
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffect of Si, Be, Al, N and S dual doping on arsenene: first-principles insights†
Muhammad Mushtaq *a,
Sumegha Godarab,
Rabah Khenatac and
Muhammad Usman Hameedd
*a,
Sumegha Godarab,
Rabah Khenatac and
Muhammad Usman Hameedd
aDepartment of Physics, Women University of AJK, 12500, Bagh, Pakistan. E-mail: mushtaq325@gmail.com
bLouisiana Tech University, Louisiana, USA
cLaboratoire de Physique Quantique de la Matière et de Modélisation Mathématique (LPQ3M), Université de Mascara, 29000 Mascara, Algeria
dDepartment of Chemistry, Women University of AJK, 12500, Bagh, Pakistan
First published on 20th July 2021
Abstract
First-principles calculations based on density functional theory (DFT) have been performed to investigate the effect of Si/Be, Si/Al, Si/N and Si/S co-doping on the geometries, electronic structure, magnetism and particularly the adsorption of CO in arsenene. The results show that the incorporation of foreign atoms slightly distorts the host lattice. All doped structures are found to be thermodynamically stable. The replacement of host As atoms with foreign atoms results in some interesting changes in the electronic and magnetic properties of arsenene. The doped arsenene systems exhibit a semiconducting character with band gaps smaller than the original value of 1.59 eV due to the emergence of defect states within the actual band gap. Besides, arsenene remains nonmagnetic (NM) upon Si/Be or Si/S dual doping, whereas both Si/Al and Si/N dopings induce magnetism with a total magnetic moment of 1 μB. Finally, the adsorption of CO molecules over pristine arsenene (p-As) and dual doped arsenene systems is investigated in terms of adsorption energy, adsorption height, charge transfer, charge density difference (CDD), work function, electronic band structures and density of states. It is observed that CO molecule has physisorption over p-As, SiAl–As, SiN–As and SiS–As systems, whereas chemisorption is reported for the SiBe–As system. Our study suggests that chemically modifying arsenene with suitable dopants might extend its applications in spintronic and gas sensing applications.
1. Introduction
In the last five years, arsenene, a monolayer of bulk gray arsenic, has received significant attention in the scientific community.1–3 Arsenene has a stable buckled honeycomb structure and an indirect band gap. Within the structure, each arsenic As atom is bonded to three neighboring As atoms through covalent bonding. In an early attempt, successful synthesis of multilayer arsenene on an InAs substrate using plasma inspires further studies on exploring the structure and related properties.4 Very recently, monolayer arsenene is experimentally evidenced by Shah et al.5 The existence of a large band gap, high carrier mobility, good thermoelectric performance with low thermal conductivity, and excellent and tunable absorption in the visible-ultraviolet region makes arsenene a superior material for the next generation nanoscale devices.6–8 In particular, arsenene shows enormous interesting features when subjected to external strain, electric field or chemical alternation, expanding its technical scope. For example, arsenene makes indirect–direct band gap transition with strain.9 Similarly, cutting arsenene into zigzag edged nanoribbons also leads to a direct band gap.10 Besides, fully hydrogenated arsenene exhibits the quantum spin Hall effect with 193 meV gap at the Dirac cone.11 In the same way, halogenated arsenene is verified to be a quantum spin Hall insulator.12 Furthermore, alloyed or doped arsenene also shows properties different from those of the pristine one.13However, being non-magnetic in its pristine form, arsenene may not be a potential material to be used in spin-devices or gas-sensors. To acquire magnetism in arsenene, various techniques have been established, with particular emphasis on defect formation, foreign atom doping, and nanohole patterning.14–16 For instance, single vacancies induce spin-polarized states in the band gap of arsenene.17 While V, Cr and Fe doping makes arsenene a magnetic semiconductor, Ti and Mn doping leads arsenene to exhibit half-metallic characteristics.18 Likewise, dilute magnetism is demonstrated for 3d metal adatom adsorbed arsenene.19 Besides, non-magnetic elements (C or O) also have the ability to turn arsenene into a spin-polarized material.20 Moreover, the incorporation of Ge into arsenene decorates the system with dilute magnetism.21 All these studies promise the exploitation of arsenene in the spintronic field. In addition, like other 2D materials, arsenene can be employed for the adsorption of gas molecules. For example, the adsorption of CO, CO2, N2, NH3, NO and NO2 molecules on the pristine arsenene monolayer investigated by first-principles revealed a moderate interaction between arsenene and molecules (between physisorption and chemisorption).22 In another study, chemisorption was observed in Al-, B- and Si-doped arsenene for NO molecules, while S-doped arsenene showed physisorption.23 Furthermore, Mao et al. investigated the adsorption of NH3, CO, CO2, NO, and NO2 on black arsenene (also known as puckered arsenene) and found that the adsorption strength could be modulated via vertical electrical field due to variation in charge transfer.24
Energetically feasible dual doping i.e. replacing two atoms in the host structure with two external atoms can alter the electronic properties and improve the functionality of two-dimensional (2D) materials. It is evidenced that BeX (X = B, N, and O) dual doping in graphene enhances the adsorption of Li and Na ions.25 The introduction of one atom from 2p-elements (B, N, and O) and other from 3p-elements (Al, Si, P and S) into graphene witnesses promising catalytic activities.26 More importantly, experimentally achieved nitrogen and sulfur dual doped graphene structures have been used for oxygen reduction reactions.27 It is well understood that so far dual doping in 2D systems has been mainly limited to graphene, and no such study is found for arsenene. Therefore, it becomes necessary to investigate the effect of simultaneous doping with two different atoms on the fundamental properties of arsenene. In this paper, Si and X (X = Be, Al, N and S) dual doped arsenene (SiX–As) is studied using first-principles calculations to provide insights into the local geometry, electronic structure, magnetism and the effect of dual doping on the adsorption of CO gas molecules.
2. Computational methods
Density functional theory (DFT), incorporated in the Vienna Ab initio Simulation Package (VASP), is used as known to be very reliable to predict the ground-state properties such as the adsorption of molecules on surfaces. The method relies on expanding atomic orbitals in the plane-wave basis set using pseudopotentials to solve the Kohn–Sham equations and estimate the total energy.28 The exchange–correlation potential is presented using the generalized gradient approximation (GGA) of Perdew–Burke–Ernzerhof (PBE) formalism.29 Initially the unit cell structure of p-As consisting of two atoms is fully relaxed for atomic positions and lattice vectors. Then a supercell of 5 × 5 size (having 50 As atoms) is relaxed. The supercell lattice vectors a and b lie in the xy plane, whereas the c-vector lies along the z-direction. The size of the supercell along the c-axis is chosen sufficiently large with a vacuum space of 15 Å to ensure the insulation and isolation of the As monolayer and to avoid any interactions between the system and its periodic images. The geometries are fully relaxed by giving freedom to both motions of the lattice vectors and the atomic positions until the convergence of total energy with a tolerance of less than 10−4 eV. The Brillouin-zone sampling is performed using the known Monkhorst–Pack algorithm [MP]. A grid of 8 × 8 × 1 is used for calculating the charge density and a more refined grid of 12 × 12 × 1 is used for the density of states (DOS) calculation. An energy cut-off of 500 eV is employed for the expansion of the plane wave basis set. Besides, the long-range van der Waals like interactions are described within the scheme of the dispersion of Grimme (i.e. DFT-D3).30 The reliability of the computational parameters is checked by calculating the lattice vectors, As–As bond length, electronic band structure and density of states of the p-As. It is found that p-As has a lattice constant of 3.60 Å, As–As bond length of 2.50 Å, an indirect band gap of 1.59 eV, and valence and conduction band edge states dominated by p-states (see Fig. S1†). These results are in agreement with our previous study on arsenene, confirming the reliability of the methodology.16In order to investigate the effect of co-doping on arsenene, we consider five types of dopants, including Be, Al, Si, N and S. Four kinds of doped arsenene structures are built by substituting two As atoms by two dopant atoms: (1) Si and Be substituted arsenene, SiBe–As, (2) Si and Al substituted arsenene, SiAl–As, (3) Si and N substituted arsenene, SiN–As, and (4) Si and S substituted arsenene, SiS–As. For each SiX–As (hereafter X = Be, Al, N, or S) system the doping concentration is fixed, i.e. 4% but various doping configurations are included by varying the dopant sites.
