
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of spiro[4.4]thiadiazole derivatives via double 1,3-dipolar cycloaddition of hydrazonyl chlorides with carbon disulfide†
Kai-Kai Wang *a,
Yan-Li Lib,
Dong-Guang Guoc,
Peng-Tao Panb,
Aili Sun*a and
Rongxiang Chen*a
*a,
Yan-Li Lib,
Dong-Guang Guoc,
Peng-Tao Panb,
Aili Sun*a and
Rongxiang Chen*a
aSchool of Pharmacy, Key Laboratory of Nano-carbon Modified Film Technology Engineering of Henan Province, Xinxiang 453000, P. R. China. E-mail: chenhlmei@163.com; sunailifly@126.com; Fax: +86-373-3682674
bMedical College, Xinxiang University, Xinxiang 453000, P. R. China
cSchool of Life Sciences and Basic Medicine, Xinxiang University, Xinxiang 453000, P. R. China
First published on 21st May 2021
Abstract
An operationally simple and convenient synthesis method toward a series of diverse spiro[4.4]thiadiazole derivatives via double [3 + 2] 1,3-dipolar cycloaddition of nitrilimines generated in situ from hydrazonyl chlorides with carbon disulfide has been achieved under mild reaction conditions.
The spirocyclic compounds having cyclic structures connected through just one carbon atom have attracted much interest from synthetic chemists and medicinal chemists because of their ubiquitous presence in the core of a plethora of natural products and non-natural products, many of which display a broad range of pharmacological and biological activities.1 Moreover, spiro-compounds are unique because of their rigidity and distinctly three-dimensional structure and have proved to be very interesting for medicinal chemistry or as ligand and catalyst motifs in asymmetric synthesis.2 Due to the importance of the spirocyclic architectures, the methods for synthesis of the spirocyclic moiety are too many to enumerate. Some common strategies to afford spirocyclic scaffolds include radical cyclizations,3 Diels–Alder reactions,4 cycloaddition,5 and ring expansion,6 among others. Many of the known methods for synthesizing spiro structures are through constructing a new ring of which the substrates include a carbo- or heterocycle structure.7 To the best of our knowledge, a few approaches have offered efficient ways on the formation of two rings through a double 1,3-dipolar cycloaddition in one pot for constructing spirocyclic scaffold.8 Therefore, the design and development of innovative and efficient methodologies via double 1,3-dipolar cycloaddition under mild reaction conditions for synthesizing bioactive content spirocyclic scaffolds from readily available precursors is in great demand in both organic and medicinal chemistry.
On the other hand, the 1,3-dipolar cycloaddition reaction (1,3-DCs) has been one of the most prominent reactions to build five- or six-membered heterocycle in one step in the field of organic synthesis.9 In particular, nitrilimines generated in situ from the corresponding hydrazonyl chloride in the presence of a base are highly active intermediates in organic synthesis and have been widely used as useful synthons for preparing bioactive nitrogen heterocyclic derivatives and spirocyclic compounds through the [3 + 2],10 [3 + 3]11 and [3 + 4]12 cycloaddition reactions. In addition, the Lu group reported 1,3-dipolar cycloaddition of nitrilimines with carbon dioxide (CO2), providing elegant access to 1,3,4-oxadiazole-2(3H)-ones derivatives.13 Meanwhile, carbon disulfide (CS2) that it is an analogue of CO2, has been used for the synthesis of various sulfur-containing heterocyclic compounds for agricultural, medicinal, and pharmaceutical applications.14 Based on the above literatures and in continuation of our interest in synthesis of heterocycles, we disclose a novel protocol for the synthesis of spiro[4.4]thiadiazole derivatives double 1,3-dipolar cycloaddition of nitrilimines generated in situ from hydrazonyl chlorides with carbon disulfide under mild conditions.
