DOI:
10.1039/D1RA03233J
(Paper)
RSC Adv., 2021,
11, 21397-21404
Wide-bandgap donor polymers based on a dicyanodivinyl indacenodithiophene unit for non-fullerene polymer solar cells†
Received
25th April 2021
, Accepted 10th June 2021
First published on 16th June 2021
Abstract
A wide-bandgap polymer donor with improved efficiency plays an important role in improving the photovoltaic performance of polymer solar cells (PSCs). In this study, two novel wide-bandgap polymer donors, PBDT and PBDT-S, were designed and synthesized based on a dicyanodivinyl indacenodithiophene (IDT-CN) moiety, in which benzo[1,2-b:4,5-b′]dithiophene (BDT) building blocks and IDT-CN are used as electron-sufficient and -deficient units, respectively. In our study, the PBDT and PBDT-S polymer donors exhibited similar frontier-molecular-orbital energy levels and optical properties, and both copolymers showed good miscibility with the widely used narrow-bandgap small molecular acceptor Y6. Non-fullerene polymer solar cells (NF-PSCs) based on PBDT:Y6 exhibited an impressive power conversion efficiency of 10.04% with an open circuit voltage of 0.88 V, a short-circuit current density of 22.16 mA cm−2 and a fill factor of 51.31%, where the NF-PSCs based on PBDT-S:Y6 exhibited a moderate power conversion efficiency of 6.90%. The enhanced photovoltaic performance, realized by virtue of the improved short-circuit current density, can be attributed to the slightly enhanced electron mobility, higher exciton dissociation rates, more efficient charge collection and better nanoscale phase separation of the PBDT-based device. The results of this work indicate that the IDT-CN unit is a promising building block for constructing donor polymers for high-performance organic photovoltaic cells.
Introduction
Polymer solar cells (PSCs) with bulk heterojunctions (BHJs) have attracted tremendous research attention due to their great potential for fabricating large-area and flexible devices using a roll-to-roll solution process.1–4 Recently, significant progress has been made with non-fullerene PSCs (NF-PSCs) based on extensive investigations of non-fullerene acceptors (NFAs). NFAs have several distinct advantages, including easy tuning of the absorption and electronic energy levels, and their high morphological stability, etc.5–16 One of the most significant recent breakthroughs in NFAs is the narrow-bandgap small-molecular acceptor Y6 and its derivatives,17–22 which boost the power conversion efficiencies (PCEs) of single-junction PSCs by more than 18%, thereby mitigating their performance gap relative to other emerging photovoltaic technologies.
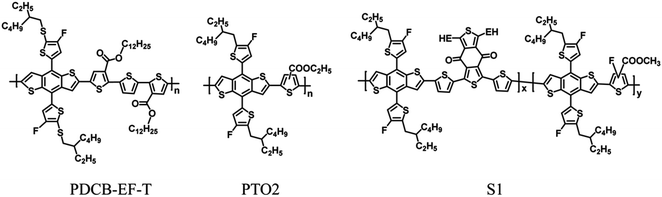
High-performance NFAs have broad absorption profiles with edges extending into the near-infrared region and also the increase the short-circuit current density (JSC).1,6 However, the open-circuit voltages (VOC) of these devices are relatively low because of the low-lying lowest unoccupied molecular orbital (LUMO) levels of the NFAs.23,24 To further improve the efficiency of NF-PSCs and overcome the fundamental compromise between the JSC and VOC output, it is important to develop an ideal wide-bandgap (WBG) polymer with complementary absorption in the short wavelength region and a deeper highest occupied molecular orbital (HOMO) energy level.25,26 Enabling them to fully utilize the solar spectrum, WBG polymer donors better match the NFAs, thus maximizing the JSC of the devices.27–30 To date, just a few high-performance conjugated polymer donors, i.e., PM6,7 PTQ10,31 P2F-EHp,8 D18,10 PTO2,32 and S1,33 have been reported to match NFAs to achieve more efficient NF-PSCs. Thus, it remains a demanding task to synthesize well-designed high-efficiency donor materials with deep HOMO levels that suitably match state-of-the-art low-bandgap NFAs to obtain high-performance NF-PSCs.
Recently research had demonstrated that introducing a suitable electron-withdrawing groups onto the main and the side chain is an effective strategy to downshift the HOMO level while keeping the WBG. For instance, several donor polymer with fluoro-substituted thiophene group or ester-substituted thiophene groups on the main chains show wide-bandgap and low-lying HOMO levels (Fig. 1),28,32,33 which could promote the VOC and maintain the high JSC. Besides fluorine and ester group, another functional group that deserves attention is the cyano group. Cyano (–CN) group-substituted units have been widely used in the construction of donor–acceptor conjugated polymers for optoelectronic devices, because the electron-withdrawing –CN substituents increase the electron affinity to promote efficient charge transport and it can endow the D–A copolymers with deep-lying HOMO levels.34–38 Several polymers with –CN groups on the main chains show large bandgaps and low-lying HOMO levels, for example, Park et al. reported that the polymer PBDCS based on β-dicyanodistyrylbenzene and benzodithiophene (BDT) with thiophene-conjugated side chains exhibited a deep HOMO of −5.59 eV and a band-gap of 1.75 eV.39 In previous work, we reported that a series of copolymers containing a dicyanodistyrylbenzene unit exhibited a deep HOMO.34 These findings indicate that –CN substituents on the electron-acceptor moiety can be used to tune the properties of donor polymers toward those of a WBG.