To explore the stability of the SiX–As systems, the binding energy (Ebind) is calculated using the following expression:
| Ebind = ESiX–As + 2 μAs − μSi − μX + EAs | (1) |
The adsorption energy of CO molecules on the SiX-doped arsenene monolayer (ML) is defined as
| Eads = EML–CO − EML − ECO | (2) |
3. Results and discussion
We first discuss the energetics and geometries of the SiBe–As, SiAl–As, SiN–As and SiS–As co-doped systems. For each co-doped system, five different doping configurations are considered and their optimized geometries are presented in Fig. 1 along with relative energy ΔE (where ΔE = Etotal − Etotal−lowest). The total energy of the doped systems is compared with that of p-As, shown in Fig. S2.† It can be seen that, in comparison with p-As, four systems SiZ–As (Z = Be, Al, and S) have higher energy, whereas SiN–As has lower energy. Accordingly, SiN–As has the lowest binding energy. In addition, for three systems namely SiBe–As, SiN–As and SiS–As, the most stable configuration (i.e. having lowest energy) is obtained when the initial distance between Si and X atoms, dSi–X, is 2.50 Å. Similarly for SiAl–As energetically most favorable is a dSi–Al of 6.25 Å. Thereafter, we discuss the properties of the systems in their most stable doping configuration.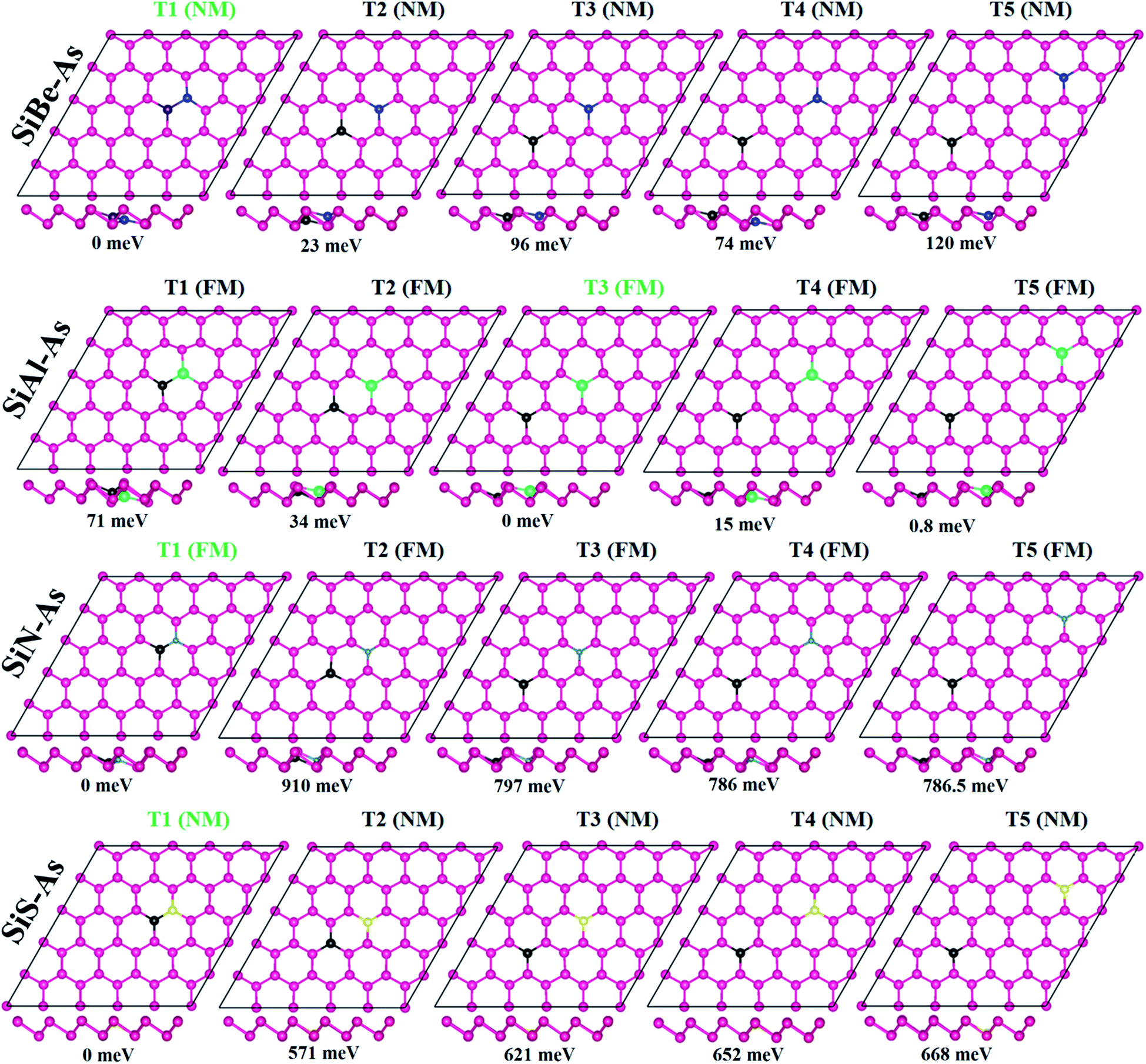 | ||
| Fig. 1 Optimized geometries of SiX (X = Be, Al, N, and S) dual doped arsenene (SiX–As) under different doping configurations. The relative total energy (RE) in meV is calculated by subtracting the total energy from the lowest total energy. The most stable configuration is shown by green letters. | ||
The incorporation of foreign atoms into the host lattice may induce distortion, which can be explored from the geometric parameters such as bond lengths, bond angles and buckling heights. The bond lengths between dopants and neighboring As atoms are shown in Fig. S3.† We observe that Si–As, X–As and Si–X (where X = Be, Al, N, and S) bond length is somewhat smaller than the actual As–As bond length of 2.50 Å. In particular, the N–As bond length has the smallest value of 1.98 Å, being 20% smaller than the As–As bond length. The decrease of bond length is often associated with electronegativity or atomic size of the atoms.13 Moreover, the other two structural parameters such as bond angles and buckling height at the dopant sites are also found slightly changed as some dopants move downward after relaxation (Fig. 1, side view). Despite these minor changes the geometry of arsenene is overall well maintained after doping.