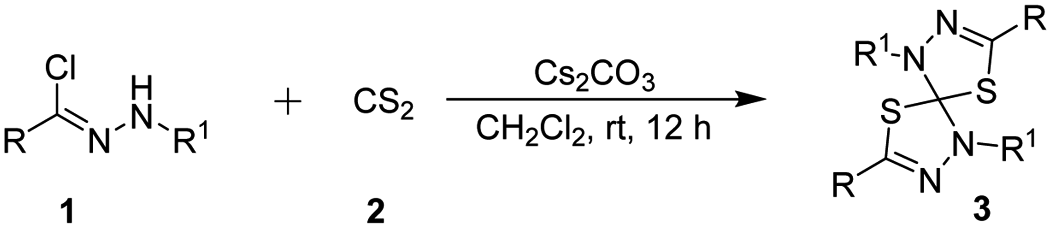
In our initial investigation, we chose the hydrazonyl chloride as the nitrilimine precursor with CS2 as the model reaction to optimize the reaction conditions. The results of these experiments are summarized in Table 1. No product was observed when the reaction was performed in the absence of base (Table 1, entry 1). Usually, the active nitrilimine is generated in situ by dehydrohalogenation of the corresponding hydrazonyl halide in the presence of an equivalent base. Pleasingly, with the use of TEA as the base, the double 1,3-dipolar cycloaddition reaction proceeded very smoothly in CH2Cl2 to give spirothiadiazole product 3a with 60% yield at room temperature (Table 1, entry 2). The product 3a was obtained with 56% yield in the presence of DABCO (entry 3). However, the DBU base turned out to be inactive in the double 1,3-dipolar cycloaddition reaction (entry 4). Subsequently, we screened a series of inorganic base to improve the yield, such as Na2CO3, K2CO3, Cs2CO3, NaOH and KOH (entries 5–9). In comparison, the product 3a was gave in moderate yields in the presence of TEA, Na2CO3 or K2CO3 (entries 2, 5 and 6). To our delight, the yield of the product 3a was increased to 92% when used Cs2CO3 as the base (entry 7). Nevertheless, the yield was slightly reduced with the use of NaOH and KOH as the base (entries 8 and 9). Encouraged by this result, the reaction was examined with different solvents to improve the yield of 3a. The reaction can be carried out in CHCl3 and DCE to provide the expected product in 80% yield and 85% yield, respectively (entries 10 and 11). Whereas no the desired product formation take place upon using THF and Et2O as the solvent (entries 14 and 15). The reactions run with either dioxane or MeCN resulted in moderate conversions (entries 16 and 17). It was found that CH2Cl2 was the best solvent for this transformation compared to CHCl3, DCE, EtOAc, toluene, THF, Et2O, dioxane and MeCN (entries 10–17). Moreover, when decreased the amounts of CS2, the reaction time was prolonged and the yield of the product was lowered (entry 18). Whereas, the yield was not further improved when using 5.0 equiv. of CS2 (entry 19). Therefore, the optimal reaction conditions have been determined which using Cs2CO3 as the base in CH2Cl2 at room temperature for 12 h (Table 2).
