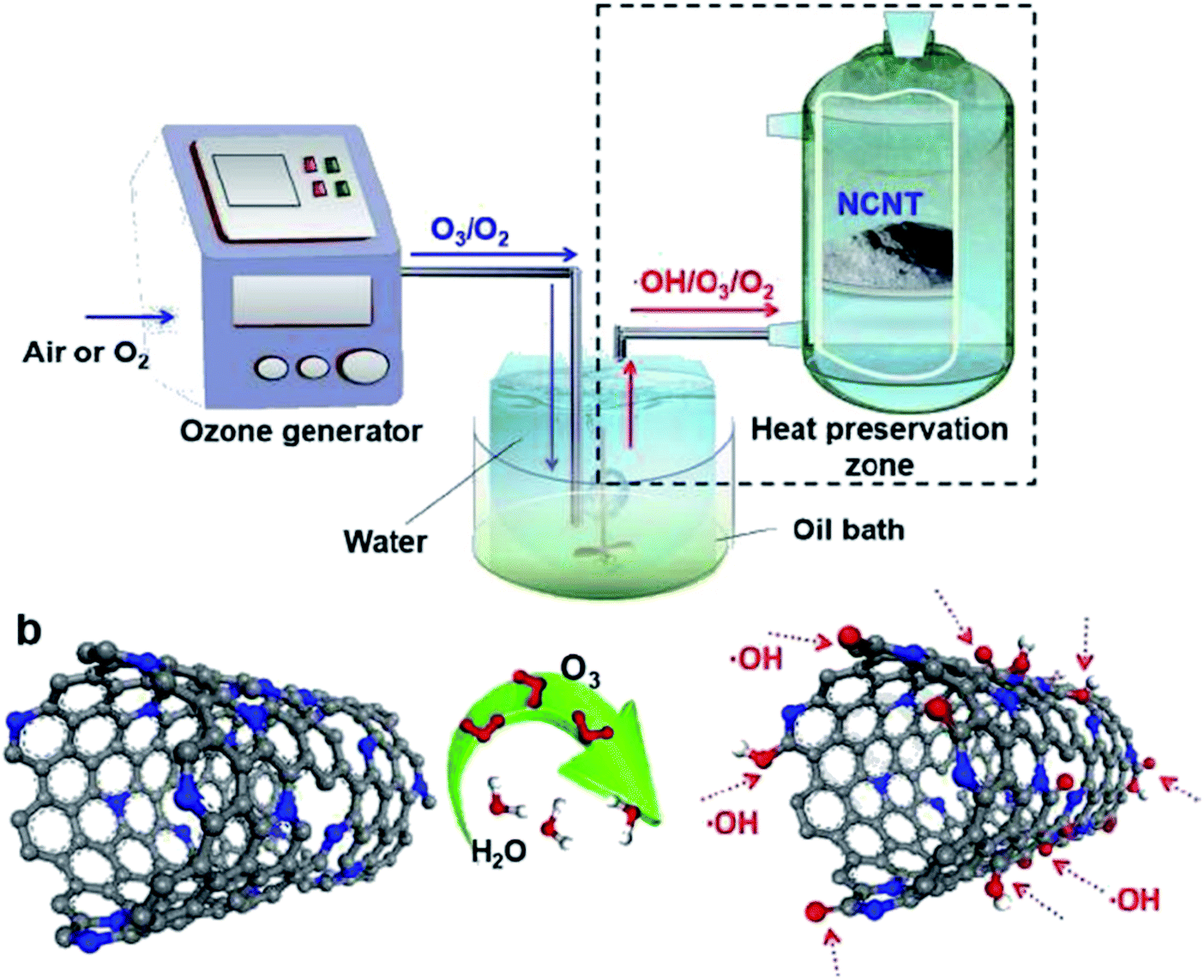 | ||
| Fig. 38 Preparation of oNCNT. Reprinted with permission from ref. 248. Copyright 2020 Elsevier. | ||
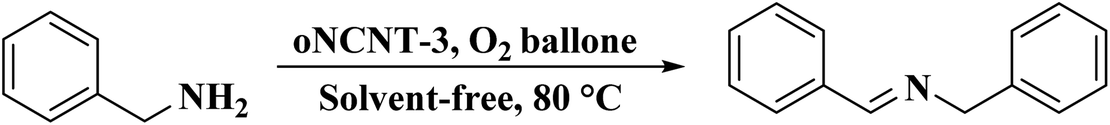 | ||
| Scheme 34 Oxidative coupling of benzylamine in the presence of oNCNT-3. | ||
An efficient and simple route to manufacture multi-heteroatoms (nitrogen, phosphorus, and sulfur) doped ultrathin carbon nanosheets was offered by Cao and co-workers249 in 2016 (Fig. 39). The synthesis procedure involved three steps and started with the synthesis of g-C3N4 nanosheets with the thermal decomposition of urea. g-C3N4 nanosheets functioned as porogen, extra N doping source, and morphology templates. Next, the g-C3N4@PZS nanosheets were formed by coating highly cross-linked poly(cyclotriphosphazene-co-4,4′-sulfonyldiphen-ol) (PZS) as C, N, P, S sources on g-C3N4 nanosheets. Finally, the calcination of the g-C3N4@PZS nanosheets under flowing argon atmosphere at desired temperature led to the decomposition of g-C3N4 nanosheets, carbonization of PZS layer, and formation of NPS-CNS. During this process, N, P, and S heteroatoms in the PZS were homogenously incorporated into the carbon nanosheets. The catalytic ability of the resulting material was assessed in selective oxidation of aromatic alkanes in aqueous media (Table 10). Also, NPS-CNS was applied as an electrochemical catalyst for oxygen reduction reaction in alkaline solution.
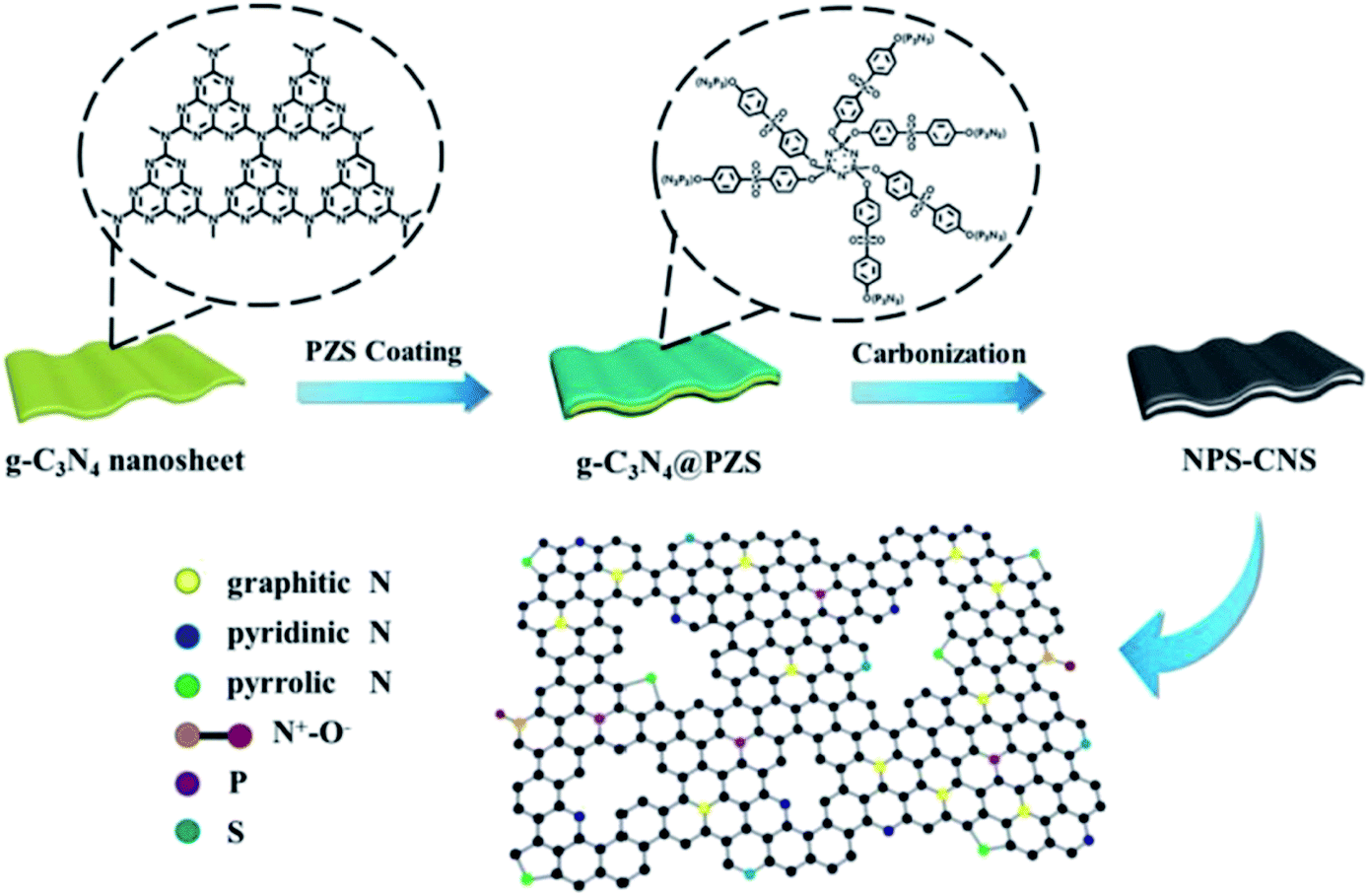 | ||
| Fig. 39 Preparation of NPS-CNS. Reprinted with permission from ref. 249. Copyright 2016 Royal Society of Chemistry. | ||
|
|||
|---|---|---|---|
| Substrate | Product | Con. (%) | Sel. (%) |
 |
 |
96 | 99 |
 |
 |
84 | 5.22:1 |
 |
 |
92 | 99 |
 |
 |
>99 | >99 |
 |
 |
84 | 1:3.4 |
 |
 |
>99 | >99 |
 |
 |
>99 | >99 |

In a separate study, the same group reported250 the synthesis of nitrogen, phosphorus, and sulfur co-doped hollow carbon shells (NPS-HCS) (Fig. 40) and utilized the prepared material as a non-metallic carbocatalyst in the selective oxidation of aromatic alkanes in water (Table 11). In this study, the PZS polymer as a shell was first coated onto a ZIF-67 core. Subsequently, the resultant ZIF-67@PZS composite was pyrolyzed under flowing argon to generate the Co2P@NPS-HCS composite. Ultimately, after acid etching to remove Co2P nanoparticles, N, P, and S co-doped hollow carbon shells were prepared. The presence of ZIF-67 was essential for the preparation of NPS-HCS and led to the following beneficial results: (1) ZIF-67 acted as a structural template for PZS coating and therefore helped to maintain the structural integrity of the hollow carbon shells during the carbonization process; (2) the MOF helped to form a mesoporous structure, enhancing the surface area of catalyst through the release of gases after the ZIF-67 decomposition; (3) the MOF increased the total content of nitrogen atoms in the NPS-HCS; (4) the ZIF-67 improved the wettability of the catalyst in H2O; (5) the Co species in the MOF promoted the graphitization of the C, which was crucial for excellent selectivity. This result was further confirmed by an additional test which indicated that if the cores of the catalysts comprise mixed metal MOFs (ZnCo-ZIFs) or only Zn (like ZIF-8), the resultant carbons could not obtain the catalytic efficiency similar to NPS-HCS because of limited graphitization.251
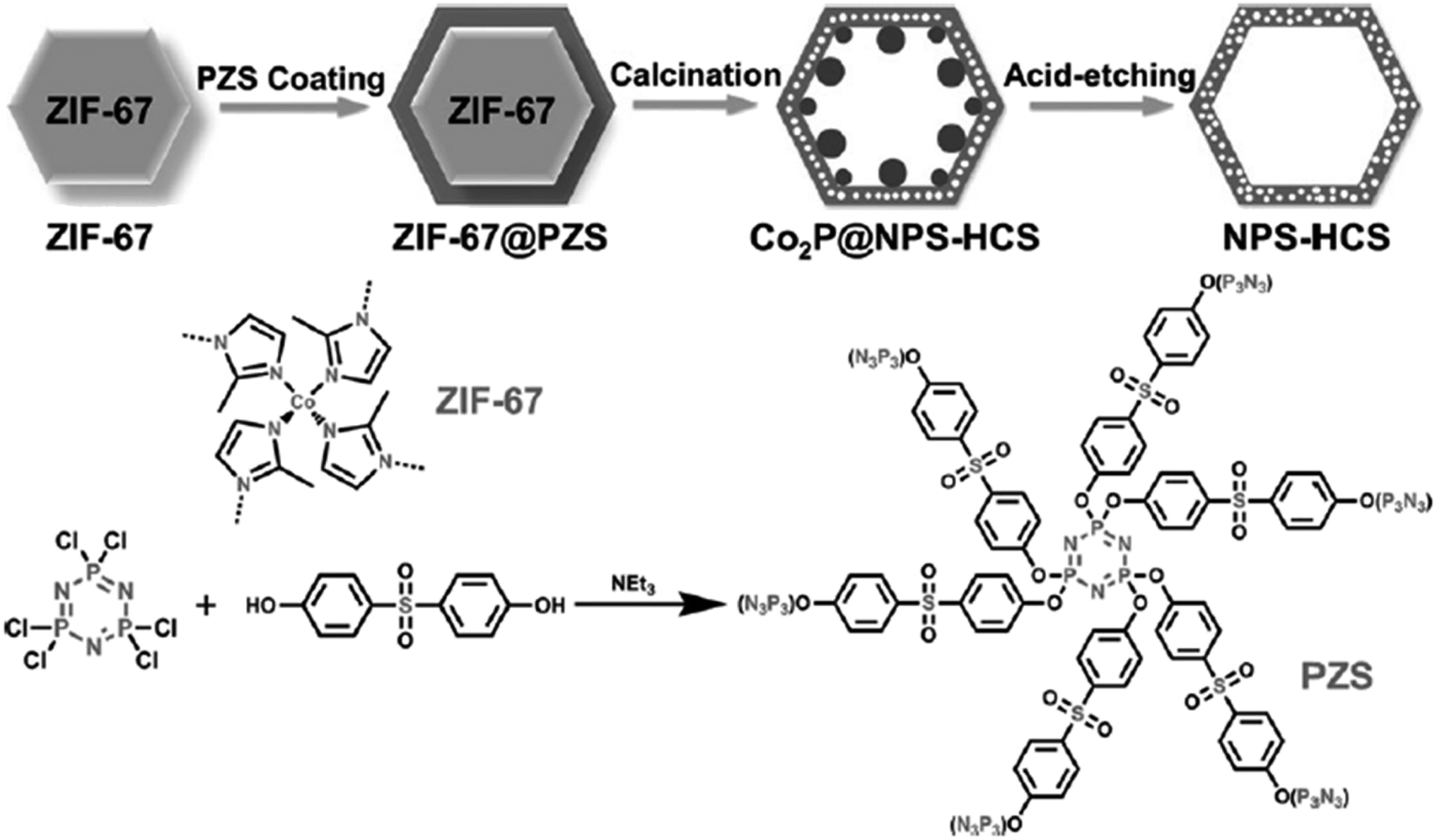 | ||
| Fig. 40 Preparation of NPS-HCS. Reprinted with permission from ref. 250. Copyright 2016 John Wiley and Sons. | ||
|
|||
|---|---|---|---|
| Substrate | Product | Con. (%) | Sel. (%) |
 |
 |
>99 | 99 |
 |
 |
>99 | 99 |
 |
 |
95 | 99 |
 |
 |
>99 | 99 |
 |
 |
90 | 1.0:3.3 |
 |
 |
>99 | 99 |
 |
 |
>99 | 99 |
 |
 |
57 | 1.0:1.3 |