To verify the stability of SiX–As systems, the binding energy Eb is collected in Table 1. All systems exhibit negative Eb, signifying that selected co-doping in arsenene is energetically favorable. In particular, the binding strength follows an order Eb(SiN–As) > Eb(SiS–As) > Eb(SiAl–As) > Eb(SiBe–As), similar to the electronegativity trend (N > S > Al > Be). Thus, in SiN–As, higher energy would be required to break the lattice into constituent atoms. Furthermore, the Eb for SiN–As is larger (in magnitude) than the cohesive energy reported for N doped arsenene, expecting a superior character of hetero-atom doping over single ones in arsenene.13 Also the co-doped arsenene structures have lower Eb in comparison with the formation energy reported for single IV and VI atom doped arsenene, suggesting that co-doped arsenene is expected to be thermodynamically more stable than the corresponding single atom doped counterpart.31 However, for the present systems, Ebs are smaller than those reported for transition metal atom (Ti, V, Cr, Fe, and Ni) doped arsenene.32
| System | Eb | q (Si, As) | q (X, As) | M | Eg |
|---|---|---|---|---|---|
| p-As | — | — | — | 0 | 1.60 |
| SiBe–As | −2.9714 | 0.226, −0.320 | 1.403, −0.591 | 0 | 0.44 |
| SiAl–As | −3.3819 | 0.584, −0.215 | 1.672, −0.577 | 1 | 0.66 |
| SiN–As | −4.4701 | 1.068, −0.184 | −1.511, 0.415 | 1 | 0.63 |
| SiS–As | −3.7476 | 0.532, −0.198 | −0.658, 0.241 | 0 | 1.19 |
The nature of bonding between dopants and connected As atoms is examined with Bader charge analysis which provides a net partial charge on Si, X and As atoms, shown in Table 1. The negative (positive) sign indicates charge accumulation (donation). Clearly, in SiBe–As and SiAl–As, the Si, Be and Al systems act as charge donors and As atoms as charge acceptors, whereas in the other two systems, Si and As atoms are charge donors and N and S atoms behave as charge acceptors. The large charge transfer between dopants and host As atoms indicates strong bonding between them. The magnitude of charge on Si with q = 1.068e and N with q = −1.511e in SiN–As is in agreement with its largest binding energy. The exchange of charge indicates the formation of strong bonding between dopants and host atoms.
3.1 Electronic and magnetic properties
The replacement of As atoms with Si/X atoms is expected to destroy the sub-lattice symmetry and charge distribution in arsenene and bring a significant change in electronic properties. To get insights into the electronic properties of SiX–As systems, the electronic band structures and density of states for non-magnetic SiBe–As/SiS–As and magnetic SiAl–As/SiN–As are illustrated in Fig. 2 and 3(a and b). For reference, the electronic band structure of p-As is also calculated and shown in Fig. S1.† It is found that p-As is an indirect-band semiconductor with a band gap of 1.59 eV between the valence band maximum (VBM) located at the Γ point and conduction band maximum (CBM) along the Γ–M direction, agreeing with the literature.9 Fig. 2 shows that each system has a unique band structure with the emergence of some flat bands (localized defect states) in the fundamental gap of p-As, which reduce the actual band gap of the material. The computed band gaps shown in Table 1 predict that the actual band gap is reduced by 72%, 