|
|||
|---|---|---|---|
| Entry | Base | Solvent | Yield of 3ab (%) |
| a Unless noted otherwise, reactions were performed with hydrazonyl chloride 1a (0.2 mmol), carbon disulfide (0.3 mmol, 1.5 equiv.), base (0.2 mmol, 1 equiv.) in solvent (1.0 mL) at rt for 12 h.b Isolated yield by chromatography on silica gel.c Reaction was performed with carbon disulfide (0.2 mmol, 1 equiv.) for 24 h.d Carbon disulfide (1.0 mmol, 5 equiv.). | |||
| 1 | None | CH2Cl2 | 0 |
| 2 | TEA | CH2Cl2 | 60 |
| 3 | DABCO | CH2Cl2 | 56 |
| 4b | DBU | CH2Cl2 | Trace |
| 5 | Na2CO3 | CH2Cl2 | 62 |
| 6 | K2CO3 | CH2Cl2 | 70 |
| 7 | Cs2CO3 | CH2Cl2 | 92 |
| 8 | NaOH | CH2Cl2 | 90 |
| 9 | KOH | CH2Cl2 | 88 |
| 10 | Cs2CO3 | CHCl3 | 80 |
| 11 | Cs2CO3 | DCE | 85 |
| 12 | Cs2CO3 | EtOAc | 70 |
| 13 | Cs2CO3 | Toluene | 61 |
| 14 | Cs2CO3 | THF | Trace |
| 15 | Cs2CO3 | Et2O | Trace |
| 16 | Cs2CO3 | Dioxane | 56 |
| 17 | Cs2CO3 | MeCN | 62 |
| 18c | Cs2CO3 | CH2Cl2 | 80 |
| 19d | Cs2CO3 | CH2Cl2 | 92 |
|
|---|
| a Unless noted otherwise, reactions were performed with hydrazonyl chloride 1 (0.2 mmol), carbon disulfide (0.3 mmol, 1.5 equiv.), base (0.2 mmol, 1 equiv.) in solvent (1.0 mL) at rt for 12 h. Isolated yield by chromatography on silica gel. |
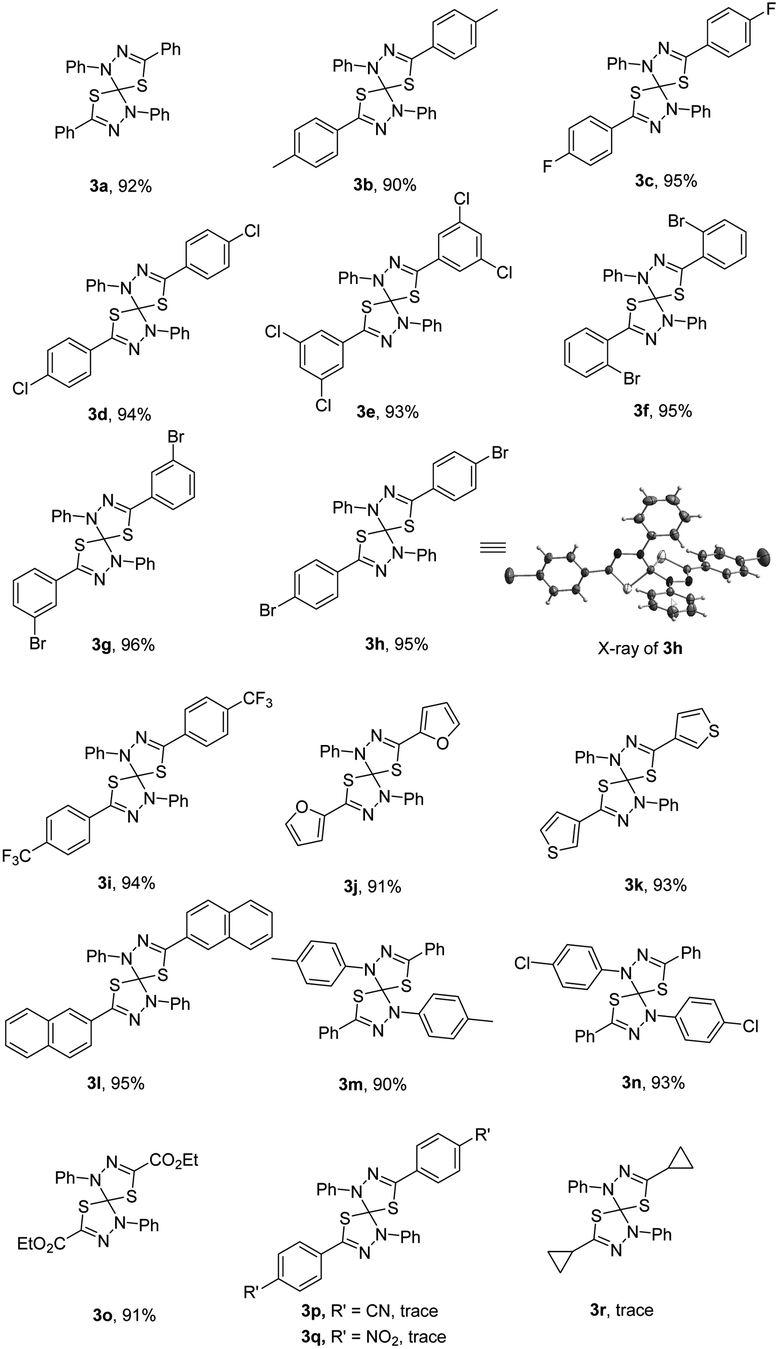 |
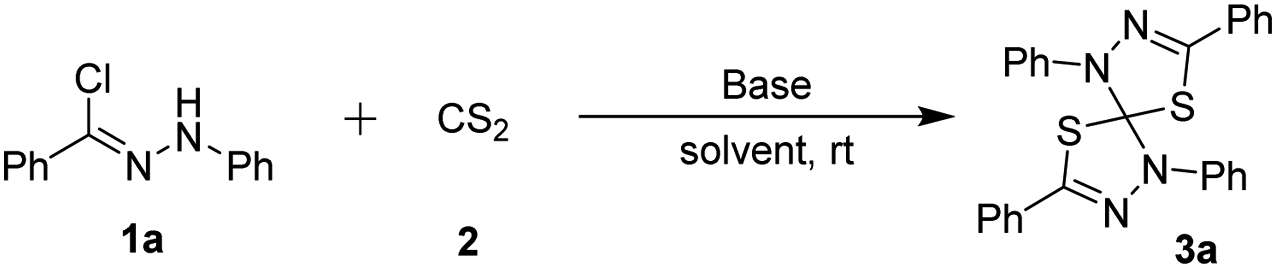
With the optimal reaction conditions in hand, we subsequently investigated the substrate scope and limitation of the nitrilimine precursors. The results are shown in Table 1. Under the optimized conditions, the double 1,3-dipolar cycloaddition reaction could tolerate a variety of hydrazonyl chlorides bearing different substituents, regardless of the electronic properties (such as electron-donating or electron-withdrawing) or