 O groups, 1.3% of surface nitrogen content, and enhanced structural defects compared to the prepared H-CNT. Also, the combination of the nitrogen adsorption–desorption measurement results and the reaction results suggested that the direct dehydrogenation reaction was mainly performed on the HN-CNT external surface.
O groups, 1.3% of surface nitrogen content, and enhanced structural defects compared to the prepared H-CNT. Also, the combination of the nitrogen adsorption–desorption measurement results and the reaction results suggested that the direct dehydrogenation reaction was mainly performed on the HN-CNT external surface.
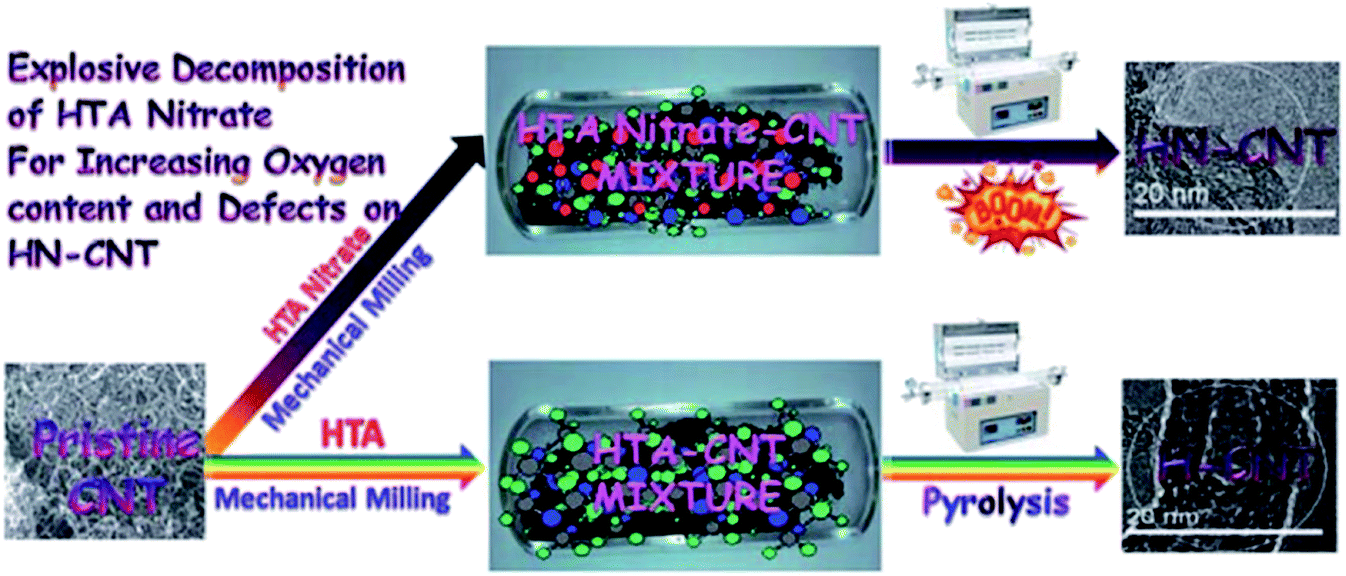 | ||
| Fig. 41 Preparation of HN-CNT and H-CNT. Reprinted with permission from ref. 252. Copyright 2015 Royal Society of Chemistry. | ||
Tsubaki and colleagues253 used nitrogen-doped graphenes as efficient catalysts in the dehydrogenation of ethanol and acetaldehyde was achieved as the sole product in all cases. These materials with various N contents were prepared through a one-pot hydrothermal method by using graphene oxide and urea as precursor and N source, respectively. XPS results revealed that three nitrogen atom types including graphitic, pyrrolic, and pyridinic were doped on the carbon sheet and their contents were different. Besides, the authors found that both reaction temperatures and N-doped contents significantly influenced the catalytic performance of nitrogen-doped graphenes.
Zhao and co-workers254 employed scalable and facile physical dry milling and pyrolysis methods to manufacture nitrogen-doped carbon nanotubes (CNTs) (Fig. 42). For the preparation of these samples, first, CNT was ground with melamine in the attendance of guanidine nitrate with various dosages, and next, pyrolyzed in diverse temperatures under flowing nitrogen. Dehydrogenation of ethylbenzene was performed for the evaluation of the catalytic activity of the synthesized N-doped CNTs. The authors stressed that the addition of guanidine nitrate possessed a significant influence on the catalytic performance, surface properties, and structure. The optimized N-doped CNTs demonstrated higher activity toward the formation rates of styrene than those of the commercially available K–Fe catalyst, the established nanodiamond, and parent CNTs. The superiority in the catalytic performance of N-doped CNTs was attributed to the smaller graphitic carbon crystallites, larger pore volume, and surface area, the basic properties originated from N-doping, and the defect- and CO group-rich surface nature.
 | ||
| Fig. 42 Preparation of N-doped carbon nanotubes. Reprinted with permission from ref. 254. Copyright 2015 John Wiley and Sons. | ||
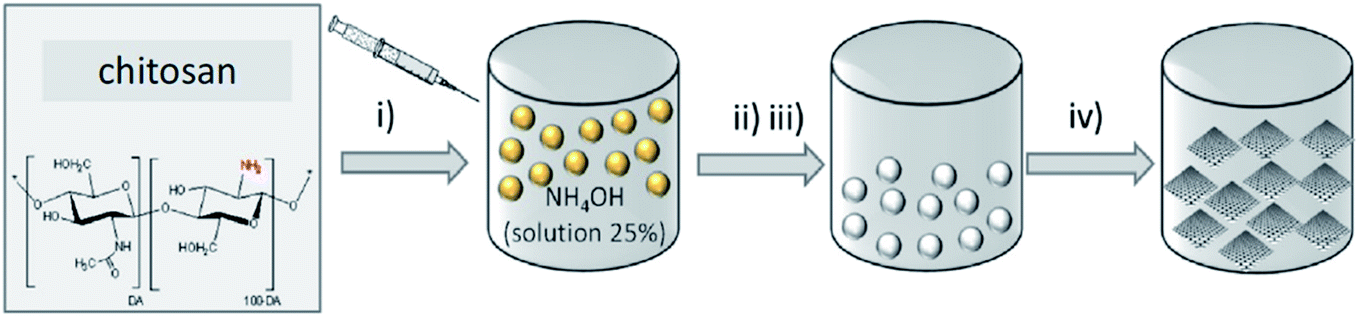 | ||
| Fig. 43 Preparation of (N)G. Reprinted with permission from ref. 255. Copyright 2019 Elsevier. | ||
 | ||
| Scheme 35 Henry (1) and Michael (2a and 2b) reaction. | ||
Another strategy for the preparation of hierarchically porous N-doped carbon nanotubes was reported by Liu and co-workers.103 The resulting materials were prepared using morphology-preserved carbonization of carbazole- and pyridine bifunctionalized conjugated microporous polymers in various temperatures (400, 600, and 800 °C) under flowing nitrogen and without adding any template (Fig. 44). The N-CNTs-800 functioned as a superior catalyst for C–H arylation of benzene with good durability and simple recyclability for five cycles. This catalyst was also applicable for aryl iodides containing electron-releasing and electron-attracting groups, providing the products in good to high yields (Scheme 36). Besides, N-CNTs-800 displayed high catalytic activity for the hydrogenation of nitrobenzene to aniline. Furthermore, the selective oxidation of aromatic alkanes in aqueous media was successfully carried out over N-CNTs-800. The origin of these superior catalytic activities can be related to high BET surface area, the unique nanotubular structures, and the N doping.
