
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePreparative separation of gallic acid from Fallopia aubertii using middle-pressure chromatogram isolated gel coupled with reversed-phase chromatography with hydrophilic groups
Dijun Jiabc,
Qi Wangabc,
Huan Wangabc,
Qian Maabc,
Min Wangabc and
Yongchang Lu *abc
*abc
aKey Laboratory for Tibet Plateau Phytochemistry of Qinghai Province, Qinghai Nationalities University, Xining, Qinghai, P. R. China. E-mail: qhlych@126.com; Tel: +86-13997239611
bModern Tibetan Medicine Creation Engineering Technology Research Center of Qinghai Province, China
cCollege of Pharmacy, Qinghai Nationalities University, Xining, Qinghai, China
First published on 10th August 2021
Abstract
Fallopia aubertii (L. Henry) Holub is a traditional Tibetan medicine to treat gout, but the research on its chemical composition is limited, probably due to the complex purification process. In this study, the chemical constituents of F. aubertii were isolated and identified by medium pressure chromatography and high-pressure preparative liquid chromatography. The general separation process was as follows: first, the extraction part of ethyl acetate collected seven components according to the chromatographic peaks of the sample through the medium pressure chromatographic separation system. Next, the separation effects of Fr2 on five different preparation columns were compared. A reversed-phase chromatographic column with hydrophilic groups was selected for separation. Finally, the principal component compound (gallic acid) in Fr2 was isolated and identified. The results show that the combination of medium pressure chromatographic separation gel column and reversed phase chromatographic column with hydrophilic groups is an effective separation method for the separation of gallic acid from F. aubertii.
Introduction
Fallopia aubertii (L. Henry) Holub, also known as polygonum, is a traditional Tibetan medicine (TTM) belonging to the genus Polygonum multiflorum of the Polygonaceae family.1 It is mainly found in hillside grasslands and valley thickets, and therefore is produced in places such as Inner Mongolia (Helan Mountain), Shanxi, Henan, Gansu, Qinghai, Sichuan, Yunnan and Tibet.2 It has also been reported that the herb has good anti-HIV-1 virus activity, analgesic effect, anti-inflammatory effect, uric acid-lowering effect and anti-gout effect in vitro.3–6 However, these bioactivity reports were based on the study of crude extracts, while their material basis and pharmacological activities have not been explored. Therefore, it is of great significance to separate and identify the chemical constituents of this herb.Tibetan medicine generally contains hundreds of different polarity and structural types of chemical compositions. Therefore, it has always been an arduous scientific research task to separate and describe the chemical components of Tibetan medicine on a global scale. At present, general separation and purification methods of natural products mainly rely on column chromatography using traditional silica gel and macroporous resins.7–10 This method has been widely employed by researchers because of its simple equipment and large separation capacity. However, the serious nonspecific adsorption of fillers makes it difficult to be used for the separation of compounds with low content and similar polarity. What is more worth mentioning is that they are time-consuming and tedious.
The advent of preparative high-performance liquid chromatography (prep-HPLC) provides considerable convenience for the separation of complex chemical components in TTM and has become one of the fastest and most effective separation tools at present.11,12 It has the characteristics of efficient separation, on-line detection and a high degree of automation, particular analytical liquid chromatography, ultra-high performance liquid chromatography and liquid chromatography-mass spectrometry, which are widely used to characterize the chemical composition of TTM.13–15 When compounds are separated and purified from TTM by preparative liquid chromatography, a lot of useful information can be obtained from the analysis. Most commercial chromatographic columns can be used for the separation and purification of TTM, which enables the wide application of preparative liquid chromatography.
In general, the reversed-phase chromatography column uses a non-polar stationary phase that uses silica gel as the matrix to bond different groups (such as C18 and C8), which is suitable for the separation of non-polar or weak polar compounds.16,17 C18 column is the most commonly used and necessary universal column in every laboratory. The filler is octadecyl bonded on a silica gel matrix, which has high carbon content and good hydrophobicity, and therefore, it is suitable for most compounds, including non-polar, polar small molecules, and some peptides and proteins. Different types of C18 columns have different separation effects. For example, compounds with large polar properties can choose columns containing hydrophilic groups, which can use 100% aqueous phase (C18-AQ) as the mobile phase, while columns without hydrophilic groups cannot use pure water as the mobile phase as it will cause the column to collapse and lead to low column efficiency.18,19
However, the direct injection of the crude extract will cause irreparable damage to the preparation column and shorten the service life of the preparation column; and the separation effect of the sample will be affected after multiple injections of the crude extract. Therefore, it is very necessary to find a suitable sample pretreatment method.
Middle chromatogram isolated (MCI) gel, usually combined with column chromatography, has been widely used in the purification and enrichment of complex components in TTM.20–22 Herein, we used a mid-pressure chromatographic tower containing the MCI gel as the preparation column to connect with the preparation liquid to construct a medium pressure preparation chromatographic separation system for the pretreatment of the crude extract. Next, five different types of preparation columns, namely HT-ODS-P, ZORBAX SB-C18, ZORBAX SB-CN, ZORBAX SB-AQ and Xamide, were selected to analyze the samples, and finally the ZORBAX SB-AQ column with the best separation effect was selected for the final preparation and separation. The peak shape and sample loads for the sample were excellent (the specific separation process is shown in Fig. 1). The large-scale purification of gallic acid from F. aubertii extracts can be achieved using a middle-pressure chromatogram isolated gel column coupled with reversed-phase chromatography with hydrophilic groups that would facilitate the future study on the in vivo pharmacological activities of F. aubertii.
 | ||
| Fig. 1 Separation mechanism diagram. | ||
Materials and methods
Equipment and chemicals
The enrichment and preparation experiments were performed using a chromatography system consisting of sample pumps, a workstation manufactured by Soochow high-tech chromatography Co., Ltd (Suzhou, China) and a UV detector. Chromatographic analysis was performed using an Agilent Technologies 1200 system (Santa Clara, America) containing a G1311A degassing unit, two G1311A pumps, a G1316A thermostat, a G1376B autosampler, a G1365B ultraviolet detector and an Agilent workstation.HT-ODS-P (4.6 × 250 mm, 5 μm) analytical column and HT-ODS-P (20 × 250 mm, 5 μm) preparative column were purchased from Soochow high-tech chromatography Co., LTD. ZORBAX SB-CN (21.2 × 250 mm, 7 μm), ZORBAX SB-AQ (21.2 × 250 mm, 7 μm), and ZORBAX SB-C18 (21.2 × 250 mm, 7 μm) preparative columns were obtained from Agilent Technologies. HILIC (4.6 × 250 mm, 5 μm), PolyPak C18 (5 μm, 4.6 × 250 mm) analytical columns and XAmide (20 × 250 mm, 10 μm) preparative column were purchased from Zhejiang Fuli Analytical Instruments Corp. MCI GEL (CHP20P) used in the middle-pressure chromatographic tower was purchased from Mitsubishi Chemical Holdings (Tokyo, Japan).