58%, 60% and 25%, respectively after Si/Be, Si/Al, Si/N and Si/S doping, confirming high sensitivity of arsenene toward the dopants. To gain insight into the formation of the band structure and emergence of defect states, the calculated orbital projected density of states shown in Fig. 3(a and b) is considered. For the SiBe–As system, the valence band edge is formed by the p-orbitals of Be and first neighbored As atoms. The defect states emerging in the 0 to 0.5 eV energy interval above the Fermi level (EF) originate from the p-orbitals of Si, Be and As atoms. In the SiS–As system, the presence of p-orbitals of Si, S and As atoms in the VBM and CBM is obvious. Furthermore, the spin-polarized PDOS of SiAl–As shows that the VBM is formed by the py-orbitals of the constituent atoms. The defect states rising around 0.5 eV above the EF are governed by the pz-states of the Si and As atoms. In the SiS–As system, the defect states appearing just below the EF are characterized by the pz-orbitals. This concludes that electronic properties are effectively diluted by the presence of Si and X (X = Be, Al, N, and S) atoms in arsenene.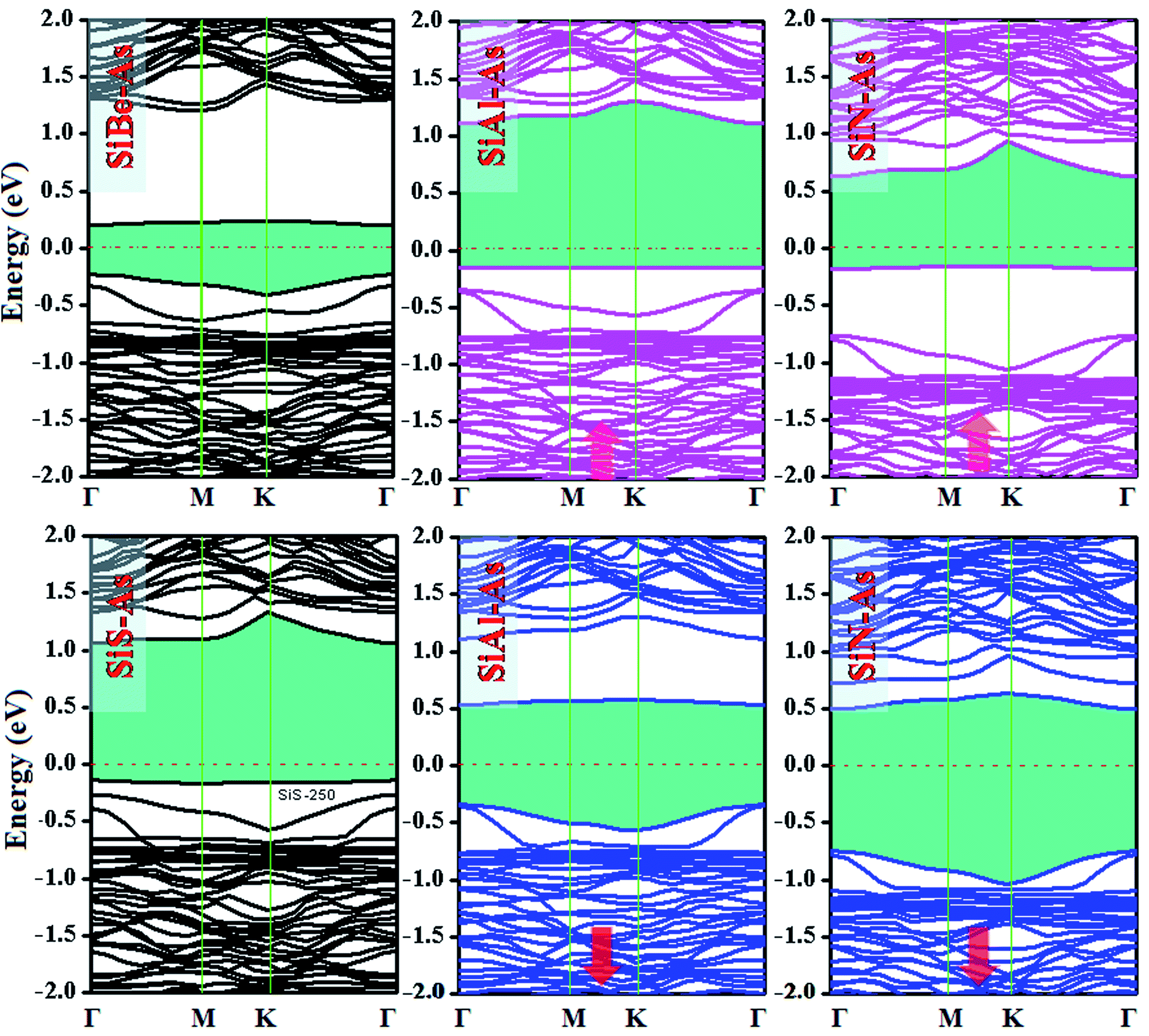 | ||
| Fig. 2 Ground state electronic band structures for SiX (X = Be, Al, N, and S) dual doped arsenene systems. | ||
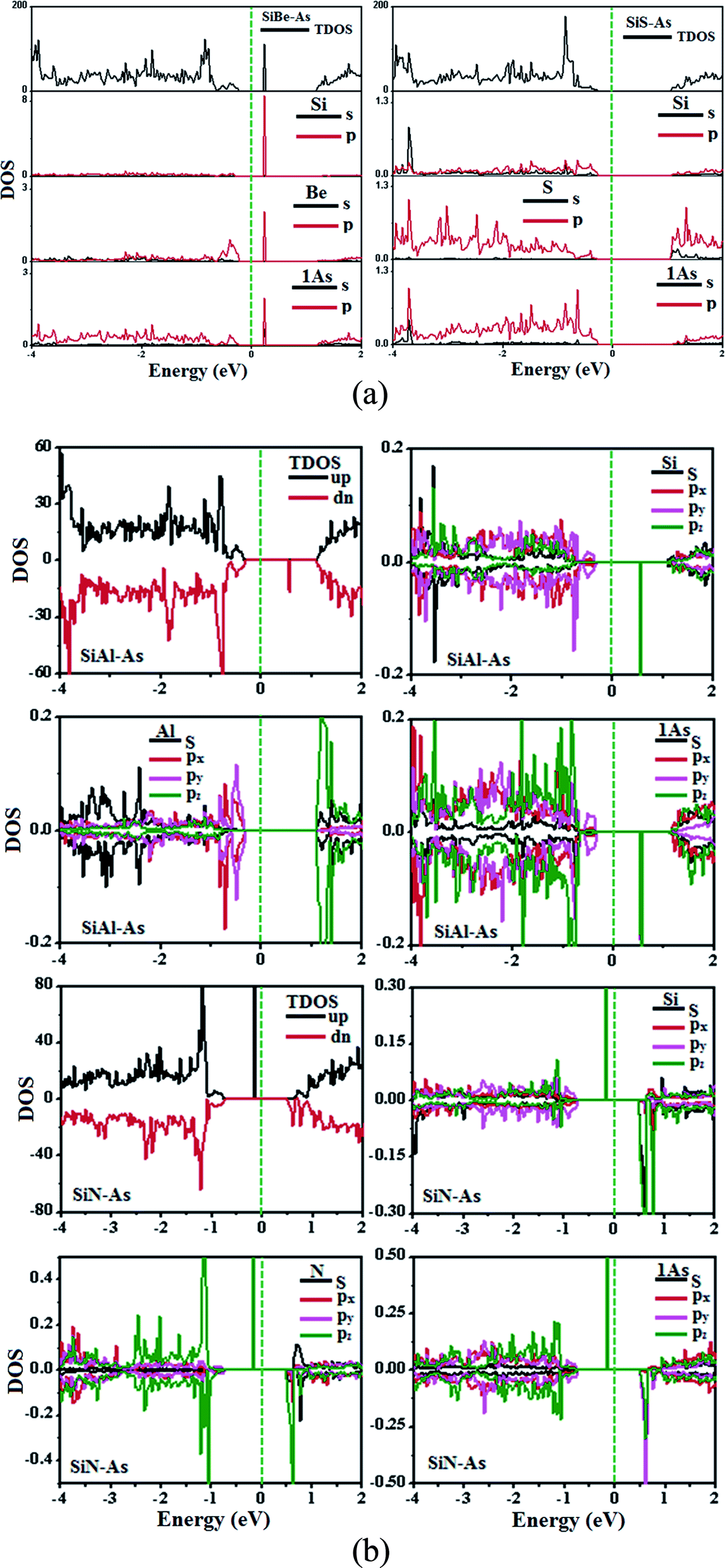 | ||
| Fig. 3 Ground state density of states (DOS) for (a) non-magnetic SiBe–As and SiS–As systems, (b) magnetic SiAl–As and SiN–As systems. | ||
Next, we discuss the effect of doping on the magnetic properties of arsenene. For p-As being nonmagnetic in the pristine form, the introduction of external atoms might bring magnetism. In the present case, Si/Be and Si/S co-doping did not induce any magnetic moment in arsenene; however, SiAl–As and SiN–As systems exhibit magnetism in the ground state. The calculated total magnetic moment for these systems is integer, shown in Table 1. To explore the source of magnetism, the spin-polarized PDOS for the two systems is presented in Fig. 3b. Evidently, the orbital resolved DOS displays asymmetric distribution between spin-up and spin-down electronic states over the given energy range, certifying spin-polarization in the constituent atoms. In particular, the Si atom is more spin-polarized than Al and As atoms. Moreover, among p-orbitals of the Si atom, the occupied part of the py-orbital has a clear asymmetric character in two spin channels. Accordingly, the Si py-orbital has a major contribution to the magnetic moment of the systems. Next, focusing on the orbital resolved DOS of Si, N and As atoms in the SiN–As system, it proves that pz-orbitals are remarkably spin-polarized, particularly 100% spin-polarized just below the EF, leading to a distinguished role in the observed magnetism. To further elucidate the witnessed magnetism in SiAl–As and SiN–As systems, spin-density (ρ↑–ρ↓) plots are provided in Fig. 4. Clearly, spin density is mainly located at the Si atom and bonded As atoms in SiAl–As. On the other hand, for SiN–As, the spin density resides at both Si and N atoms, and connected As atoms also have spin density. This concludes that magnetism in SiAl–As is mainly due to the presence of Si atoms, while both Si and N atoms play a leading role in the emergence of magnetism in the SiN–As system.
