
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructure and absolute configuration assignments of ochracines F–L, chamigrane and cadinane sesquiterpenes from the basidiomycete Steccherinum ochraceum HFG119†
Zhen-Zhu Zhao a,
Qi-Lu Zhaoa,
Wei-Sheng Fenga,
Hai-Rong Hea,
Meng Lia,
Gui-Min Xuea,
He-Ping Chen*b and
Ji-Kai Liu*b
a,
Qi-Lu Zhaoa,
Wei-Sheng Fenga,
Hai-Rong Hea,
Meng Lia,
Gui-Min Xuea,
He-Ping Chen*b and
Ji-Kai Liu*b
aSchool of Pharmacy, Henan University of Chinese Medicine, Zhengzhou 450046, China
bSchool of Pharmaceutical Sciences, South-Central University for Nationalities, Wuhan 430074, China. E-mail: chenhp@mail.scuec.edu.cn; liujikai@mail.scuec.edu.cn
First published on 24th May 2021
Abstract
Ochracines F–L (1–7), seven previously undescribed chamigrane and cadinane sesquiterpenoids, together with four known chamigranes were isolated from cultures of the wood-decaying fungus Steccherinum ochraceum HFG119. Ochracines F–L were structurally characterized by extensive analysis of HRMS and NMR spectroscopic data. The relative configurations were assigned through a combination of NOE correlations and J-based configuration analysis (JBCA), while the absolute configurations were determined by X-ray single-crystal diffraction, and calculated methods (ECD, [α], 13C NMR). All the new isolates were evaluated for their cytotoxicity against five human cancer cell lines HL-60, SMMC-7721, A549, MCF-7, and SW-480, and inhibitory activity on NO production in RAW 264.7 macrophages.
Introduction
Mushroom-derived sesquiterpenes are recognized as the most structural among the reported mushroom natural products.1 Chamigrane sesquiterpenoids featuring a spiro backbone, are rare among higher fungi.2,3 Most reported chamigranes are generally produced by the red alga of the genus Laurencia (Family Rhodomelaceae)4–8 and endophytic fungi.9–14Our previous chemical study on Steccherinum ochraceum resulted in the isolation of four rare endoperoxide type chamigranes, namely steperoxides A–D, of which steperoxides C and D exhibited antimicrobial activity.2,3 Since natural endoperoxides are of special interest, they have attracted and continue to attract considerable attention for their bioactivities.15–17 To explore additional peroxy natural products from the cultures of S. ochraceum, we scaled up the fermentation, leading to the isolation of five chamigrane-related norsesquiterpene derivatives, ochracines A–E,18 and 9 chamigrane sesquiterpenes including five new ones, namely ochracines F–J (1–5), and two cadinane sesquiterpenes, namely ochracines K (6) and L (7) (Fig. 1).
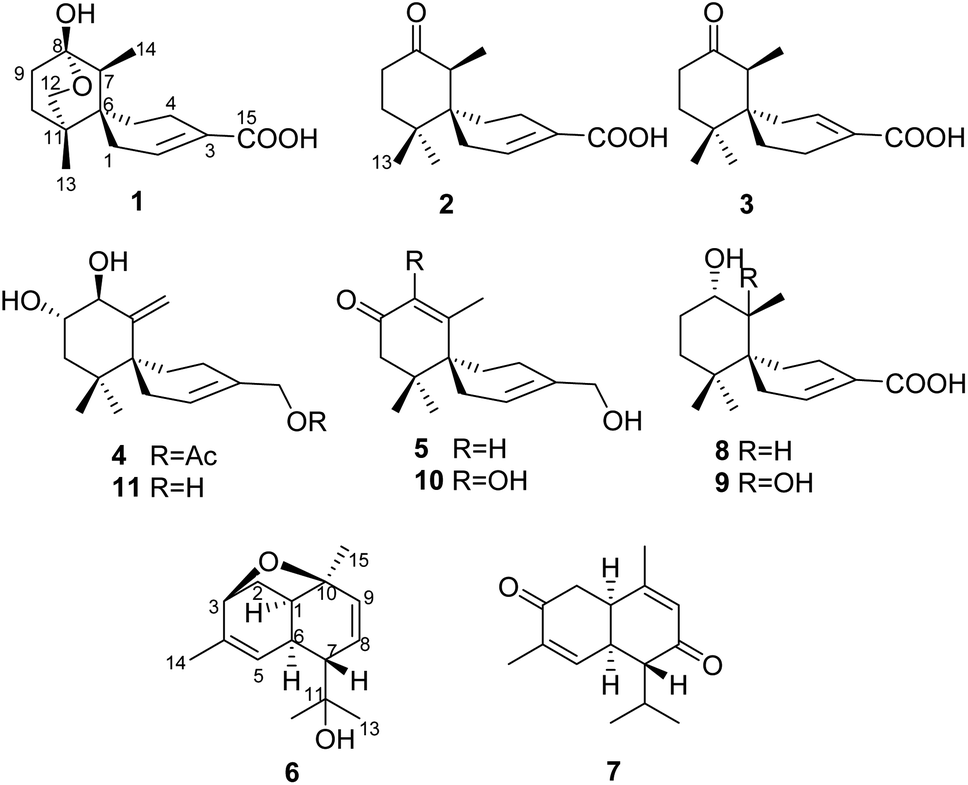 | ||
| Fig. 1 Structures of compounds 1–11. | ||
As some studies have shown that the production of secondary metabolites is highly dependent on fermentation conditions and modes,19–21 the fermentation conditions for producing endoperoxide chamigranes might be special.
On the other hand, the assignment of absolute configurations of small organic natural molecules sometimes is challenging due to the lack of chromophores which result in non-appropriate CD spectrum, and the overlapped proton signals in NMR spectrum causes problems of interpreting the NOE correlations, etc. Here, we described the isolation, structure elucidation, and more importantly, the absolute configuration assignment of the sesquiterpenoids from the cultures of the basidiomycete S. ochraceum by using combinatorial methods, including J-based configuration analysis (JBCA), calculated ECD, [α] and NMR. Moreover, the fermentation conditions for production endoperoxide type are discussed.