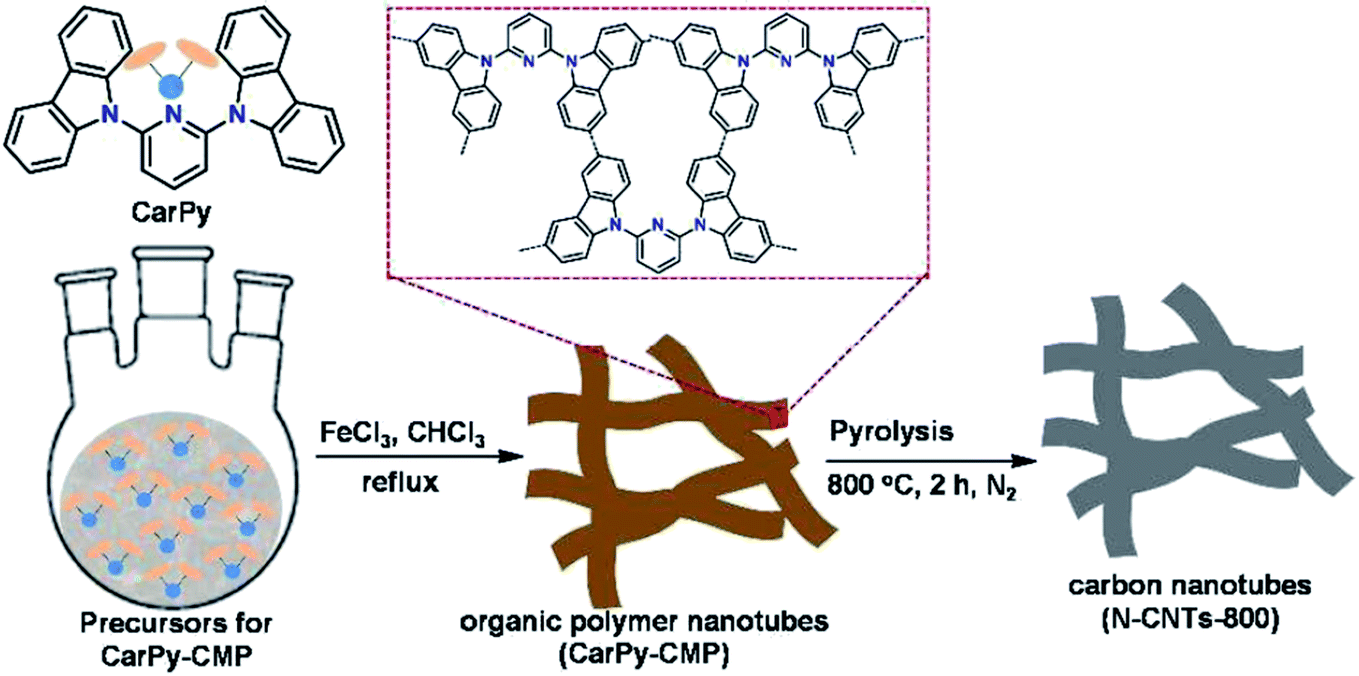 | ||
| Fig. 44 Preparation of N-CNTs-t. Reprinted with permission from ref. 103. Copyright 2017 Royal Society of Chemistry. | ||
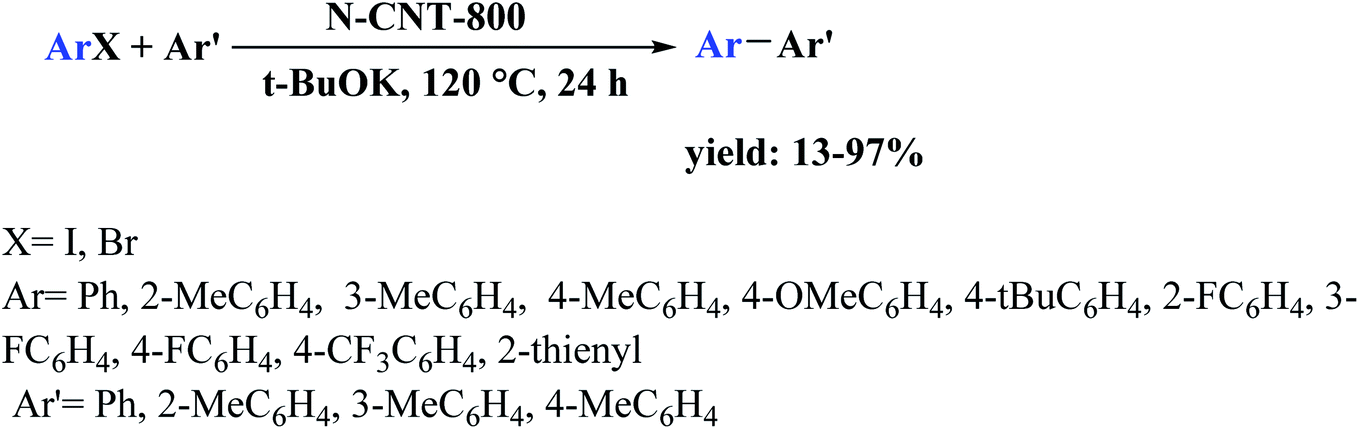 | ||
| Scheme 36 C–H arylation in the presence of N-CNTs-800. | ||
 | ||
| Scheme 37 Isomerization of glucose catalyzed by MCN-2-DH. | ||
In 2019, Tsang and co-workers257 developed a solid base N-doped biochar catalyst through two-step pyrolysis of spent coffee grounds and melamine as C and N sources, which was found to catalyze the isomerization of glucose to fructose (Fig. 45). This catalyst displayed higher selectivity compared with homogeneous base catalysts including amines and aqueous hydroxides and comparable catalytic efficiency. XPS results revealed the predominant formation of pyridinic N species on the surface of the biochar, which were mainly responsible for the strong basic properties in the catalyst. The authors also assessed the influence of co-solvent on base-catalyzed conversion and found that application of acetone enhances the overall basic properties using stabilizing protonated H2O clusters by hydrogen bonding, thereby leading faster glucose conversion and superior fructose selectivity in comparison to H2O.
 | ||
| Fig. 45 Preparation of N-doped biochar and catalytic application in isomerization of glucose. Reprinted with permission from ref. 257. Copyright 2018 American Chemical Society. | ||
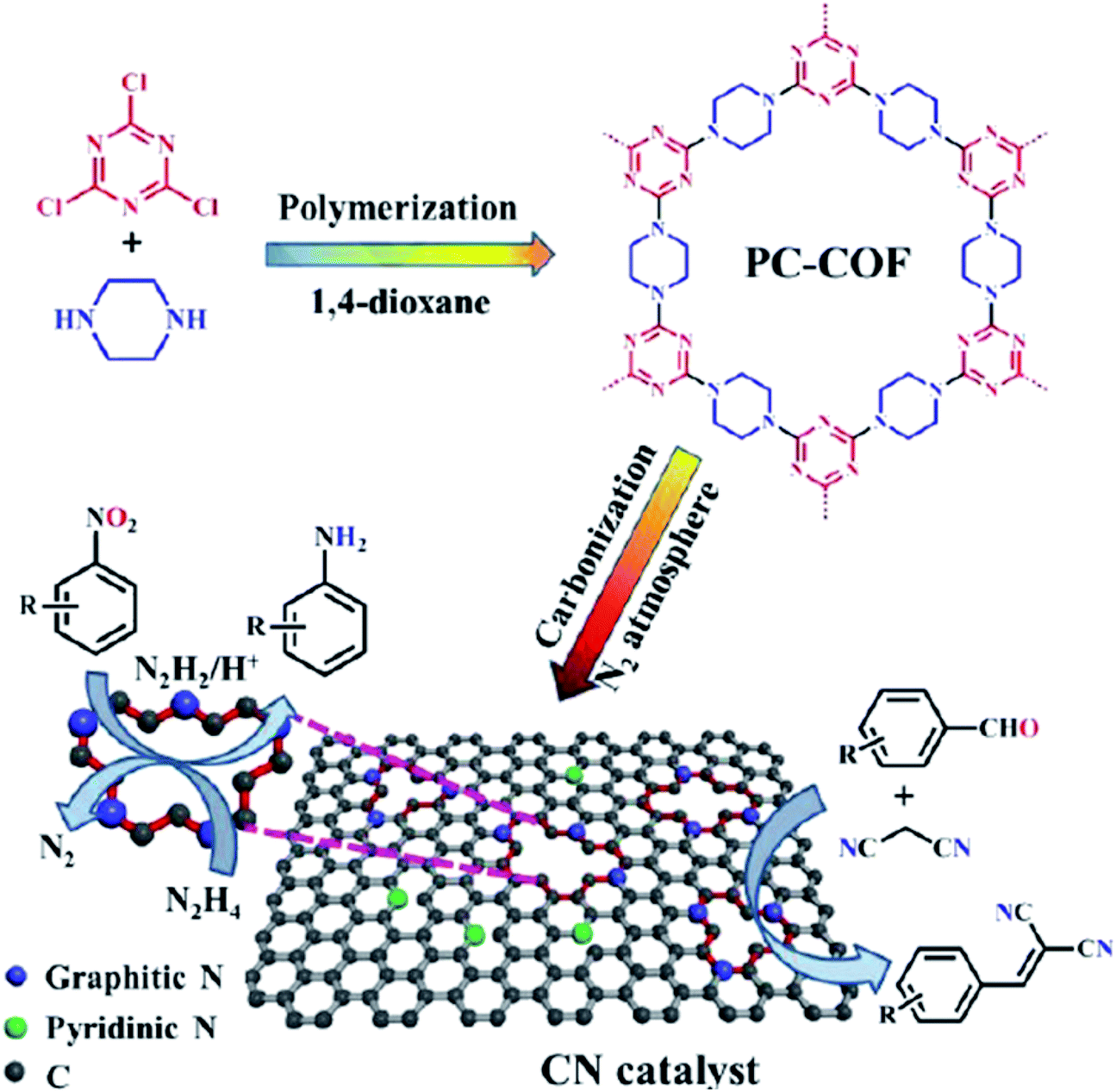 | ||
| Fig. 46 Preparation of NC. Reprinted with permission from ref. 30. Copyright 2019 Elsevier. | ||
 | ||
| Scheme 38 Knoevenagel condensation of aromatic aldehydes with NC-700. | ||
In 2015, Arai and co-workers258 employed N-doped carbon materials for the Knoevenagel condensation of ethyl cyanoacetate and benzaldehyde (Scheme 39). These catalysts were prepared through the calcination of commercial polyacrylonitrile (PAN) and then ammoxidation for doping nitrogen (ammoxidation of the calcined PAN). The calcined PAN showed very low catalytic activity for the reaction, but it was significantly increased by the ammoxidation (but the ammoxidation remarkably increased its catalytic activity) and the improvement depended on the calcination temperature as well as ammoxidation temperature. XPS tests displayed the presence of pyridine-type and pyrrole-/pyridone-type nitrogen atoms and the content of the pyridine N atoms were important factors to determine the catalytic performance. The N-carbon catalysts synthesized from PAN were much more effective compared to reported solid-based catalysts such as metal oxide-based catalysts, N-doped carbon nanotubes, and N carbon derived from activated carbon.