HPLC-grade acetonitrile (ACN) and methanol were purchased from Shandong Yuwang Hetian New Materials Co., Ltd (Dezhou, China). Analytical grade methanol and ethanol used for extraction and enrichment were obtained from Hongyan Chemical Reagent Factory (Tianjin, China). Chromatographic grade water was prepared using a Moore laboratory water system (Chongqing Moore Water Treatment Equipment, Chongqing, China).
Experimental samples
F. aubertii was collected from North Mountain in Huzhu, Qinghai, and was validated by Prof. Yongchang Lu of Qinghai Nationalities University.The air-dried stems of F. aubertii (2.0 kg) were extracted by ultrasonication with 10 L of 95% ethanol solution three times, one hour each time. The above process was repeated with 75% and 60% of the ethanol solution. 90 L of three different ethanol concentrations were combined and filtered with a gauze and then concentrated on a rotary evaporator at 55 °C to obtain the extract. Total extract (∼180.0 g) was suspended in water and partitioned successively with petroleum ether PE, EtOAc and n-BuOH, followed by concentrating it in a vacuum to yield the PE portion, EtOAc portion, n-BuOH portion and aqueous portion, respectively. After the EtOAc portion (∼38.0 g) was dried and dissolved in methanol solution, the solution was delaminated by a 0.45 μm filter to obtain the crude sample solution. After the sample solution was concentrated to 0.3 L, it is separated in a medium pressure chromatographic tower. Samples were eluted in methanol/water. Linear gradient elutions were performed for 0–120 min (0 to 100% methanol) and 120–150 min (100% methanol) at a flow rate of 60 mL min−1. Seven components were obtained after preparation and separation at the absorption wavelength of 210 nm. The samples were injected five times, and each component was dried separately. Fraction two (representative sample, 2.3 g) was chosen for subsequent separation.
Chromatographic conditions
Fraction 2 (2.3 g) was dissolved in methanol/water (20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :80 v/v) and filtered through a 0.45 μm filter to separate the sample solution.
:80 v/v) and filtered through a 0.45 μm filter to separate the sample solution.
Analytical chromatography was carried out with the HT-ODS-P (4.6 × 250 mm, 5 μm) column using chromatographic grade water (A) and methanol (B). Gradient elutions of 5–30% B and isometric elutions of 5–5% B were performed for 30 min, respectively.
Preparation chromatography was performed on HT-ODS-P (20 × 250 mm, 5 μm), ZORBAX SB-C18 (21.2 × 250 mm, 7 μm), and ZORBAX SB-CN (21.2 × 250 mm, 7 μm) columns in 0.1% formic acid/water (A) and methanol (B). Isometric elutions were performed for 5–5% B on these columns. Preparation chromatography was performed on the ZORBAX SB-AQ (21.2 × 250 mm, 7 μm) column, where mobile phase A was 0.1% formic acid/water, and mobile phase B was methanol, gradient elutions of 5–30% B. For XAmide (20 × 250 mm, 10 μm) column, mobile phase A was 0.1% formic acid/water, and mobile phase B was ACN, the linear gradient was 95–70% B. No change in other chromatographic conditions: analytical time was 40 min, monitoring wavelength was 280 nm, flow rate was 20 mL min−1, and the sample analysis was carried out at room temperature.
Two different chromatographic columns were used to evaluate the purity of the target peak: PolyPark C18 column and HILIC column. The PolyPark C18 column and the ZORBAX SB-AQ column used the same mobile phase, while the HILIC column and XAmide column used the same mobile phase. Experimental conditions for PolyPark C18 column: Gradient program: 0–60 min, 5–90% B. Experimental conditions for HILIC column: gradient: 0–60 min, 95–50% B. The other experimental variables were the same: flow rate was 1.0 mL min−1 and monitoring wavelength was 280 nm.