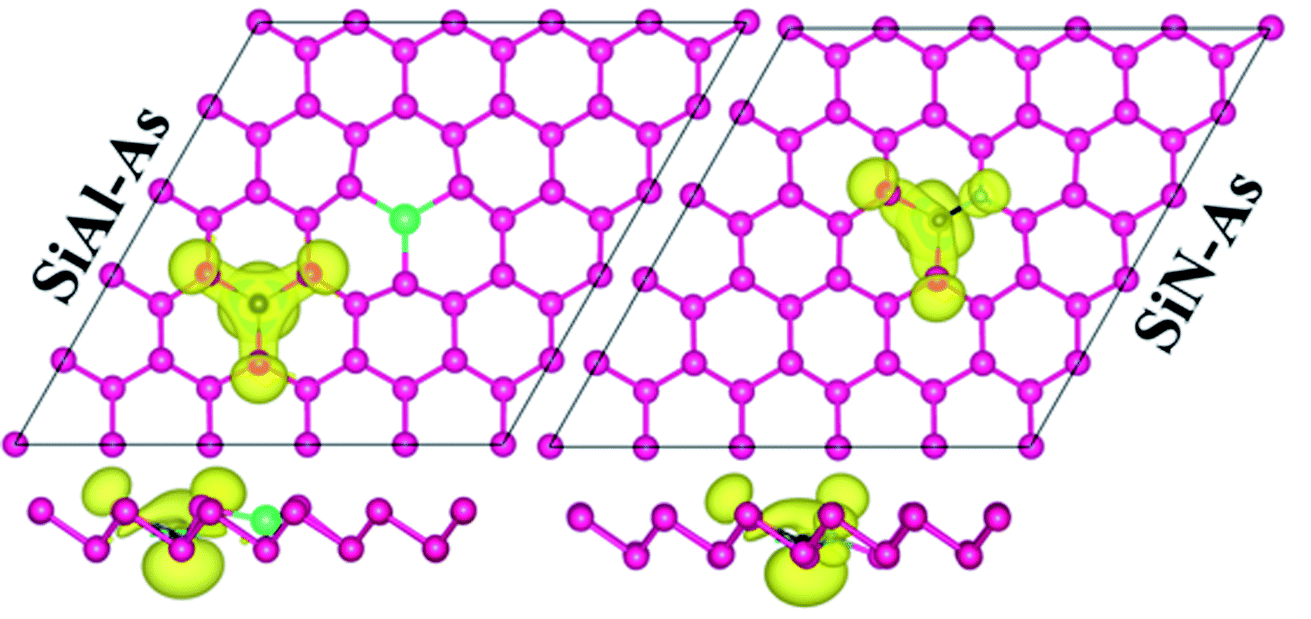 | ||
| Fig. 4 Spin density plots for SiAl–As and SiN–As systems. | ||
3.2 Adsorption of CO
The adsorption of CO over p-As and co-doped arsenene is studied in terms of adsorption energy (Ea), charge transfer (ΔQ), adsorption height, the vertical distance between the adsorbent and gas molecule (h), smallest atom to atom distance between the adsorbent and gas molecule dmin, Fermi energy EF, and work function. Besides, the amount and direction of the charge transfer is revealed through charge density difference (CDD) plots. Moreover, to examine the origin of adsorbent–molecule interaction, the electronic band structures and density of states are also presented.In p-As, there are typically four different adsorption sites namely the bridge site (B), hollow site (H), top of the As atom in the upper plane (T) and top of the As atom in the lower plane called the valley site (V), shown in Fig. 5. Meanwhile, as shown in Fig. 6, four different orientations of the molecule are considered including (1) x-orientation (the C–O bond length is along the x-axis and parallel to the surface), (2) y-orientation (the C–O bond length is along the y-axis and parallel to the surface), C-vertical (where the C–O bond length is normal to the surface and the C atom is directed towards the surface), and (4) O-vertical (where the C–O bond length is normal to the adsorbent and the O atom points towards the surface).
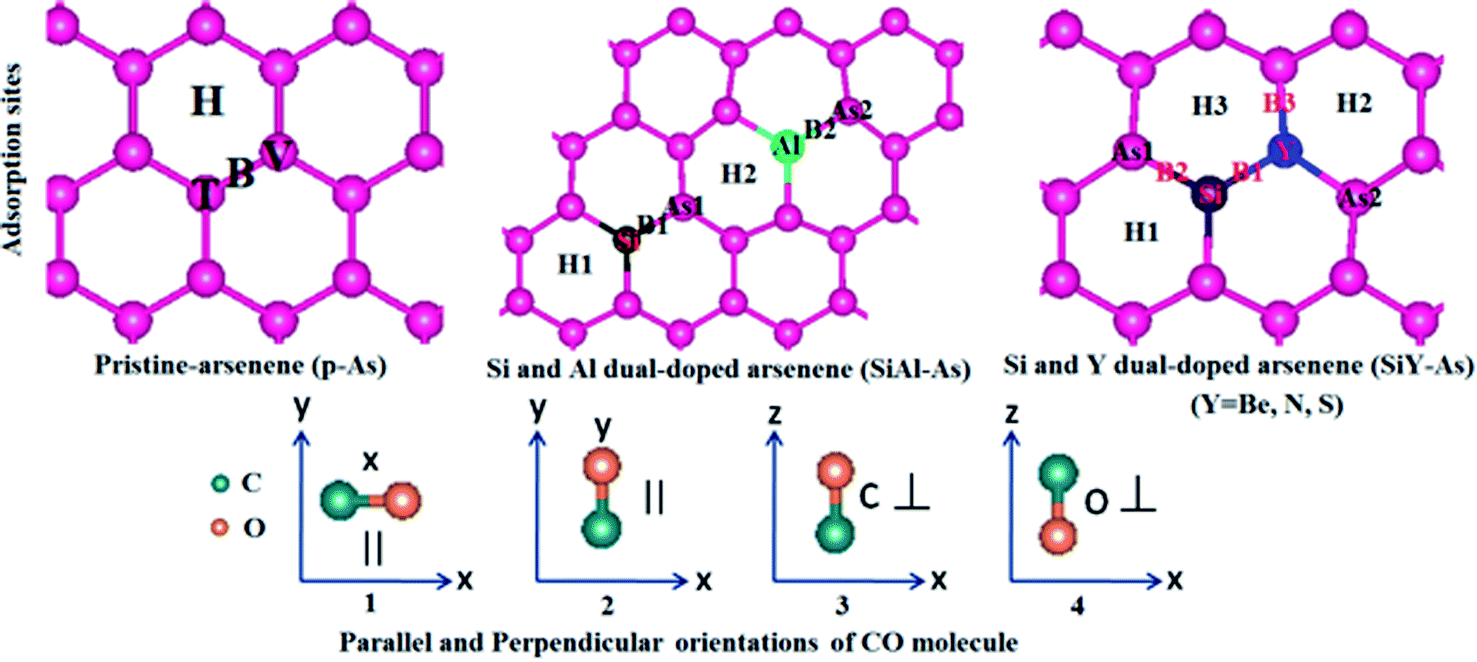 | ||
| Fig. 5 Initial adsorption configurations of CO over p-As and SiX–As systems. | ||
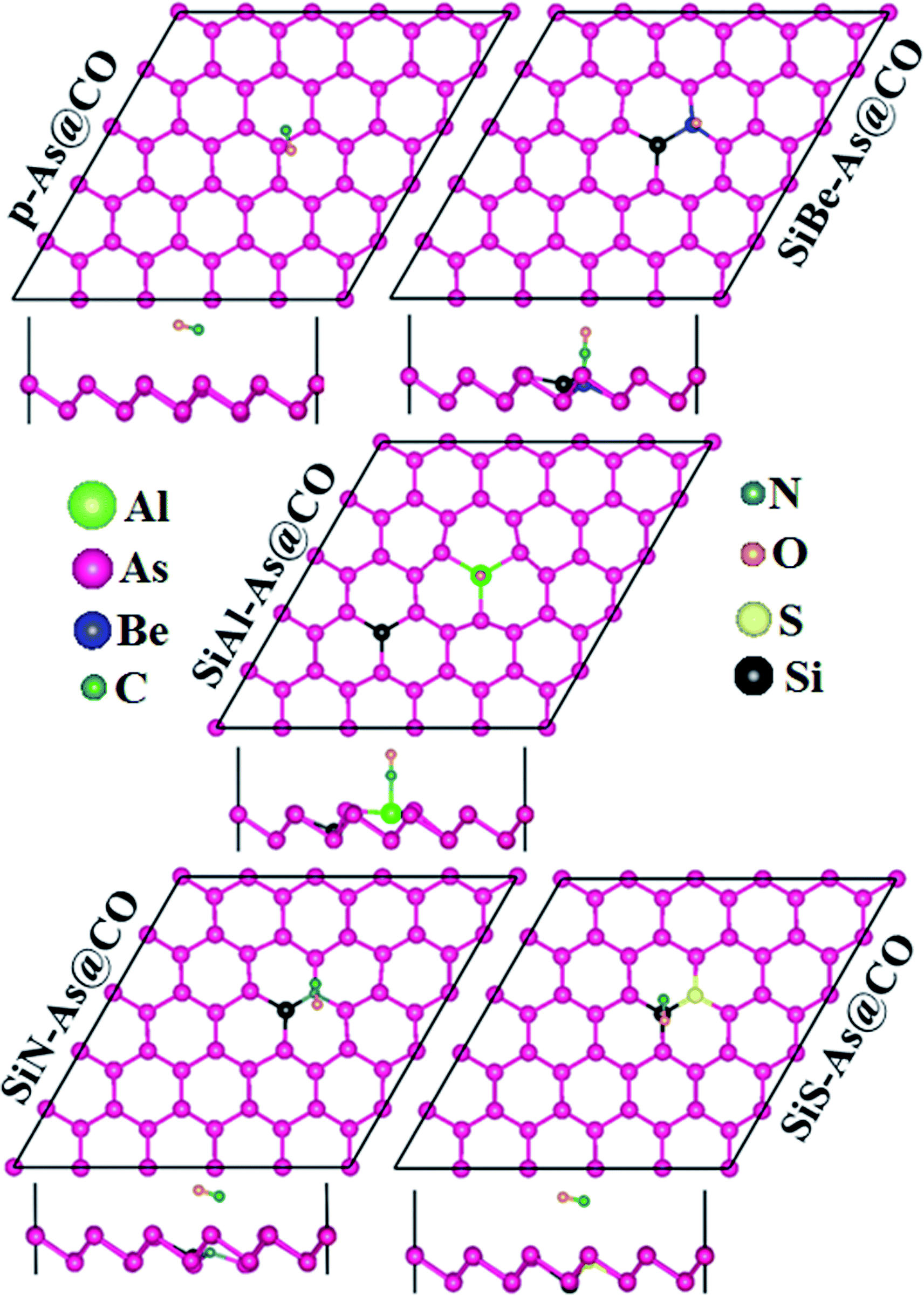 | ||
| Fig. 6 Most stable adsorption configurations of CO adsorbed on p-As and SiX–As systems. | ||
To find the most stable adsorption site, each system (adsorbent + molecule) is fully relaxed in spin-polarized calculations and its Ea is calculated and resulting stable adsorption configurations are presented in Fig. 6. It is found that the system with molecules adsorbed at the B site has the lowest total energy which means the lowest Ea. The calculated values of Ea, ΔQ, h, dmin, EF, and Φ are presented in Table 2. The negative value of Ea indicates that adsorption of CO over p-As might be energetically favorable. The magnitude of the adsorption energy i.e. |Ea| can be used to estimate the strength of the interaction between the adsorbent and molecule. In our case, |Ea| = 0.135 eV is very small and thus proves that p-As weakly interacts with CO gas molecules. Similarly, the calculated values of h (2.952 Å), dmin (3.546 Å), and ΔQ (−0.0279e) also confirm weak interaction. From Fig. 6, one can find that the CO molecule is slightly tilted with the C atom pointing towards the adsorbent. The observed dmin between As and C atoms is larger than the sum of covalent radius (As = 1.19 Å and C = 0.76 Å).33 Thus, the two sub-systems are assumed to have no chemical bonding except weak van der Waals interactions