 | ||
| Scheme 39 Knoevenagel condensation catalyzed by N-doped carbon catalysts. | ||
 | ||
| Scheme 40 Synthesis of 1,5-benzodiazepine derivatives in the presence of S-doped graphene. | ||
:1 was ball-milled. Then, the B,N-C materials with high contents of boron and nitrogen up to 7 atom% and 10 atom%, respectively were obtained by pyrolysis of Glu/H3BO3 composites in various temperatures. By tuning the molar ratio of H3BO3 to Glu, the surface chemical states of B and N could be simply modulated. Also, with increasing H3BO3 amount, the pore size of samples could be adjusted ranging from micropores to mesopores. The catalytic activity of as-designed B,N-Cs materials was greater than those of the physically mixed catalyst of NC-900 and B4C and the pure N doped carbon catalyst (NC-900) in the cycloaddition of CO2 with epoxides (Table 12). The enhanced catalytic performance was related to the positive cooperative effect between N and B sites.
 | ||
| Fig. 47 Preparation of porous B,N-Cs. Reprinted with permission from ref. 260. Copyright 2020 John Wiley and Sons. | ||
|
|---|
 |
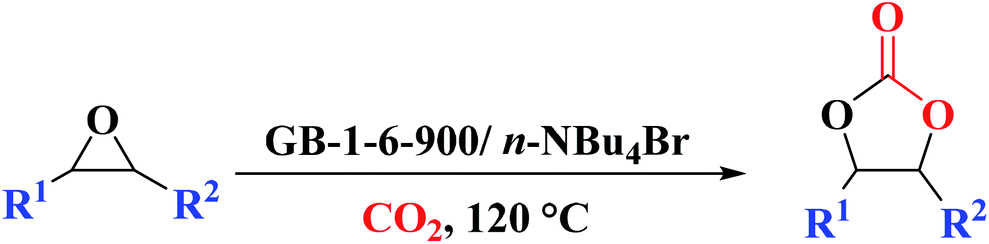
XPS data revealed that BN3 in these catalysts served as a critical factor to facilitate the ring-opening process of epoxides.
3. Conclusions and future perspectives
Metals and metal oxides have been broadly applied as catalysts in different organic reactions. Heteroatom-doped carbon materials have outstanding advantages such as earth abundance, low-cost, high specific surface areas, pore volumes, large amounts of surface defects, strong tolerance to acidic/alkaline media, and structure tenability at the molecular and morphological levels and hence can be considered as ideal alternatives to the metal catalysts. Unlike metal alloys that often suffer from separation problems, good operational durability can be obtained for most of the heteroatom-doped carbons because of the covalent chemical bonds between the carbon and heteroatom.In addition, due to their wide accessibility, environmental friendliness, and no heavy metal pollution, carbon catalysts are good candidates for green and sustainable chemistry.
In this review, we summarized recent advances in the development of metal-free heteroatom-doped carbon materials by focusing on their preparation and catalytic applications in organic transformations (Table 13).
| Catalyst | Dopant | Application | Yield or conversion% | Ref. |
|---|---|---|---|---|
| NC-950 | N | Reduction of nitro compounds | C = 87.5–100 | 187 |
| NGU/K-900 | N | N-Formylation of amines | Y = 69 to >99 | 188 |
| NCNTs-800 | N | Reduction of nitroarenes | C = 70.3–100 | 189 |
| N-CNTs | N | Hydrogenation of nitrobenzene | C = 60 | 190 |
| m-NCNTs | N | Reduction of nitroarenes | C = 63.7–100 | 16 |
| NHG | N | Hydrogenation of nitrobenzenes | Y = 93.6–99.1 | 191 |
| NC-2 | N | Hydrogenation of nitrobenzene | — | 192 |
| 3D-NGF | N | Reduction of p-nitrophenol | — | 193 |
| N-RGO mesh | N | Reduction of 4-nitrophenol | C = 99.5 | 194 |
| NC-800 | N | Reduction of 4-nitrophenol | C = 100 | 195 |
| SG | S | Hydrogenation of p-nitrophenol | C = 100 | 196 |
| P-CNT | P | Reduction of nitrobenzenes | C = 90 to >99 | 197 |
| PV-900 | P | Reduction of nitroarene compounds | C = 72.1–99.2 | 198 |
| B-OLC-2 | B | Hydrogenation of nitroarenes | C > 99 | 199 |
| B-CNTs-2 | ||||
| S,N-CNT | S,N | Hydrogenation of 4-aminophenol | C = 100 | 200 |
| PDNSC-800 | S,N | Hydrogenation of nitrobenzenes | C = 92.7–99.9 | 201 |
| BNG-800 | B,N | Hydrogenation of nitroarenes | Y = 85–99 | 202 |
| B-NPC-1200 | B,N | Reduction of p-nitrophenol | C = 94 | 203 |
| NPG | P,N | Hydrogenation of nitroarenes | Y = 83.9–98.5 | 204 |
| NPC | P,N | Reduction of p-nitrophenol | — | 186 |
| ONPC | O,N | Hydrogenation of nitroarenes | C = 5.5–100 | 205 |
| NC | N | Hydrochlorination of acetylene | Y = 81 | 167 |
| N@CBC-FE | N | Hydrochlorination of acetylene | C = 75 | 206 |
| NC-800 | N | Hydrochlorination of acetylene | C = 98 | 207 |
| Z4M1 | N | Hydrochlorination of acetylene | C = 60 | 208 |
| ZIF-8/SAC | N | Hydrochlorination of acetylene | C = 81 | 209 |
| NC | N | Hydrochlorination of acetylene | C = 92 | 210 |
| S/B-SAC | S | Hydrochlorination of acetylene | C = 50 | 211 |
| B,N-G | B,N | Hydrochlorination of acetylene | C = 95 | 212 |
| NP-C600 | P,N | Hydrochlorination of acetylene | C > 99.2% | 213 |
| S,N-carbon | S,N | Hydrochlorination of acetylene | C = 82.44 | 214 |
| NSC | S,N | Hydrochlorination of acetylene | C = 90 | 215 |
| N-CNTs | N | Epoxidation of styrene | C = 85 | 216 |
| NBND | N | Oxidation of phenols | C = 93.5–99.5 | 217 |
| NBND | N | Oxidation of alcohols | C = 32.4–94.2 | 217 |
| NG | N | Oxidation of p-hydroxylbenzoic acid | — | 218 |
| NG | N | Epoxidation of styrene | C = 7.9 | 219 |
| N-rGO | N | Oxidation of phenol | — | 220 |
| M-G-1.5-800 | N | Oxidation of alkanes | Y = 9.8 to >99 | 221 |
| 67-CN-600 | N | Oxidative coupling of amines to imines | Y = 78 to >99 | 222 |
| 67-CN-600 | N | Oxidation of cyclohexane | C = 48 | 222 |
| 67-CN-600 | N | Oxidation of toluene | C = 6 | 222 |
| NH2-rGO | N | Oxidation of glucose to succinic acid | C = 100 | 223 |
| NCNF | N | Oxidation of arylalkanes | C = 78–99 | 224 |
| NC-800 | N | Oxidation of amines | Y = 20–99.9 | 225 |
| N-AC | N | Oxidation of alcohols | C = 21–24 | 226 |
| meso-N/C-900 | N | Synthesis of nitriles | Y = 0–93 | 228 |
| N-doped carbon foam | N | Oxidation of ethylbenzene and pyridines | C = 24 to >99 | 229 |
| NPC-700 | N | Aerobic oxidation of HMF | C = 95.3 | 230 |
| ALPC | N | Oxidation of aromatic alkanes | C = 15–93 | 231 |
| PCNx | N | Oxidation of HMF | Y = 83 | 232 |
| NC-950 | N | Oxidation of HMF | Y = 95.1 | 233 |
| NG | N | Oxidation of phenol | — | 89 |
| NG | N | Oxidation of HMF | Y > 99 | 234 |
| N-CNTs | N | Oxidation of glycerol | C = 36.5 | 235 |
| NC-700 | N | Oxidation of 5-HMF | Y = 83 | 236 |
| NC-800-5 | N | Oxidation of D-xylose | Y = 57.4 | 237 |
| SEC | N | Oxidative coupling of amines | Y = 21–98 | 238 |
| (S)G | S | Aerobic oxidation of styrene | C = 13 | 92 |
| ACS-800 | S | Oxidation of 4-chlorophenol | — | 239 |
| SG | S | Oxidation of benzylic alcohol | C = 20.86 | 240 |
| MCel-PC4(800) | P | Oxidation of benzyl alcohol | Y = 99.7 | 241 |
| PC-700 | P | Oxidation of benzyl alcohols | C < 1–99.8 | 242 |
| PG | P | Aerobic oxidative coupling of amines | C < 10–98 | 243 |
| PGc | P | Aerobic oxidation of benzyl alcohols | C < 2–98.9 | 244 |
| B-C | B | Oxidative coupling of amines | Y = 68–89 | 245 |
| BGs | B | Oxidation of benzyl alcohol | C = 21.5 | 246 |
| SNG | S,N | Oxidation of phenol | — | 87 |
| BxCN | B,N | Oxidative dehydrogenation of propane | Y = 84.6 | 247 |
| oNCNT-3 | O,N | Oxidative coupling of benzylamine | Y = 66 | 248 |
| NPS-CNS-300-1000 | N,P,S | Oxidation of aromatic alkanes | C = 84 to >99 | 249 |
| NPS-HCS | N,P,S | Oxidation of aromatic alkanes | C = 57 to >99 | 250 |
| HN-CNT | N | Dehydrogenation of ethylbenzene | — | 252 |
| NG | N | Dehydrogenation of ethanol | C = 9.11 | 253 |
| N-CNTs | N | Dehydrogenation of ethylbenzene | — | 254 |
| (N)G | N | Henry & Michael addition | Y = 80.2 & C = 41.2–100 | 255 |
| N-CNTs-800 | N | C–H arylation | Y = 13–97 | 103 |
| MCN-2-DH | N | Isomerization of glucose | Y = 31.6 | 256 |
| N-doped biochar | Isomerization of glucose | C = 22 | 257 | |
| NC-700 | N | Knoevenagel condensation | C = 98.6 to >99 | 30 |
| NC | N | Knoevenagel condensation | Y = 99 | 258 |
| S-doped graphene | S | Synthesis of 1,5-benzodiazepines | Y = 65–94 | 259 |
| B,N-Cs | N,B | Cycloaddition reaction | Y = 51–98 | 260 |
By utilizing suitable post-treatment or direct synthesis strategies, heteroatoms (mainly N, B, P, and S) can incorporate into the framework of carbon materials to substitute for carbon atoms. The choice of synthetic conditions, including temperature, source of heteroatoms, and catalyst can modulate the arrangement and distribution of heteroatoms, which further alters the chemical and electronic properties of carbons. These improved chemical and electronic properties lead to a superb performance of heteroatom-doped carbons in the various organic transformations compared to un-doped carbon materials.
The future challenges and perspectives are as follows:
(1) New and suitable starting materials and synthetic strategies are demanded to design and fabricate these carbocatalysts with a large number of active sites and high density of heteroatoms which in turn can provide the materials with increased catalytic performance.
(2) From industrial and commercial perspectives, it is also necessary to develop simple and highly efficient methods for the large-scale production of new carbon materials with a well-defined structure at low cost.
(3) Though single heteroatom-doped carbon catalysts have been received much attention, further studies on the design and synthesis of dual-, tri-, and multi-heteroatom-doped carbons and investigate their application in various organic reactions would be of great interest.
(4) Despite the improvements achieved so far, it is still very difficult to control the content of heteroatom, distributional uniformity, bonding formats, and some other features. This is especially valid for co-doping cases. All of these features directly or indirectly determine the properties of the doped carbons in catalytic reactions.