Results and discussion
Sample pretreatment using a mid-pressure chromatography tower
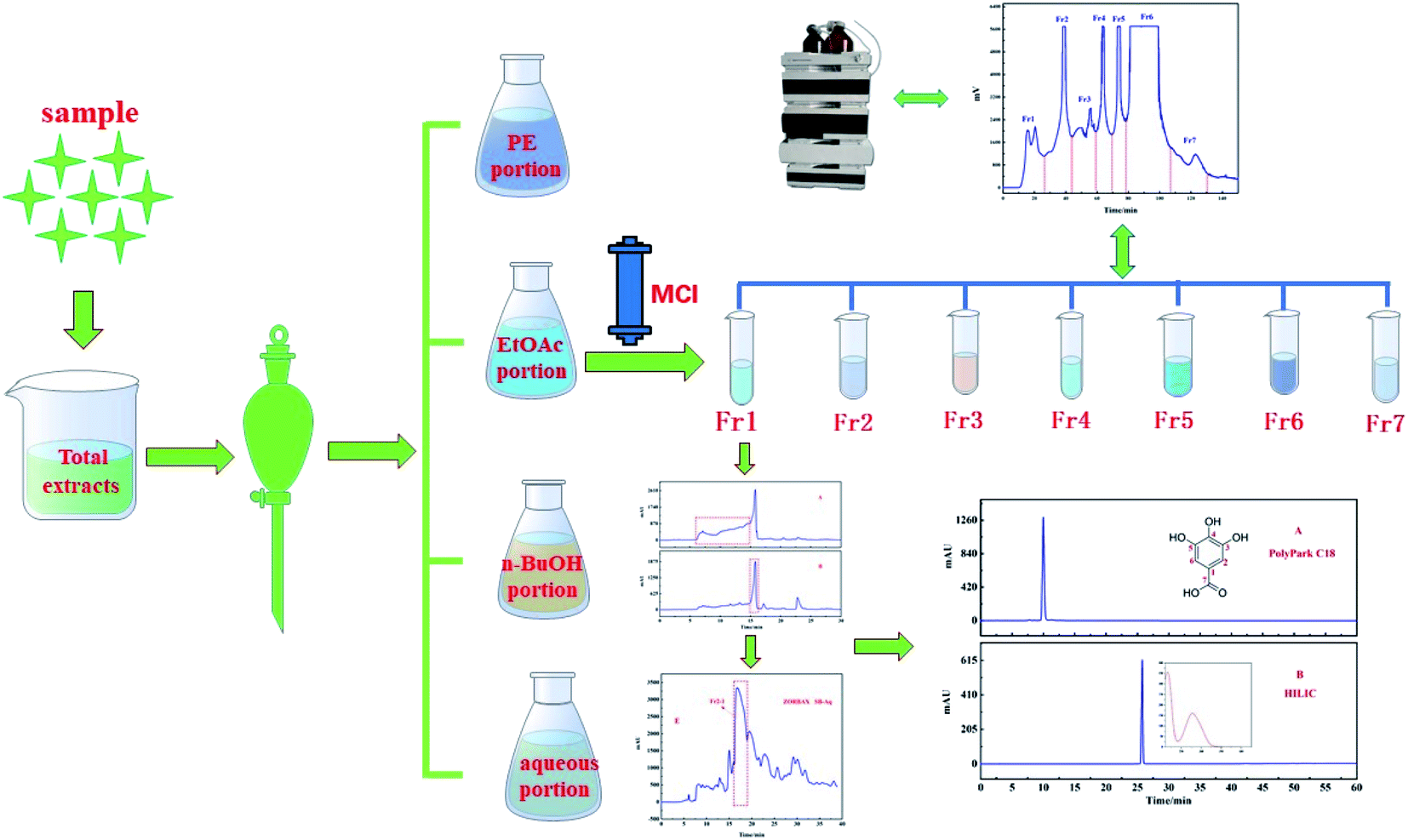
The med-pressure chromatographic tower containing the MCI gel was used as a preparation column to connect with the preparation liquid to construct a medium pressure preparation chromatographic separation system for the pretreatment of the EtOAc portion. The extracted part of ethyl acetate was dissolved in the methanol solution and then injected into a medium pressure chromatographic column to obtain the separation of MCI. A total of 7 components were obtained after 5 repeated samplings. The separation chromatogram is shown in Fig. 2. Next, 2.3 g of Fr2 (representative sample) was dissolved in 30 mL of methanol/water and filtered through a 0.45 μm membrane for subsequent isolation.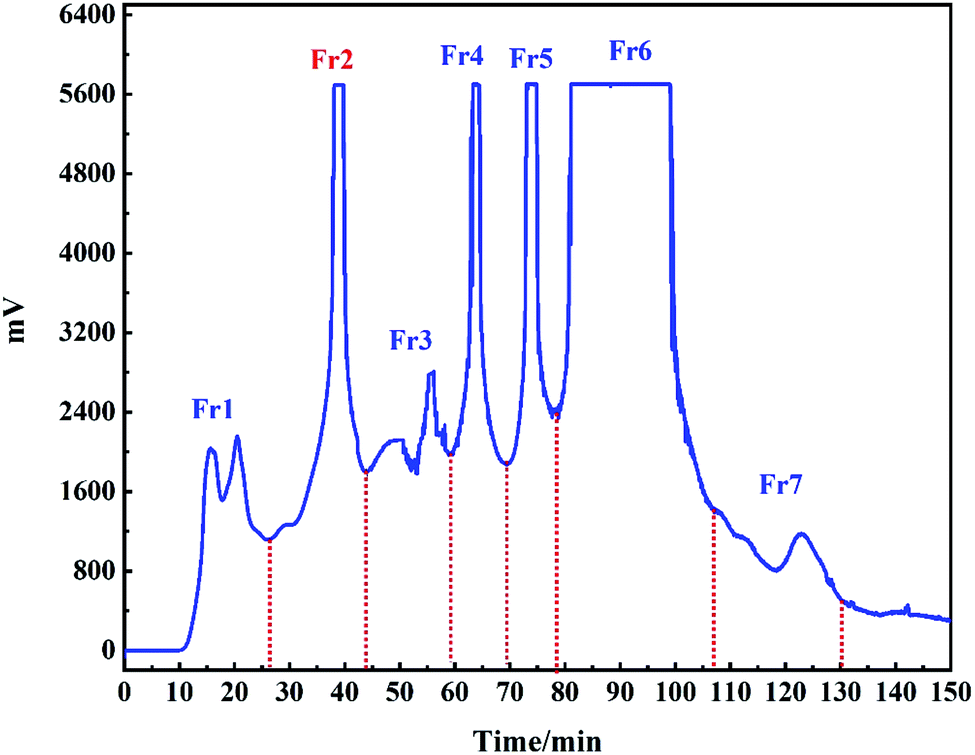 | ||
| Fig. 2 Pretreatment chromatograms of the ethyl acetate extract crude part of F. aubertii with MCI middle-pressure chromatographic tower. | ||
As shown in Fig. 2, the EtOAc portion was preliminarily separated in the medium pressure chromatographic separation system. According to the displayed chromatographic peak on the detector of the medium pressure preparation separation system, the separation of the sample in MCI packing could be directly understood, and the samples could be collected more pertinently. Column silica gel chromatography and macroporous resin are the most commonly used methods for the separation of monomer compounds from TTM, which require less equipment and separation training. However, gallic acid cannot be easily separated by these methods because of its poor reproducibility.23,24 Therefore, in order to avoid this issue, high-speed CCC has been employed for isolation. However, this method involves complex partition coefficient calculation and low separation efficiency.25,26
Analysis of fraction 2 on various chromatographic columns