 |
| | Fig. 1 Chemical structures of representative high-performance polymer donors containing fluorine and ester group for NF-PSCs. | |
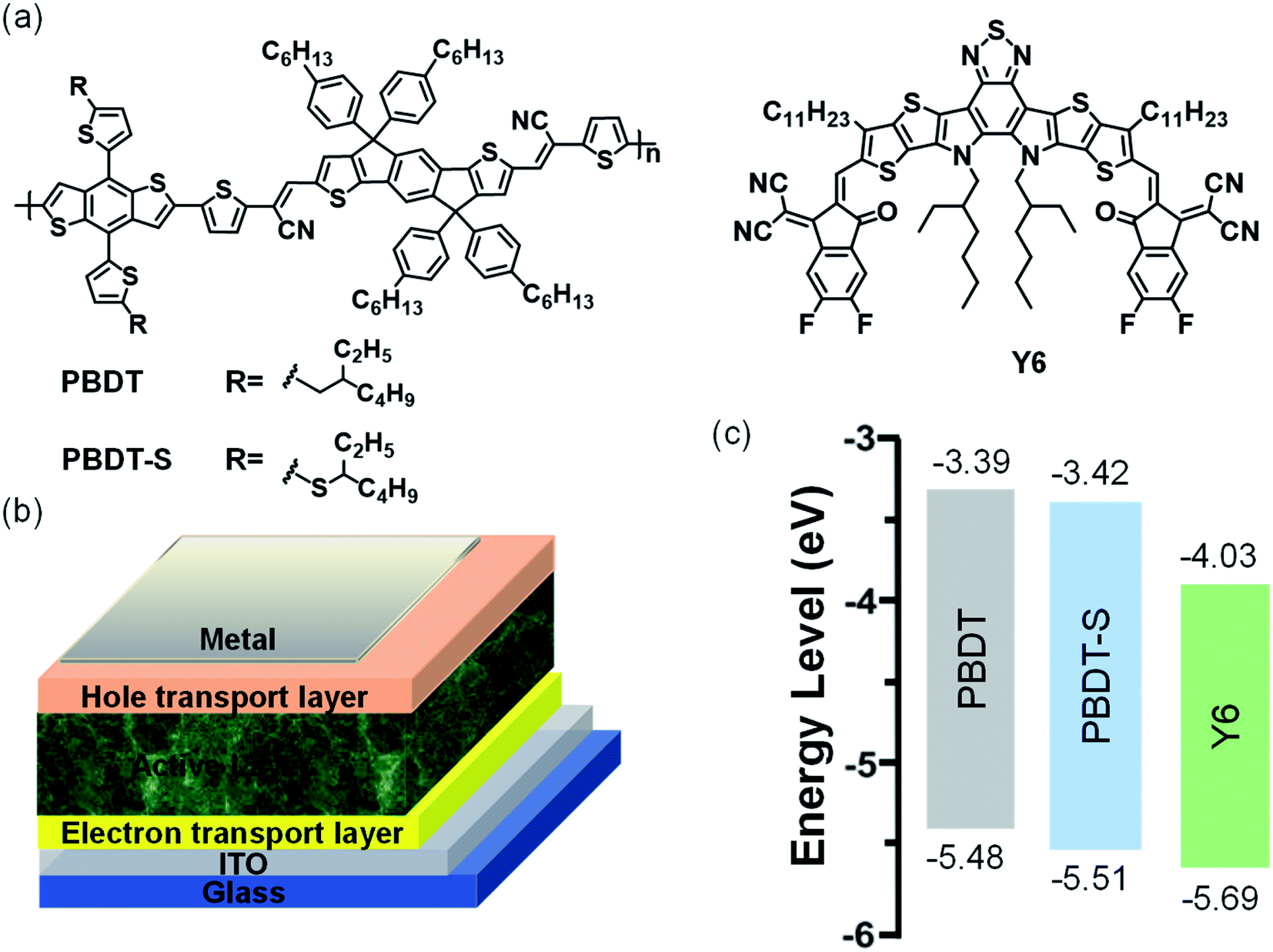
Hence, in this work, we designed and synthesized two new CN-substituted conjugated polymers with a typical D–A architecture through the Stille coupling reaction. Two electron-donating BDT units with an alkyl-thienyl or alkylthio-thienyl side-chain were linked to the electron withdrawing IDT-CN unit to generate the target polymers PBDT and PBDT-S, respectively (Fig. 2a). From the chemical structure we could find that the polymer consisting of BDT and IDT building block connected by the strong electron withdrawing group cyanovinylene unit, which is considered an effective strategy for constructing donor polymer with wide-bandgap and deep HOMO level. We show that these two copolymers present almost the same absorption spectra in the wavelength range of 450–670 nm, whereas Y6 forms a complementary absorption spectrum in the film state longer wavelength range of 600–900 nm, which is favorable for the efficient harvesting of solar light. These copolymers also exhibit low-lying HOMO levels of with −5.48 eV and −5.51 eV for PBDT and PBDT-S, respectively, which is beneficial for matching with NFAs to achieve the desired VOC. Using Y6 as an electron acceptor, the PBDT-based PSCs device delivered a PCE of 10.04% with a VOC of 0.88 V, a JSC of 22.16 mA cm−2 and an FF of 51.31%, whereas the PBDT-S-based PSCs device obtained a moderate PCE of 6.90% with a VOC of 0.90 V, a JSC of 16.18 mA cm−2 and an FF of 46.99%. The enhanced performance of the PBDT-based device was attributable to the more balanced hole/electron mobility, lesser charge recombination and more favorable morphology. The results of our comparative research indicate that the incorporating of a strong electron withdrawing group linker unit into donor and donor unit provide an interesting platform for the design of donor polymer with wide-bandgap and deep HOMO level, which would be helpful for the rational molecular design of organic semiconducting materials for PSCs.
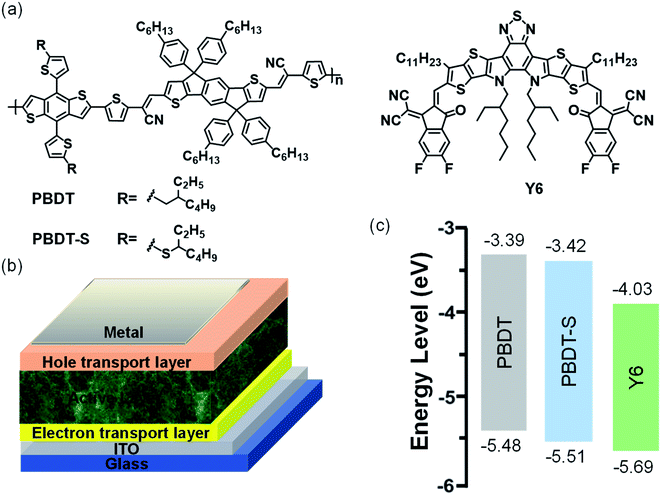 |
| | Fig. 2 (a) Chemical structure of donors and acceptor; (b) device structure of polymer solar cells; (c) energy-level diagram of donors and acceptor. | |
Results and discussion
Material synthesis
Fig. 1 shows the molecular structures of the copolymers PBDT and PBDT-S, each of which has Y6 as a small molecular acceptor. Details regarding the procedures for synthesizing these copolymers are provided in the ESI (Scheme S1†). Compound 3 was synthesized according to our previously reported procedure.34 The copolymers were synthesized using a typical Stille-coupling reaction with chlorobenzene as solvent and Pd(PPh3)4 as catalyst. The copolymers exhibited good solubility in common solvents, including chloroform, chlorobenzene (CB), and o-dichlorobenzene, and thus provide good solution-processing condition for a producing high-quality blend film. The molecular weights of the copolymers were determined by high-temperature gel permeation chromatography in 1,2,4-trichlorobenzene, using polystyrene as the standard. The number-average molecular weights (Mn) for PBDT and PBDT-S were estimated to be 37.6 kDa and 36.8 kDa, with polydispersity indices (PDI) are 2.29 and 2.83, respectively.