between them. Besides, the CDD plot shown in Fig. 8 further confirms that there is no obvious exchange of charge between the adsorbent and molecule. The work function Φ of a material (the energy taken by the electrons to leave the metal surface) might be sensitive to the presence of gas molecules. In the present case, Φ is almost unchanged upon molecule adsorption. To explore the origin of the observed interaction, total, atom projected, and orbital projected density of states (DOS) are calculated and shown in Fig. 8. Clearly, the states near the EF mainly originate from the As atoms, while occupied states of C and O atoms appear together around −8.8 eV, −3.8 eV in the valence band and unoccupied states around 3 eV in the conduction band. These regions indicate strong hybridization between C-p and O-p states. To establish a better understanding, the DOS of the isolated CO gas molecule is also computed and shown in Fig. S4.† Besides, such regions show that C-p and O-p states weakly overlap with As-p states which results in a minor charge transfer. This insignificant charge transfer does not induce any significant change in the electronic band structure of the p-As and the calculated Eg = 1.67 eV is almost close to the actual value (1.59 eV).
| System | Ea | ΔQ | h | dmin | dC–O | EF | Φ | M | Eg |
|---|---|---|---|---|---|---|---|---|---|
| p-As@CO | −0.135 | −0.0279 | 2.952 | 3.546 | 1.144 | −2.072 | 4.588 | 0 | 1.67 |
| SiBe–As@CO | −1.275 | −0.426 | 1.235 | 1.671 | 1.151 | −2.448 | 4.944 | 0 | 0.656 |
| SiAl–As@CO | −0.488 | −0.2368 | 2.058 | 2.071 | 1.148 | −2.142 | 4.724 | 1 | 0.677 |
| SiN–As@CO | −0.161 | −0.0351 | 2.053 | 3.041 | 1.144 | −1.927 | 4.413 | 1 | 0.645 |
| SiS–As@CO | −0.1195 | −0.02594 | 2.881 | 3.637 | 1.144 | −2.008 | 4.529 | 0 | 1.174 |
Next we discuss the adsorption of CO gas molecules over SiX–As systems. The various adsorption sites for these systems are shown in Fig. 6. The most stable adsorption sites and molecule orientations determined after geometry optimization are shown in Fig. 7 and related calculated parameters (Ea, h, dmin, ΔQ, Φ, Eg, etc.) are mentioned in Table 2. In the case of the SiBe–As system, the most stable adsorption site is found to be the top of the Be atom with the C atom of the CO molecule pointing towards the adsorbent. The small Ea, h and dmin, and large ΔQ values reveal that, unlike p-As, there exists strong interaction (chemisorption) between the gas molecule and substrate. The calculated dmin (1.67 Å) between Be and C atoms is fairly close to the sum of covalent radii (Be = 0.96 Å, C = 0.76 Å), ensuring the formation of a strong chemical bond between Be and C atoms. The formation of such a chemical bond is associated with a considerable amount of charge transfer (−0.426e) from the substrate to the molecule. Consequently, the work function changes by about 0.356 eV, from the value of 4.588 eV found in the p-As@CO system.