In order to observe the chromatographic peak of Fr2, the sample was first eluted by gradient elution on the chromatographic column HT-ODS-P (Fig. 3A). It can be seen in Fig. 3A that there was a obvious principal component peak in fraction 2, but there was a cluster of small peaks with similar polarity in front of the principal component peak (the part marked with a red-dashed line), which were not well separated. This may be the result of the use of gradient elution conditions, under which all chromatographic peaks appeared within 15 min, indicating that the compounds in Fr2 have a greater polarity. Subsequently, the sample was analyzed with a 5% iso-elution condition on the same chromatographic column. Fig. 3B shows the analytical chromatogram of Fr2 under the condition of 5% iso-elution. Although not all the chromatographic peaks had good separation, according to previous preparation experience, the main peak of Fr2 (the part marked with a red dashed line) can be separated by preparative liquid chromatography. The sample was first run on an HT-ODS-P (20 × 250 mm, 5 μm) preparation column because it has exactly the same stationary phase as the analytical column (4.6 × 250 mm, 5 μm). The preparative chromatogram of the Fr2 target peak is shown in Fig. 4A.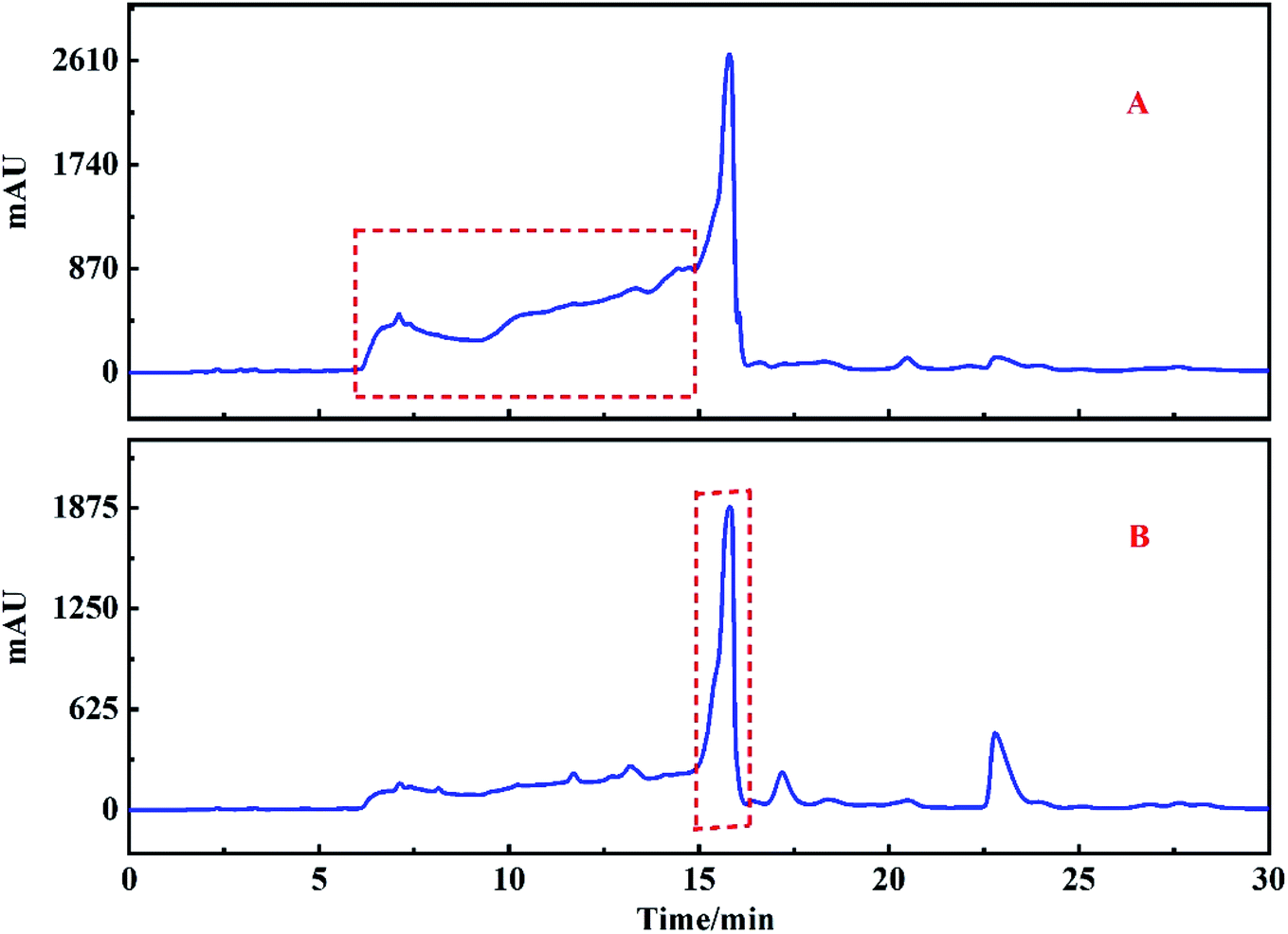 | ||
| Fig. 3 Gradient elution chromatogram (A) and isometric elution chromatogram (B) of Fr 2 on the HT-ODS-P analytical column. | ||
 | ||
| Fig. 4 HPLC analysis of Fr2 on HT-ODS-P (A), ZORBAX SB-C18 (B), ZORBAX SB-CN (C), XAmide (D) and ZORBAX SB-AQ (E) preparation columns. | ||
Unfortunately, the sample was not well separated on the preparation column, and all the chromatographic peaks were gathered together. In order to determine the optimal stationary phase, ZORBAX SB-C18 preparation columns, ZORBAX SB-CN preparation columns, ZORBAX SB-AQ and XAmide preparation columns were selected to test the samples. Separation chromatograms of the Fr2 target peak are shown in Fig. 4B–E, respectively. Similar phenomena were observed with the ZORBAX SB-C18 and HT-ODS-P columns, as shown in Fig. 4A and B, respectively, in which all the chromatographic peaks were are squeezed together. Fr2 was then analyzed on ZORBAX SB-CN preparation columns. In Fig. 4C, some irregular chromatographic peaks are observed, which could not be separated in the next step. Similar phenomena were observed with the XAmide preparation columns, as shown in Fig. 4C, which shows anomalous tailing chromatographic peaks.