Results and discussion
Ochracine F (1), colourless needles (MeOH), afforded the molecular formula C15H22O4 deduced from the HRESIMS ion at m/z 289.1408 [M + Na]+, requiring five degrees of unsaturation. Combined analysis of 1H, 13C NMR data (Tables 1 and 2), and the HSQC spectroscopic correlations disclosed the presence of one singlet methyl, one doublet methyl, six sp3 methylenes [one oxygenated (δC/H 73.3/4.07, 3.57)], one sp3 methine, one trisubstituted double bond (δC/H 142.4/7.07; δC 131.7), and four quaternary carbons, including one hemiketal carbon (δC 99.3) and a conjugated carboxylic carbon (δC 170.8). The above assignments (an α,β-unsaturated carboxyl) account for two of five hydrogen deficiencys, indicating that 1 is a tricyclic compound. Further analysis of the 1H–1H COSY spectrum (Fig. 3) established five spin systems of H2-1/H-2, H2-4/H2-5, H3-14/H-7, and H2-9/H2-10. The HMBC spectrum (Fig. 3) showed correlations from H3-13 to C-6/C-10/C-11/C-12, H3-14 to C-6/C-7/C-8, and H-7 to C-9, establishing a partial structure of a six-membered ring with a hemiketal (C-8), 12-CH2OH, 13-CH3 and 14-CH3, and correlations from H2-4 to C-3/C-6, and H-2 to C-15, confirming the presence of a cyclohexene ring with a carboxylic group (15-COOH) substituted at C-3. These two rings share C-6 as the spiro carbon judging from the HMBC correlations of H3-13/H3-14/H2-4/H-2 to C-6. The molecular formula, C15H22O4, and the HMBC correlations from H2-12 to C-8 suggested that C-8 and C-12 are linked through an ether bond, which rendered compound 1 a novel chamigrane sesquiterpene featuring a 6/6/6 tricyclic system. As shown in Fig. 4, the relative configuration was determined by NOE correlations. Fortunately, single crystal X-ray analysis using CuKα radiation not only confirmed the structure validity of 1, but also established its absolute configuration (Fig. 2). Moreover, in order to provide integrated data for the structure of this type, experimental CD and calculated ECD spectra of (6R,7S,8S,11R)-1 were carried out (Fig. 5) (ESI†).| No. | 1a | 2b | 3b | 4b | 5b |
|---|---|---|---|---|---|
| a Measured in CD3OD.b Measured in CDCl3.c Interchangeable assignments. | |||||
| 1 | 2.33, brd (20.0), 2.16, brd (20.0) | 2.11, brd (17.9), 2.06, brd (17.9) | 2.38, brd (19.3), 2.00, brd (19.3) | 2.18, brd (18.5), 2.07, brd (18.5) | 2.29, brd (18.5), 2.11, brd (18.5, 2.0) |
| 2 | 7.07, brs | 7.12, brs | 7.11, brs | 5.70, brs | 5.82, brs |
| 4 | 2.49, brd (18.0), 2.27, brd (18.0) | 2.45, overlapped, 2.30, m | 2.31, m, 2.25, m | 1.95, brd (18.0), 1.71, brd (18.0) | 2.19, m, 1.94, overlapped |
| 5 | 1.87, overlapped, 1.50, overlapped | 1.72, overlapped, 1.67, overlapped | 1.67, ddd (16.0, 10.0, 6.2), 1.58, ddd (16.0, 4.5, 4.5) | 1.90, brd (13.0), 1.54, brd (13.0) | 1.90, m, 1.85, m |
| 7 | 1.74, q (7.2) | 2.67, q (7.0) | 2.61, q (7.0) | ||
| 8 | 4.08, brd (9.3) | 5.88, s | |||
| 9 | 1.90, overlapped, 1.69, overlapped | 2.42, overlapped, 2.40, overlapped | 2.51, ddd (14.7, 6.0, 6.0), 2.38, ddd (14.7, 8.5, 6.5) | 3.46, ddd (11.5, 9.3, 5.0) | 2.64, d (17.0), 2.08, d (17.0) |
| 10 | 1.87, overlapped, 1.46, overlapped | 1.81, m, 1.71, overlapped | 1.81, ddd (13.7, 8.5, 6.0), 1.75, ddd (13.7, 6.5, 6.0) | 1.81, dd (13.3, 11.5), 1.59, dd (13.3, 5.0) | |
| 12 | 4.07, d (9.0), 3.57, d (9.0) | 1.16, s | 1.10c, s | 0.93, s | 0.96c, s |
| 13 | 0.70, s | 0.98, s | 1.11c, s | 0.83, s | 1.04c, s |
| 14 | 0.91, d (7.2) | 1.04, d (7.0) | 1.07, d (7.0) | 5.39, d (1.7), 4.78, d (1.7) | 1.98, s |
| 15 | 4.40, d (12.0), 4.36, d (12.0) | 4.07, d (13.4), 4.03, d (13.4) | |||
| 15-OAc | 2.05, s | ||||
| No. | 1a | 2b | 3b | 4b | 5b | 6b | 7a |
|---|---|---|---|---|---|---|---|
| a Measured in CD3OD.b Measured in CDCl3. | |||||||