Photophysical and electrochemical properties
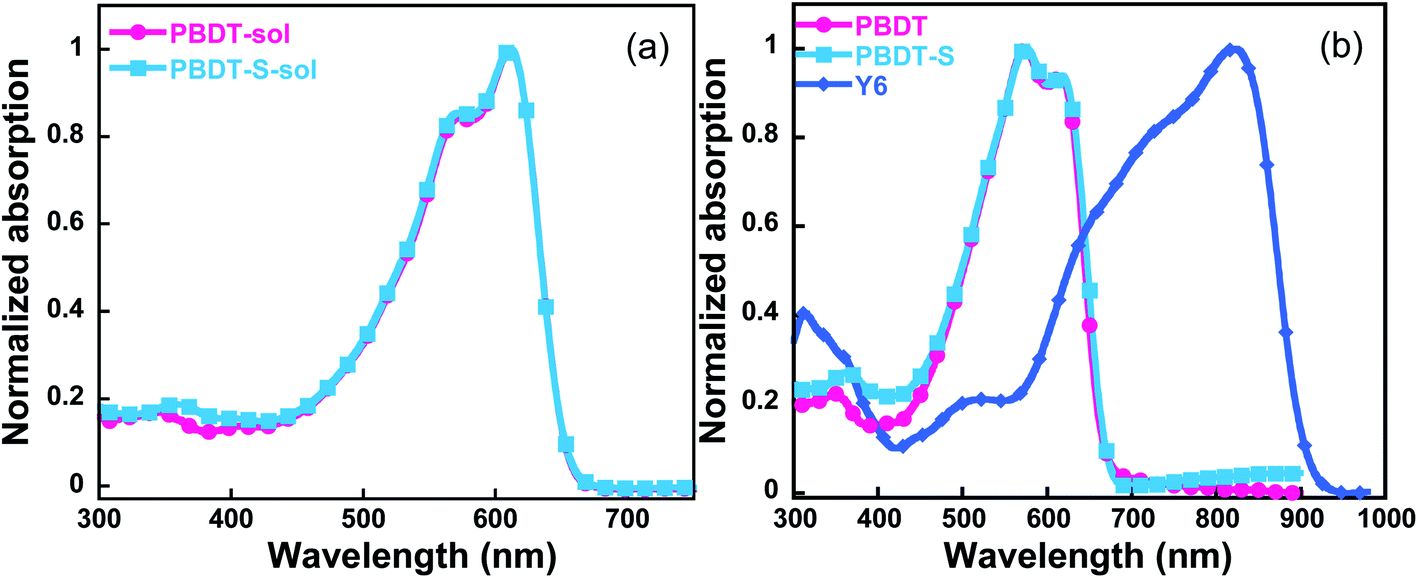
The normalized UV-vis absorption spectra of the copolymers in both CB solution and a thin-film state were investigated (Fig. 3), and the corresponding data are presented in Table 1. These two copolymers have similar absorption spectra in both the solution and thin film. In solution, the copolymers exhibited absorption profiles ranging from 300 to 680 nm, with the short wavelength range attributed to the π–π* transition and the long wavelength absorption peak possibly correlated with intra-molecular charge-transfer characteristics.28 In the corresponding thin films, both copolymers exhibited vibronic shoulder peaks, which indicated strong intermolecular packing in the solid state. The PBDT film spectrum shows two absorption peaks at 575 nm and 617 nm, r with an absorption edge at 664 nm, corresponding to an optical bandgap of 1.87 eV. The absorption spectra spectrum of PBDT-S film shows an absorption edge at 669 nm (corresponding to an optical bandgap of 1.85 eV), which indicates a slightly red shift in comparison to that of PBDT film. Therefore, both copolymers demonstrated complementary absorption with the Y6 acceptor in the vis-NIR region, which effectively broadens the absorption range of the blended film and facilitates light harvesting by PSCs devices.
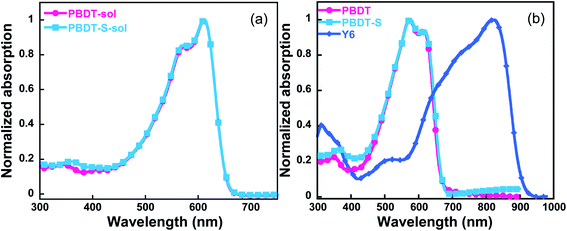 |
| | Fig. 3 Absorption spectra of the copolymers (a) in CB solution and (b) as thin films. | |
Table 1 Molecular weights, absorption and electrochemical properties for copolymers
| Copolymers |
Mna (kDa) |
PDIa |
λsolmax (nm) |
λfilmmax (nm) |
EHOMOb (eV) |
ELUMOb (eV) |
Eoptgc (eV) |
| Determined by GPC (1,2,4-trichlorobenzene) against PS standards. Measured by cyclic voltammetry. Calculated from the absorption edge of the film. |
| PBDT |
37.6 |
2.29 |
611 |
617 |
−5.48 |
−3.39 |
1.87 |
| PBDT-S |
36.8 |
2.83 |
611 |
617 |
−5.51 |
−3.42 |
1.85 |
The HOMO and the LUMO energy levels of the copolymers were measured by electrochemical cyclic voltammetry (Fig. S1, ESI†). The corresponding HOMO/LUMO energy levels of the copolymers were −5.48/−3.39 eV and −5.51/−3.42 eV for PBDT and PBDT-S, respectively (Fig. 2c). The HOMO energy levels of PBDT-S copolymers were slightly lower than those of the PBDT copolymers, which promotes a higher VOC of the PSCs that use these polymers as donor. The low-lying HOMO levels of the copolymers matched with the acceptor Y6.