the positions of substituents on the benzoyl chloride moiety, to generate the corresponding products 3 (3a–i) in high yields (92–96%). In addition, the structure of product 3h (CCDC 2041891)15 was further determined by single-crystal X-ray crystallography analysis. Furthermore, when using the fused aromatic and heteroaromatic hydrazonyl chlorides as the substrate reacted with CS2, the reactions were also found to be compatible and gave the products (3j–l) in high yields. On the other hand, the hydrazonyl chlorides containing different substituents (such as methyl and chloro) on phenylhydrazone moiety also worked well in the reaction successfully to obtain the desired cycloadducts 3m and 3n in 90% and 93% yield, respectively. Nevertheless, the hydrazonyl chlorides bearing cyano or nitro group on the benzoyl chloride moiety were not suitable and the expected cycloadduct 3p and 3q were not formed. Moreover, the double 1,3-dipolar cycloaddition reaction also didn't work with aliphatic group (3r) at the hydrazonyl chloride.
To further exhibit the synthetic utility for spirocyclic compounds, under the optimized conditions, a gram scale experiment between 4 mmol of hydrazonyl chloride 1a and 6 mmol of CS2 proceeded smoothly to afford the desired product 3a without a significant loss of efficiency (1.670 g, in 90% yield) (Scheme 1). The easy scale-up of this process shows the reaction to be a practical tool for the synthesis of structurally diversified natural product-like molecules possessing privileged scaffold for potential application in biomedical research and other research fields.
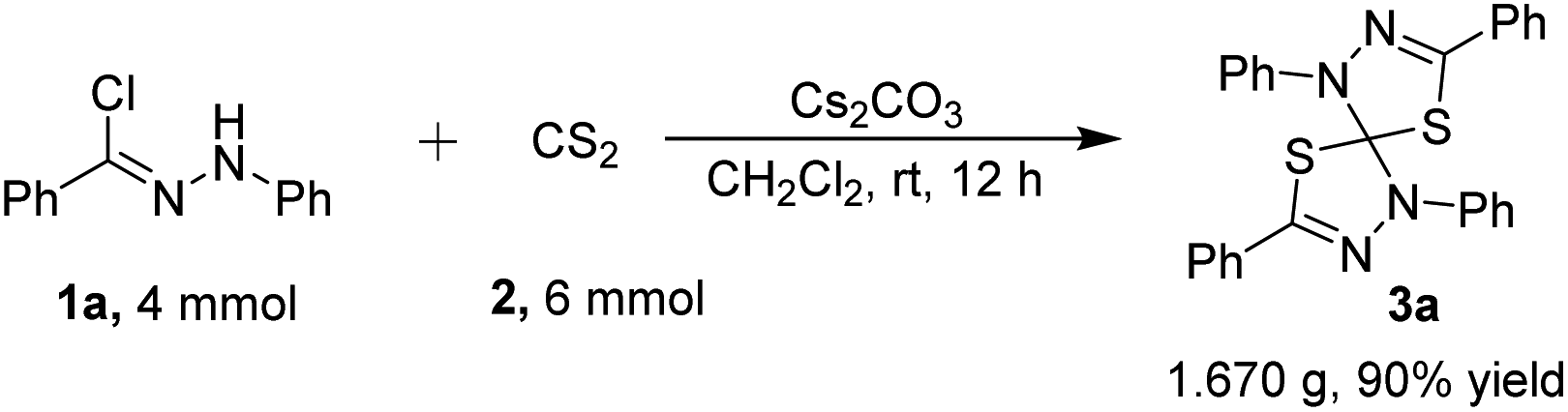 | ||
| Scheme 1 Scaled-up version of synthesis of 3a. | ||
As shown in Scheme 2, a plausible mechanism was proposed. Firstly, the nitrilimine intermediate 4 generated in situ from the corresponding hydrazonyl halide 1 via eliminating of HCl in the presence of a base. Then, the nitrilimine 4 reacts with CS2 through the double 1,3-dipolar cycloaddition reaction to give the desired product 3.