Fraction 2 was analyzed on ZORBAX SB-AQ preparation columns and the result is shown in Fig. 4E. A good separation effect was obtained using ZORBAX SB-AQ preparation columns, and the main component peak appeared obviously on the chromatogram. This phenomenon indicated that the ZORBAX SB-AQ preparation column had a higher separation effect compared to the other columns tested. Therefore, it was possible to separate the peaks of the main components in Fr2 using ZORBAX SB-AQ preparation columns.
Preparation of fraction 2 on the ZORBAX SB-AQ preparation column
Based on the analysis above, Fr2 was injected repeatedly on the ZORBAX SB-AQ preparation column 12 times, and the target peak (the part marked with a red dashed line in Fig. 4E) was collected simultaneously. In order to prepare high purity compounds, good repeatability of chromatographic analysis was important. This factor minimizes the cross contamination of chromatographic peaks during the repeated injection. In this study, the chromatogram of the target peak was recorded only after 12 repeated injections to evaluate the repeatability of the process. The chromatograms (Fig. 5) of injections 1, 6 and 12 showed that the system has good repeatability. Therefore, the target score can be increased by repeated collection. After 12 runs of preparative chromatography, the target peak was collected and dried, followed by redissolving in 20% methanol/aqueous solution.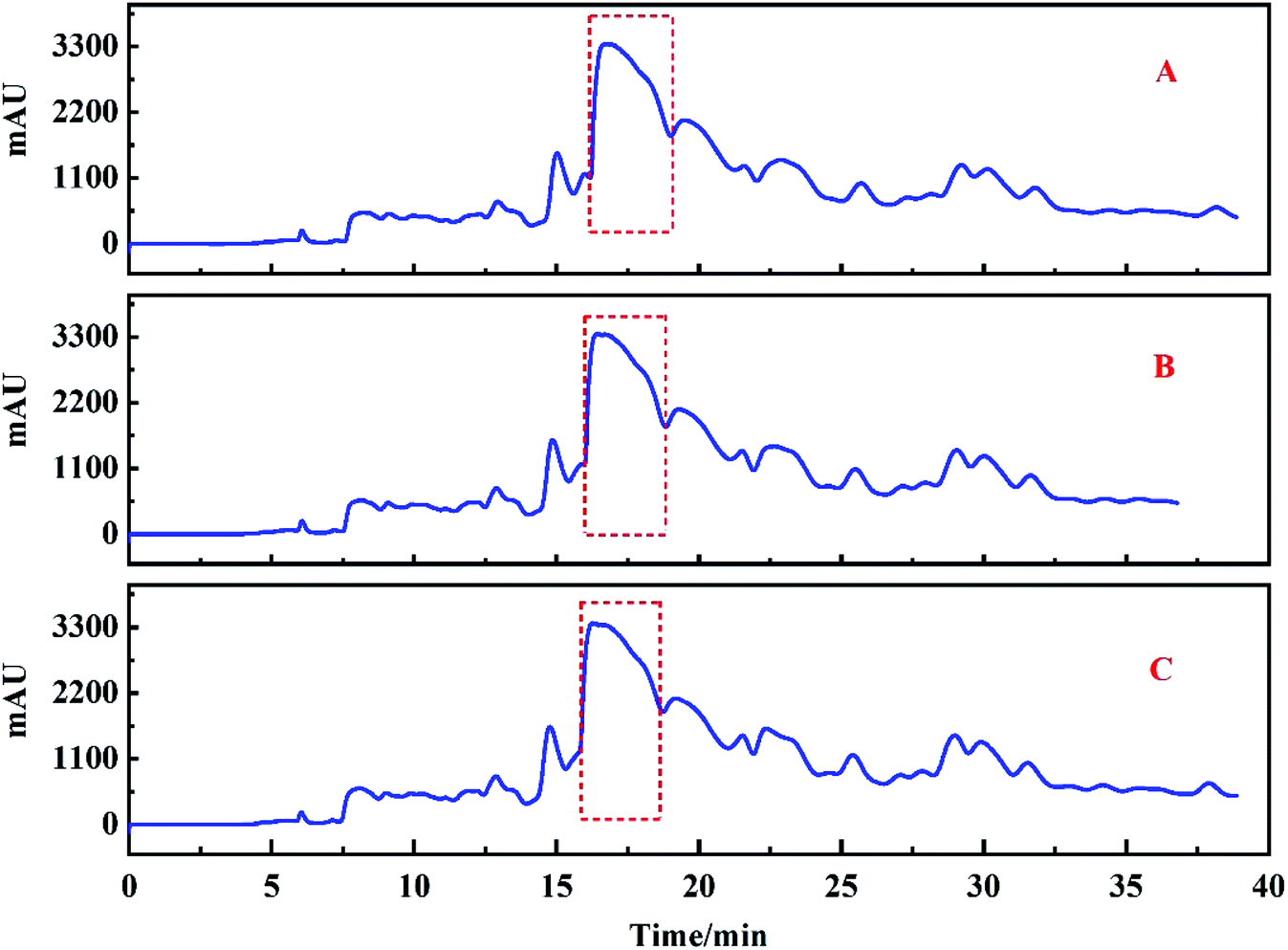 | ||