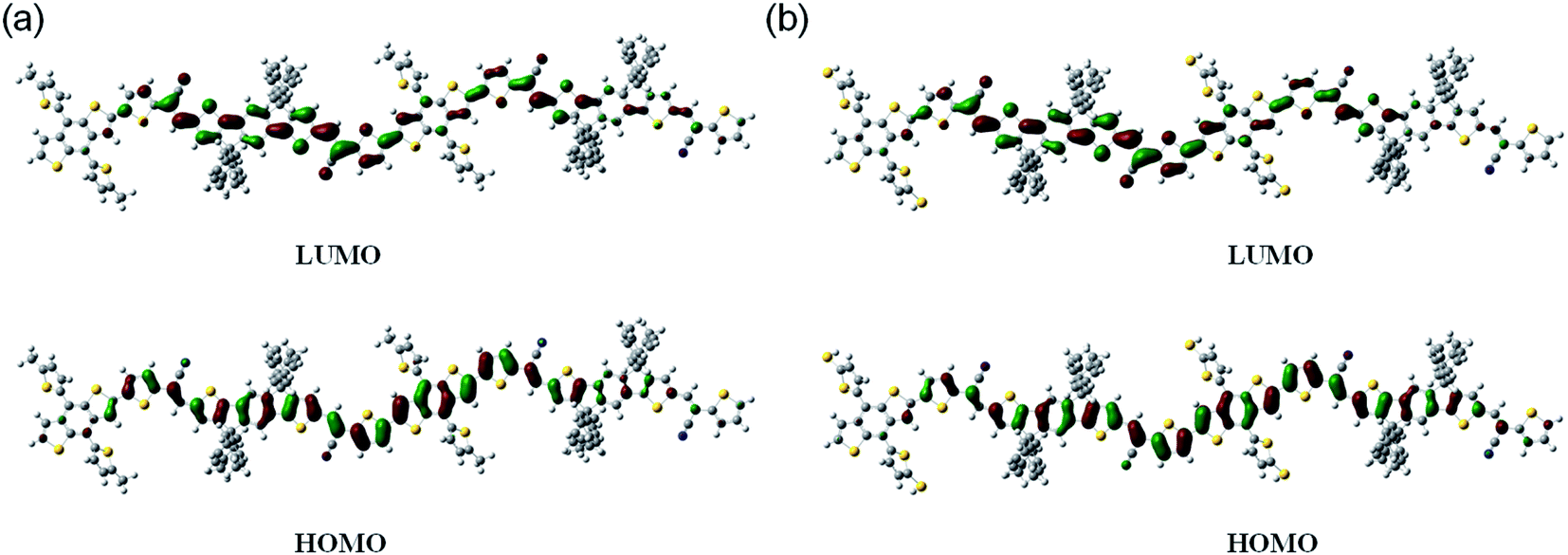
To demonstrate the optimal molecular geometry and electronic properties, we performed theoretical calculations using density functional theory at the B3LYP level with the 6-31G* basis set.34 Fig. 4 shows the optimized molecular geometries of the models and their calculated LUMO and HOMO frontier orbitals. These two copolymers exhibited similar electron density distributions both in the LUMO and HOMO levels. The HOMO surfaces of the resulting copolymers were well delocalized along their backbones, whereas their LUMO surfaces were mainly localized on the electron-deficient units.
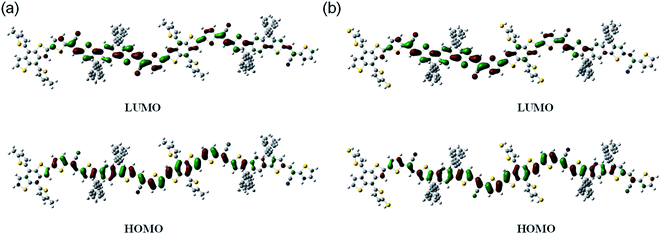 |
| | Fig. 4 The HOMO and LUMO orbital distributions of (a) PBDT and (b) PBDT-S. | |
Photovoltaic properties
To investigate their photovoltaic properties, we fabricated NF-PSCs based on copolymers with Y6 using an ITO/ZnO/active layer/MoO3/Al with an inverted structure. The initial optimization of the photovoltaic performances of these devices was performed using PBDT:Y6. The photovoltaic properties of the devices were systematically optimized in terms of their D–A thermal annealing temperature, weight ratios and solvent additive (Tables S1–S3, ESI†).
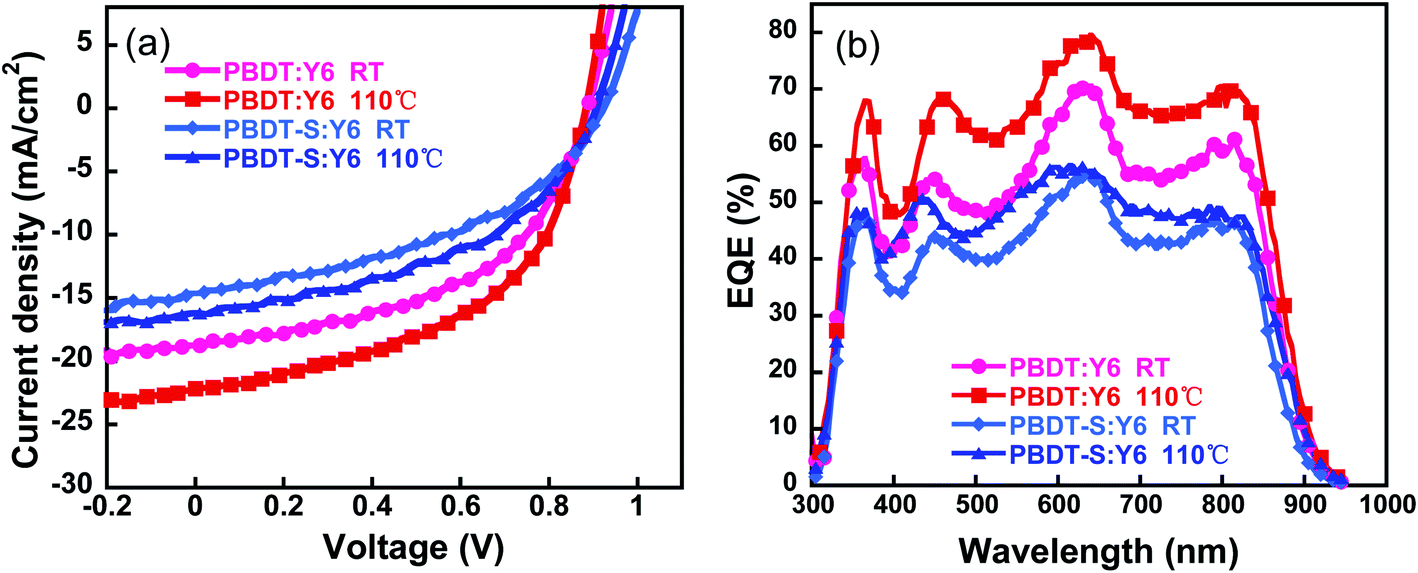
The optimized active layer was spin-coated from chloroform solution with an optimized donor/acceptor (D/A) weight ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.2 and without any additive, the corresponding performance parameters of which are listed in Table 2. The as-cast NF-PSCs based on PBDT:Y6 obtained a PCE value of 8.41% with a VOC of 0.89 V, a JSC of 18.74 mA cm−2, and an FF of 50.60%. The device based on the PBDT-S copolymer obtained a low PCE of 5.86% with a VOC of 0.92 V, a JSC of 14.12 mA cm−2, and an FF of 43.38%. To further improve the efficiency of the devices, we performed a thermal annealing treatment to optimize the morphology of the active layer. With thermal annealing treatment at 110 °C for 5 min, significant improvements in PCE were obtained along with an increase in the FF and JSC for the devices (Fig. 5a and Table 2). A PCE of 10.04% was obtained for the PBDT:Y6 device, with a VOC of 0.88 V, a JSC of 22.16 mA cm−2, and an FF of 51.31%, whereas a PCE of 6.90% was obtained for the PBDT-S:Y6 device. The improvement in the PCE values of the NF-PSCs with thermal annealing was mainly due to the increase of JSC, which is likely due to the optimization of the active-layer morphology. The effect of thermal annealing on the morphology is discussed later. Moreover, we also used non-halogen solvents tetramethylene oxide (THF) to fabricate PSCs. However, the resulting devices presented moderate photovoltaic performances with PCE of 8.4% (Tables S5 and S6, ESI†), which is slightly lower than those spin-coated from chloroform solution.