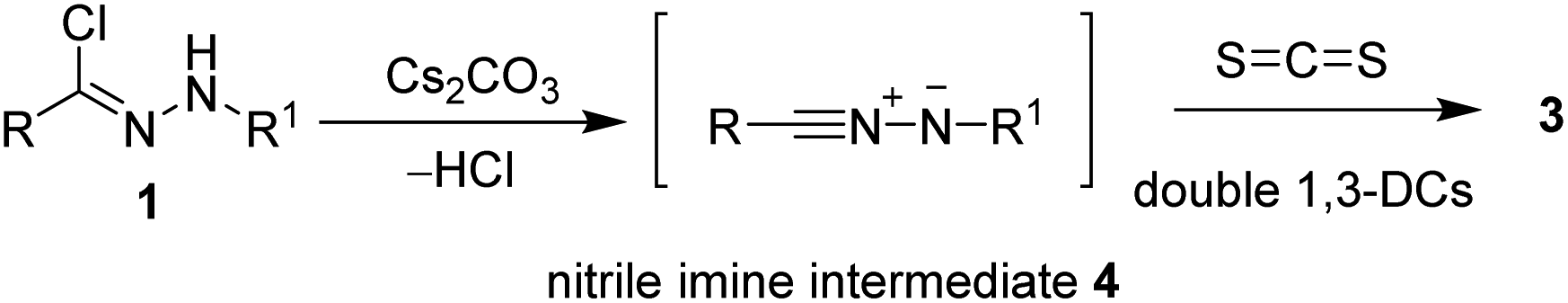 | ||
| Scheme 2 Proposed mechanism of the double [3 + 2] cycloaddition. | ||
In conclusion, we have developed an efficient and simple method to synthesize a broad range of diverse spiro[4.4]thiadiazole derivatives in high yields (up to 96%) through the double 1,3-dipolar cycloaddition of nitrilimines generated in situ with CS2. This reaction proceeds with readily available starting materials, transition-metal free, the experimental simplicity, easy purification, and mild reaction conditions make this procedure highly appropriate for the synthesis of spiro[4.4]thiadiazole derivatives.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
We are grateful for the financial support from the National Natural Science Foundation of China (No. 21801214 and 81702074), Key Scientific Research Project of Colleges and Universities in Henan Province of China (No. 18A150014, 19A230009 and 20B150019), Key Project of Science and Technology of Henan Province (No. 202102310298 and 192102110066), the Natural Science Foundation of Henan Province (No. 202300410016).Notes and references
- (a) Y.-S. Chong, S. Anwar and K. Chen, Mini-Rev. Org. Chem., 2018, 15, 364 CrossRef; (b) D. Jo and S. Han, Chem. Commun., 2018, 54, 6750 RSC; (c) A. Quintavalla, Curr. Med. Chem., 2018, 25, 917 CrossRef PubMed; (d) E. Chupakhin, O. Babich, A. Prosekov, L. Asyakina and M. Krasavin, Molecules, 2019, 24, 4165 CrossRef CAS PubMed; (e) S. Mandal and B. Thirupathi, Org. Biomol. Chem., 2020, 18, 5287 RSC.