| Fig. 5 The repeatability of separation of Fr2 with the ZORBAX SB-AQ column. (A) The 1st injection, (B) the 6th injection, and (C) the 12th injection. | ||
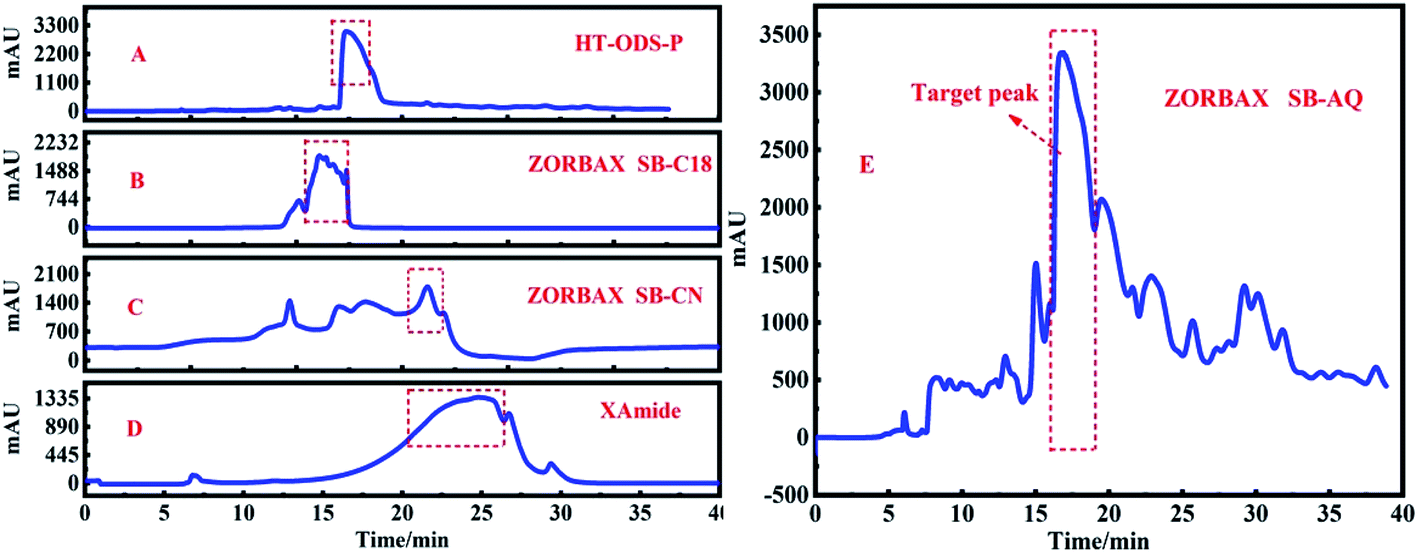
After the target peak was re-dissolved, the HPLC analysis was carried out on the HT-ODS-P analysis column, and the chromatographic conditions were consistent with the Fr2. The chromatogram of Fr2 and the target fraction peak obtained using the HT-ODS-P analytical column are shown in Fig. 6A and B, respectively. The dotted lines in Fig. 6A and B clearly show that through the one-dimensional separation of the ZORBAX SB-AQ preparation column, the chromatogram of the target peak was greatly simplified even though no high purity monomer compounds were obtained, and this provided a firm basis for the two-dimensional separation of the target fraction peak.
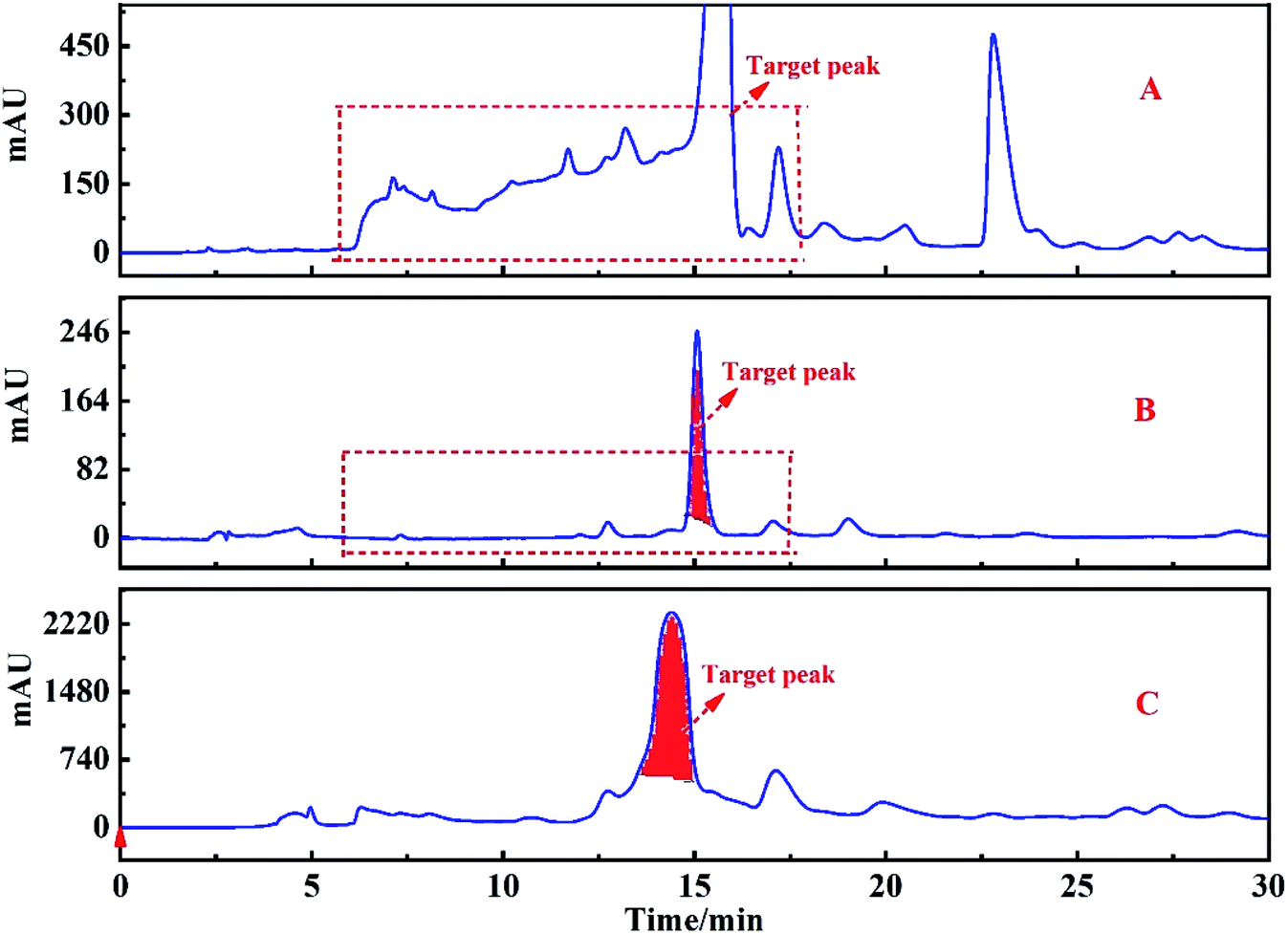 | ||
| Fig. 6 The chromatogram for Fr2 before pretreatment (A), and the target peak of Fr2 after pretreatment (B) on the HT-ODS-P analytical column. The chromatogram of the target peak re-purified (C) on the HT-ODS-P preparation column. | ||
Because the target peak has a high purity after one-dimensional separation (Fig. 6B), the two-dimensional separation was carried out on the HT-ODS-P preparation column, and the two-dimensional separation chromatogram of the target peak is shown in Fig. 6C. After 10 cycles of chromatography, sample concentration and drying, 1.03 g of an unknown compound corresponding to the target peak was obtained.