:1.2 and without any additive, the corresponding performance parameters of which are listed in Table 2. The as-cast NF-PSCs based on PBDT:Y6 obtained a PCE value of 8.41% with a VOC of 0.89 V, a JSC of 18.74 mA cm−2, and an FF of 50.60%. The device based on the PBDT-S copolymer obtained a low PCE of 5.86% with a VOC of 0.92 V, a JSC of 14.12 mA cm−2, and an FF of 43.38%. To further improve the efficiency of the devices, we performed a thermal annealing treatment to optimize the morphology of the active layer. With thermal annealing treatment at 110 °C for 5 min, significant improvements in PCE were obtained along with an increase in the FF and JSC for the devices (Fig. 5a and Table 2). A PCE of 10.04% was obtained for the PBDT:Y6 device, with a VOC of 0.88 V, a JSC of 22.16 mA cm−2, and an FF of 51.31%, whereas a PCE of 6.90% was obtained for the PBDT-S:Y6 device. The improvement in the PCE values of the NF-PSCs with thermal annealing was mainly due to the increase of JSC, which is likely due to the optimization of the active-layer morphology. The effect of thermal annealing on the morphology is discussed later. Moreover, we also used non-halogen solvents tetramethylene oxide (THF) to fabricate PSCs. However, the resulting devices presented moderate photovoltaic performances with PCE of 8.4% (Tables S5 and S6, ESI†), which is slightly lower than those spin-coated from chloroform solution.
Table 2 Photovoltaic performances of copolymer:Y6 devices with or without thermal annealing under the illumination of AM 1.5 G, 100 mW cm−2
| Copolymers |
Annealing (°C) |
VOC (V) |
JSC (mA cm−2) |
JSC(cal.)b (mA cm−2) |
FF (%) |
PCEa (%) (max.) |
| Statistical data obtained from 10 independent devices. The JSC calculated from the EQE spectrum. |
| PBDT |
W/o |
0.89 |
18.74 |
18.02 |
50.60 |
8.41 |
| PBDT |
110 |
0.88 |
22.16 |
21.50 |
51.31 |
10.04 |
| PBDT-S |
W/o |
0.92 |
14.63 |
14.12 |
43.38 |
5.86 |
| PBDT-S |
110 |
0.90 |
16.18 |
15.78 |
46.99 |
6.90 |
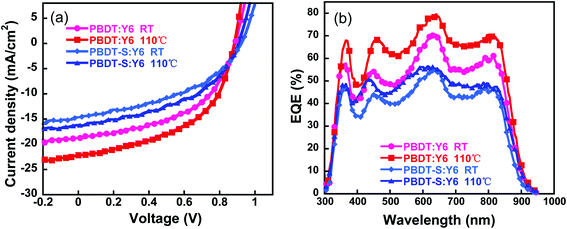 |
| | Fig. 5 (a) Current–voltage characteristics of polymer solar cells for each copolymer with (annealing) or without (as-cast) thermal annealing at 110 °C, under simulated AM 1.5 G illumination (100 mW cm−2). (b) External quantum efficiency curves of the devices for each copolymer. | |
Fig. 5b shows the external quantum efficiency (EQE) values of the NF-PSCs with and without thermal annealing at 110 °C for 5 min. It can be seen that charge generation benefits from the complementary absorption of the donors and acceptors. The optimized devices based on PBDT:Y6 or PBDT-S:Y6 exhibit a broad photo-response from 300 nm to 900 nm with the maximum values reaching 78.8% and 57.8% at 640 nm, respectively. The current densities obtained integrated from the EQE spectra are in good agreement with those obtained from J–V measurements (Table 2), for which the mismatch between the two calculation methods is less than 5%. The EQE responses of the PBDT-based PSCs were stronger than those of the PBDT-S-based device in the region of 350–850 nm, which resulted in a higher JSC value.
We note that the obtained PCE based on PBDT is not as high as the reported most highly efficient moieties, such as PCE12 or PM6. However, the polymer PBDT designed through incorporating cyanovinylene linker unit into donor–donor unit, provided an interesting strategy for constructing donor polymer with wide-bandgap and deeper HOMO energy level, which is beneficial to enhance the JSC and VOC values of PSCs. Moreover, it is worth pointing out that the PCE essentially correlates to various parameters, such as the miscibility of donor/acceptor, film thickness and morphology of the blend film, the geometry of solar cell devices and so forth. Our current work is mainly devoted to demonstrating a electron-withdrawing linker introduced onto the backbone is a good strategy to downshift the HOMO while keeping the large Eg of donor polymer. Meanwhile, we fabricated large area devices (1 cm2) by chloroform as processing solvent obtained a PCE of 6.93% (Table S7, ESI†).
Charge generation and transport properties
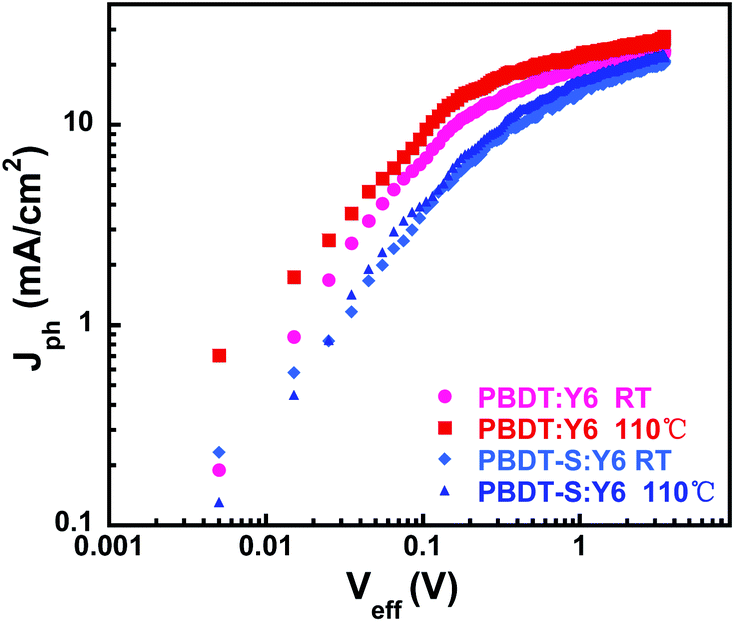
To determine the exciton dissociation and charge collection properties of the devices, we investigated the relationship of the photocurrent density (Jph) with the effective voltage (Veff) of the devices, the results of which are shown in Fig. 6. The photocurrent density Jph is defined as Jph = JL − JD, where JL and JD are the light and dark current densities, respectively, and the effective voltage Veff is defined as Veff = V0 − Vbias, where V0 is the voltage when Jph is zero and Vbias is the applied bias voltage.8 At a high value of Veff (>2 V), all of the photogenerated excitons dissociated into free charges and were collected by the electrodes, with the Jph value reaching saturation (Jsat). Thus, the maximum exciton generation rate (Gmax) can be defined as Gmax = Jsat/qL, where Jsat is the saturated photocurrent density, q is the electronic charge and L is the thickness of the active layer in the devices. The calculated Gmax values of the devices are presented in Table S8.† For the device based on PBDT:Y6, the annealed devices presented slightly higher Gmax values (1.55 × 1027) than those of the as-cast devices (1.34 × 1027), which indicates that the exciton generation of the annealed devices is faster than that of the as-cast device and agrees with the corresponding JSC values. The device based on PBDT-S:Y6 exhibited the same trend, with Gmax values of 1.21 × 1027 and 1.14 × 1027 for the annealed and as-cast devices.
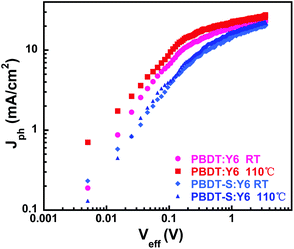 |
| | Fig. 6 Jph versus Veff of the devices without and with thermal annealing at 110 °C. | |
To understand the effect of thermal annealing on the charge transfer and transport performance, we used the space-charge-limited current method to evaluate the electron and hole mobilities of the blended films.40 The carrier mobilities were calculated from the slope of the J1/2–V curves (Fig. S2, ESI†), and are presented in Table 3. A high and balanced carrier mobility is a crucial factor affecting the JSC and FF values of PSCs. The as-cast device obtained hole mobility (μh) and electron mobility (μe) values as high as 7.63 × 10−4 and 1.19 × 10−4 for PBDT:Y6 and 4.84 × 10−4 and 3.03 × 10−5 for PBDT-S:Y6, respectively. After annealing, the μe value of these two devices increased significantly, with the PBDT:Y6 obtaining higher μh and μe values of 9.18 × 10−3 and 2.30 × 10−4 cm2 V−1 s−1, respectively. Thus, the blended film with thermal annealing exhibited higher and more balanced carrier mobility than the as-cast film, which was responsible for the higher JSC and FF values of the thermally annealed devices.
Table 3 Carrier mobilities of copolymer:Y6 devices with or without thermal annealing
| Copolymer:Y6 |
Annealing |
μh (cm2 V−1 s−1) |
μe (cm2 V−1 s−1) |
μh/μe |
| PBDT |
W/o |
7.63 × 10−4 |
1.19 × 10−4 |
6.41 |
| PBDT |
110 °C |
9.18 × 10−3 |
2.30 × 10−4 |
3.99 |
| PBDT-S |
W/o |
4.48 × 10−4 |
3.03 × 10−5 |
15.97 |
| PBDT-S |
110 °C |
7.41 × 10−4 |
8.10 × 10−5 |
9.15 |
Charge extraction, and recombination
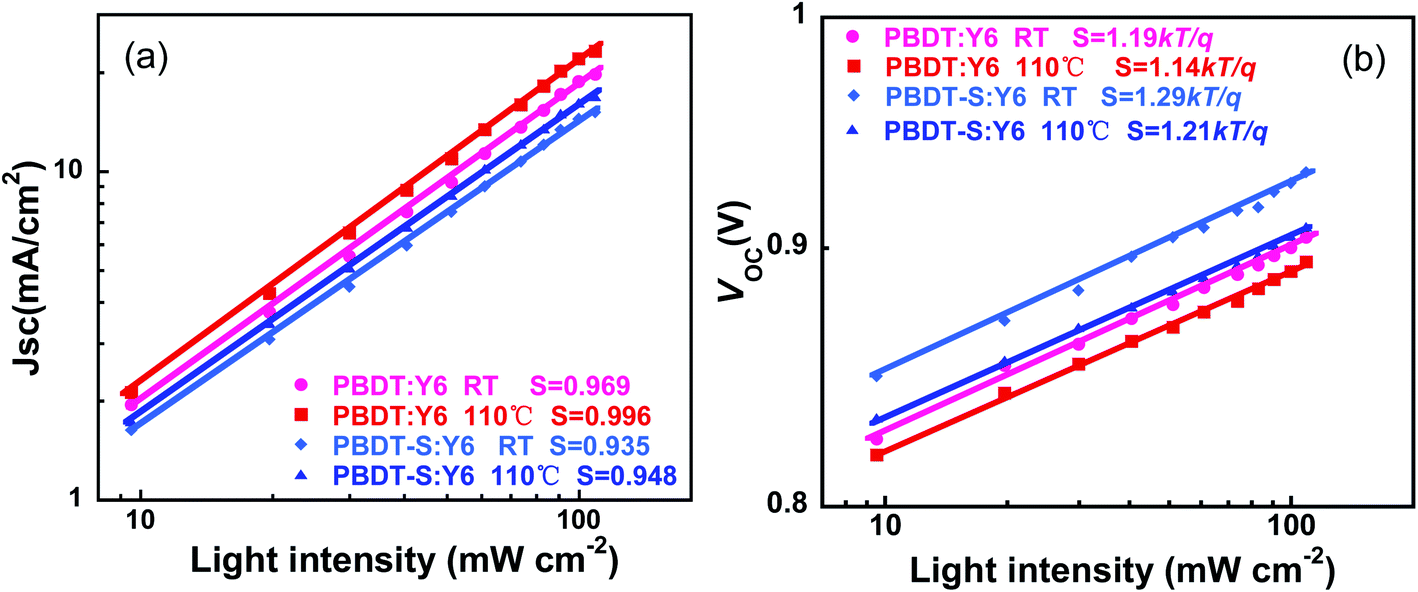
To understand the charge recombination process of NF-PSCs, we measured the dependence of JSC on the light intensity (Plight). The relationship between JSC and Plight can be expressed as JSC ∝ (Plight)S, in which Plight is light intensity and S is the exponential factor.41,42 When the bimolecular recombination of the charge carriers is weak, JSC shows a linear dependence on Plight, with an S value close to 1. In Fig. 7a, for as-cast device, the logarithmic plots of JSC versus light intensity have slopes of 0.969 and 0.935 for PBDT and PBDT-S, respectively. After thermal annealing, the logarithmic plots of the slopes of JSC versus light intensity increased to 0.996 and 0.948 for PBDT and PBDT-S, respectively, which indicates that in comparison to as-cast devices, thermally annealed devices can sweep out charge carriers most efficiently with negligible bimolecular recombination, which is consistent with the higher JSC and FF values obtained by the thermally annealed devices. As showed in Fig. 7b, the plots of the dependence of VOC on the light intensity for PBDT:Y6 and PBDT-S:Y6 with thermal annealing have slope of 1.14kT/q and 1.21kT/q, repectively, compared with the 1.19kT/q and 1.29kT/q for the as-cast devices, which also implies a lower rate of trap-assisted recombination in the devices upon thermal annealing.
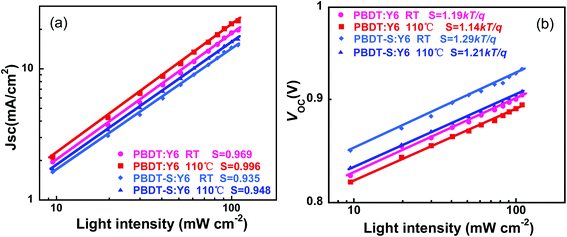 |
| | Fig. 7 Light intensity dependence of (a) JSC and (b) VOC of as-cast and annealed devices. | |
Morphological characterization
Because the performance of these devices is closely related to the morphology of the blended film, we used tapping-mode atomic force microscopy (AFM) to investigate the effect of the thermal annealing on the morphology of the blended films, the results of which are shown in Fig. 8. For the copolymer PBDT, the as-cast blended film showed a rough surface with a root mean square (RMS) roughness of 1.61 nm, whereas the thermally annealed blended film exhibited a uniform and smooth surface with an RMS of 1.33 (Fig. 8a and b, respectively). Similarly, for the copolymer PBDT-S, the thermally annealed blended film showed a smaller RMS (1.48 nm) than the as-cast blended film (Fig. 8c and d). These results indicated that thermally annealed blended films exhibited a smooth surface morphology, which favors exciton dissociation and charge transport, resulting in a higher JSC and FF values.24 We used transmission electron microscopy (TEM) to further characterize the morphology of copolymer:Y6 blended films obtained under the same conditions. As shown in Fig. 8e–h, no clear phase separation is evident in the as-cast blended films, whereas the thermally annealed blended films (Fig. 8f and h) show clear fibrous structures and phase separation, which provide effective pathways for exciton dissociation and charge transport, which are consistent with the improved JSC and FF values.43
 |
| | Fig. 8 AFM height images (5 × 5 mm2) and TEM images of (a and e) PBDT:Y6 as-cast, (b and f) PBDT:Y6 with thermal annealing, (c and g) PBDT-S:Y6 as-cast, and (d and h) PBDT-S:Y6 with thermal annealing. | |
Conclusion
In summary, we synthesized two novel copolymers, PBDT and PBDT-S, as donor materials for NF-PSCs based on the dicyanodivinyl indacenodithiophene (IDT-CN) moiety and benzo[1,2-b:4,5-b′]dithiophene (BDT) units, and investigated their optical and electrochemical properties. These two copolymers exhibited similar absorption profiles and low-lying HOMO levels. Using the narrow-bandgap acceptor Y6, the NF-PSCs based on PBDT and PBDT-S as-cast blended films obtained low PCEs of 8.41% and 5.86%, respectively. After thermal annealing, the phase-separated morphology of the blended films is enhanced in these copolymers, which promotes efficient charge dissociation and collection, weaker bimolecular recombination and weaker trap-assisted recombination, resulting in increased JSC and FF values for the devices. The NF-PSCs obtained high PCE values of 10.04% and 6.90% for PBDT-based and PBDT-S-based blend films, respectively. These results demonstrate that the IDT-CN-based copolymer is a promising donor material for the construction of high-performance NF-PSCs.
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
This work was financially supported by the Guangdong Basic and Applied Basic Research Foundation (Grant No. 2019A1515011141), the characteristic Innovation Project for Higher Education Institution of Guangdong Province (Grant No. 2019KTSCX170), the Project of Educational Commission of Guangdong Province (Grant No. 2019GCZX010), and the Natural Science Foundation of Hunan Province, China (2020JJ5544).
References
- C. Yan, S. Barlow, Z. Wang, H. Yan, A. K. Y. Jen, S. R. Marder and X. Zhan, Nat. Rev. Mater., 2018, 3, 18003 CrossRef CAS.
- F. Huang, Z. Bo, Y. Geng, X. Wang, L. Wang, Y. Ma, J. Hou, W. Hu, J. Pei, H. Dong, S. Wang, Z. Li, Z. Shuai, Y. Li and Y. Cao, Acta Polym. Sin., 2019, 50, 988–1046 CAS.
- Y. Li, G. Xu, C. Cui and Y. Li, Adv. Energy Mater., 2018, 8, 1701791 CrossRef.
- N. Li, I. McCulloch and C. J. Brabec, Energy Environ. Sci., 2018, 11, 1355–1361 RSC.
- P. Cheng, G. Li, X. Zhan and Y. Yang, Nat. Photonics, 2018, 12, 131–142 CrossRef CAS.
- J. Hou, O. Inganäs, R. H. Friend and F. Gao, Nat. Mater., 2018, 17, 119–128 CrossRef CAS PubMed.
- J. Yuan, Y. Zhang, L. Zhou, G. Zhang, H. L. Yip, T. K. Lau, X. Lu, C. Zhu, H. Peng, P. A. Johnson, M. Leclerc, Y. Cao, J. Ulanski, Y. Li and Y. Zou, Joule, 2019, 3, 1–12 CrossRef.
- B. Fan, D. Zhang, M. Li, W. Zhong, Z. Zeng, L. Ying, F. Huang and Y. Cao, Sci. China: Chem., 2019, 62, 746–752 CrossRef CAS.
- X. Xu, K. Feng, Z. Bi, W. Ma, G. Zhang and Q. Peng, Adv. Mater., 2019, 31, 1901872 CrossRef PubMed.
- Q. Liu, Y. Jiang, K. Jin, J. Qin, J. Xu, W. Li, J. Xiong, J. Liu, Z. Xiao, K. Sun, S. Yang, X. Zhang and L. Ding, Sci. Bull., 2020, 65, 272–275 CrossRef CAS.
- S. Holliday, R. S. Ashraf, A. Wadsworth, D. Baran, S. A. Yousaf, C. B. Nielsen, C.-H. Tan, S. D. Dimitrov, Z. Shang, N. Gasparini, M. Alamoudi, F. Laquai, C. J. Brabec, A. Salleo, J. R. Durrant and I. McCulloch, Nat. Commun., 2016, 7, 11585 CrossRef CAS.
- G. Zhang, J. Zhao, P. C. Y. Chow, K. Jiang, Z. Zhu, J. Zhang, F. Huang and H. Yan, Chem. Rev., 2018, 118, 3447–3507 CrossRef CAS PubMed.
- L. Meng, Y. Zhang, X. Wan, C. Li, X. Zhang, Y. Wang, X. Xe, Z. Xiao, L. Ding, R. Xia, H. L. Yip, Y. Cao and Y. Chen, Science, 2018, 361, 1094–1098 CrossRef CAS PubMed.
- H. Huang, Q. Guo, S. Feng, C. Zhang, Z. Bi, W. Xue, J. Yang, J. Song, C. Li, X. Xu, Z. Tang, W. Ma and Z. Bo, Nat. Commun., 2019, 10, 3038 CrossRef.
- Z. P. Yu, Z. X. Liu, F. X. Chen, R. Qin, T. K. Lau, J. L. Yin, X. Kong, X. Lu, M. Shi, C. Z. Li and H. Chen, Nat. Commun., 2019, 10, 2152 CrossRef PubMed.
- X. Liu, Y. Wei, X. Zhang, L. Qin, Z. Wei and H. Huang, Sci. China: Chem., 2021, 64, 228–231 CrossRef CAS.
- Y. Cui, H. Yao, J. Zhang, K. Xian, T. Zhang, L. Hong, Y. Wang, Y. Xu, K. Ma, C. An, C. He, Z. Wei, F. Gao and J. Hou, Adv. Mater., 2020, 32, 1908205 CrossRef CAS PubMed.
- Z. Luo, R. Ma, T. Liu, J. Yu, Y. Xiao, R. Sun, G. Xie, J. Yuan, Y. Chen, K. Chen, G. Chai, H. Sun, J. Min, J. Zhang, Y. Zou, C. Yang, X. Lu, F. Gao and H. Yan, Joule, 2020, 4, 1236–1247 CrossRef CAS.
- L. Liu, Y. Kan, K. Gao, J. Wang, M. Zhao, H. Chen, C. Zhao, T. Jiu, A. K. Jen and Y. Li, Adv. Mater., 2020, 32, 1907604 CrossRef CAS PubMed.
- S. Liu, J. Yuan, W. Deng, M. Luo, Y. Xie, Q. Liang, Y. Zou, Z. He, H. Wu and Y. Cao, Nat. Photonics, 2020, 14, 300–305 CrossRef CAS.
- J. Zhao, C. Yao, M. U. Ali, J. Miao and H. Meng, Mater. Chem. Front., 2020, 4, 3487–3504 RSC.
- Z. Luo, T. Liu, R. Ma, Y. Xiao, L. Zhan, G. Zhang, H. Sun, F. Ni, G. Chai, J. Wang, C. Zhong, Y. Zou, X. Guo, X. Lu, H. Chen, H. Yan and C. Yang, Adv. Mater., 2020, 2005942 CrossRef CAS PubMed.
- X. Li, H. Huang, H. Bin, Z. Peng, C. Zhu, L. Xue, Z. G. Zhang, Z. Zhang, H. Ade and Y. Li, Chem. Mater., 2017, 29, 10130 CrossRef CAS.
- W. Zhao, S. Zhang, Y. Zhang, S. Li, X. Liu, C. He, Z. Zheng and J. Hou, Adv. Mater., 2018, 30, 1704837 CrossRef PubMed.
- C. An, Z. Zheng and J. Hou, Chem. Commun., 2020, 56, 4750–4760 RSC.
- J. You, L. Dou, Z. Hong, G. Li and Y. Yang, Prog. Polym. Sci., 2013, 38, 1909 CrossRef CAS.
- H. Fu, Z. Wang and Y. Sun, Angew. Chem., Int. Ed., 2019, 58, 4442–4453 CrossRef CAS PubMed.
- S. Li, L. Ye, W. Zhao, H. Yan, B. Yang, D. Liu, W. Li, H. Ade and J. Hou, J. Am. Chem. Soc., 2018, 140, 7159–7167 CrossRef CAS PubMed.
- S. Chen, Y. Liu, L. Zhang, P. C. Y. Chow, Z. Wang, G. Zhang, W. Ma and H. Yan, J. Am. Chem. Soc., 2017, 139, 6298–6301 CrossRef CAS PubMed.
- C. Xue, Y. Tang, S. Li, H. Feng, S. Li and D. Xia, New J. Chem., 2020, 44, 13100–13107 RSC.
- C. Sun, F. Pan, H. Bin, J. Zhang, L. Xue, B. Qiu, Z. Wei, Z. G. Zhang and Y. Li, Nat. Commun., 2018, 9, 743 CrossRef PubMed.
- H. Yao, Y. Cui, D. Qian, C. S. Ponseca Jr, A. Honarfar, Y. Xu, J. Xin, Z. Chen, L. Hong, B. Gao, R. Yu, Y. Zu, W. Ma, P. Chabera, T. Pullerits, A. Yartsev, F. Gao and J. Hou, J. Am. Chem. Soc., 2019, 141, 7743–7750 CrossRef CAS PubMed.
- H. Sun, T. Liu, J. Yu, T. K. Lau, G. Zhang, Y. Zhang, M. Su, Y. Tang, R. Ma, B. Liu, J. Liang, K. Feng, X. Lu, X. Guo, F. Gao and H. Yan, Energy Environ. Sci., 2019, 12, 3328–3337 RSC.
- B. He, Q. Yin, X. Yang, L. Liu, X.-F. Jiang, J. Zhang, F. Huang and Y. Cao, J. Mater. Chem. C, 2017, 5, 8774–8781 RSC.
- B. He, Q. Yin, B. Xie, J. Zhang, R. Xie, Z. Hu, X. Peng, F. Huang and Y. Cao, Polym. Chem., 2020, 11, 1653–1662 RSC.
- H. J. Yun, S. J. Kang, Y. Xu, S. O. Kim, Y. H. Kim, Y. Y. Noh and S. K. Kwon, Adv. Mater., 2014, 26, 7300–7307 CrossRef CAS PubMed.
- O. K. Kwon, J.-H. Park, S. K. Park and S. Y. Park, Adv. Energy Mater., 2015, 5, 1400929 CrossRef.
- N. S. Cho, D. Hwang, B. J. Jung, E. Lim, A. Jaemin Lee and H. K. Shim, Macromolecules, 2004, 37, 5265–5273 CrossRef CAS.
- J. M. Park, D. W. Kim, H. Y. Chung, J. E. Kwon, S. H. Hong, T. L. Choi and S. Y. Park, J. Mater. Chem. A, 2017, 5, 16681–16688 RSC.
- G. G. Malliaras, J. R. Salem, P. J. Brock and C. Scott, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 13411–13414 CrossRef.
- M. M. Mandoc, F. B. Kooistra, J. C. Hummelen, B. de Boer and P. W. M. Blom, Appl. Phys. Lett., 2007, 91, 263505 CrossRef.
- A. K. Kyaw, D. H. Wang, D. Wynands, J. Zhang, T. Q. Nguyen, G. C. Bazan and A. J. Heeger, Nano Lett., 2013, 13, 3796 CrossRef CAS PubMed.
- N. Qiu, H. Zhang, X. Wan, C. Li, X. Ke, H. Feng, B. Kan, H. Zhang, Q. Zhang, Y. Lu and Y. Chen, Adv. Mater., 2017, 29, 1604964 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra03233j |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Yulin Chena,
Jinglong Chenb,
Songxi Chena,
Manjun Xiao*b,
Guiting Chen
*a,
Yulin Chena,
Jinglong Chenb,
Songxi Chena,
Manjun Xiao*b,
Guiting Chen