| 1 | 29.4, CH2 | 27.4, CH2 | 32.7, CH2 | 29.6, CH2 | 27.9, CH2 | 40.0, CH | 39.5, CH |
| 2 | 142.4, CH | 142.4, CH | 141.8, CH | 125.0, CH | 123.5, CH | 35.6, CH2 | 40.9, CH |
| 3 | 131.7, C | 129.3, C | 129.4, C | 132.1, C | 137.7, C | 77.3, CH | 199.6, C |
| 4 | 22.2, CH2 | 22.7, CH2 | 22.1, CH2 | 23.8, CH2 | 24.0, CH2 | 138.4, C | 137.4, C |
| 5 | 31.0, CH2 | 28.3, CH2 | 23.9, CH2 | 26.6, CH2 | 30.6, CH2 | 126.1, CH | 149.2, CH |
| 6 | 39.0, C | 44.5, C | 43.6, C | 45.4, C | 44.0, C | 37.1, CH | 40.2, CH |
| 7 | 47.2, CH | 49.9, CH | 50.5, CH | 146.7, C | 170.9, C | 51.4, CH | 59.1, CH |
| 8 | 99.3, C | 214.3, C | 214.2, C | 74.5, CH | 127.3, CH | 123.8, CH | 202.6, C |
| 9 | 28.0, CH2 | 37.7, CH2 | 37.0, CH2 | 73.3, CH | 49.1, CH2 | 136.5, CH | 128.1, CH |
| 10 | 30.6, CH2 | 38.2, CH2 | 37.1, CH2 | 43.1, CH2 | 198.8, C | 77.4, C | 162.1, C |
| 11 | 35.8, C | 37.5, C | 37.5, C | 37.5, C | 40.6, C | 72.9, C | 28.1, CH |
| 12 | 73.3, CH2 | 23.7, CH3 | 25.2, CH3 | 24.7, CH3 | 25.0, CH3 | 28.3, CH3 | 21.0, CH3 |
| 13 | 17.7, CH3 | 26.2, CH3 | 24.6, CH3 | 24.1, CH3 | 24.0, CH3 | 29.8, CH3 | 21.5, CH3 |
| 14 | 13.8, CH3 | 11.8, CH3 | 12.5, CH3 | 109.8, CH2 | 24.5, CH3 | 21.5, CH3 | 15.7, CH3 |
| 15 | 170.8, C | 170.4, C | 170.9, C | 68.3, CH2 | 67.0, CH2 | 28.4, CH3 | 21.9, CH3 |
| 15-OAc | 171.1, C | ||||||
| 15-OAc | 21.2, CH3 | ||||||
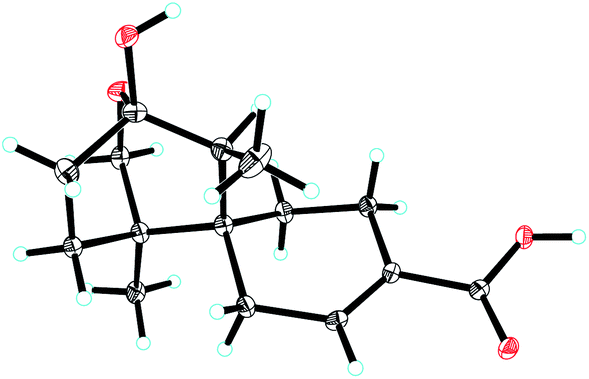 | ||
| Fig. 2 ORTEP drawing of compound 1. | ||
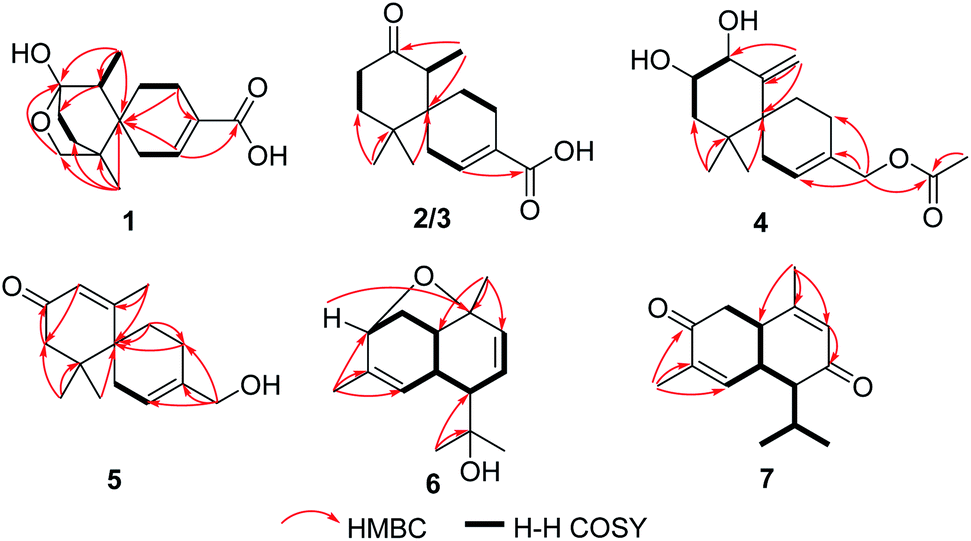 | ||
| Fig. 3 Selected HMBC and 1H–1H COSY correlations of 1–7. | ||
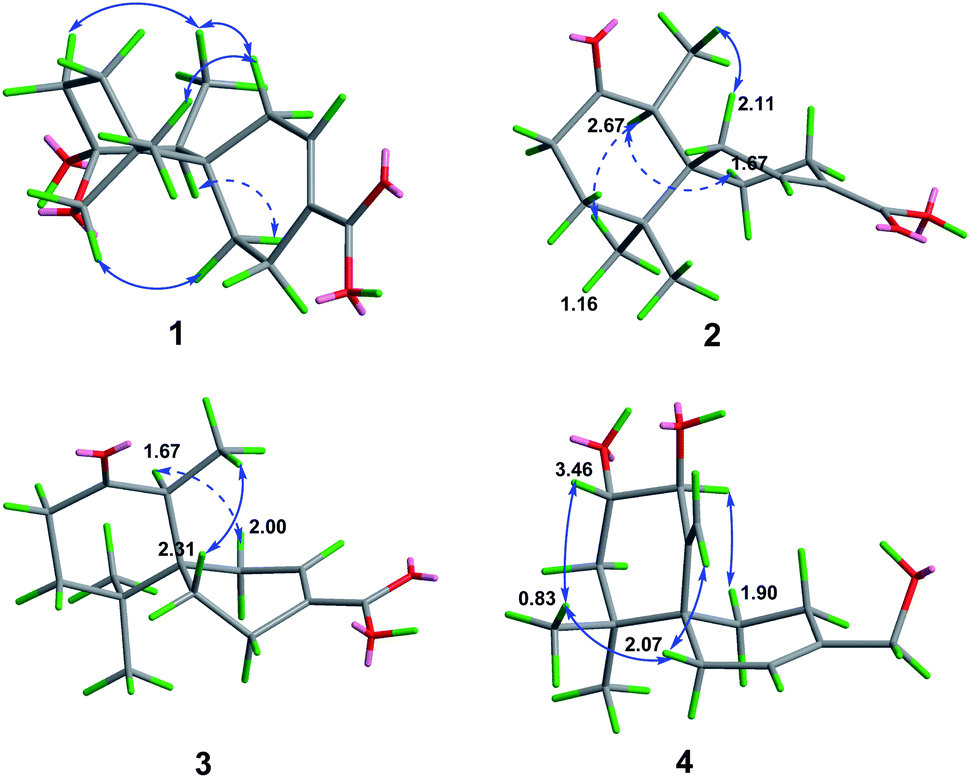 | ||
| Fig. 4 Selected NOE correlations of 1–4. | ||
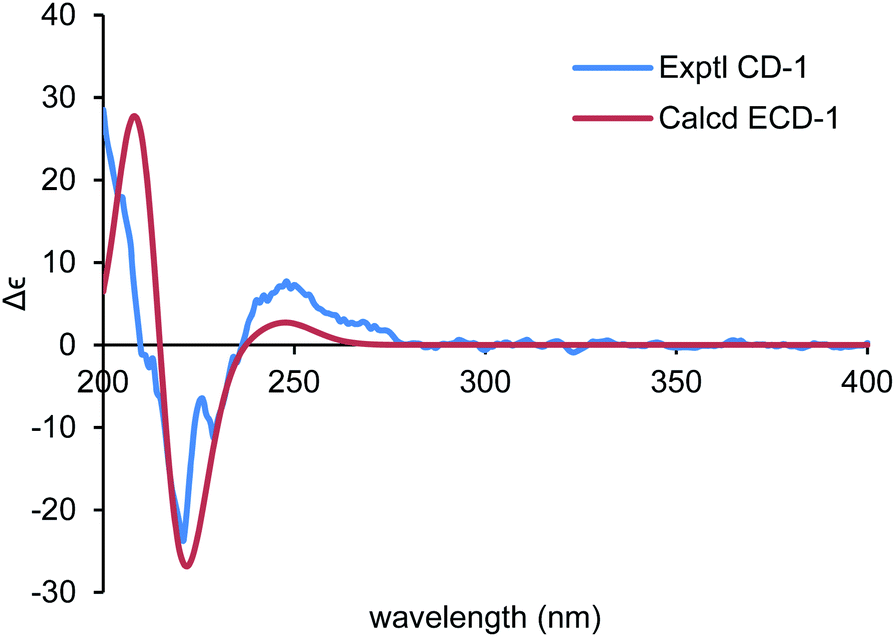 | ||
| Fig. 5 Comparison of the calculated ECD spectrum for (6R,7S,8S,11R)-1 (σ = 0.22 eV, UV shift 0 nm) with the experimental spectrum of 1 in MeOH. | ||
Ochracine G (2), C15H22O3, had comparable 1D NMR data (Tables 1 and 2) with those of co-isolate merulinol (8),11 implying that 2 is a chamigrane sesquiterpene. Analysis of the spectroscopic data showed that 8-CHOH in 8 was oxygenated to a ketone (δC 214.3) in 2, which was corroborated by HMBC correlations from H3-14 to C-8 (Fig. 3) and the HRMS result. The relative configuration of 2 was deduced by NOE correlations of H3-12 (δH 1.16)/H-7 (δH 2.67)/Heq-5 (δH 1.67), and H3-14 (δH 1.04)/Hax-1 (δH 2.11) (Fig. 4), showing that the relative stereochemistries of 2 are 6R* and 7S*.
The molecular formula of ochracine H (3), C15H22O3, is identical with that of 2, implying that they are analogues. The quite similar 1D NMR spectra of 2 and 3 (Tables 1 and 2), except for minor differences of chemical shifts of C-1, C-5, and C-6, suggested that they share the same planar structures, but with different configurations, which was further verified by 1H–1H COSY and HMBC spectra (Fig. 3). The configuration difference of C-6 between 2 and 3 led to changes of the chemical shifts at C-1, C-5 and C-6, suggesting the chiral carbon C-6 should be S* in 3 rather than the R* in 2. In the ROESY experiment (Fig. 4), cross peaks of H-7 (δH 2.61) with Heq-1 (δH 2.00), and H3-14 (δH 1.07) with Hax-5 (δH 1.67) observed revealed that relative configurations of 2 are 6S* and 7S*.
To establish the absolute configurations of 2 and 3, calculated electronic circular dichroisms (ECDs) for (6R,7S)-2 and (6S,7S)-3 were carried out (ESI†), respectively. As shown in Fig. 6 and 7, comparison with the experimental of (6R,7S)-2, and (6S,7S)-3 with the calculated ones, the calculated ECD spectra of presented good fitting, which are in accord with octant rule to the Cotton effect near 290 nm observed in the ECD spectra. Thus, the absolute configurations of 2 and 3 were determined with no doubt as suggested above.