Purity verification and structure identification of the target peak
The PolyPark C18 analysis column was used to verify the purity of the compounds separated by the target peak. It can be seen from Fig. 7A that although the purity of the target peak was more than 95%, the target peak was eluted within 10 min. Considering that the short retention time of the target peak on the chromatographic column may affect the purity of the monomer, the HILIC column was used to verify it again (Fig. 7B). The results show that the purity of the target peak was still more than 95%.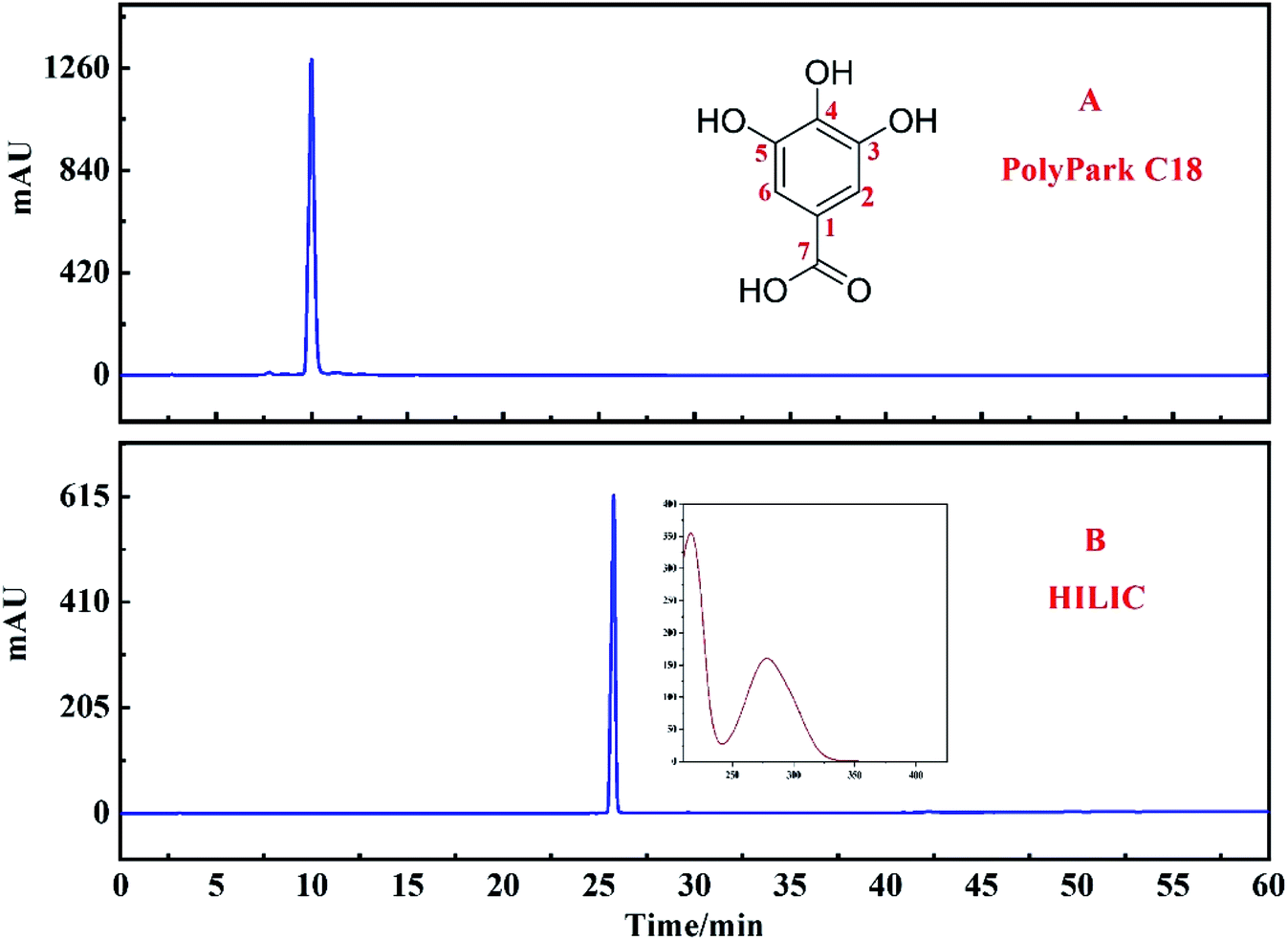 | ||
| Fig. 7 Two different chromatographic columns were used to evaluate the purity of the target peak: PolyPark C18 column (A) and HILIC column (B). | ||
In order to elucidate the structure of the target compound, HR ESI-MS, 1H and 13C NMR spectra were recorded and compared with the published literature data. It can be concluded from the spectral data that the target peak value was gallic acid, and this compound was reported for the first time in F. aubertii.