- For selected examples, see: (a) K. Ding, Z. Han and Z. Wang, Chem.–Asian J., 2009, 4, 32 CrossRef CAS PubMed; (b) X.-Y. Ye, Z.-Q. Liang, C. Jin, Q.-W. Lang, G.-Q. Chen and X. Zhang, Chem. Commun., 2021, 57, 195 RSC; (c) W. Guo, Q. Liu, J. Jiang and J. Wang, Org. Lett., 2020, 22, 3110 CrossRef CAS PubMed; (d) X. Xie, W. Huang, C. Peng and B. Han, Adv. Synth. Catal., 2018, 360, 194 CrossRef CAS.
- (a) J. Sperry, Y.-C. Liu and M. A. Brimble, Org. Biomol. Chem., 2010, 8, 29 RSC; (b) R. Pawlowski, P. Skorka and M. Stodulski, Adv. Synth. Catal., 2020, 362, 4462 CrossRef CAS.
- M. A. Rizzacasa and A. Pollex, Org. Biomol. Chem., 2009, 7, 1053 RSC.
- (a) A. Natarajan, K. Raju Suresh, I. A. Abdulrahman and P. Subbu, Curr. Org. Chem., 2013, 17, 1929 CrossRef; (b) G. P. Savage, Curr. Org. Chem., 2010, 14, 1478 CrossRef CAS.
- T. Sarkar, B. K. Das, K. Talukdar, T. A. Shah and T. Punniyamurthy, ACS Omega, 2020, 5, 26316 CrossRef CAS PubMed.
- (a) J. Bariwal, L. G. Voskressensky and E. V. Van der Eycken, Chem. Soc. Rev., 2018, 47, 3831 RSC; (b) K. P. Melnykov and S. V. Ryabukhin, Chem. Heterocycl. Compd., 2020, 56, 1411 CrossRef.
- (a) H. Liu, H. Jia, B. Wang, Y. Xiao and H. Guo, Org. Lett., 2017, 19, 4714 CrossRef CAS PubMed; (b) M. A. Arai, T. Arai and H. Sasai, Org. Lett., 1999, 1, 1795 CrossRef CAS.
- (a) R. Narayan, M. Potowski, Z.-J. Jia, A. P. Antonchick and H. Wadmann, Acc. Chem. Res., 2014, 47, 1296 CrossRef CAS PubMed; (b) C. Najera, J. M. Sansano and M. Yus, Org. Biomol. Chem., 2015, 13, 8596 RSC; (c) K. Martina, S. Tagliapietra, V. V. Veselov and G. Cravotto, Front. Chem., 2019, 7, 1 CrossRef PubMed; (d) T. Hashimoto and K. Maruoka, Chem. Rev., 2015, 115, 5366 CrossRef CAS PubMed; (e) N. Thanh Binh, A. Martel, C. Gaulon-Nourry, R. Dhal and G. Dujardin, Org. Prep. Proced. Int., 2012, 44, 1 CrossRef; (f) C. Najera and J. M. Sansano, Org. Biomol. Chem., 2009, 7, 4567 RSC.