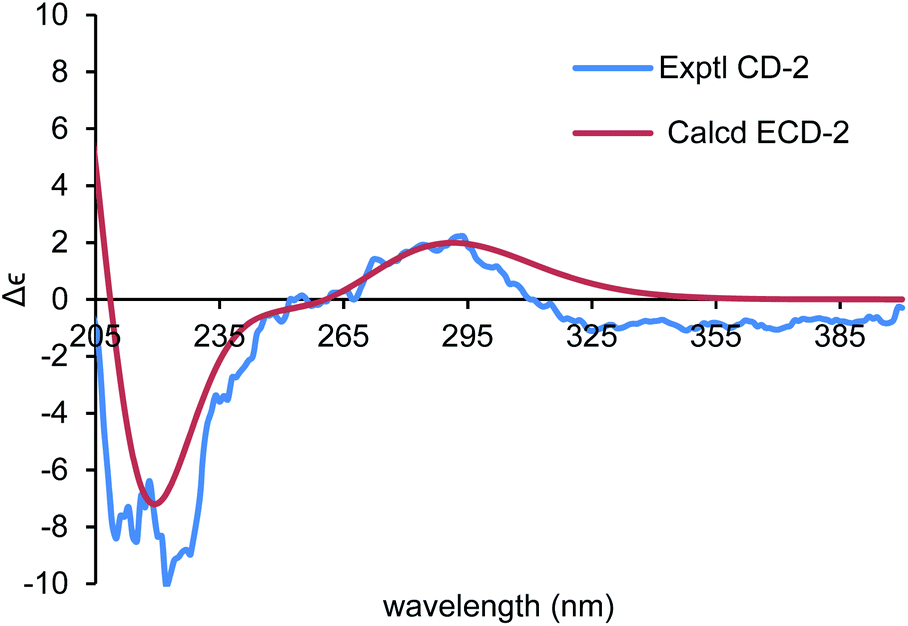 | ||
| Fig. 6 Comparison of the calculated ECD spectrum for (6R,7S)-2 (σ = 0.40 eV, UV shift −1 nm) with the experimental spectrum of 2 in MeOH. | ||
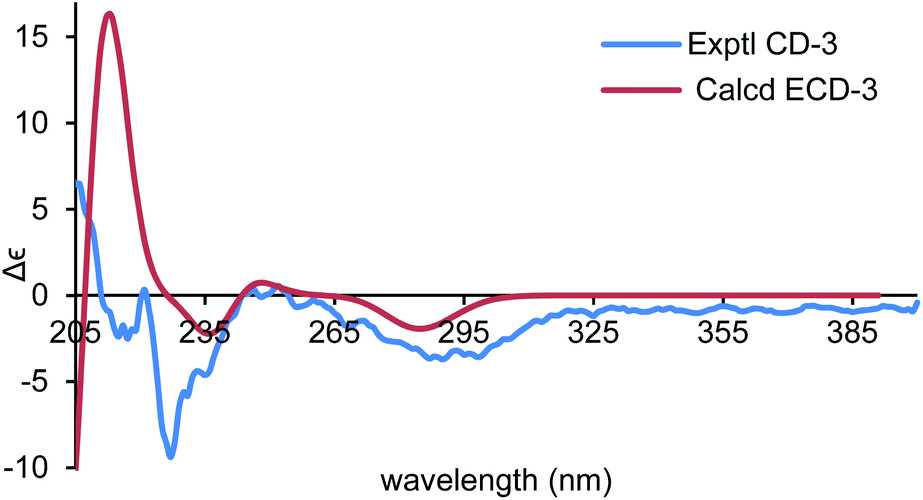 | ||
| Fig. 7 Comparison of the calculated ECD spectra for (6S,7S)-3 (σ = 0.16 eV, UV shift −8 nm) with the experimental spectrum of 3 in MeOH. | ||
Ochracine I (4) had a molecular formula C17H26O4 with five degrees of unsaturation. Its 1H and 13C NMR (Tables 1 and 2) data showed a close similarity to those of the co-isolate compound, acaciicolinol F (11).13 Analysis of these data indicated that the hydroxyl at C-15 in 11 was changed into an acetate in 4, which was supported by HMBC correlations from H2-15 to carboxyl carbon (Fig. 3). Similar to 11, a large coupling constant (J = 9.3 Hz) between H-8 and H-9 observed for 4, together with NOE cross peaks of H-8 (δH 4.08)/Heq-5 (δH 1.90), and H-9 (δH 3.46)/H3-13 (δH 0.83)/Heq-1 (δH 2.07)/H-14 (δH 4.78) (Fig. 4), suggested that compound 4 is the acetyl derivative of 11, and they share the same relative configuration at C-6, C-8, and C-9. In addition, the optical rotation of 4 showed positive sign ([α]20 D + 45.5) similar to of 11 ([α]26 D + 79.7),13 leading to the assignment of the absolute stereochemistry of 4 as 6S, 8S, 9S (Fig. 1).
The molecular formula of ochracine J (5), is C15H22O3 deduced by HREIMS ion at m/z 250.1567 [M]+. The 1D NMR (Tables 1 and 2) and HSQC spectra displayed signals for three methyl singlets, five methylenes (one oxygenated), an α,β-unsaturated ketone (δC 170.9; δC/H 127.3/5.88, s; 198.8), one pair of trisubstituted double bond carbons, and two sp3 quaternary carbons. These data shared close similarity to those of acaciicolinol L (10)13 which was also isolated from in this study, the only difference between them being the absence of hydroxyl at C-8 in 5. This difference was confirmed by the presence of a singlet olefinic proton (δH 5.88) and a long-range correlation from H3-14 to C-8 in the HMBC experiment (Fig. 3). Since there is only one chiral carbon, the absolute stereochemistry of C-6 was defined S by comparison of the experimental value of optical rotation ([α]20 D + 39.4) with both the reported data of 10 ([α]27 D + 159.6) and the calculated data ([α]D + 22.3) (ESI†). Hence, the structure of 5 was identified as shown in Fig. 1.
Ochracine K (6), presented a molecular formula C15H22O2 based on the HRESIMS (observed m/z 257.1512 [M + Na]+), requiring for five indices of double bond equivalents. The 13C NMR and DEPT spectra of 6 (Table 2) showed 15 carbon signals corresponding to four methyls (δC 21.5, 28.3, 28.4, and 29.8), one sp3 methylene (δC 35.6), three sp2 methines (δC 123.8, 126.1, and 136.5), four sp3 methines (δC 37.1, 40.0, 51.4, and 77.3), and three quaternary carbons (one sp2 δC 138.4, and two oxygenated δC 72.9, 77.4). The 1H NMR spectrum of 6 (Table 3) displayed signals of four singlet methyl groups at δH 1.21 (H3-15), 1.26 (H3-13), 1.31 (H3-12), and 1.67 (H3-14), one oxymethine at δH 4.04 (d, J = 5.0 Hz, H-3), three olefinic protons at 4.86 (brs, H-5), 5.66 (dd, J = 10.3, 6.2 Hz, H-8), 5.99 (d, J = 10.3 Hz, H-9). These described NMR characteristic signals and MS data suggested 6 should be a sesquiterpene. Starting from obvious HMBC correlations of H3-12/13 to C-7/11, H3-14 to C-3/4/5, H3-15 to C-1/9/10, coupled with the successive 1H–1H COSY correlations of H-3/H2-2/H-1/H-6/H-7/H-8/H-9 and H-5/H-6 (Fig. 3) suggested that 6 is a cadinane with two pair of double bonds at C-4(5) and C-8(9), and three oxygen baring carbons (C-3, C-10, and C-11). However, the MS result and the above assignments indicated that 6 is tricyclic. Further analysis of HMBC, a key correlation from H-3 to C-10 implied that the third ring is formed between 3-OH and 10-OH via a dehydration reaction. The relative stereochemistry of 6 (1S*, 3R*, 6R*, 7R*, 10R*) was determined by a combination of JBCA and ROESY spectrum (Fig. 8). Firstly, dihedral angles of H-3–C-3–C-2–Hax-2 and Hax-2–C-2–C-1–H-1 close to 90° deduced by the JBCA (H-3, δH 4.04, d, J = 5.0 Hz; Hax-2, δH 1.87, d, J = 11 Hz), together with the correlations of H-3/H2-2/H-1 (δH 2.60), H3-15/Heq-2 (δH 2.21) in the ROESY spectrum (Fig. 8), suggested that the orientations of H-1, H-3, and H3-15 are β. The broad singlet signal for H-6 indicative of 90° dihedral angles between H-6 and other protons at adjacent carbons, coupled NOE cross peaks of H-1/H-6 (δH 2.93)/H3-12 (δH 1.31), and H-7 (δH 1.99)/H-5 (δH 4.86), illustrated that the orientation of H-6 is β, and the orientation of H-7 is α.