Target peak: (gallic acid, white powder, HR ESI-MS m/z 169.06, calcd for C7H6O5 m/z 170.0215): 1H NMR (600 MHz, MeOH-d4) 7.06 (2H, s, H-2, 6); 13C NMR (151 MHz, MeOH-d4) 170.3 (C-7), 146.4 (C-3, 5), 139.6 (C-4), 121.9 (C-1), 110.3 (C-2, 6).
Conclusions
In this study, the ethyl acetate extraction part of F. aubertii was pretreated first by a medium pressure chromatographic separation gel column. After 5 MCI treatments, 7 components were obtained, and Fr2 was selected as the next separation component. Because of the complexity of the Fr2 chromatogram, we selected the principal component compounds of Fr2 as the target peak and then used five different stationary phases for analysis. By comparing the separation effect of the Fr2 target peak on different stationary phases, the separated stationary phase (ZORBAX SB-AQ preparation column) was determined. Finally, the target peak with satisfactory purity was isolated and identified. The results show that this purification method is quite effective for the separation of gallic acid from F. aubertii. It also showed that the medium pressure chromatography separation system combined with high-pressure preparative chromatography can separate and purify monomer compounds from other complex Tibetan medicine components.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the Natural Science Foundation of Qinghai Province in China (Grant No. 2019-ZJ-915), Qinghai Provincial Thousand Talents Program of High-level Innovation Talent and Academic Leader of Natural Science and Engineering Technology in Qinghai Province, China, High-level Talent Project of the Qinghai Nationality University of Qinghai Province in China (Grant No. 2018XJG05) and The Innovation Platform for the Development and Construction of Special Project of Key Laboratory for Tibet Plateau Phytochemistry of Qinghai Province (Grant No. 2017-ZJ-Y19).References
- L. Q. Zhao, X. P. Zhang and C. gaF, Food Sci., 2012, 7, 1–5 Search PubMed.
- J. Wang, G. Ma and J. J. B. S. Hu, Ecology, 2019, 85, 50–53 CAS.
- L. N. Gao, R. Q. Jia, P. Ai, Q. Hu and P. C. Lin, Chin. J. Exp. Tradit. Med. Formulae, 2017, 23, 171–176 Search PubMed.
- X. Li, Y. C. Lu, M. R. Ren and Q. X. Xia, J. Nat. Sci. Hunan Norm. Univ., 2019, 03, 007 Search PubMed.
- J. Gengzang, C. Gazang and D. Z. Gazang, Journal of Medicine and Pharmacy of Chinese Minorities, 2012, 18, 16–17 Search PubMed.
- Q. Ma, P. C. Zhao and Y. C. Lu, Chinese Journal of Ethnomedicine and Ethnopharmacy, 2020, 29, 16–20 Search PubMed.
- Y. Mimaki, M. Kuroda, A. Kameyama, A. Yokosuka and Y. J. P. Sashida, Phytochemistry, 2019, 48, 485–493 CrossRef.
- S. Duan, Q. Huang, X. Shen, J. Hu, X. Yi, Z. Li and B. J. F. S. Ding, Nutrition, 2020, 8, 1 Search PubMed.
- N. H. Lu, H. Q. Zhao, M. Jing, X. Liu, C. Z. Ren, X. F. Liu, J. J. Liu and Y. X. J. I. I. Zhang, Int. Immunopharmacol., 2017, 46, 163–169 CrossRef CAS.
- K. Esmaiili and S. V. Shetab-Boushehri, J. Anal. Sci. Technol., 2017, 8, 22 CrossRef.
- N. Jagadeesh, K. Muralidharan, K. R. Babu, G. S. Bai and R. C. J. Swamy, J. Chromatogr. Sci., 2019, 9, 9 Search PubMed.
- Y. Zhang, S. Wang, J. Luo, Y. Lin, X. Xu, C. Han and L. J. Kong, J. Chromatogr. A, 2018, 1575, 122–127 CrossRef CAS.
- M. D. Joshi and J. L. Anderson, RSC Adv., 2012, 2, 5470–5484 RSC.
- H. F. Chen, C. Zhang, Y. Yao, J. M. Li, W. D. Du, M. L. Li, B. Wu, S. L. Yang, Y. L. Feng and W. G. P. Zhang, Analysis, 2019, 165, 213–223 CAS.
- C. Eckers, D. S. Skrabalak and J. J. Henion, Clin. Chem., 2019, 9, 9 Search PubMed.
- D. D. Bankson, R. M. Russell and J. A. Sadowski, Clin. Chem., 2020, 1, 1 Search PubMed.
- L. Castilho, V. Gama, A. Santos and A. Faria, J. Liq. Chromatogr. Relat. Technol., 2020, 1–17 Search PubMed.
- K. Liu, P. Aggarwal, J. S. Lawson, H. D. Tolley and M. L. J. Lee, J. Sep. Sci., 2013, 36, 2767–2781 CrossRef CAS.
- E. Loeser and P. J. Drumm, J. Sep. Sci., 2015, 29, 2847–2852 CrossRef.
- P. Li, C. Sun, Y. Wang, S. Wang, C. Yan, S. Deng, X. Huo, L. Feng, C. Wang and Y. J. Tian, Catal. Commun., 2017, 97, 106–110 CrossRef CAS.
- W. Liu, Z. W. Li, H. Wang, F. L. Yang and C. P. Wan, Mater. Sci. Technol., 2018, 34, 263–267 Search PubMed.
- K. Esmaiili and S. V. Shetab-Boushehri, J. Anal. Sci. Technol., 2017, 8, 22 CrossRef.
- D. Nguyen, D. J. Seo, H. B. Lee, I. S. Kim, K. Y. Kim, R. D. Park and W. J. J. Jung, Microb. Pathog., 2013, 56, 8–15 CrossRef CAS.
- Y. Shen, H. Yang, G. Xia, J. Wang, B. Cai and X. J. Jia, Acta Chromatogr., 2013, 25, 687–701 CrossRef.
- J. J. Lu, Y. Wei and Q. P. Yuan, Sep. Purif. Technol., 2007, 55, 40–43 CrossRef CAS.
- G. Tian, T. Zhang, F. Yang and Y. J. Ito, J. Chromatogr. A, 2000, 886, 309–312 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |