
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTobacco mosaic virus for the targeted delivery of drugs to cells expressing prostate-specific membrane antigen†
Sourabh Shukla‡
 a,
Isaac Marks‡a,
Derek Churcha,
Soo-Khim Chana,
Jonathan K. Pokorskiaef and
Nicole F. Steinmetz*abcdef
a,
Isaac Marks‡a,
Derek Churcha,
Soo-Khim Chana,
Jonathan K. Pokorskiaef and
Nicole F. Steinmetz*abcdef
aDepartment of NanoEngineering, University of California San Diego, La Jolla, CA 92093, USA. E-mail: nsteinmetz@ucsd.edu
bDepartment of Bioengineering, University of California San Diego, La Jolla, CA 92093, USA
cDepartment of Radiology, University of California San Diego, La Jolla, CA 92093, USA
dMoores Cancer Center, University of California San Diego, La Jolla, CA 92093, USA
eCenter for Nano-ImmunoEngineering, University of California San Diego, La Jolla, CA 92093, USA
fInstitute for Materials Discovery and Design, University of California San Diego, La Jolla, CA 92093, USA
First published on 6th June 2021
Abstract
Prostate-specific membrane antigen (PSMA) is a membrane-bound protein that is preferentially expressed in the prostate gland and induced in many prostate cancers, making it an important target for new diagnostics and therapeutics. To improve the efficacy of nanoparticle formulations for the imaging and/or eradication of prostate cancer, we synthesized the PSMA-binding glutamic acid derivative DUPA and conjugated it to the external surface of tobacco mosaic virus (TMV) particles. DUPA-targeted TMV was subsequently loaded with the antineoplastic agent mitoxantrone (MTO) or conjugated internally with the fluorescent dye cyanine 5 (Cy5). We found that TMV particles could be efficiently decorated with DUPA and loaded with MTO or Cy5 while maintaining structural integrity. DUPA-targeted TMV particles were able to bind more efficiently to the surface of PSMA+ LNCaP cells compared to non-targeted TMV; but there was little difference in binding efficiency between targeted and untargeted TMV when we tested PSMA− PC3 cells (both cell lines are prostate cancer cell lines). DUPA-targeted TMV particles were internalized by LNCaP cells enabling drug delivery. Finally, we loaded the DUPA-targeted TMV particles and untargeted control particles with MTO to test their cytotoxicity against LNCaP cells in vitro. The cytotoxicity of the TMV-MTO particles (IC50 = 10.2 nM) did not differ significantly from that of soluble MTO at an equivalent dose (IC50 = 12.5 nM) but the targeted particles (TMV-DUPA-MTO) were much more potent (IC50 = 2.80 nM). The threefold increase in cytotoxicity conferred by the DUPA ligand suggests that MTO-loaded, DUPA-coated TMV particles are promising as a therapeutic strategy for PSMA+ prostate cancer and should be advanced to preclinical testing in mouse models of prostate cancer.
Introduction
Prostate cancer is the second most commonly diagnosed cancer in males worldwide, and the most commonly diagnosed cancer in males in the USA.1,2 The incidence of this disease is increasing, particularly in developed countries.3 Androgen deprivation therapy is the standard of care to slow the progression of the disease, but more targeted approaches are required to eradicate later-stage castration-resistant prostate cancer and metastatic prostate cancer.4 Several promising targets have been identified, including prostate-specific membrane antigen (PSMA), otherwise known as glutamate carboxypeptidase II, N-acetyl-L-aspartyl-L-glutamate peptidase I or folate hydrolase 1.5 PSMA is approximately 100-fold more abundant in the human prostate gland than other tissues, and is induced a further 10-fold or more in many prostate cancers6 with higher expression levels indicating worse prognosis.7 Reagents that target PSMA can therefore be used for diagnostic prognostic or longitudinal imaging of disease progression and response to treatment. For example, 68Ga-labeled small-molecule reagents that bind PSMA can be detected by positron emission tomography and computed tomography (PET/CT)8,9 and the first such reagent was approved by the FDA in December 2020.10 Clinical trials are also underway using PSMA-binding molecules conjugated to radiotherapy sources such as 177Lu allowing the development of novel therapeutic and theranostic approaches.11,12The delivery of imaging reagents and drugs can be facilitated by nanoparticles that encapsulate the active ingredient to enhance its pharmacology profiles through targeted delivery to tumors via passive effects or active targeting strategies.13 The development pipeline of nanoparticle platform technologies continues to grow and many advances have been made using inorganic nanoparticles,14 synthetic or natural polymers15 lipid-based nanoparticles,16 as well a viral vectors and viral nanoparticles.17
Our work has focused on viral nanoparticles, specifically those based on plant viruses, which offer the benefits of biocompatibility, biodegradability, an inability to infect humans, and the ability both to carry drug/imaging reagent payloads within an internal cavity and to display targeting molecules on the external surface.18 For example, tobacco mosaic virus (TMV) naturally exists as a 300 × 18 nm hollow nanorod with an internal channel 4 nm wide. While native TMV exists as a hollow nanorod, programmed self-assembly allows the production of longer or shorter tubes, spherical particles, stellate bio-inorganic hybrid colloids and falcate structures.19 The internal and external surface chemistries of TMV are distinct, allowing the placement of contrast agents or drugs in the channel and targeting ligands on the exterior.20 In previous work we have demonstrated utility of TMV as a contrast agent for optical and magnetic resonance imaging applications21,22 and drug delivery.23,24 Here we built on these design concept and developed a PSMA-targeted TMV to enhance drug delivery targeting prostate cancer.
Specifically, we made use of T158K mutant of TMV,25 which presents 2130 reactive lysine residues on the external surface and 4260 glutamic acid residues lining the internal channel. This allows the primary amines of external lysine residues to be used for N-hydoxysuccinimide (NHS)-mediated bioconjugation while the carboxylate groups of internal glutamic acid can be addressed via 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) crosslinking or the native glutamic acid chains can be used to define a negatively charged cavity for the confinement of a positively charged molecular cargo. Using a two-step bioconjugation protocol (installation of an azide by conjugation of an NHS reactive linker to the surface lysines, followed by conjugation of an alkyne-terminated targeting ligand using copper-free click chemistry), we conjugated and displayed (((S)-5-amino-1-carboxypentyl)carbamoyl)-L-glutamic acid (DUPA), a small targeting ligand that binds specifically to PSMA.26 The TMV channel was covalently labeled with the near-infrared fluorophore Cy5 to enable imaging and tracking of the nanoparticles or loaded with the antineoplastic agent mitoxantrone (MTO), a type II topoisomerase inhibitor.27 We tested the targeting specificity and the efficiency of drug delivery in vitro using PSMA+ and PSMA− prostate cancer cells.
Materials and methods
Synthesis of the PSMA-targeting small molecule DUPA and its derivative DBCO-PEG4-DUPA
Glassware was flame-dried, cooled under vacuum and backfilled with nitrogen before use. DUPA was synthesized in two steps (Fig. 1) via the intermediate tri-tert-butyl (10S,14S)-2,2-dimethyl-4,12-dioxo-3-oxa-5,11,13-triazahexadecane-10,14,16-tricarboxylate (1). First, we mixed 2.5 g L-glutamate di-tert-butyl ester hydrochloride (8.45 mmol, 1.0 eq.) with 100 mL dichloromethane and cooled the reaction to −65 °C. We then added 827 mg triphosgene (2.79 mmol, 0.33 eq.) and 2.5 mL triethylamine (17.79 mmol, 2.1 eq.) and increased the reaction temperature to 10 °C for 5 h before cooling again to −65 °C. Finally, we added 2.98 g N-boc-L-lysine tert-butyl ester hydrochloride (9.04 mmol, 1.07 eq.) and 1.5 mL triethylamine (10.67 mmol, 1.26 eq.) and warmed the reaction mixture to room temperature over 18 h. The reaction was then quenched with 40 mL 1 M HCl. The biphasic solution was transferred to a separatory funnel and the aqueous layer was discarded. The organic layer was sequentially washed with water (50 mL) and saturated NaCl solution (50 mL) before drying over Na2SO4. The crude intermediate (1) was purified by flash chromatography using 50 μm silica gel (Luknova, Mansfield, MA, USA) and eluted in a gradient of 20–30% ethyl acetate in hexanol under nitrogen pressure.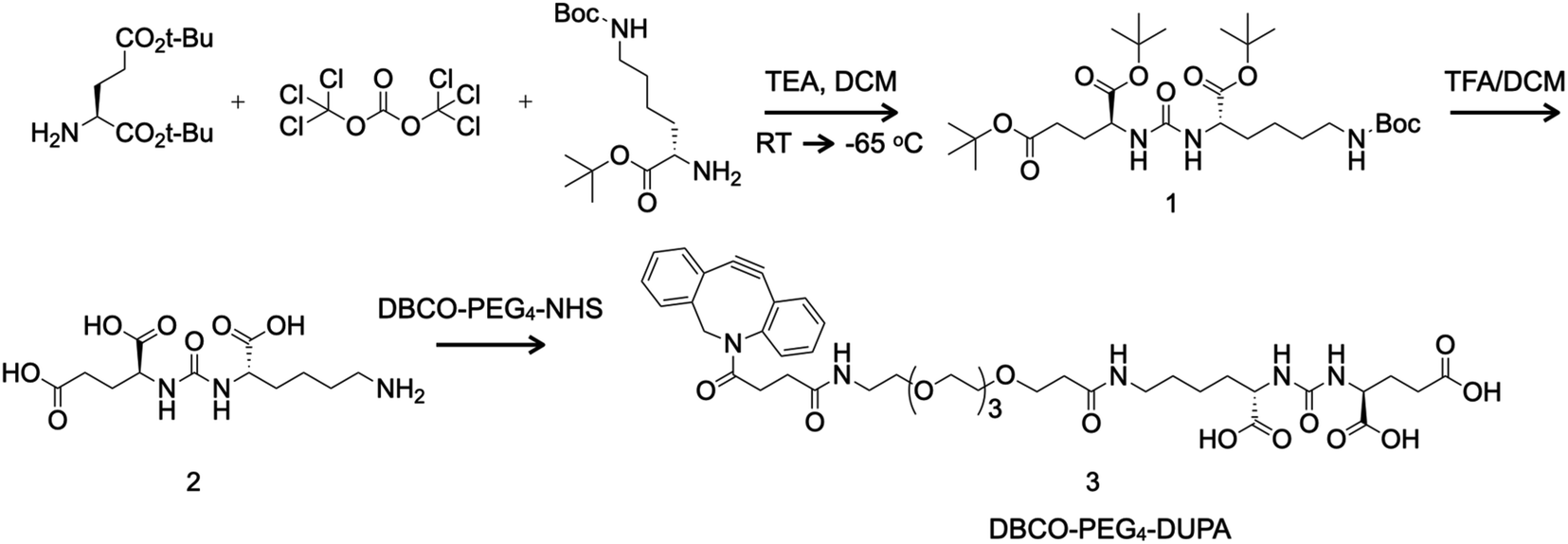 | ||
| Fig. 1 Synthesis of the PSMA-targeting small molecule DUPA (2) via a boc-protected amine intermediate (1) and derivation of the linker DBCO-PEG4-DUPA (3) for bioconjugation mediated by click chemistry. DCM = dichloromethane; TEA = triethylamine; TFA = trifluoroacetic acid; RT = room temperature. | ||
Next we mixed 87.1 mg (0.148 mmol) of the boc-protected intermediate (1) with 2 mL dichloromethane and 0.4 mL trifluoroacetic acid and stirred for 4 h at room temperature before removing liquids under reduced pressure. Without further purification, we mixed 14.7 mg (0.046 mmol, 1.2 eq.) of the product DUPA (2) with 1 mL dimethylsulfoxide and 0.034 mL N,N-diisopropylethylamine (0.20 mmol, 5 eq.) for 5 min before adding 12.5 mg (0.039 mmol, 1 eq.) DBCO-PEG4-NH2 (Click Chemistry Tools, Scottsdale, AZ, USA). The reaction was allowed to proceed for 2 h and the product DBCO-PEG4-DUPA (3) was purified by reversed-phase HPLC on a Shimadzu Prominence system using an Agilent Technologies (Santa Clara, CA, USA) Eclipse XDB-C18 column and recovered as a white solid (14.3 mg, 43% yield).
The boc-protected intermediate (1) was analyzed by proton nuclear magnetic resonance (1H NMR) spectroscopy in CDCl3 on a 300 MHz device (Bruker, Billerica, MA, USA). Analysis by 1H NMR yielded the following data: δ 5.15 (s, 2H), 4.75 (s, 1H), 4.34 (s, 2H), 3.09 (s, 2H), 2.30 (m, 3H), 2.16–1.29 (m, 43H). Exact mass [M] calculated for C29H53N3O9 was 587.38 and protonated mass [M + H]+ was 588.42 (Fig. S1†). The intermediate (1) and final product (3) were analyzed by electrospray ionization mass spectrometry (ESI-MS) using a Micromass Quattro Ultima triple quadrupole mass spectrometer (Waters, Milford, MA, USA). Exact mass [M] calculated for C42H55N5O14 was 853.37; and [M + H]+ was 854.26 (Fig. S2†).
Preparation of TMV particles, conjugation and drug loading
The T158K mutant TMV-lys (described as TMV for simplicity) was propagated in Nicotiana benthamiana plants by mechanical inoculation and purified as previously described.25 TMV-Cy5 particles were prepared by conjugating cyanine 5 (Cy5) to the internal glutamic acid residues using a combination of carbodiimide coupling and click chemistry.28 Internal alkyne handles were introduced by mixing 2 mg mL−1 TMV with 0.1 M propargylamine (120 eq. per coat protein), 0.1 M hydroxybenzotriazole and 0.1 M EDC (30 eq. per coat protein in three steps, t = 0, t = 6 h and t = 18 h, making 90 eq. per coat protein in total) in 0.1 M 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) pH 7.4. After incubation at room temperature for 24 h, the modified TMV particles were purified by centrifugation at 112![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × g for 1 h on a 40% (w/v) sucrose cushion. Click chemistry was then used to conjugate sulfo-Cy5-azide (Lumiprobe, Hunt Valley, MD, USA) at 5 eq. per coat protein to the re-dispersed TMV-alkyne in the presence of 2 mM aminoguanidine, 2 mM ascorbic acid and 1 mM copper sulfate in ice-cold 10 mM KP buffer (pH 7.4). After 30 min, the reaction was stopped by adding EDTA to a final concentration of 5 mM. The TMV-Cy5 particles were purified by ultracentrifugation as described above.
000 × g for 1 h on a 40% (w/v) sucrose cushion. Click chemistry was then used to conjugate sulfo-Cy5-azide (Lumiprobe, Hunt Valley, MD, USA) at 5 eq. per coat protein to the re-dispersed TMV-alkyne in the presence of 2 mM aminoguanidine, 2 mM ascorbic acid and 1 mM copper sulfate in ice-cold 10 mM KP buffer (pH 7.4). After 30 min, the reaction was stopped by adding EDTA to a final concentration of 5 mM. The TMV-Cy5 particles were purified by ultracentrifugation as described above.
DBCO-PEG4-DUPA was conjugated to the external lysine residues of TMV or TMV-Cy5 by mixing the particles with a 20000 molar excess of NHS-PEG4-N3 linker overnight at room temperature in 10 mM KP buffer (pH 7.4). Excess unreacted NHS-PEG4-N3 was removed using 100 kDa cutoff spin filters and DBCO-PEG4-DUPA (10000 eq. per coat protein) was added to the TMV-N3 or TMV-Cy5-N3 particles overnight to yield DUPA-TMV and DUPA-TMV-Cy5 particles, respectively. For drug loading, we mixed TMV or DUPA-TMV particles (1 mg mL−1) with a 10000-fold molar excess of MTO (Millipore-Sigma, Burlington, MA, USA) in 10 mM KP buffer (pH 7.4) containing 10% DMSO for 18 h at 4 °C. The particles were purified by ultracentrifugation as above and the pellet was resuspended in 10 mM KP buffer (pH 7.4).
Characterization of TMV nanoparticles
The particles were characterized by transmission electron microscopy (TEM), size exclusion chromatography (SEC), SDS-PAGE, and UV-vis spectroscopy. For TEM, particle samples (0.5 mg mL−1) were mounted on Formvar-coated 400-mesh copper grids and stained with 2% (w/v) uranyl acetate prior to analysis on a Tecnai Spirit G2 BioTWIN microscope (FEI, Hillsboro, OR, USA). For SEC analysis, particles were loaded onto a Superose6 column mounted on the ÄKTA Explorer system (GE Healthcare, Marlborough, MA, USA). The external and internal modification of the TMV particles was confirmed by SDS-PAGE using 12% NuPAGE gels in 1× MOPS running buffer (Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA) followed by densitometry with ImageJ software (https://imagej.net/Fiji). UV-vis spectroscopy was used to determine TMV concentration as well as Cy5 or MTO loading using the extinction coefficients ε260 nm = ∼3.0 mL mg−1 cm−1 (TMV), ε647 nm = 271000 M−1 cm−1 (sulfo-Cy5) and ε622 nm = 25000 M−1 cm−1 (MTO). MTO also absorbs at 260 nm and therefore, the MTO contribution at 260 nm was subtracted when determining the TMV concentration.
Cell binding, uptake and cytotoxicity assays
Human prostate cancer cell line LNCaP (ATCC, Manassas, VA, USA) was cultured in RPMI-1640 medium supplemented with 10% (v/v) fetal bovine serum (FBS) and 1% (w/v) penicillin and streptomycin (all reagents from Thermo Fisher Scientific) at 37 °C in a 5% CO2 atmosphere. The expression of PSMA was confirmed by incubating 100000 LNCaP cells per well in 100 μL of the above medium with an AlexaFluor 488-conjugated mouse anti-human PSMA antibody (Invitrogen, Thermo Fisher Scientific; clone GCP-05) on ice for 1 h. Antibody binding to the cell surface was then measured by flow cytometry using an Accuri C6 Plus device (BD Biosciences, Franklin Lakes, NJ, USA). TMV-Cy5 and DUPA-TMV-Cy5 particles were used for cell binding and uptake assays. Briefly, 50000 LNCaP cells were incubated with 2 μg of TMV-Cy5 particles (with and without the targeting ligand) for 2 h at 4 °C (for cell binding) or for 24 h at 37 °C (for cell uptake). The cells were then washed twice with PBS containing 3% (v/v) FBS and analyzed by flow cytometry as above. Data were analyzed using FlowJo v10.6.1. The comparative cytotoxicity of MTO-loaded TMV particles (with and without the targeting ligand) was determined using the substrate 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide in MTT assays (R&D Systems, Minneapolis, MN, USA). Briefly, 15000 LNCaP cells per well were cultured overnight in 96-well plates and exposed for 24 h to 10-fold incremental concentrations of soluble MTO (100 pM to 10 μM) or equivalent doses of TMV-MTO and DUPA-TMV-MTO. Cytotoxicity was then determined by measuring the conversion of MTT its insoluble formazan derivative using a Tecan (Zürich, Switzerland) microplate reader followed by analysis using GraphPad Prism v8 (GraphPad Software, San Diego, CA, USA).
Results and discussion
Preparation of DUPA and DBCO-PEG4-DUPA
DUPA was prepared using the two-step chemical reaction shown in Fig. 1. Compound 1 was purified by flash chromatography, resulting in a colorless oil (1.18 g, 24% yield). Analysis by 1H NMR yielded the following data: δ 5.15 (s, 2H), 4.75 (s, 1H), 4.34 (s, 2H), 3.09 (s, 2H), 2.30 (m, 3H), 2.16–1.29 (m, 43H). Exact mass [M] calculated for C29H53N3O9, 587.38; [M + H]+, 588.42 (Fig. S1†). Compound 1 was subjected to a TFA mediated global deprotection step to yield DUPA (2) which was used without further purification in the next step. Following the reaction of compound 2 with DBCO-PEG4-NHS ester, DBCO-PEG4-DUPA 3 was purified by reversed-phase HPLC and was recovered as a white solid (14.3 mg, 43% yield). Exact mass [M] calculated for C42H55N5O14, 853.37; found [M + H]+, 854.26 (Fig. S2†).Characterization of TMV particles after bioconjugation and drug loading
TMV was modified externally with the PSMA-targeting molecule DUPA using a two-step bioconjugation strategy. First, external lysine residues were reacted with NHS-PEG4-N3 to generate an azide conjugation handle, which was in turn conjugated to DBCO-PEG4-DUPA using a copper-free alkyne–azide click reaction (Fig. 2A). Next, MTO was loaded into the internal cavity of non-targeted TMV and PSMA-targeted TMV-DUPA by exploiting the electrostatic interaction between the positively charged drug molecule and the negatively charged carboxylate groups lining the internal cavity (Fig. 2B). The protocols were as we previously reported;24 under physiologic conditions stable TMV-MTO complexes are formed and drug release is triggered under acidic conditions (e.g. in the tumor microenvironment or endolysosome) by protonation of the carboxylates. To enable tracking of the formulations in tissue culture, we also prepared Cy5-TMV particles by first placing alkyne conjugation handles in the internal cavity by reacting the carboxylate groups with propargylamine. A subsequent copper-catalyzed alkyne–azide crosslinking (CuCAAC) click reaction was then used for the conjugation of sulfo-Cy5-N3 (Fig. 2C). Cy5-TMV particles were then modified to create fluorescent PSMA-targeted Cy5-TMV-DUPA particles as described above.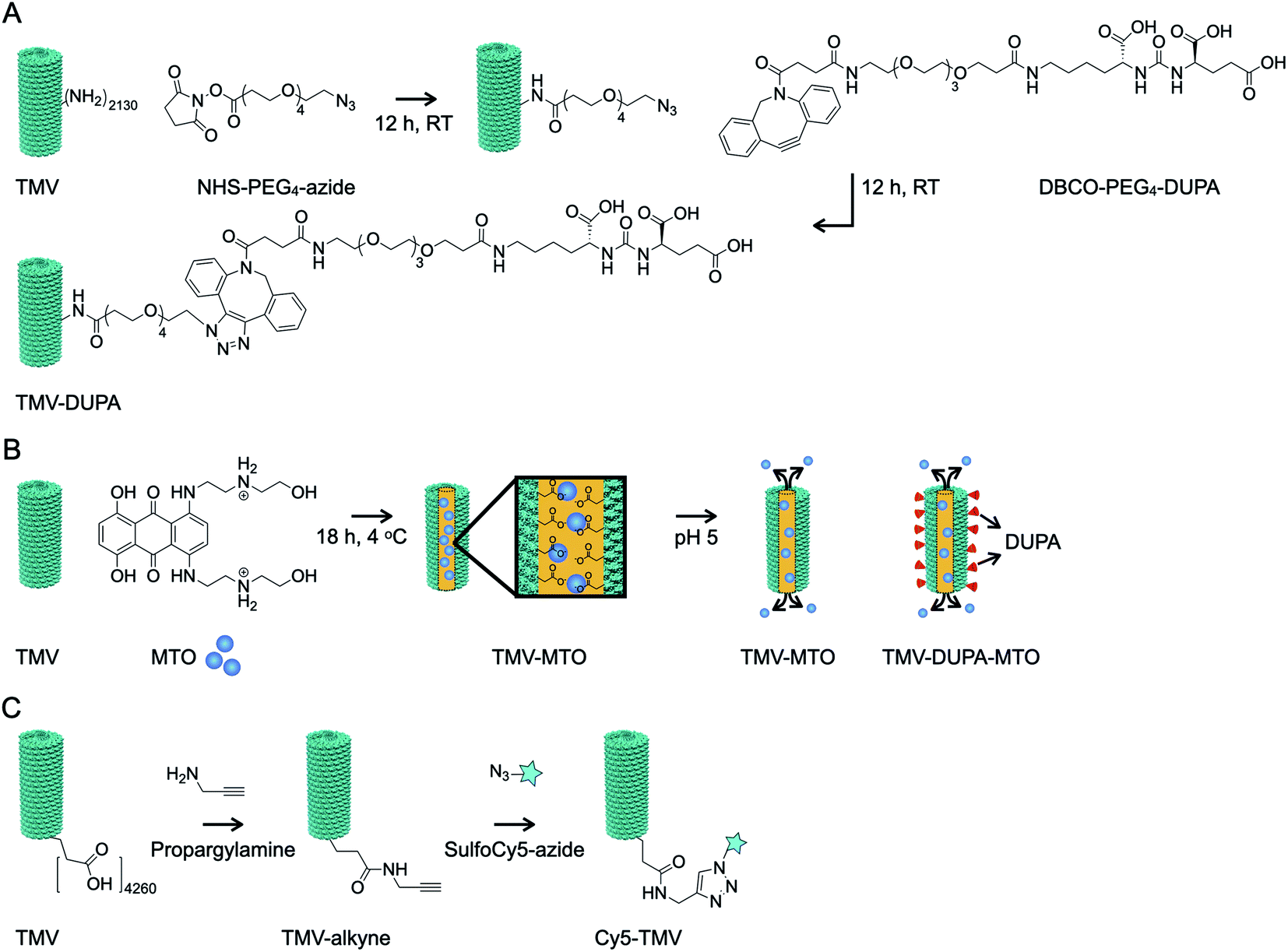 | ||
| Fig. 2 Bioconjugation of TMV particles. (A) TMV was modified with DUPA, a PSMA-targeting small molecule, in a two-step process by first adding a conjugation handle to external lysine residues and then conjugating the linker molecule DBCO-PEG4-DUPA via click chemistry. (B) TMV and TMV-DUPA particles were loaded internally with MTO by exploiting electrostatic interactions with carboxylate groups lining the cavity. (C) TMV particles were labeled with Cy5 by conjugating sulfo-Cy5 to internal glutamic acid residues using EDC chemistry. The resulting Cy5-TMV particles were also conjugated to DUPA as described above (not shown). | ||
The successful modification of the TMV particles was confirmed by UV-vis spectroscopy (Fig. 3A), SDS-PAGE (Fig. 3B) and TEM (Fig. 3C). UV-vis spectroscopy confirmed that a 10000 molar excess of MTO resulted in formation of TMV and TMV-DUPA particles loaded with ∼1270 MTO molecules (Fig. 3A); drug loading was calculated based on the absorbance using the Beer–Lambert law and the respective extinction coefficient of MTO and TMV (see Methods section). Similarly, the analysis of fluorescent particles showed that the Cy5-TMV and Cy5-TMV-DUPA particles carried ∼215 Cy5 molecules per particle (Fig. 3A). SDS-PAGE analysis under denaturing conditions revealed a single ∼18 kDa band representing the unmodified TMV coat protein (Fig. 3B, lane 1) whereas all the DUPA-modified TMV particles, namely TMV-DUPA (lane 2), TMV-DUPA-MTO (lane 4) and Cy5-TMV-DUPA (lane 6), featured an additional higher-molecular-weight band (at ∼20 kDa; DUPA has a mass of 853.37 and a protonated mass [M + H]+ of 854.26, see Fig. S2†) corresponding to DUPA-conjugated coat proteins with ∼1000 DUPA molecules per particle, respectively. It should be noted that a CP dimer is also apparent for any of the conjugated TMV nanoparticles; this phenomenon is generally observed for chemically modified TMV coat proteins and may be explained by enhanced non-covalent interactions or proteins that entangle through the PEG chains. Fluorescence imaging of the Cy5-labeled Cy5-TMV and Cy5-TMV-DUPA particles in the gel was also consistent with the presence of both DUPA-modified and unmodified TMV coat proteins (Fig. 3B, lanes 5 and 6, comparing white light and fluorescence images). Lastly, TEM revealed that the modification with DUPA and/or MTO did not affect the structural integrity of the particles (Fig. 3C); high aspect ratio TMV nanoparticles were observed on the negative-stained grids. The appearance of longer TMV nanoparticles, as is observed for any preparation (native or modified TMV) can be explained by end-to-end assembly which may occur under certain bathing conditions or solvent evaporation.29
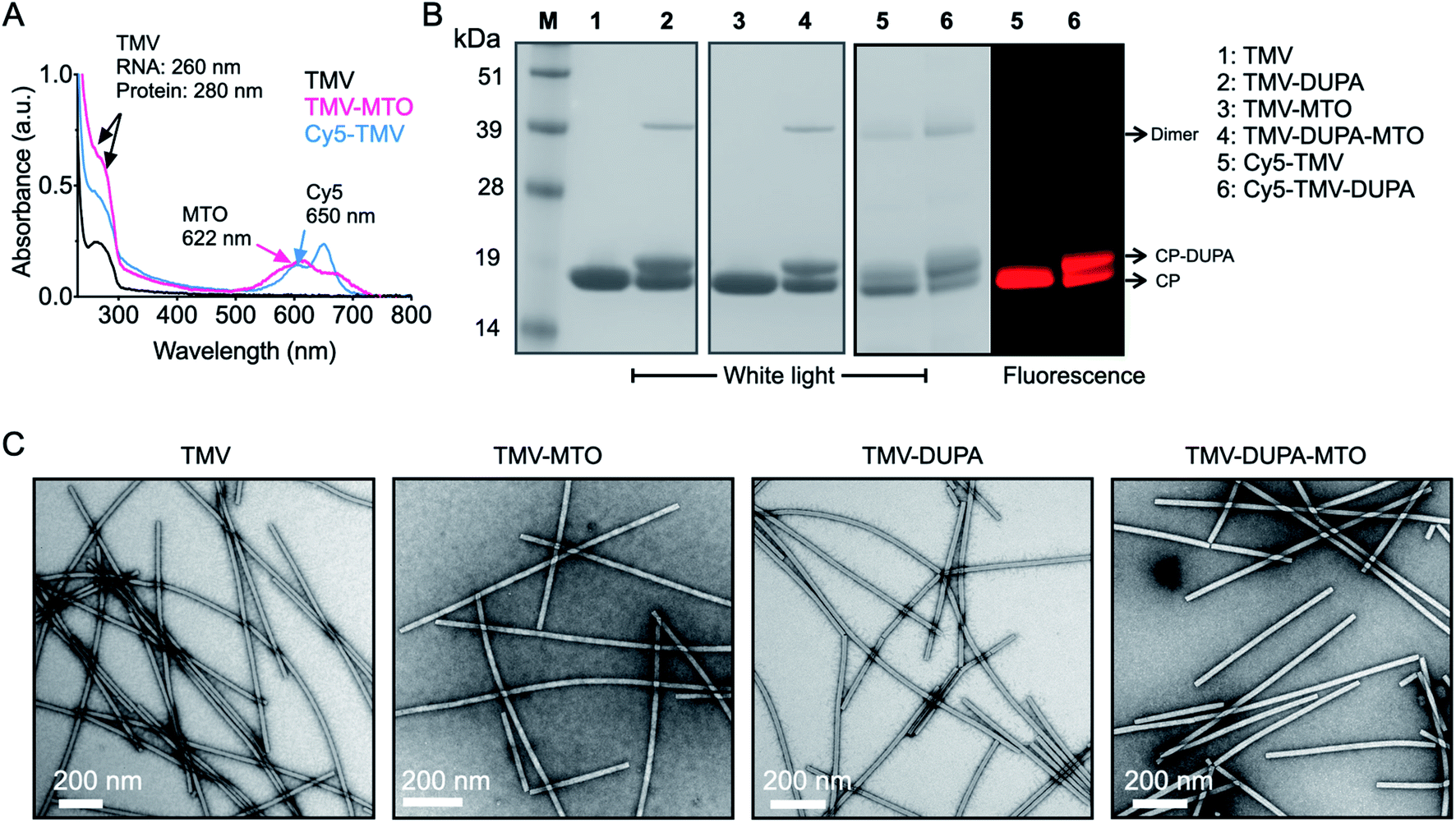 | ||
| Fig. 3 Characterization of TMV-MTO formulations. (A) Drug and dye loading on TMV was quantified by UV-vis spectroscopy, allowing the number of drug and dye molecules per particle to be calculated using Beer–Lambert law and the extinction coefficient of TMV and MTO. (B) SDS PAGE analysis under white light and fluorescence imaging was used to confirm the modification of the TMV with DUPA and Cy5. (C) TEM images confirmed structural integrity of native TMV and particles conjugated to DUPA and/or loaded with MTO. | ||
Analysis of cell binding and uptake
The expression of PSMA on the surface of PSMA+ LNCaP cells was confirmed by flow cytometry following incubation with a fluorescent PSMA-specific monoclonal antibody. The mean fluorescence intensity (MFI) of the LNCaP cells increased 25-fold when probed with the antibody, relative to untreated controls (Fig. 4A). In contrast, negligible background binding was observed when PSMA− PC3 cells were probed with the antibody (Fig. 4A).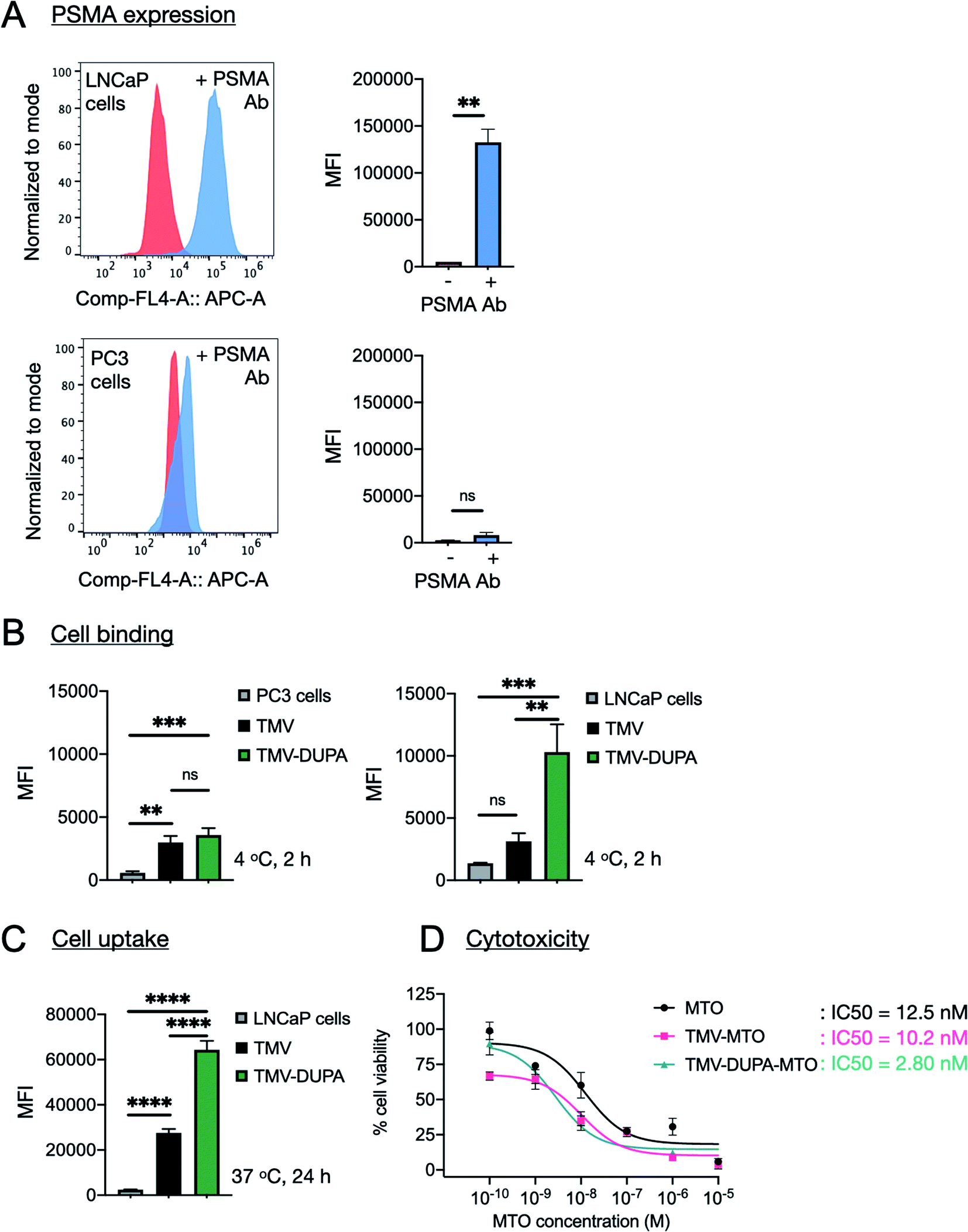 | ||
| Fig. 4 PSMA-specific cell binding, uptake and cytotoxicity of different TMV formulations. (A) Confirmation of the presence of PSMA on the surface of LNCaP cells by flow cytometry using a fluorescent anti-PSMA antibody. (B and C) Comparison of the cellular binding (2 h incubation at 4 °C with LnCAP and PC3 cells) and uptake (24 h incubation at 37 °C with LNCaP cells) of TMV-Cy5 and TMV-Cy5-DUPA particles. (D) Cytotoxicity of soluble MTO and equivalent MTO concentrations delivered by non-targeted TMV particles (TMV-MTO) and corresponding formulations displaying a PSMA-specific ligand (TMV-DUPA-MTO) in LNCaP cells was determined using MTT assay. TMV was used as a control and cytotoxicity was not apparent. The data was analyzed using GraphPad Prism and log(inhibitor) vs. response (three parameters) was used to determine the IC50 values; statistical analysis was performed by unpaired t-test for PSMA expression (LNCaP: **p = 0.006, n = 3) and ordinary one-way ANOVA for cell binding (n = 3; LNCaP: ***p = 0.0004, **p = 0.0015; PC3: ***p = 0.0004, **p = 0.0013) and cellular uptake (LNCaP: ****p < 0.0001) using GraphPad Prism. | ||
The binding and uptake of non-targeted TMV and PSMA-targeted TMV was compared by flow cytometry using the Cy5-TMV and Cy5-TMV-DUPA particles. Binding assays were carried out at 4 °C for 2 h to limit the uptake of particles into the cells. Under these conditions, the TMV-DUPA particles bound to the LNCaP cells with 3-fold higher efficiency than the non-targeted TMV particles, confirming the improved binding specificity achieved by conjugation to DUPA (Fig. 4B). There was no significant difference in binding efficiency between TMV and TMV-DUPA particles when the same assay was applied to PC3 cells, confirming that the interaction between DUPA and PSMA underpins the binding specificity observed in LNCaP cells (Fig. 4B). The assay with LNCaP cells was repeated under conditions that allow the uptake of particles into cells (24 h at 37 °C). We observed a ∼4-fold difference between the TMV and TMV-DUPA particles, with the latter taken up by the cells more efficiently (Fig. 4C). These results confirm that, although many cancer cells take up nanoparticles passively, the addition of targeting ligands can achieve a significant increase in cellular uptake.
Analysis of cytotoxicity
Finally, we evaluated the ability of targeted and non-targeted TMV particles loaded with MTO to kill LNCaP cells relative to the potency of the free drug (Fig. 4D). LNCaP cells were incubated with different concentrations of soluble MTO ranging from 100 pM to 10 μM and equivalent concentrations of the MTO-loaded particles (TMV-MTO and TMV-DUPA-MTO) for 24 h before the cell viability was determined using an MTT assay. The IC50 values indicated comparable efficacy of TMV-MTO particles (IC50 = 10.2 nM) and the free drug (IC50 = 12.5 nM) but the PSMA-targeted TMV-DUPA-MTO particles were ∼4-fold more potent (IC50 = 2.8 nM) (Fig. 4D). These results mirror the enhanced uptake of PSMA-targeted TMV particles observed using flow cytometry (Fig. 4C).We have previously reported cytotoxicity of non-targeted MTO loaded TMV formulation against breast, colon and prostate cancer cells.24 In these studies, the cellular uptake and cytotoxicity of non-targeted TMV-MTO formulation was comparable to free MTO with IC50 values ranging from 641–472 nM.24 These results indicated that even in absence of targeting, TMV-based drug carriers are taken up by cancer cells and are efficient drug delivery platforms as evident from observed efficacy in a murine breast cancer model. LNCaP cells display higher sensitivity to MTO than PC3 cells resulting in improved IC50 values of even the non-targeted TMV-MTO formulation (IC50 = 10.2 nM in LNCaP cells vs. 472 nM in PC3 cells24). However, a further improvement in the cytotoxicity of the targeted formulation in LNCaP cells is attributed to the enhanced cellular uptake of the TMV-DUPA particles and validates the advantages of targeting strategies in drug delivery applications. The in vivo efficacy of the non-targeted TMV-MTO formulation observed in the breast cancer models in earlier study is attributed to enhanced permeability and retention (EPR) mediated accumulation of the anisotropic TMV nanoparticles in the tumor tissue. However, targeted formulations can further improve tumor accumulation and retention of drug delivery platforms.21,22
Conclusions
We have developed a novel nanoparticle formulation based on TMV carrying the PSMA-targeting ligand DUPA, allowing selective binding and internalization by PSMA+ prostate cancer cells and the efficient killing of such cells in vitro. The particles can be loaded with imaging reagents for the specific detection of PSMA+ cells (as demonstrated here with Cy5) or with drugs for targeted cell killing (as demonstrated here with MTO). The TMV particles retain their structural integrity when simultaneously decorated with DUPA and loaded with dyes or drugs, and the DUPA-conjugated particles show a 3-fold increase in binding efficiency and a concomitant 3-fold reduction in IC50 value compared to untargeted particles containing the same cargo. DUPA-coated TMV particles could therefore be promising as both imaging reagents and drug-delivery vehicles that can now be tested for the ability to detect and eradicate prostate cancer cells in vivo.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by a grant from the National Cancer Institute R01-CA202814. We thank Dr Wege for providing the T158K mutant of TMV.References
- F. Bray, J. Ferlay, I. Soerjomataram, R. L. Siegel, L. A. Torre and A. Jemal, Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries, Ca-Cancer J. Clin., 2018, 68(6), 394–424 CrossRef PubMed.