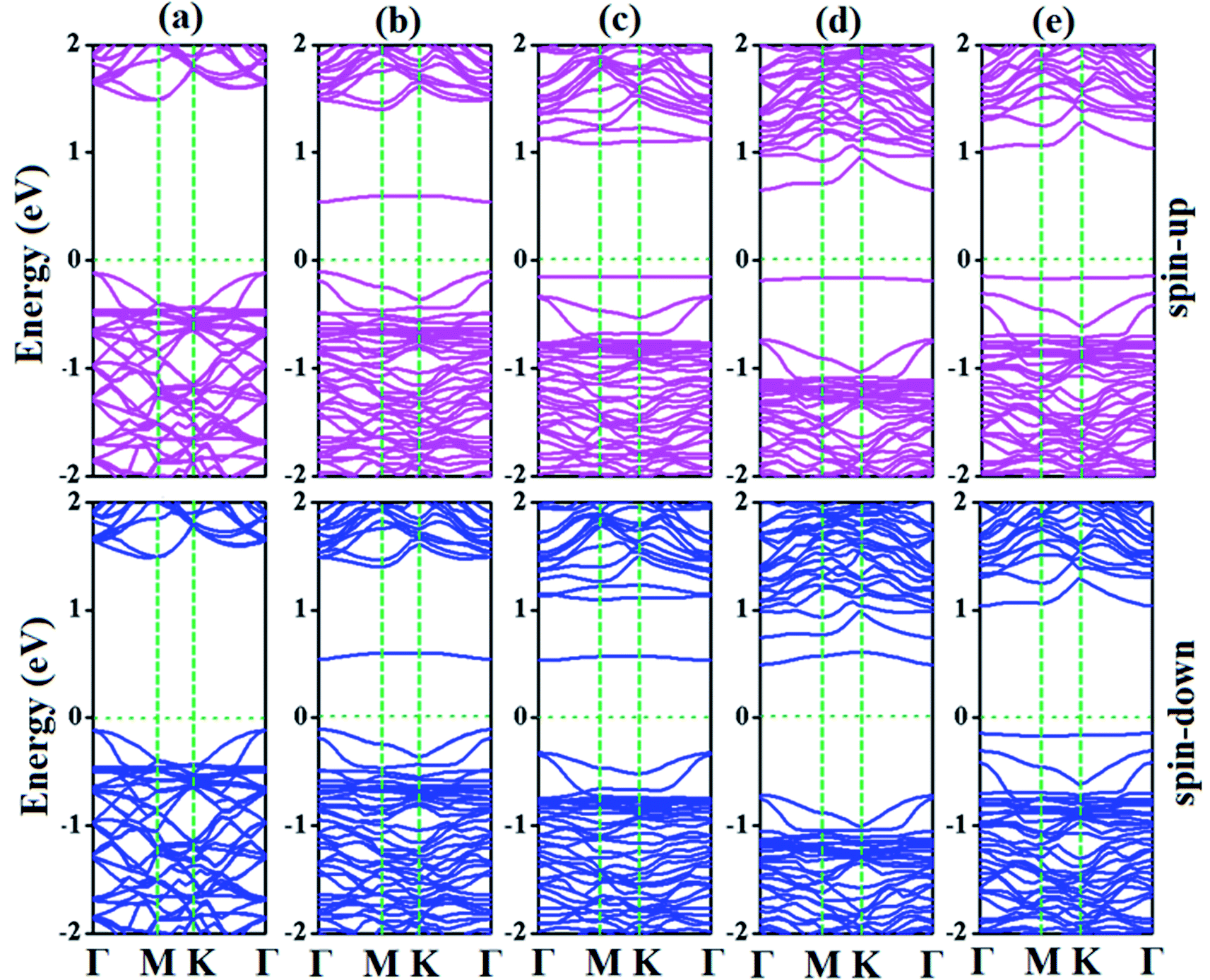 | ||
| Fig. 7 Electronic band structures of (a) p-As@CO, (b) SiBe–As@CO, (c) SiAl–As@CO, (d) SiN–As@CO, and (e) SiS–As@CO systems. | ||
For the SiAl–As@CO system, the energetically favorable adsorption site is the top of the Al atom with adsorption parameters presented in Table 2. The data show that the interaction between the gas molecule and the adsorbent is somewhat stronger than that of p-As@CO but weaker than that found in the SiBe–As@CO system. We may identify this interaction as physisorption. Similarly, in the SiN–As system, the stable adsorption place is the top of the N atom and for the SiS–As system, the molecule prefers to occupy the top of the Si atom. Recalling Table 2, we found that both these systems have weak interaction with CO gas molecules. Finally, to examine the effect of adsorption on the electronic properties of SiX–As systems, the corresponding band structures and DOS plots are shown in Fig. 7 and 8. Table 2 shows that the CO adsorption has negligible effect on the band gap of the systems, except for the SiBe–As@CO system where the band gap slightly increases by about 0.216 eV. The DOS plots show that in physisorbed SiAl/N/S–As@CO systems, the states of C/O atoms lie far away from the Fermi level EF, whereas in the chemisorbed SiBe–As@CO system, some of the states emerge from C/O atoms.
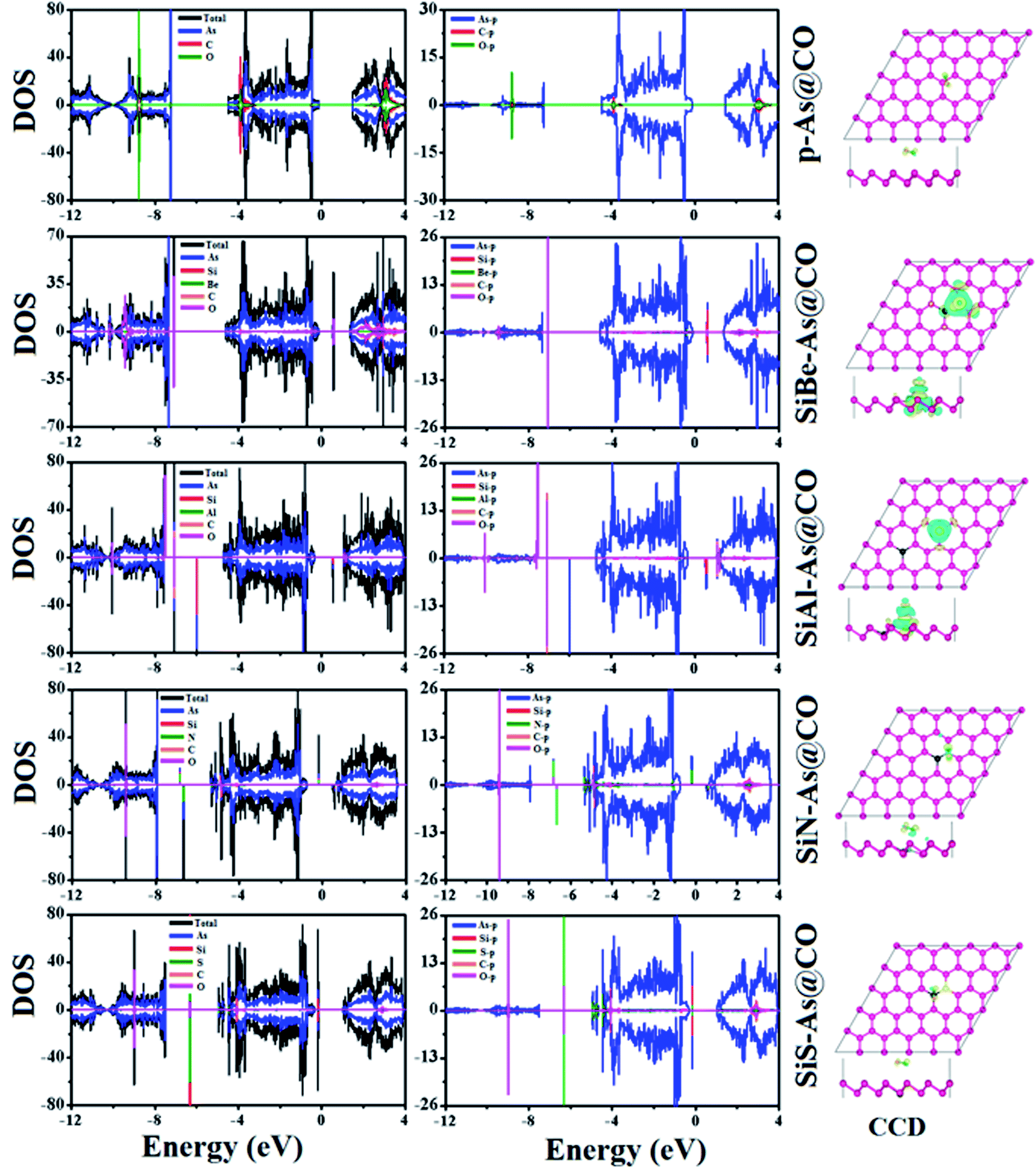 | ||
| Fig. 8 Density of states (DOS) and charge density difference (CDD) plots for SiX–As@CO systems. Yellow (blue) colors indicate charge accumulation (depletion), with the isosurface value set at 0.0005 e Bohr−3. | ||
The performance of a gas sensor is found to be dependent on recovery time τ which is related to Ea through eqn (3).
| τ = υ0−1e(−Ea/kBT) | (3) |
The above discussion demonstrates that the CO gas molecule has physisorption over p-As, SiAl–As, SiN–As and SiS–As systems, while SiBe–As shows chemisorption, which might be used in the CO gas sensing devices.
4. Conclusions
In this paper, the effect of dual doping with Si and X (X = Be, Al, N, and S) atoms on the structural, electronic, magnetic and adsorption properties of arsenene is investigated within the theoretical framework. The results show that dual doping (DD) slightly alters the local atomic structure of p-As and forms strong chemical bonds with the neighboring As atoms. The doped systems show a reduced band gap due to the emergence of defect states within the actual band gap. Interestingly, while SiBe–As and SiS–As systems retain the non-magnetic character, the SiAl–As and SiN–As systems carry a magnetic moment of 1 μB, mainly caused by the spin-polarization of the dopants. Finally, to predict the potential of the doped systems for gas sensing purpose, the adsorption of CO gas is investigated. The obtained adsorption properties (adsorption energy Ea, adsorption height h, minimum atom-to-atom distance dmin, charger transfer ΔQ, work function Φ, and band gap Eg) reveal that the CO gas molecule is physically adsorbed over pristine As (p-As), SiAl–As, SiS–As and SiS–As systems, whereas chemical adsorption is observed for the SiBe–As system. Our calculations predict that the interaction of arsenene with environmentally hazardous CO gas may be enhanced via Si and Be dual doping, which might extend the potential of the material in gas sensing applications.Conflicts of interest
The authors declare no conflict of interest.References
- G. Pizzi, M. Gibertini, E. Dib, N. Marzari, G. Iannaccone and G. Fiori, Nat. Commun., 2016, 7, 12585 CrossRef CAS PubMed.
- X. Sun, Z. Song, S. Liu, Y. Wang, Y. Li, W. Wang and J. Lu, ACS Appl. Mater. Interfaces, 2018, 10, 22363–22371 CrossRef CAS PubMed.
- Y. P. Wang, C. W. Zhang, W. X. Ji, R. W. Zhang, P. Li, P. J. Wang, M. J. Ren, X. I. Chen and M. Yuan, J. Phys. D: Appl. Phys., 2016, 49, 055305 CrossRef.
- H. S. Tsai, S. W. Wang, C. H. Hsiao, C. W. Chen, H. Ouyang, Y. L. Chueh, H. C. Kuo and J. H. Liang, Chem. Mater., 2016, 28, 425–429 CrossRef CAS.
- J. Shah, W. Wang, H. M. Sohail and R. I. G. Uhrberg, 2D Mater., 2020, 7, 025013 CrossRef CAS.
- P. Jamdagni, A. Thakur, A. Kumar, P. K. Ahluwalia and R. Pandey, Phys. Chem. Chem. Phys., 2018, 20, 29939–29950 RSC.
- S. Sharma, S. Kumar and U. Schwingenschlögl, Phys. Rev. Appl., 2017, 8, 044013 CrossRef.
- H. Shu, Y. Li, X. Niu and J. Guo, J. Mater. Chem. C, 2018, 6, 83–90 RSC.
- C. Kamal and M. Ezawa, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 91, 085423 CrossRef.
- Y. Wang and Y. Ding, Nanoscale Res. Lett., 2015, 10, 254 CrossRef PubMed.
- Y. P. Wang, W. X. Ji, C. W. Zhang, P. Li, F. Li, M. J. Ren, X. L. Chen, M. Yuan and P. J. Wang, Sci. Rep., 2016, 6, 20342 CrossRef CAS PubMed.
- D. Wang, L. Chen, C. Shi, X. Wang, G. Cui, P. Zhang and Y. Chen, Sci. Rep., 2016, 6, 28487 CrossRef CAS PubMed.
- M. Y. Liu, Y. Huang, Q. Y. Chen, C. Cao and Y. He, Sci. Rep., 2016, 6, 29114 CrossRef CAS PubMed.
- X. Sun, Y. Liu, Z. Song, Y. Li, W. Wang, H. Lin, L. Wang and Y. Li, J. Mater. Chem. C, 2017, 5, 4159–4166 RSC.
- J. Du, C. Xia, Y. An, T. Wang and Y. Jia, J. Mater. Sci., 2016, 51, 9504–9513 CrossRef CAS.
- M. Mushtaq, Y. G. Zhou and X. Xiang, J. Phys.: Condens. Matter, 2018, 30, 195305 CrossRef CAS PubMed.
- K. Iordanidou, J. Kioseoglou, V. V. Afanas'ev, A. Stesmans and M. Houssa, Phys. Chem. Chem. Phys., 2017, 19, 9862–9871 RSC.
- M. Sun, S. Wang, Y. Du, J. Yu and W. Tang, Appl. Surf. Sci., 2016, 389, 594–600 CrossRef CAS.
- M. Y. Liu, Q. Y. Chen, Y. Huang, Z. Y. Li, C. Cao and Y. He, Nanotechnology, 2018, 29, 095203 CrossRef PubMed.
- Y. P. Wang, C. W. Zhang, W. X. Ji and P. J. Wang, Appl. Phys. Express, 2015, 8, 065202 CrossRef.
- M. Bai, W. X. Zhang and C. He, J. Solid State Chem., 2017, 251, 1–6 CrossRef CAS.
- C. Liu, C. S. Liu and X. Yan, Phys. Lett. A, 2017, 381, 1092–1096 CrossRef CAS.
- K. Wang, J. Li, Y. Huang, M. Lian and D. Chen, Processes, 2019, 7, 538 CrossRef CAS.
- J. Mao and Y. Chen, J. Mater. Chem. C, 2020, 8, 4073–4080 RSC.
- S. Ullah, P. A. Denis and F. Sato, New J. Chem., 2018, 42, 10842–10851 RSC.
- P. A. Denis and C. Pereyra Huelmo, Carbon, 2015, 87, 106–115 CrossRef CAS.
- C. Zhao, J. Li, Y. Chen and J. Chen, New J. Chem., 2019, 43, 9389–9395 RSC.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- J. Du, C. Xia, T. Wang, X. Zhao, X. Tan and S. Wei, Appl. Surf. Sci., 2016, 378, 350–356 CrossRef CAS.
- Y. Liu, Q. Zhou, W. Ju, J. Li and Y. Liu, Superlattices Microstruct., 2019, 132, 106163 CrossRef CAS.
- B. Cordero, V. Gómez, A. E. Platero-Prats, M. Revés, J. Echeverría, E. Cremades, F. Barragán and S. Alvarez, Dalton Trans., 2008, 21, 2832–2838 RSC.
- S. Peng, K. Cho, P. Qi and H. Dai, Chem. Phys. Lett., 2004, 387, 271–276 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra03394h |
| This journal is © The Royal Society of Chemistry 2021 |