- (a) Y. Zeng, L. Zhou, X. Gao, Y. Wu, Q. Wang, Z. Guo, J. Huang and H. Guo, J. Heterocycl. Chem., 2018, 55, 2781 CrossRef CAS; (b) I. Yavari, Z. Taheri, M. Naeimabadi, S. Bahemmat and M. R. Halvagar, Synlett, 2018, 29, 918 CrossRef CAS; (c) G. Wang, X. Liu, T. Huang, Y. Kuang, L. Lin and X. Feng, Org. Lett., 2013, 15, 76 CrossRef CAS PubMed; (d) V. V. Voronin, M. S. Ledovskaya, E. G. Gordeev, K. S. Rodygin and V. P. Ananikov, J. Org. Chem., 2018, 83, 3819 CrossRef CAS PubMed; (e) Y. Su, Y. Zhao, B. Chang, X. Zhao, R. Zhang, X. Liu, D. Huang, K.-H. Wang, C. Huo and Y. Hu, J. Org. Chem., 2019, 84, 6719 CrossRef CAS PubMed; (f) H. Gazzeh, S. Boudriga, M. Askri, A. Khatyr, M. Knorr, C. Strohmann, C. Golz, Y. Rousselin and M. M. Kubicki, RSC Adv., 2016, 6, 49868 RSC; (g) A. Alizadeh, L. Moafi and L.-G. Zhu, Synlett, 2016, 27, 595 CrossRef CAS; (h) A. Alizadeh and L. Moafi, Helv. Chim. Acta, 2016, 99, 457 CrossRef CAS; (i) A. Alizadeh and A. Roosta, Synlett, 2016, 27, 2455 CrossRef CAS; (j) S.-E. Tsai, W.-P. Yen, Y.-T. Li, Y.-T. Hu, C.-C. Tseng and F. F. Wong, Asian J. Org. Chem., 2017, 6, 1470 CrossRef CAS; (k) M. P. Sibi, L. M. Stanley and T. Soeta, Adv. Synth. Catal., 2006, 348, 2371 CrossRef CAS; (l) F. Rouatbi, C. Mhiri, M. Askri, M. Knorr, Y. Rousselin and M. M. Kubicki, J. Heterocycl. Chem., 2017, 54, 1152 CrossRef CAS; (m) S. S. Mykhaylychenko, N. V. Pikun, E. B. Rusanov, A. B. Rozhenko and Y. G. Shermolovich, Chem. Heterocycl. Compd., 2017, 53, 1268 CrossRef CAS; (n) R. S. Jasass, F. Alshehrei and T. A. Farghaly, J. Heterocycl. Chem., 2018, 55, 2099 CrossRef CAS; (o) T. A. Farghaly, S. M. Gomha, E. K. Mousa and M. Elaasser, J. Chem. Res., 2016, 467 CrossRef CAS; (p) Ref. 8a.
- H.-W. Zhao, Y.-D. Zhao, Y.-Y. Liu, L.-J. Zhao, X.-Q. Song, X.-Q. Chen, H.-L. Pang, J. Du and N.-N. Feng, RSC Adv., 2017, 7, 55106 RSC.
- (a) W. Long, S. Chen, X. Zhang, L. Fang and Z. Wang, Tetrahedron, 2018, 74, 6155 CrossRef CAS; (b) M. G. Badrey, S. M. Gomha, W. A. A. Arafa and M. M. Abdulla, J. Heterocycl. Chem., 2017, 54, 1215 CrossRef CAS.
- C.-X. Guo, W.-Z. Zhang, N. Zhang and X.-B. Lu, J. Org. Chem., 2017, 82, 7637 CrossRef CAS.
- (a) L. Zhang, B. Sun, Q. Liu and F. Mo, J. Org. Chem., 2018, 83, 4275 CrossRef CAS PubMed; (b) T. Aghaalizadeh and F. Nasiri, J. Sulfur Chem., 2017, 38, 635 CrossRef CAS; (c) S. Ghrab, L. Aroua and M. Beji, J. Sulfur Chem., 2016, 37, 580 CrossRef CAS; (d) A. Samzadeh-Kermani, J. Sulfur Chem., 2019, 40, 554 CrossRef CAS; (e) N. Aoyagi and T. Endo, Synlett, 2020, 31, 92 CrossRef CAS; (f) B. K. Min, G. Kim, H. J. Roh, D. Y. Seo and J. N. Kim, Tetrahedron Lett., 2018, 59, 1674 CrossRef CAS; (g) N. R. Mohamed and M. Younis, Egypt. J. Chem., 2006, 49, 671 CAS; (h) M. El-Saidi, A. A. El-Sayed, E. B. Pedersen, M. A. Tantawy and W. A. Gad, Indones. J. Chem., 2020, 20, 1163 CrossRef CAS; (i) A. O. Abdelhamid, M. Sallam and S. A. Amer, Heteroat. Chem., 2001, 12, 468 CrossRef CAS.
- CCDC 2041891 for 3h contains the ESI crystallographic data for this paper..
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, structural proof, CIF file of 3h. CCDC 2041891. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra03229a |
| This journal is © The Royal Society of Chemistry 2021 |