| No. | 6b | 7a |
|---|---|---|
| a Measured in CD3OD.b Measured in CDCl3. | ||
| 1 | 2.60, dd (4.8, 4.8) | 3.24, m |
| 2 | 2.21, ddd (11.0, 5.0, 4.8), Heq, 1.87, d (11.0), Hax | 2.86, dd (16.3, 5.3), 2.78, dd (16.3, 5.6) |
| 3 | 4.04, d (5.0) | |
| 5 | 4.86, brs | 6.55, brs |
| 6 | 2.93, brs | 3.32, overlapped |
| 7 | 1.99, d (6.2) | 2.17, dd (8.5, 4.3) |
| 8 | 5.66, dd (10.3, 6.2) | |
| 9 | 5.99, d (10.3) | 5.81, brs |
| 11 | 2.02, m | |
| 12 | 1.31, s | 1.14, d (7.0) |
| 13 | 1.26, s | 0.92, d (7.0) |
| 14 | 1.67, s | 1.72, s |
| 15 | 1.21, s | 1.92, s |
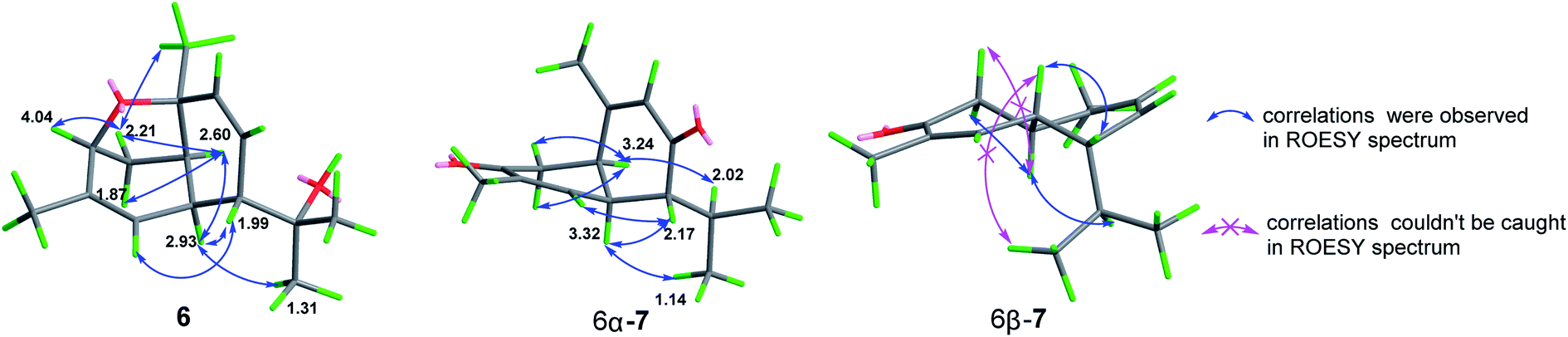 | ||
| Fig. 8 Selected NOE correlations of 6 and 7. | ||
By using of the same quantum chemistry mechanic methods as 1–3, ECD of (1S,3R,6R,7R,10R)-6 (6a) and 13C NMR of (1S*,3R*,6R*,7R*,10R*)-6 were calculated (ESI†) to establish its absolute configurations and prove its structure validity, which shared an opposite curve and a highly comparable linear regression (R2 = 0.9993) compared with its experimental CD/NMR spectra (Fig. 9 and 10), respectively, confirming the absolute configurations of 6 are 1R,3S,6S,7S,10S. And the structure of 6 is (1R,3S,6S,7S,10S)-amorpha-5α,10α-epoxy-4(5),8(9)-dien-11-ol.
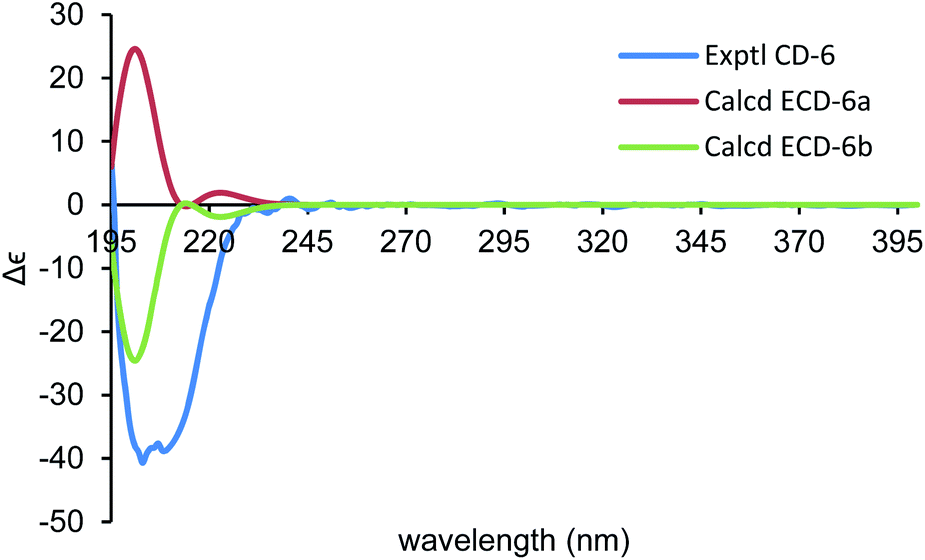 | ||
| Fig. 9 Comparison of the calculated ECD spectra for (1S,3R,6R,7R,10R)-6 (6a) (σ = 0.22 eV, UV shift 0 nm) and (1R,3S,6S,7S,10S)-6 (6b) with the experimental spectrum of 6 in MeOH. | ||
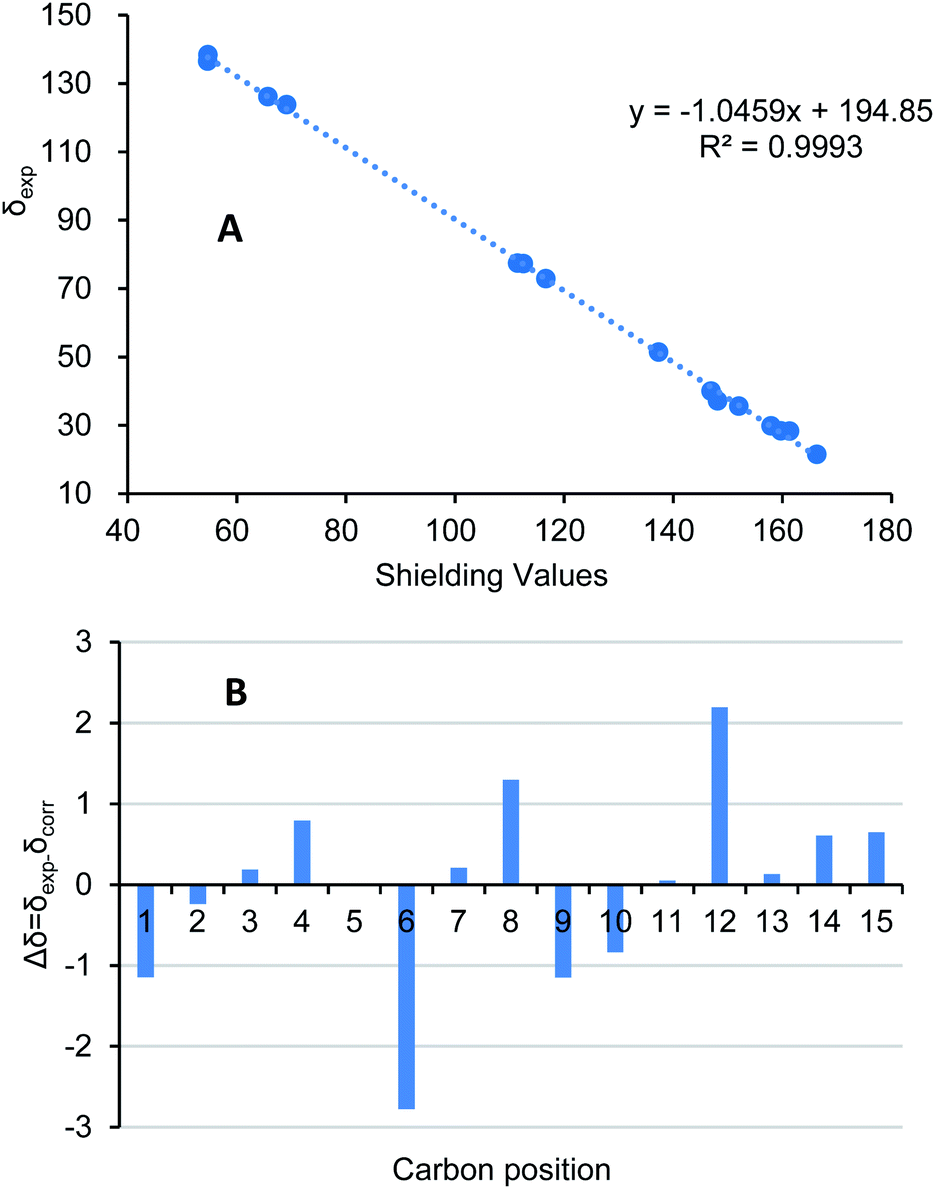 | ||
| Fig. 10 (A) Regression analysis of experimental versus calculated 13C NMR chemical shifts of 6 at GIAO B3LYP/6-31G(d) level; linear fitting was shown as a line. (B) Relative chemical shift errors between scaled 13C NMR and experimental 13C NMR for 6. | ||
Ochracine L (7), was obtained as a colourless oil, and its molecular formula, C15H20O2, was determined from the HREIMS (observed m/z 232.1466 [M]+), requiring six degrees of unsaturation. The UV spectrum of 7 presented maximal absorption at 230.5, and 248.0 nm implying a conjugated group. Combined analyses of 1H, 13C, and HSQC data of 7 (Tables 2 and 3) revealed the presence of two down-shifted methyl singlets and two secondary methyls, one sp3 methylene, two sp2 and four sp3 methines, five quaternary carbons (two conjugated ketones, two olefinic carbons). Since the two ketones and two double bonds takes up 4 degrees of hydrogen deficiency, 7 should be bicyclic. The existence of a propyl unit supported by 1H–1H COSY (Fig. 3) correlations between H3-12/H-11/H3-13 indicated that this bicyclic molecule is a cadinane-type sesquiterpene, and shared close similarity to α-amorphene-3-one,22 except the presence of an additional conjugated ketone (δC 202.6) at C-8 in 7 rather than 8-CH2 in α-amorphene-3-one. The relative configuration of 7 was mainly assigned by ROESY spectrum. Correlations of H-1 (δH 3.24)/H-11 (δH 2.02) suggested that H-1 and H-7 are in opposite side. It was difficult to deduce the relative configuration of H-6 by NOE correlation and JBCA analysis due to the close chemical shifts of H-1 and H-6, and the partially overlapped signal of H-6. Therefore, NOE correlations of the two possible stereoisomers, 6α-/6β-7, were compared. As shown in Fig. 8, only the 6α-7 can satisfy these NOE correlations of H2-2/H-1, and H3-12/H-6/H-7, so the relative configurations of 7 are 1R*,6S*,7R* (6α-7). To further ensure this deduction that the structure of 7 is 6α-7 other than 6β-7, their 13C NMR were calculated as shown in Fig. 11 (ESI†). Obviously, 6β-7 could be excluded by the R2 value (6α-7, R2 = 0.9995; 6β-7, R2 = 0.9990).
Considering the biosynthetic pathway and the calculated optical rotation ([α]D + 83.2) of 6α-7 (ESI†), the absolute configurations of 7 were assigned as 1R,6S,7R. Thus, the structure of 7 is (1R,6S,7R)-amorpha-4,9(10)-diene-3,8-dione.