(5) The exact location of heteroatoms, the nature of their active sites in carbon materials, and their doping mechanism are still not clear. A combination of experimental studies, state-of-the-art characterizations, and powerful computational calculations are indeed highly required for the better understanding of the structure, mechanism, and kinetics of the catalytic center.
(6) Although N-doped carbon materials have been examined widely as catalysts in various organic transformations, it is evident from this review that there are other dopants such as S, P, and B beyond the N, with suitable physicochemical properties for the introduction into the carbon matrix. In addition, the use of heteroatoms such as selenium, tellurium, etc., can be also studied in the next investigations.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are thankful to Alzahra University research Council. We are also grateful to Iran National Science Foundation (INSF) for the financial support provided by the post-doctoral project (99006750).References
- H.-U. Blaser, A. Indolese, A. Schnyder, H. Steiner and M. Studer, J. Mol. Catal. Chem., 2001, 173, 3–18 CrossRef CAS.
- Y. Gong, M. Li, H. Li and Y. Wang, Green Chem., 2015, 17, 715–736 RSC.
- J. Hagen, Industrial catalysis: a practical approach, John Wiley & Sons, 2015 Search PubMed.
- J. A. Dumesic, G. W. Huber and M. Boudart, Handbook of Heterogeneous Catalysis, 2008 Search PubMed.
- Y. Rangraz, F. Nemati and A. Elhampour, Appl. Surf. Sci., 2020, 507, 145164 CrossRef CAS.
- B. Vellaichamy and P. Periakaruppan, New J. Chem., 2017, 41, 7123–7132 RSC.
- M. Li, F. Xu, H. Li and Y. Wang, Catal. Sci. Technol., 2016, 6, 3670–3693 RSC.
- M. Antonietti, N. Lopez-Salas and A. Primo, Adv. Mater., 2019, 31, 1805719 CrossRef PubMed.
- J. Oliver-Meseguer, J. R. Cabrero-Antonino, I. Domínguez, A. Leyva-Pérez and A. Corma, Science, 2012, 338, 1452–1455 CrossRef CAS PubMed.
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS PubMed.
- A. Corma, A. Leyva-Pérez and M. J. Sabater, Chem. Rev., 2011, 111, 1657–1712 CrossRef CAS PubMed.
- S. K. Kaiser, R. Lin, S. Mitchell, E. Fako, F. Krumeich, R. Hauert, O. V. Safonova, V. A. Kondratenko, E. V. Kondratenko and S. M. Collins, Chem. Sci., 2019, 10, 359–369 RSC.
- J. Zhong, Y. Xu and Z. Liu, Green Chem., 2018, 20, 2412–2427 RSC.
- H. Xu and G. Luo, J. Ind. Eng. Chem., 2018, 65, 13–25 CrossRef CAS.
- X.-L. Xu, J. Zhao, C.-S. Lu, T.-T. Zhang, X.-X. Di, S.-C. Gu and X.-N. Li, Chin. Chem. Lett., 2016, 27, 822–826 CrossRef CAS.
- J. Shan, X. Sun, S. Zheng, T. Wang, X. Zhang and G. Li, Carbon, 2019, 146, 60–69 CrossRef CAS.
- K. Wang, P. Jiang, M. Yang, P. Ma, J. Qin, X. Huang, L. Ma and R. Li, Green Chem., 2019, 21, 2448–2461 RSC.
- M. R. Benzigar, S. N. Talapaneni, S. Joseph, K. Ramadass, G. Singh, J. Scaranto, U. Ravon, K. Al-Bahily and A. Vinu, Chem. Soc. Rev., 2018, 47, 2680–2721 RSC.
- H. Huang and X. Wang, J. Mater. Chem. A, 2014, 2, 6266–6291 RSC.
- D. Haag and H. H. Kung, Top. Catal., 2014, 57, 762–773 CrossRef CAS.
- C. J. Shearer, A. Cherevan and D. Eder, Adv. Mater., 2014, 26, 2295–2318 CrossRef CAS.
- X.-K. Kong, C.-L. Chen and Q.-W. Chen, Chem. Soc. Rev., 2014, 43, 2841–2857 RSC.
- P. Veerakumar, P. Thanasekaran, T. Subburaj and K.-C. Lin, Journal of Carbon Research, 2018, 4, 54 CrossRef CAS.
- A. K. Geim and K. S. Novoselov, in Nanoscience and technology: a collection of reviews from nature journals, World Scientific, 2010, pp. 11–19 Search PubMed.
- C. E. N. E. R. Rao, A. E. K. Sood, K. E. S. Subrahmanyam and A. Govindaraj, Angew. Chem., Int. Ed., 2009, 48, 7752–7777 CrossRef CAS.
- Y. Zhu, S. Murali, W. Cai, X. Li, J. W. Suk, J. R. Potts and R. S. Ruoff, Adv. Mater., 2010, 22, 3906–3924 CrossRef CAS PubMed.
- Y.-J. Wang, D. P. Wilkinson and J. Zhang, Chem. Rev., 2011, 111, 7625–7651 CrossRef CAS.
- Y. Gao, D. Ma, C. Wang, J. Guan and X. Bao, Chem. Commun., 2011, 47, 2432–2434 RSC.
- A. Primo, F. Neatu, M. Florea, V. Parvulescu and H. Garcia, Nat. Commun., 2014, 5, 1–9 Search PubMed.
- X. Hu, Y. Long, M. Fan, M. Yuan, H. Zhao, J. Ma and Z. Dong, Appl. Catal., B, 2019, 244, 25–35 CrossRef CAS.
- R. Gao, L. Pan, J. Lu, J. Xu, X. Zhang, L. Wang and J. J. Zou, ChemCatChem, 2017, 9, 4287–4294 CrossRef CAS.
- J. Song, Z.-F. Huang, L. Pan, K. Li, X. Zhang, L. Wang and J.-J. Zou, Appl. Catal., B, 2018, 227, 386–408 CrossRef CAS.
- K. Gong, F. Du, Z. Xia, M. Durstock and L. Dai, Science, 2009, 323, 760–764 CrossRef CAS PubMed.
- S. Ni, Z. Li and J. Yang, Nanoscale, 2012, 4, 1184–1189 RSC.
- L. Yang, S. Jiang, Y. Zhao, L. Zhu, S. Chen, X. Wang, Q. Wu, J. Ma, Y. Ma and Z. Hu, Angew. Chem., Int. Ed., 2011, 50, 7132–7135 CrossRef CAS.
- D. Wei, Y. Liu, Y. Wang, H. Zhang, L. Huang and G. Yu, Nano Lett., 2009, 9, 1752–1758 CrossRef CAS PubMed.
- Z. Li, H. Yu, T. Bian, Y. Zhao, C. Zhou, L. Shang, Y. Liu, L.-Z. Wu, C.-H. Tung and T. Zhang, J. Mater. Chem. C, 2015, 3, 1922–1928 RSC.
- D. Xiong, X. Li, L. Fan and Z. Bai, Catalysts, 2018, 8, 301 CrossRef.
- S. G. Peera, H.-J. Kwon, T. G. Lee and A. M. Hussain, Ionics, 2020, 26, 1563–1589 CrossRef.
- J. Quílez-Bermejo, E. Morallón and D. Cazorla-Amorós, Carbon, 2020, 165, 434–454 CrossRef.
- D.-S. Yang, D. Bhattacharjya, S. Inamdar, J. Park and J.-S. Yu, J. Am. Chem. Soc., 2012, 134, 16127–16130 CrossRef CAS PubMed.
- B. Li and D. Su, J. Phys. Chem. C, 2013, 117, 17485–17492 CrossRef CAS.
- K. Chizari, A. Deneuve, O. Ersen, I. Florea, Y. Liu, D. Edouard, I. Janowska, D. Begin and C. Pham-Huu, ChemSusChem, 2012, 5, 102–108 CrossRef CAS.
- M. Latorre-Sánchez, A. Primo and H. García, Angew. Chem., Int. Ed., 2013, 52, 11813–11816 CrossRef PubMed.
- E. J. Biddinger and U. S. Ozkan, J. Phys. Chem. C, 2010, 114, 15306–15314 CrossRef CAS.
- C. Tang, H. F. Wang, X. Chen, B. Q. Li, T. Z. Hou, B. Zhang, Q. Zhang, M. M. Titirici and F. Wei, Adv. Mater., 2016, 28, 6845–6851 CrossRef CAS.
- X. Wang, G. Sun, P. Routh, D.-H. Kim, W. Huang and P. Chen, Chem. Soc. Rev., 2014, 43, 7067–7098 RSC.
- K. N. Wood, R. O'Hayre and S. Pylypenko, Energy Environ. Sci., 2014, 7, 1212–1249 RSC.
- E. K. Rideal and W. M. Wright, J. Chem. Soc., 1926, 129, 1813–1821 RSC.
- S. K. Kaiser, Z. Chen, D. Faust Akl, S. Mitchell and J. Pérez-Ramírez, Chem. Rev., 2020, 120, 11703–11809 CrossRef CAS PubMed.
- S. Dang, ChemSusChem, 2010, 3, 169–180 CrossRef PubMed.
- L. Liu, Y. P. Zhu, M. Su and Z. Y. Yuan, ChemCatChem, 2015, 7, 2765–2787 CrossRef CAS.
- R. Mirsafaei, M. M. Heravi, S. Ahmadi, M. H. Moslemin and T. Hosseinnejad, J. Mol. Catal. Chem., 2015, 402, 100–108 CrossRef CAS.
- M. M. Heravi, S. Sadjadi, H. A. Oskooie, R. H. Shoar and F. F. Bamoharram, Catal. Commun., 2008, 9, 470–474 CrossRef CAS.
- M. M. Heravi, E. Hashemi, Y. S. Beheshtiha, K. Kamjou, M. Toolabi and N. Hosseintash, J. Mol. Catal. Chem., 2014, 392, 173–180 CrossRef CAS.
- M. M. Heravi, V. Zadsirjan, K. Bakhtiari, H. A. Oskooie and F. F. Bamoharram, Catal. Commun., 2007, 8, 315–318 CrossRef CAS.
- M. M. Heravi, K. Bakhtiari, A. Fatehi and F. F. Bamoharram, Catal. Commun., 2008, 9, 289–292 CrossRef CAS.
- S. Sadjadi, M. M. Heravi and M. Malmir, Carbohydr. Polym., 2018, 186, 25–34 CrossRef CAS PubMed.
- S. Sadjadi, M. Malmir and M. Heravi, RSC Adv., 2017, 7, 36807–36818 RSC.
- S. Sadjadi, T. Hosseinnejad, M. Malmir and M. M. Heravi, New J. Chem., 2017, 41, 13935–13951 RSC.
- R. Mirsafaei, M. M. Heravi, T. Hosseinnejad and S. Ahmadi, Appl. Organomet. Chem., 2016, 30, 823–830 CrossRef CAS.
- F. G. Kahangi, M. Mehrdad, M. M. Heravi and S. Sadjadi, Sci. Rep., 2020, 10, 1–11 CrossRef PubMed.
- S. Sadjadi, M. Akbari, F. G. Kahangi and M. M. Heravi, Appl. Clay Sci., 2020, 192, 105640 CrossRef CAS.
- M. M. Heravi, S. Asadi, S. M. Hoseini Chopani and E. Jaderi, Appl. Organomet. Chem., 2020, 34, e5805 CrossRef CAS.
- S. Hosseinzadeh-Baghan, M. Mirzaei, H. Eshtiagh-Hosseini, V. Zadsirjan, M. M. Heravi and J. T. Mague, Appl. Organomet. Chem., 2020, 34, e5793 CAS.
- R. Zoghi, M. M. Heravi, N. Montazeri, M. M. Zeydi and T. Hosseinnejad, Appl. Organomet. Chem., 2020, 34, e5435 CrossRef CAS.
- S. Sadjadi, M. Akbari and M. M. Heravi, ACS Omega, 2019, 4, 19442–19451 CrossRef CAS PubMed.
- S. Sadjadi, G. Lazzara, M. Malmir and M. M. Heravi, J. Catal., 2018, 366, 245–257 CrossRef CAS.
- M. M. Heravi, B. Heidari, V. Zadsirjan and L. Mohammadi, RSC Adv., 2020, 10, 24893–24940 RSC.
- N. Lotfian, M. M. Heravi, M. Mirzaei and B. Heidari, Appl. Organomet. Chem., 2019, 33, e4808 CrossRef.
- S. Sadjadi and M. M. Heravi, Curr. Org. Chem., 2016, 20, 1404–1444 CrossRef CAS.
- M. M. Heravi, B. Heidari, M. Ghavidel and T. Ahmadi, Curr. Org. Chem., 2017, 21, 2249–2313 CAS.
- S. Sadjadi and M. M. Heravi, RSC Adv., 2017, 7, 30815–30838 RSC.
- M. M. Heravi and T. Alishiri, Heterocycles, 2012, 85, 545–586 CrossRef CAS.
- M. M. Heravi and S. Sadjadi, J. Iran. Chem. Soc., 2009, 6, 1–54 CrossRef CAS.
- L. Sun, L. Wang, C. Tian, T. Tan, Y. Xie, K. Shi, M. Li and H. Fu, RSC Adv., 2012, 2, 4498–4506 RSC.
- Y. Wang, Y. Shao, D. W. Matson, J. Li and Y. Lin, ACS Nano, 2010, 4, 1790–1798 CrossRef CAS.
- E. Yoo, J. Kim, E. Hosono, H.-s. Zhou, T. Kudo and I. Honma, Nano Lett., 2008, 8, 2277–2282 CrossRef CAS PubMed.
- D. R. Kauffman and A. Star, Analyst, 2010, 135, 2790–2797 RSC.
- S. Wang, L. Zhang, Z. Xia, A. Roy, D. W. Chang, J.-B. Baek and L. Dai, Angew. Chem., Int. Ed., 2012, 51, 4209–4212 CrossRef CAS PubMed.
- D. S. Su, J. Zhang, B. Frank, A. Thomas, X. Wang, J. Paraknowitsch and R. Schlögl, ChemSusChem, 2010, 3, 169–180 CrossRef CAS PubMed.
- D. R. Dreyer, H.-P. Jia and C. W. Bielawski, Angew. Chem., Int. Ed., 2010, 49, 6686 CrossRef.
- H. Sun, Y. Wang, S. Liu, L. Ge, L. Wang, Z. Zhu and S. Wang, Chem. Commun., 2013, 49, 9914–9916 RSC.
- C. Biswas and Y. H. Lee, Adv. Funct. Mater., 2011, 21, 3806–3826 CrossRef CAS.
- D. Haag and H. H. Kung, Top. Catal., 2014, 57, 762–773 CrossRef CAS.
- C. Huang, C. Li and G. Shi, Energy Environ. Sci., 2012, 5, 8848–8868 RSC.
- X. Duan, K. O'Donnell, H. Sun, Y. Wang and S. Wang, Small, 2015, 11, 3036–3044 CrossRef CAS PubMed.
- D. Geng, S. Yang, Y. Zhang, J. Yang, J. Liu, R. Li, T.-K. Sham, X. Sun, S. Ye and S. Knights, Appl. Surf. Sci., 2011, 257, 9193–9198 CrossRef CAS.
- C. Wang, J. Kang, H. Sun, H. M. Ang, M. O. Tadé and S. Wang, Carbon, 2016, 102, 279–287 CrossRef CAS.
- C. Su, M. Acik, K. Takai, J. Lu, S.-j. Hao, Y. Zheng, P. Wu, Q. Bao, T. Enoki, Y. J. Chabal and K. Ping Loh, Nat. Commun., 2012, 3, 1298 CrossRef PubMed.
- Y. Gao, P. Tang, H. Zhou, W. Zhang, H. Yang, N. Yan, G. Hu, D. Mei, J. Wang and D. Ma, Angew. Chem., Int. Ed., 2016, 55, 3124–3128 CrossRef CAS PubMed.
- A. Dhakshinamoorthy, M. Latorre-Sanchez, A. M. Asiri, A. Primo and H. Garcia, Catal. Commun., 2015, 65, 10–13 CrossRef CAS.
- Y. Zhai, Z. Zhu and S. Dong, ChemCatChem, 2015, 7, 2806–2815 CrossRef CAS.
- M. F. L. De Volder, S. H. Tawfick, R. H. Baughman and A. J. Hart, Science, 2013, 339, 535–539 CrossRef CAS PubMed.
- V. Campisciano, M. Gruttadauria and F. Giacalone, ChemCatChem, 2019, 11, 90–133 CrossRef CAS.
- S. Reich, C. Thomsen and J. Maultzsch, Carbon nanotubes: basic concepts and physical properties, John Wiley & Sons, 2008 Search PubMed.
- D. M. Guldi and N. Martín, Carbon nanotubes and related structures, VCH-Wiley, Weinheim, Germany, 2010 Search PubMed.
- A. C. Dillon, Chem. Rev., 2010, 110, 6856–6872 CrossRef CAS PubMed.
- L. Wang, H. Liu, R. M. Konik, J. A. Misewich and S. S. Wong, Chem. Soc. Rev., 2013, 42, 8134–8156 RSC.
- S. A. Miners, G. A. Rance and A. N. Khlobystov, Chem. Soc. Rev., 2016, 45, 4727–4746 RSC.
- D. Tasis, N. Tagmatarchis, A. Bianco and M. Prato, Chem. Rev., 2006, 106, 1105–1136 CrossRef CAS PubMed.
- Y. Zhang, J. Zhang and D. S. Su, ChemSusChem, 2014, 7, 1240–1250 CrossRef CAS PubMed.
- Z. Yang, Z. Liu, H. Zhang, B. Yu, Y. Zhao, H. Wang, G. Ji, Y. Chen, X. Liu and Z. Liu, Chem. Commun., 2017, 53, 929–932 RSC.
- S. K. Movahed, Z. Piraman and M. Dabiri, J. Photochem. Photobiol., A, 2018, 351, 208–224 CrossRef CAS.
- H. Li, Prog. Chem., 2016, 28, 1462 CAS.
- Y. Zheng, J. Liu, J. Liang, M. Jaroniec and S. Z. Qiao, Energy Environ. Sci., 2012, 5, 6717–6731 RSC.
- S. M. Saufi and A. F. Ismail, Carbon, 2004, 42, 241–259 CrossRef CAS.
- C. West, C. Elfakir and M. Lafosse, J. Chromatogr., A, 2010, 1217, 3201–3216 CrossRef CAS PubMed.
- J. L. Figueiredo, J. Mater. Chem. A, 2013, 1, 9351–9364 RSC.
- K. Zhang, Z. Hu and J. Chen, J. Eng. Chem., 2013, 22, 214–225 CrossRef CAS.
- S. De, A. M. Balu, J. C. van der Waal and R. Luque, ChemCatChem, 2015, 7, 1608–1629 CrossRef CAS.
- Y. Xia, Z. Yang and R. Mokaya, Nanoscale, 2010, 2, 639–659 RSC.
- B. Sakintuna and Y. Yürüm, Ind. Eng. Chem. Res., 2005, 44, 2893–2902 CrossRef CAS.
- J. H. Khan, F. Marpaung, C. Young, J. Lin, M. T. Islam, S. M. Alsheri, T. Ahamad, N. Alhokbany, K. Ariga, L. K. Shrestha, Y. Yamauchi, K. C. W. Wu, M. S. A. Hossain and J. Kim, Microporous Mesoporous Mater., 2019, 274, 251–256 CrossRef CAS.
- T. Kesavan, T. Partheeban, M. Vivekanantha, M. Kundu, G. Maduraiveeran and M. Sasidharan, Microporous Mesoporous Mater., 2019, 274, 236–244 CrossRef CAS.
- B. Li, H. Xiong and Y. Xiao, Int. J. Electrochem. Sci., 2020, 15, 1363–1377 CrossRef CAS.
- J. Lee, J. Kim and T. Hyeon, Adv. Mater., 2006, 18, 2073–2094 CrossRef CAS.
- R. C. Bansal, J.-B. Donnet and F. Stoeckli, Active carbon, 1988 Search PubMed.
- T. R. Gaffney, Curr. Opin. Solid State Mater. Sci., 1996, 1, 69–75 CrossRef CAS.
- X. Q. Zhang and A. H. Lu, Materials for Carbon Capture, 2020, pp. 29–95 Search PubMed.
- R. Ryoo, S. H. Joo and S. Jun, J. Phys. Chem. B, 1999, 103, 7743–7746 CrossRef CAS.
- P. Zhang, H. Zhu and S. Dai, ChemCatChem, 2015, 7, 2788–2805 CrossRef CAS.
- T. Asefa, Acc. Chem. Res., 2016, 49, 1873–1883 CrossRef CAS PubMed.
- M. Enterría and J. Figueiredo, Carbon, 2016, 108, 79–102 CrossRef.
- Y. Cao, S. Mao, M. Li, Y. Chen and Y. Wang, ACS Catal., 2017, 7, 8090–8112 CrossRef CAS.
- D. Salinas-Torres, M. Navlani-García, K. Mori, Y. Kuwahara and H. Yamashita, Appl. Catal. Gen., 2019, 571, 25–41 CrossRef CAS.
- T. Zhang and T. Asefa, Adv. Mater., 2019, 31, 1804394 CrossRef PubMed.
- B. Wang, T. P. Ang and A. Borgna, Microporous Mesoporous Mater., 2012, 158, 99–107 CrossRef CAS.
- D. S. Su, S. Perathoner and G. Centi, Chem. Rev., 2013, 113, 5782–5816 CrossRef CAS PubMed.
- P. Zhang, J. Zhang and S. Dai, Chem.–Eur. J., 2017, 23, 1986–1998 CrossRef CAS PubMed.
- W. Zhang, F. Wang, X. Li, Y. Liu and J. Ma, RSC Adv., 2016, 6, 27313–27319 RSC.
- L. Wang and X. Hu, Chem. Asian J., 2018, 13, 1518–1529 CrossRef CAS PubMed.
- Z. Luo, S. Lim, Z. Tian, J. Shang, L. Lai, B. MacDonald, C. Fu, Z. Shen, T. Yu and J. Lin, J. Mater. Chem., 2011, 21, 8038–8044 RSC.
- X. Wang, X. Li, L. Zhang, Y. Yoon, P. K. Weber, H. Wang, J. Guo and H. Dai, Science, 2009, 324, 768–771 CrossRef CAS PubMed.
- D. H. Lee, W. J. Lee and S. O. Kim, Nano Lett., 2009, 9, 1427–1432 CrossRef CAS PubMed.
- H. Gao, Z. Liu, L. Song, W. Guo, W. Gao, L. Ci, A. Rao, W. Quan, R. Vajtai and P. M. Ajayan, Nanotechnology, 2012, 23, 275605 CrossRef PubMed.
- J. Xu, G. Dong, C. Jin, M. Huang and L. Guan, ChemSusChem, 2013, 6, 493–499 CrossRef CAS PubMed.
- E. Cruz-Silva, D. A. Cullen, L. Gu, J. M. Romo-Herrera, E. Muñoz-Sandoval, F. López-Urías, B. G. Sumpter, V. Meunier, J.-C. Charlier and D. J. Smith, ACS Nano, 2008, 2, 441–448 CrossRef CAS PubMed.
- S. Some, J. Kim, K. Lee, A. Kulkarni, Y. Yoon, S. Lee, T. Kim and H. Lee, Adv. Mater., 2012, 24, 5481–5486 CrossRef CAS PubMed.
- L. Panchakarla, K. Subrahmanyam, S. Saha, A. Govindaraj, H. Krishnamurthy, U. Waghmare and C. Rao, Adv. Mater., 2009, 21, 4726–4730 CAS.
- Z. Wang, C. Yu, D. Ba and J. Liang, Vacuum, 2007, 81, 579–582 CrossRef CAS.
- S. K. Kaiser, K. S. Song, S. Mitchell, A. Coskun and J. Pérez-Ramírez, ChemCatChem, 2019, 12, 1922–1925 CrossRef.
- Y. Xue, B. Wu, L. Jiang, Y. Guo, L. Huang, J. Chen, J. Tan, D. Geng, B. Luo and W. Hu, J. Am. Chem. Soc., 2012, 134, 11060–11063 CrossRef CAS PubMed.
- Y. Luo, Z. Yang, W. Guo, H. Chen, T. Wang, Y. Liu, Y. Lyu, H. Luo and S. Dai, J. Mater. Chem. A, 2020, 8, 4740–4746 RSC.
- L. Zhao, P.-W. Xiao and B.-H. Han, Porous Carbon Materials from Sustainable Precursors, Green Chem., 2015, 32, 191–224 CAS.
- G. Wu, A. Santandreu, W. Kellogg, S. Gupta, O. Ogoke, H. Zhang, H.-L. Wang and L. Dai, Nano Energy, 2016, 29, 83–110 CrossRef CAS.
- J. Zhang, Z. Xia and L. Dai, Sci. Adv., 2015, 1, e1500564 CrossRef PubMed.
- R. Ma, Y. Ma, Y. Dong and J.-M. Lee, Nano Adv., 2016, 1, 50–61 CrossRef.
- Y. Deng, Y. Xie, K. Zou and X. Ji, J. Mater. Chem. A, 2016, 4, 1144–1173 RSC.
- G. Wu, C. M. Johnston, N. H. Mack, K. Artyushkova, M. Ferrandon, M. Nelson, J. S. Lezama-Pacheco, S. D. Conradson, K. L. More and D. J. Myers, J. Mater. Chem., 2011, 21, 11392–11405 RSC.
- K. Chizari, A. Vena, L. Laurentius and U. Sundararaj, Carbon, 2014, 68, 369–379 CrossRef CAS.
- S. Wang, C. Han, J. Wang, J. Deng, M. Zhu, J. Yao, H. Li and Y. Wang, Chem. Mater., 2014, 26, 6872–6877 CrossRef CAS.
- Z. Xia, L. An, P. Chen and D. Xia, Adv. Energy Mater., 2016, 6, 1600458 CrossRef.
- T. Schiros, D. Nordlund, L. Pálová, D. Prezzi, L. Zhao, K. S. Kim, U. Wurstbauer, C. Gutiérrez, D. Delongchamp and C. Jaye, Nano Lett., 2012, 12, 4025–4031 CrossRef CAS PubMed.
- L. Dong, X. Chen, J. Ma, Q. Shao, A. Li, W. Yan and J. Zhang, Russ. J. Electrochem., 2019, 55, 1098–1109 CrossRef.
- J. Quílez-Bermejo, E. Morallón and D. Cazorla-Amorós, Carbon, 2020, 165, 434–454 CrossRef.
- L. Xu, G. Pan, X. Liang, G. Luo, C. Zou and G. Chen, J. Energy Chem., 2014, 23, 498–506 CrossRef.
- L. Lai, J. R. Potts, D. Zhan, L. Wang, C. K. Poh, C. Tang, H. Gong, Z. Shen, J. Lin and R. S. Ruoff, Energy Environ. Sci., 2012, 5, 7936–7942 RSC.
- Z. Huang, Z. Liao, W. Yang, H. Zhou, C. Fu, Y. Gong, L. Chen and Y. Kuang, Electrochim. Acta, 2017, 245, 957–966 CrossRef CAS.
- D. A. Bulushev, M. Zacharska, A. S. Lisitsyn, O. Y. Podyacheva, F. S. Hage, Q. M. Ramasse, U. Bangert and L. G. Bulusheva, ACS Catal., 2016, 6, 3442–3451 CrossRef CAS.
- Y.-H. Li, T.-H. Hung and C.-W. Chen, Carbon, 2009, 47, 850–855 CrossRef CAS.
- R. Arrigo, M. E. Schuster, Z. Xie, Y. Yi, G. Wowsnick, L. L. Sun, K. E. Hermann, M. Friedrich, P. Kast and M. Hävecker, ACS Catal., 2015, 5, 2740–2753 CrossRef CAS.
- M. Zacharska, L. G. Bulusheva, A. S. Lisitsyn, S. Beloshapkin, Y. Guo, A. L. Chuvilin, E. V. Shlyakhova, O. Y. Podyacheva, J. J. Leahy and A. V. Okotrub, ChemSusChem, 2017, 10, 720–730 CrossRef CAS PubMed.
- D. Guo, R. Shibuya, C. Akiba, S. Saji, T. Kondo and J. Nakamura, Science, 2016, 351, 361–365 CrossRef CAS PubMed.
- Y. Zhao, J. Wan, H. Yao, L. Zhang, K. Lin, L. Wang, N. Yang, D. Liu, L. Song and J. Zhu, Nat. Chem., 2018, 10, 924–931 CrossRef CAS PubMed.
- J. Long, X. Xie, J. Xu, Q. Gu, L. Chen and X. Wang, ACS Catal., 2012, 2, 622–631 CrossRef CAS.
- R. Lin, S. K. Kaiser, R. Hauert and J. Pérez-Ramírez, ACS Catal., 2018, 8, 1114–1121 CrossRef CAS.
- Z. Li, J. Li, J. Liu, Z. Zhao, C. Xia and F. Li, ChemCatChem, 2014, 6, 1333–1339 CAS.
- R. Nie, M. Miao, W. Du, J. Shi, Y. Liu and Z. Hou, Appl. Catal., B, 2016, 180, 607–613 CrossRef CAS.
- Y. Yang, L. Gu, S. Guo, S. Shao, Z. Li, Y. Sun and S. Hao, Front. Chem., 2019, 7, 761 CrossRef CAS PubMed.
- X. Xu, Y. Li, Y. Gong, P. Zhang, H. Li and Y. Wang, J. Am. Chem. Soc., 2012, 134, 16987–16990 CrossRef CAS PubMed.
- Z. Li, J. Liu, C. Xia and F. Li, ACS Catal., 2013, 3(11), 2440–2448 CrossRef CAS.
- Y. Gao, G. Hu, J. Zhong, Z. Shi, Y. Zhu, D. S. Su, J. Wang, X. Bao and D. Ma, Angew. Chem., Int. Ed., 2013, 52, 2109–2113 CrossRef CAS PubMed.
- M. N. Groves, A. S. W. Chan, C. Malardier-Jugroot and M. Jugroot, Chem. Phys. Lett., 2009, 481, 214–219 CrossRef CAS.
- C. Petit, G. W. Peterson, J. Mahle and T. J. Bandosz, Carbon, 2010, 48, 1779–1787 CrossRef CAS.
- Y. Li, J. Wang, X. Li, D. Geng, M. N. Banis, Y. Tang, D. Wang, R. Li, T.-K. Sham and X. Sun, J. Mater. Chem., 2012, 22, 20170–20174 RSC.
- M. Seredych, K. Singh and T. J. Bandosz, Electroanalysis, 2014, 26, 109–120 CrossRef CAS.
- Y. Yan, Y.-X. Yin, S. Xin, Y.-G. Guo and L.-J. Wan, Chem. Commun., 2012, 48, 10663–10665 RSC.
- X. Zhao, Q. Zhang, C.-M. Chen, B. Zhang, S. Reiche, A. Wang, T. Zhang, R. Schlögl and D. Sheng Su, Nano Energy, 2012, 1, 624–630 CrossRef CAS.
- F. Li, D. Yang and H. Xu, Chem.–Eur. J., 2019, 25, 1165–1176 CrossRef CAS PubMed.
- X. Kou, S. Jiang, S.-J. Park and L.-Y. Meng, Dalton Trans., 2020, 49, 6915–6938 RSC.
- Q. Abbas, R. Raza, I. Shabbir and A. G. Olabi, J. Sci.: Adv. Mater. Devices, 2019, 4, 341–352 Search PubMed.
- P. Serp and B. Machado, Nanostructured carbon materials for catalysis, Royal Society of Chemistry, 2015 Search PubMed.
- R. Li, J. Zhao, D. Han and X. Li, Catal. Commun., 2017, 97, 116–119 CrossRef CAS.
- J. P. Paraknowitsch and A. Thomas, Energy Environ. Sci., 2013, 6, 2839–2855 RSC.
- X. Xie, J. Shi, Y. Pu, Z. Wang, L.-L. Zhang, J.-X. Wang and D. Wang, J. Colloid Interface Sci., 2020, 571, 100–108 CrossRef CAS PubMed.
- C. Liao, B. Liu, Q. Chi and Z. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 44421–44429 CrossRef CAS PubMed.
- Q. Shen, X. Chen, Y. Tan, J. Chen, L. Chen and S. Tan, ACS Appl. Mater. Interfaces, 2019, 11, 38838–38848 CrossRef CAS PubMed.
- G. Li, S. Zheng, L. Wang and X. Zhang, ACS Omega, 2020, 5, 7519–7528 CrossRef CAS PubMed.
- W. Xiong, Z. Wang, S. He, F. Hao, Y. Yang, Y. Lv, W. Zhang, P. Liu and H. A. Luo, Appl. Catal., B, 2020, 260, 118105 CrossRef CAS.
- Z. He, J. Liu, Q. Wang, M. Zhao, Z. Wen, J. Chen, D. Manoj, C. Xie, J. Xi, J. Yu, C. Tang, Z. Bai and S. Wang, J. Catal., 2019, 377, 199–208 CrossRef CAS.
- N. Liu, L. Ding, H. Li, M. Jia, W. Zhang, N. An and X. Yuan, J. Colloid Interface Sci., 2017, 490, 677–684 CrossRef CAS PubMed.
- J. Liu, X. Yan, L. Wang, L. Kong and P. Jian, J. Colloid Interface Sci., 2017, 497, 102–107 CrossRef CAS PubMed.
- Y. Dai, J. Zhou, C. Huang, Q. Gu, Y. Zeng, W. Xu, X. Meng, W. Fu and Y. Sun, Part. Part. Syst. Char., 2018, 35, 1700395 CrossRef.
- Z. Wang and Q. Chen, ChemistrySelect, 2018, 3, 1108–1112 CrossRef CAS.
- Z. Wang, R. Su, D. Wang, J. Shi, J.-X. Wang, Y. Pu and J.-F. Chen, Ind. Eng. Chem. Res., 2017, 56, 13610–13617 CrossRef CAS.
- X. Chen, Q. Shen, Z. Li, W. Wan, J. Chen and J. Zhang, ACS Appl. Mater. Interfaces, 2020, 12, 654–666 CrossRef CAS PubMed.