The structures of merulinol (8), acaciicolinols C (9), L (10), and F (11) were established by comparing the NMR data with the published values.11,13
All the new isolates (1–7) were tested for their cytotoxicity against five human cancer cell lines: HL-60, SMMC-7721, A549, MCF-7, and SW-480, and against nitric oxide (NO) production. Unfortunately, they were devoid of any cytotoxicity and NO-production inhibitory activity.
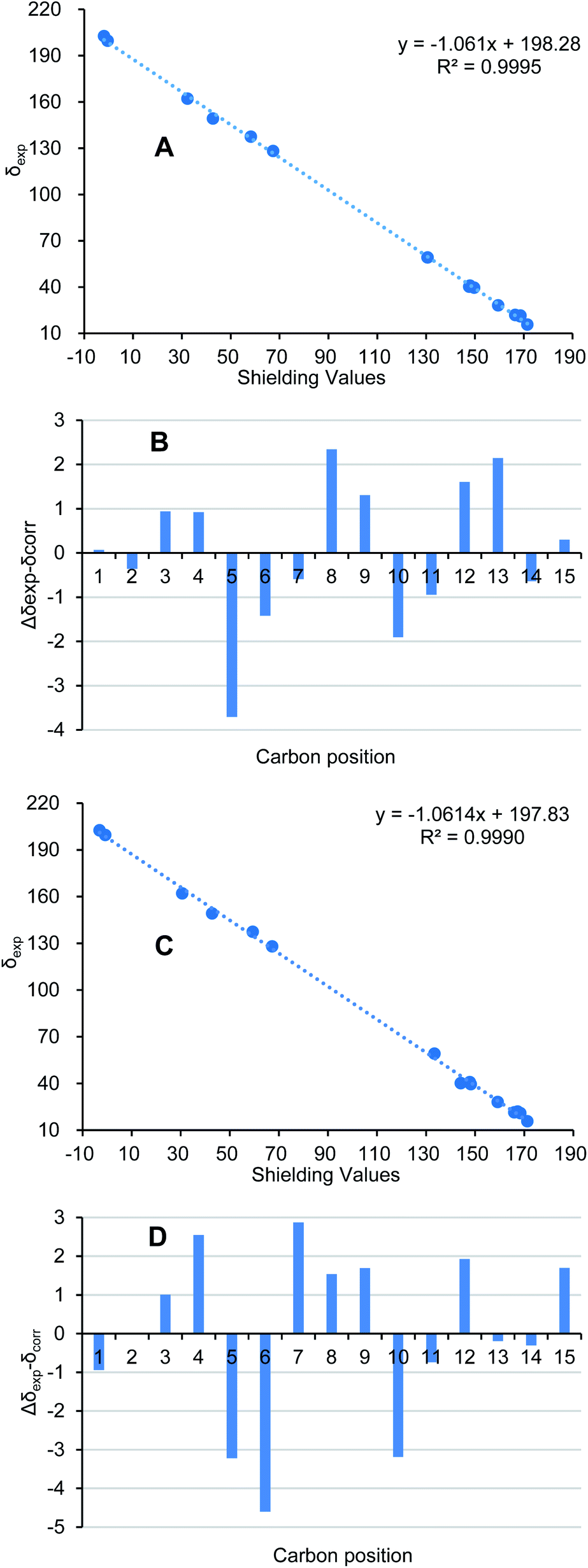 | ||
| Fig. 11 (A/C) Regression analysis of experimental versus calculated 13C NMR chemical shifts of 6α-7/6β-7 at GIAO B3LYP/6-31G(d) level. (B/D) Relative chemical shift errors between scaled 13C NMR and experimental 13C NMR for 6α-7/6β-7. | ||
Conclusions
In summary, eleven sesquiterpenes, including nine chamigrane-type ones and two cadinane-type ones, were isolated from the fermentation broth of the basidiomycete S. ochraceum. Their planar structures and absolute stereostructures were elucidated by spectroscopic analysis and calculated methods. Compounds 1 and 6, each featured by a six or five-membered ether ring with the absolute configurations established via a single X-ray diffraction experiment and/or computational methods, are quite novel tricyclic sesquiterpene in the chamigrane or cadinane families. All the new isolates were evaluated for their cytotoxicity against five cancer cell lines and inhibition against NO production. One case should be noted is that the reason fails to obtain endoperoxide chamigranes may be attributed to the culture medium used for S. ochraceum. It was suggested that potato dextrose broth (PDB)2,3,13,14 is more suitable for production of the endoperoxide chamigranes than sabouraud dextrose broth (SDB)11 or glucose peptone broth (GPB) used in this research.Experimental section
General experimental procedures
The crystal data were collected on an APEX II DUO spectrophotometer (Bruker AXS GmbH, Karlsruhe, Germany) using graphite-monochromated Cu Kα radiation (λ = 1.54178 Å) as the X-ray source. Melting point was measured on an X-4 microscopic melting point meter (Yuhua Instrument Co., Ltd, Gongyi, China). Optical rotations were obtained on a JASCOP-1020 digital polarimeter (Horiba, Kyoto, Japan). UV spectra were recorded on a Shimadzu UV-2401PC UV-visible recording spectrophotometer (Shimadzu, Kyoto, Japan). A Chiral scan circular dichroism spectrometer (Applied Photophysics Limited, Leatherhead, Surrey, UK) was used to record the CD spectra. 1D and 2D NMR spectra were obtained on a Bruker Avance III 600 MHz spectrometer (Bruker Corporation, Karlsruhe, Germany). An Agilent 6200 Q-TOF MS system (Agilent Technologies, Santa Clara, CA) was used to acquire the HRESIMS data. HREIMS were recorded on a Waters AutoSpec Premier P776 MS system. Sephadex LH-20 (Amersham Biosciences, Uppsala, Sweden) and silica gel (Qingdao Haiyang Chemical Co., Ltd) were used for column chromatography (CC). Medium pressure liquid chromatography (MPLC) was performed on a Büchi Sepacore System equipped with pump manager C-615, pump modules C-605 and fraction collector C-660 (Büchi Labortechnik AG, Flawil, Switzerland), and columns packed with Chromatorex C-18 (40–75 μm, Fuji Silysia Chemical Ltd., Kasugai, Japan). Preparative high performance liquid chromatography (prep-HPLC) was performed on an Agilent 1260 liquid chromatography system equipped with Zorbax SB-C18 columns (particle size 5 μm, dimensions 9.4 mm × 150 mm, flow rate 8 mL min−1) and a DAD detector (Agilent Technologies, Santa Clara, CA, USA).Fungal material