- R. Gao, L. Pan, J. Lu, J. Xu, X. Zhang, L. Wang and J.-J. Zou, ChemCatChem, 2017, 9, 4287–4294 CrossRef CAS.
- Y. Lin, S. Wu, W. Shi, B. Zhang, J. Wang, Y. A. Kim, M. Endo and D. S. Su, Chem. Commun., 2015, 51, 13086–13089 RSC.
- F. Wang, S. Song, K. Li, J. Li, J. Pan, S. Yao, X. Ge, J. Feng, X. Wang and H. Zhang, Adv. Mater., 2016, 28, 10679–10683 CrossRef CAS PubMed.
- X. Hu, X. Sun, Q. Song, Y. Zhu, Y. Long and Z. Dong, Green Chem., 2020, 22, 742–752 RSC.
- F. Yang, Y. Cao, Z. Chen, X. He, L. Hou and Y. Li, New J. Chem., 2018, 42, 2718–2725 RSC.
- C. Van Nguyen, S. Lee, Y. G. Chung, W.-H. Chiang and K. C. W. Wu, Appl. Catal., B, 2019, 257, 117888 CrossRef CAS.
- J. Xi, Q. Wang, J. Liu, L. Huan, Z. He, Y. Qiu, J. Zhang, C. Tang, J. Xiao and S. Wang, J. Catal., 2018, 359, 233–241 CrossRef CAS.
- Q. Wei, F. Qin, Q. Ma and W. Shen, Carbon, 2019, 141, 542–552 CrossRef CAS.
- Y. Liu, H. Zhang, X. Li, L. Wang, Y. Dong, W. Li and J. Zhang, Appl. Catal. Gen., 2021, 611, 117902 CrossRef CAS.
- F. Lu, D. Xu, Y. Lu, B. Dai and M. Zhu, Chin. J. Chem. Eng., 2021, 29, 196–203 CrossRef CAS.
- X. Li, J. Zhang and W. Li, J. Ind. Eng. Chem., 2016, 44, 146–154 CrossRef CAS.
- X. Li, J. Zhang, Y. Han, M. Zhu, S. Shang and W. Li, J. Mater. Sci., 2018, 53, 4913–4926 CrossRef CAS.
- S. Chao, F. Zou, F. Wan, X. Dong, Y. Wang, Y. Wang, Q. Guan, G. Wang and W. Li, Sci. Rep., 2017, 7, 1–7 CrossRef CAS PubMed.
- X. Qi, W. Chen and J. Zhang, RSC Adv., 2020, 10, 34612–34620 RSC.
- B. Dai, K. Chen, Y. Wang, L. Kang and M. Zhu, ACS Catal., 2015, 5, 2541–2547 CrossRef CAS.
- J. Zhao, B. Wang, Y. Yue, G. Sheng, H. Lai, S. Wang, L. Yu, Q. Zhang, F. Feng, Z.-T. Hu and X. Li, J. Catal., 2019, 373, 240–249 CrossRef CAS.
- J. Wang, F. Zhao, C. Zhang, L. Kang and M. Zhu, Appl. Catal. Gen., 2018, 549, 68–75 CrossRef CAS.
- X. Dong, S. Chao, F. Wan, Q. Guan, G. Wang and W. Li, J. Catal., 2018, 359, 161–170 CrossRef CAS.
- H. Fu, K. Huang, G. Yang, Y. Cao, H. Wang, F. Peng, Q. Wang and H. Yu, ACS Catal., 2019, 10, 129–137 CrossRef.
- Y. Lin, Z. Liu, Y. Niu, B. Zhang, Q. Lu, S. Wu, G. Centi, S. Perathoner, S. Heumann, L. Yu and D. S. Su, ACS Nano, 2019, 13, 13995–14004 CrossRef CAS PubMed.
- P. Liang, C. Zhang, X. Duan, H. Sun, S. Liu, M. O. Tade and S. Wang, ACS Sustain. Chem. Eng., 2017, 5, 2693–2701 CrossRef CAS.
- G. Wen, Q. Gu, Y. Liu, R. Schlögl, C. Wang, Z. Tian and D. S. Su, Angew. Chem., Int. Ed., 2018, 57, 16898–16902 CrossRef CAS PubMed.
- S. Indrawirawan, H. Sun, X. Duan and S. Wang, J. Mater. Chem. A, 2015, 3, 3432–3440 RSC.
- Z. Ma, H. Zhang, Z. Yang, G. Ji, B. Yu, X. Liu and Z. Liu, Green Chem., 2016, 18, 1976–1982 RSC.
- X. Wang and Y. Li, J. Mater. Chem. A, 2016, 4, 5247–5257 RSC.
- C. Rizescu, I. Podolean, J. Albero, V. I. Parvulescu, S. M. Coman, C. Bucur, M. Puche and H. Garcia, Green Chem., 2017, 19, 1999–2005 RSC.
- R. Huang, C. Cao, J. Liu, D. Sun and W. Song, Chem. Commun., 2019, 55, 1935–1938 RSC.
- K. Wang, P. Jiang, M. Yang, P. Ma, J. Qin, X. Huang, L. Ma and R. Li, Green Chem., 2019, 21, 2448–2461 RSC.
- H. Watanabe, S. Asano, S.-i. Fujita, H. Yoshida and M. Arai, ACS Catal., 2015, 5, 2886–2894 CrossRef CAS.
- S.-I. Fujita, S. Asano and M. Arai, J. Mol. Catal. Chem., 2016, 423, 181–184 CrossRef CAS.
- S. Shang, W. Dai, L. Wang, Y. Lv and S. Gao, Chem. Commun., 2017, 53, 1048–1051 RSC.
- G.-X. Qin, Y. Hao, S. Wang and Y.-B. Dong, Appl. Catal. Gen., 2020, 591, 117400 CrossRef.
- N. Teng, J.-L. Li, B.-Q. Lu, Y.-Q. Wang, S.-Y. Jia, Y.-X. Wang and X.-L. Hou, N. Carbon Mater., 2019, 34, 593–599 CrossRef.
- Y. Sun, J. Hao, X. Zhu, B. Zhang, H. Yin, S. Xu, C. Hou and K. Liu, Carbon Lett., 2020, 30, 133–141 CrossRef.
- S. Verma, M. N. Nadagouda and R. S. Varma, Sci. Rep., 2017, 7, 1–6 CrossRef PubMed.
- Y. Ren, Z. Yuan, K. Lv, J. Sun, Z. Zhang and Q. Chi, Green Chem., 2018, 20, 4946–4956 RSC.
- G. Lv, H. Wang, Y. Yang, X. Li, T. Deng, C. Chen, Y. Zhu and X. Hou, Catal. Sci. Technol., 2016, 6, 2377–2386 RSC.
- N. Gupta, O. Khavryuchenko, A. Villa and D. Su, ChemSusChem, 2017, 10, 3030–3034 CrossRef CAS PubMed.
- K. T. V. Rao, Y. Hu, Z. Yuan, Y. Zhang and C. Xu, Chem. Eng. J., 2021, 404, 127063 CrossRef CAS.
- Z. Li, Y. Huang, X. Chi, D. Li, L. Zhong, X. Li, C. Liu and X. Peng, Green Energy Environ., 2021, 1–19 Search PubMed.
- Y. Sun, C. Hou, X. Cao and K. Liu, J. Mater. Sci., 2021, 56, 6124–6134 CrossRef CAS.
- Y. Guo, Z. Zeng, Y. Li, Z. Huang and Y. Cui, Catal. Today, 2018, 307, 12–19 CrossRef CAS.
- Z. Wang, J. Shi, D. Wang, Y. Pu, J.-X. Wang and J.-F. Chen, Chem. React. Eng., 2019, 4, 507–515 RSC.
- Z. Long, L. Sun, W. Zhu, G. Chen, X. Wang and W. Sun, Chem. Commun., 2018, 54, 8991–8994 RSC.
- X. Hu, M. Fan, Y. Zhu, Q. Zhu, Q. Song and Z. Dong, Green Chem., 2019, 21, 5274–5283 RSC.
- F. Yang, X. Fan, C. Wang, W. Yang, L. Hou, X. Xu, A. Feng, S. Dong, K. Chen, Y. Wang and Y. Li, Carbon, 2017, 121, 443–451 CrossRef CAS.
- M. A. Patel, F. Luo, M. R. Khoshi, E. Rabie, Q. Zhang, C. R. Flach, R. Mendelsohn, E. Garfunkel, M. Szostak and H. He, ACS Nano, 2016, 10, 2305–2315 CrossRef CAS PubMed.
- Y. Zhai, M. Chu, C. Xie, F. Huang, C. Zhang, Y. Zhang, H. Liu, H. Wang and Y. Gao, ACS Sustain. Chem. Eng., 2018, 6, 17410–17418 CrossRef CAS.
- W. Cheng, X. Liu, N. Li, J. Han, S. Li and S. Yu, RSC Adv., 2018, 8, 11222–11229 RSC.
- R. Goyal, B. Sarkar, A. Bag, F. Lefebvre, S. Sameer, C. Pendem and A. Bordoloi, J. Mater. Chem. A, 2016, 4, 18559–18569 RSC.
- H. Wei, Y. Ma, J. Luo, K.-H. Wu, W. Xie, G. Wen, C.-L. Chiang, W. Yan, S. Perathoner, G. Centi and Y. Liu, Carbon, 2020, 170, 338–346 CrossRef CAS.
- Y.-N. Zhu, C.-Y. Cao, W.-J. Jiang, S.-L. Yang, J.-S. Hu, W.-G. Song and L.-J. Wan, J. Mater. Chem. A, 2016, 4, 18470–18477 RSC.
- S. Yang, L. Peng, P. Huang, X. Wang, Y. Sun, C. Cao and W. Song, Angew. Chem., 2016, 128, 4084–4088 CrossRef.
- S. Yang, Y. Zhu, C. Cao, L. Peng, S. Li, D. Zhai and W. Song, Nanoscale, 2017, 9, 13538–13545 RSC.
- Z. Zhao, Y. Dai, G. Ge, X. Guo and G. Wang, RSC Adv., 2015, 65, 53095–53099 RSC.
- S. Li, W. Wang, X. Liu, X. Zeng, W. Li, N. Tsubaki and S. Yu, RSC Adv., 2016, 6, 13450–13455 RSC.
- Z. Zhao, Y. Dai, G. Ge and G. Wang, ChemCatChem, 2015, 7, 1135–1144 CrossRef CAS.
- N. Candu, I. Man, A. Simion, B. Cojocaru, S. M. Coman, C. Bucur, A. Primo, H. Garcia and V. I. Parvulescu, J. Catal., 2019, 376, 238–247 CrossRef CAS.
- Y. Wang, J. Wang, Y. Zhang, F. Song, Y. Xie, M. Wang, H. Cui and W. Yi, Catal. Lett., 2020, 150, 493–504 CrossRef CAS.
- S. S. Chen, I. K. M. Yu, D.-W. Cho, H. Song, D. C. W. Tsang, J.-P. Tessonnier, Y. S. Ok and C. S. Poon, ACS Sustain. Chem. Eng., 2018, 6, 16113–16120 CrossRef CAS.
- S.-i. Fujita, A. Katagiri, H. Watanabe, S. Asano, H. Yoshida and M. Arai, ChemCatChem, 2015, 7, 2965–2970 CrossRef CAS.
- M. T. Jafari-Chermahini, H. Tavakol and W. Salvenmoser, ChemistrySelect, 2020, 5, 968–978 CrossRef CAS.
- L. Y. Zhao, X. L. Dong, J. Y. Chen and A. H. Lu, Chem.–Eur. J., 2020, 26, 2041–2050 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2021 |