The basidiomycete Steccherinum ochraceum was collected at Ailao Mountain of Yunnan Province, China, in July 2003, which was identified by Prof. Mu Zang (Kunming Institute of Botany). The voucher specimen was deposited in the Herbarium of Kunming Institute of Botany, CAS. The strain (HFG119) was isolated from the fresh fruiting bodies of S. ochraceum and preserved on potato dextrose agar (PDA) medium at 4 Celsius degree.Fermentation, extraction and isolation
The fungus S. ochraceum was cultured on PDA medium for one week in the early stage. The agar was then inoculated into 500 mL of glucose peptone broth (GPB).23 After incubation for 1 week at 25 °C, the seed liquid was then inoculated into 500 mL Erlenmeyer flasks (150 flasks in all), each containing 400 mL of GPB. After incubation for 21 days, fungal culture was filtered to separate mycelia and broth (total volume of 60 L). The culture broth of S. ochraceum (60 L) was filtered and evaporated under reduced pressure, then partitioned between ethyl acetate (EtOAc) and water four times to yield a broth extract. The mycelia were extracted by EtOH (95%) three times. The extract was evaporated and then partitioned between EtOAc and water four times to give an EtOAc layer. Totally, the weight of the crude EtOAc layer was 34 g.The crude EtOAc layer was first separated by MPLC eluted with a gradient solvent system of MeOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O (V/V 20:80, 40:60, 60:40, 80:20, 100:0), which gives twenty-one fractions (A–U). All compounds were purified by preparative HPLC (a flow rate of 8 mL min−1) with Zorbax SB-C18 column using a linear elution of MeCN:H2O under different volume ratio.
:H2O (V/V 20:80, 40:60, 60:40, 80:20, 100:0), which gives twenty-one fractions (A–U). All compounds were purified by preparative HPLC (a flow rate of 8 mL min−1) with Zorbax SB-C18 column using a linear elution of MeCN:H2O under different volume ratio.
Fraction K (210 mg) was fractionated by Sephadex LH-20 (MeOH) to produce six subfractions (K1–K6). Fractions K3 (73.5 mg), K4 (58.3 mg), and K5 (42.9 mg) were separately subjected to silica gel CC (a gradient solvent system of petroleum ether: acetone (V/V) 5:1 to 2:1) to yield thirteen fractions K3a–K3m, ten fractions K4a–K4j, and ten fractions K5a–K5j. Compounds 1 (3.5 mg, tR = 6.0 min), and 10 (5.0 mg, tR = 10.8 min) were purified by semi-preparative HPLC (MeCN:H2O (20:80 → 40:60) in 25 min) from fraction K4b. Similarly, subfractions K3g (MeCN:H2O (17:83 → 32:68) in 20 min), K3b (MeCN:H2O (17:83 → 32:68) in 20 min) and K3l (MeCN:H2O (17:83 → 28:72) in 20 min) were dealt on semi-preparative HPLC affording compounds 5 (1.4 mg, tR = 15.0 min), 7 (2.1 mg, tR = 18.9 min) and 11 (6.1 mg, tR = 14.7 min), respectively. Compound 9 (3.6 mg, tR = 7.8 min) was isolated from fraction K5b (MeCN:H2O (20:80 → 40:60) in 25 min) by preparative HPLC (a flow rate of 8 mL min−1) with Zorbax SB-C18 column.
Subsequently, fraction L (190 mg) was further purified on Sephadex LH-20 (CH3OH) affording three subfractions (L1–L3). Fraction L2 (80.9 mg) was subjected to Sephadex LH-20 (acetone) to give seventeen subfractions (L2a–L2q). By using preparative HPLC (a flow rate of 8 mL min−1) with Zorbax SB-C18 column, compound 6 (1.0 mg, tR = 11.3 min, MeCN:H2O (18:82 → 30:70) in 20 min) was purified from L2d, and compound 8 (5.4 mg, tR = 17.0 min) from L2n (MeCN:H2O (24:76 → 39:61) in 20 min).
Fractions M (160 mg) and O (113 mg) were further purified by Sephadex LH-20 twice in succession (CH3OH and then acetone). Fractions M2 and O1 gave 7 (M2a– M2g) and 8 minor fractions (O1a–O1h), among them subfractions M2f (MeCN:H2O (20:80 → 33:67) in 20 min) yielding compounds 2 (1.0 mg, tR = 17.0 min) and 3 (1.1 mg, tR = 17.5 min), and subfractions M2f (MeCN:H2O (20:80 → 35:65) in 20 min) yielding compound 4 (5.0 mg, tR = 17.0 min) by preparative HPLC (a flow rate of 8 mL min−1) with Zorbax SB-C18 column.
ε): 214.2 (3.90); 1H NMR (600 MHz CD3OD) data (Table 1); 13C NMR (150 MHz CD3OD) data (Table 2); HRESIMS m/z 289.1408 [M + Na]+ (calcd for C15H22O4Na, 289.1410).461 reflections measured 4805 independent reflections (Rint = 0.0459). The final R1 values were 0.0579 (I > 2σ(I)). The final wR(F2) values were 0.1595 (I > 2σ(I)). The final R1 values were 0.0615 (all data). The final wR(F2) values were 0.1664 (all data). The goodness of fit on F2 was 1.049. Flack parameter = 0.14(8).ε): 215.6 (3.97); 1H NMR (600 MHz CDCl3) data (Table 1); 13C NMR (150 MHz CDCl3) data (Table 2); HREIMS m/z 250.1564 [M]+ (calcd for C15H22O3 250.1569).ε): 243.8 (3.95); 1H NMR (600 MHz CDCl3) data (Table 1); 13C NMR (150 MHz CDCl3) data (Table 2); HREIMS m/z 250.1567 [M]+ (calcd for C15H22O3 250.1569).ε): 203.0 (3.62); 1H NMR (600 MHz CDCl3) data (Table 3); 13C NMR (150 MHz CDCl3) data (Table 2); HRESIMS m/z 257.1512 [M + Na]+ (calcd for C15H22O2Na 257.1512).ε): 230.5 (4.16) 248.0 (3.56); 1H NMR (600 MHz CD3OD) data (Table 3); 13C NMR (150 MHz CD3OD) data (Table 2); HREIMS m/z 232.1466 [M]+ (calcd for C15H20O2 232.1463).Bioassays
Quantum chemistry calculation details
Conformation searches on the candidate structures were performed by MMFF94s force field, giving compounds (6R,7S,8S,11R)-1, (6R,7S)-2, (6S,7S)-3 four possible conformers each, (6S)-5 seven ones, (1S,3R,6R,7R,10R)-6 (6a) three ones, 6α-7 two ones, 6β-7 three ones with distribution higher than 1%. These conformers were further optimized on B3LYP/6-31g(d) level of theory in Gaussian 16 program,25 respectively. The conformers within 3 kcal mol−1 of global minimum energy were selected for further calculations. The ECD spectra and optical rotations for the selected conformers were accomplished by B3LYP/6-31g(d) with CPCM model in methanol. The ECD calculation results were processed on SpecDis v1.71 software,26 by which the ECD spectrum of the antipode, such as (1R,3S,6S,7S,10S)-6 (6b), was generated directly by the function “enantiomeric ECD”. The calculated specific optical rotation data of these conformers were averaged according to the Boltzmann distribution theory and their relative Gibbs free energy. The NMR calculations were carried out by B3LYP/6-31g(d) level of GIAO theory. The NMR calculation were processed by Microsoft Office Excel program (ESI†).Author contributions
Z. Z. Zhao performed the experiments, data analysis, and experimental planning. H. P. Chen contributed to the calculation work. Q. L. Zhao screened the biological activities. The project was conceived, planned, and supervised by H. P. Chen. and J. K. Liu. The manuscript was written by Z. Z. Zhao, H. P. Chen, and J. K. Liu. All authors reviewed the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (grant numbers 82003607, 81903512), the Natural Science Foundation of Hubei Province (grant number 2019CFB379), the Research Project on Chinese Medicine Science in Henan Province (grant number 20-21ZY1039), and the Key Scientific Research Projects of Universities of Henan Province (grant number 21A360002).Notes and references
- H. P. Chen and J. K. Liu, Progress in the Chemistry of Organic Natural Products, ed. A. D. Kinghorn, H. Falk, S. Gibbons and J. Kobayashi, Springer Publishing AG Switzerland, 2017, vol. 106, p. 1 Search PubMed
.
- D. Z. Liu and M. H. Luo, Fitoterapia, 2010, 81, 1205 CrossRef CAS PubMed
- D. Z. Liu, Z. J. Dong, F. Wang and J. K. Liu, Tetrahedron Lett., 2010, 51, 3152 CrossRef CAS
- X. Q. Yu, C. S. Jiang, Y. Zhang, P. Sun, T. Kurtán, A. Mándi, X. L. Li, L. G. Yao, A. H. Liu, B. Wang, Y. W. Guo and S. C. Mao, Phytochemistry, 2017, 136, 81 CrossRef CAS PubMed
- I. Brito, M. Cueto, A. R. Díaz-Marrero, J. Darias and A. S. Martín, J. Nat. Prod., 2002, 65, 946 CrossRef CAS PubMed
- D. Davyt, R. Fernandez, L. Suescun, A. W. Mombrú, J. Saldaña, L. Domínguez, J. Coll, M. T. Fujii and E. Manta, J. Nat. Prod., 2001, 64, 1552 CrossRef CAS PubMed
- J. Kimura, N. Kamada and Y. Tsujimoto, Bull. Chem. Soc. Jpn., 1999, 72, 289 CrossRef CAS
- S. M. Al-Massarani, Nat. Prod. Chem. Res., 2014, 2, 1000147 Search PubMed
- S. Chokpaiboon, D. Sommit, T. Teerawatnanond, N. Muangsin, T. Bunyapaiboonsri and K. Pudhom, J. Nat. Prod., 2010, 73, 1005 CrossRef CAS PubMed
- S. Chokpaiboon, D. Sommit, T. Bunyapaiboonsri, K. Matsubara and K. Pudhom, J. Nat. Prod., 2011, 74, 2290 CrossRef CAS PubMed
- C. Siwattra, T. Thapong, M. Tohru and P. Khanitha, Mar. Drugs, 2016, 14, 132 CrossRef PubMed
- H. Li, H. Huang, C. Shao, H. Huang, J. Jiang, X. Zhu, Y. Liu, L. Liu, Y. Lu, M. Li, Y. Lin and Z. She, J. Nat. Prod., 2011, 74, 1230 CrossRef CAS PubMed
- M. Wibowo, V. Prachyawarakorn, T. Aree, C. Mahidol, S. Ruchirawat and P. Kittakoop, Phytochemistry, 2016, 122, 126 CrossRef CAS PubMed
- M. Wibowo, V. Prachyawarakorn, T. Aree, S. Wiyakrutta, C. Mahidol, S. Ruchirawat and P. Kittakoop, Eur. J. Org. Chem., 2014, 2014, 3976 CrossRef CAS
- M. Bu, B. Yang and L. Hu, Curr. Med. Chem., 2016, 23, 383 CrossRef CAS PubMed
- M. Jung, H. Kim, K. Lee and M. Park, Mini-Rev. Med. Chem., 2003, 3, 159 CrossRef CAS PubMed
- D. Z. Liu and J. K. Liu, Nat. Prod. Bioprospect., 2013, 3, 161 CrossRef CAS
- Z. Z. Zhao, W. S. Feng, X. B. Liang, G. M. Xue, Y. Y. Si, H. P. Chen and J. K. Liu, Fitoterapia, 2019, 139, 104362 CrossRef CAS PubMed
- R. N. Lawrence, Drug Discov. Today, 1999, 4, 449 CrossRef PubMed
- Y. Zhang, X. M. Li, Z. Shang, C. S. Li, N. Y. Ji and B. G. Wang, J. Nat. Prod., 2012, 75, 1888 CrossRef CAS PubMed
- X. M. Zhou, C. J. Zheng, G. Y. Chen, X. P. Song, C. R. Han, G. N. Li, Y. H. Fu, W. H. Chen and Z. G. Niu, J. Nat. Prod., 2014, 74, 2021 CrossRef PubMed
- Y. Ohta and Y. Hirose, Bull. Chem. Soc. Jpn., 1973, 46, 1535 CrossRef CAS
- Z. Z. Zhao, K. Zhao, H. P. Chen, X. Bai, L. Zhang and J. K. Liu, Phytochemistry, 2018, 147, 21 CrossRef CAS PubMed
- H. P. Chen, Z. Z. Zhao, Z. H. Li, Z. J. Dong, K. Wei, X. Bai, L. Zhang, C. N. Wen, T. Feng and J. K. Liu, ChemistryOpen, 2016, 5, 142 CrossRef CAS PubMed
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision B.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed
- T. Bruhn, A. Schaumlöffel, Y. Hemberger and G. Pecitelli. SpecDis version 1.71, Berlin, Germany, 2017, https:/specdis-software.jimdo.com Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available: The 1D & 2D NMR, HRMS, crystallographic data, and computational details of compounds 1–7. CCDC 1975662. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra03320d |
| This journal is © The Royal Society of Chemistry 2021 |