- R. L. Siegel, K. D. Miller and A. Jemal, Cancer statistics, 2020, Ca-Cancer J. Clin., 2020, 70(1), 7–30 CrossRef PubMed.
- P. D. Baade, D. R. Youlden and L. J. Krnjacki, International epidemiology of prostate cancer: geographical distribution and secular trends, Mol. Nutr. Food Res., 2009, 53(2), 171–184 CrossRef CAS PubMed.
- E. Powers, G. S. Karachaliou, C. Kao, M. R. Harrison, C. J. Hoimes, D. J. George, A. J. Armstrong and T. Zhang, Novel therapies are changing treatment paradigms in metastatic prostate cancer, J. Hematol. Oncol., 2020, 13(1), 144 CrossRef PubMed.
- D. S. O'Keefe, S. L. Su, D. J. Bacich, Y. Horiguchi, Y. Luo, C. T. Powell, D. Zandvliet, P. J. Russell, P. L. Molloy, N. J. Nowak, T. B. Shows, C. Mullins, R. A. Vonder Haar, W. R. Fair and W. D. Heston, Mapping, genomic organization and promoter analysis of the human prostate-specific membrane antigen gene, Biochim. Biophys. Acta, 1998, 1443(1–2), 113–127 CrossRef.
- D. S. O'Keefe, D. J. Bacich and W. D. Heston, Comparative analysis of prostate-specific membrane antigen (PSMA) versus a prostate-specific membrane antigen-like gene, Prostate, 2004, 58(2), 200–210 CrossRef PubMed.
- S. Perner, M. D. Hofer, R. Kim, R. B. Shah, H. Li, P. Möller, R. E. Hautmann, J. E. Gschwend, R. Kuefer and M. A. Rubin, Prostate-specific membrane antigen expression as a predictor of prostate cancer progression, Hum. Pathol., 2007, 38, 696–701 CrossRef CAS PubMed.
- Ö. Ekmekcioglu, M. Busstra, N. D. Klass and F. Verzijlbergen, Bridging the imaging gap: PSMA PET/CT has a high impact on treatment planning in prostate cancer patients with biochemical recurrence – a narrative review of the literature, J. Nucl. Med., 2019, 60, 1394–1398 CrossRef PubMed.
- M. S. Hofman, N. Lawrentschuk, R. J. Francis, C. Tang, I. Vela, P. Thomas, N. Rutherford, J. M. Martin, M. Frydenberg, R. Shakher, L. M. Wong, K. Taubman, S. Ting Lee, E. Hsiao, P. Roach, M. Nottage, I. Kirkwood, D. Hayne, E. Link, P. Marusic, A. Matera, A. Herschtal, A. Iravani, R. J. Hicks, S. Williams and D. G. Murphy, Prostate-specific membrane antigen PET-CT in patients with high-risk prostate cancer before curative-intent surgery or radiotherapy (proPSMA): a prospective, randomised, multicentre study, Lancet, 2020, 395(10231), 1208–1216 CrossRef CAS.
- FDA, FDA approves first PSMA-targeted PET imaging drug for men with prostate cancer, https://www.fda.gov/news-events/press-announcements/fda-approves-first-psma-targeted-pet-imaging-drug-men-prostate-cancer, 2020 Search PubMed.
- J. Violet, S. Sandhu, A. Iravani, J. Ferdinandus, S. P. Thang, G. Kong, A. R. Kumar, T. Akhurst, D. A. Pattison, A. Beaulieu, J. Mooi, B. Tran, C. Guo, V. Kalff, D. G. Murphy, P. Jackson, P. Eu, M. Scalzo, S. Williams, R. J. Hicks and M. S. Hofman, Long-term follow-up and outcomes of retreatment in an expanded 50-patient single-center phase II prospective trial of 177Lu-PSMA-617 theranostics in metastatic castration-resistant prostate cancer, J. Nucl. Med., 2020, 61, 857–865 CrossRef PubMed.
- H. J. Wester and M. Schottelius, PSMA-Targeted Radiopharmaceuticals for Imaging and Therapy, Semin. Nucl. Med., 2019, 49(4), 302–312 CrossRef PubMed.
- T. Lammers, L. Y. Rizzo, G. Storm and F. Kiessling, Personalized nanomedicine, Clin. Cancer Res., 2012, 18(18), 4889–4894 CrossRef CAS PubMed.
- F. Wang, C. Li, J. Cheng and Z. Yuan, Recent Advances on Inorganic Nanoparticle-Based Cancer Therapeutic Agents, Int. J. Environ. Res. Public Health, 2016, 13, 1182–1197 CrossRef PubMed.
- A. Puiggali-Jou, L. J. Del Valle and C. Aleman, Drug delivery systems based on intrinsically conducting polymers, J. Controlled Release, 2019, 309, 244–264 CrossRef CAS PubMed.
- L. Sercombe, T. Veerati, F. Moheimani, S. Y. Wu, A. K. Sood and S. Hua, Advances and Challenges of Liposome Assisted Drug Delivery, Front. Pharmacol., 2015, 6, 286 Search PubMed.
- Y. H. Chung, H. Cai and N. F. Steinmetz, Viral nanoparticles for drug delivery, imaging, immunotherapy, and theranostic applications, Adv. Drug Delivery Rev., 2020, 156, 214–235 CrossRef CAS PubMed.
- E. Sokullu, H. Soleymani Abyaneh and M. A. Gauthier, Plant/Bacterial Virus-Based Drug Discovery, Drug Delivery, and Therapeutics, Pharmaceutics, 2019, 11, 211–249 CrossRef CAS PubMed.
- G. P. Lomonossoff and C. Wege, TMV Particles: The Journey From Fundamental Studies to Bionanotechnology Applications, Adv. Virus Res., 2018, 102, 149–176 CAS.
- T. L. Schlick, Z. Ding, E. W. Kovacs and M. B. Francis, Dual-surface modification of the tobacco mosaic virus, J. Am. Chem. Soc., 2005, 127(11), 3718–3723 CrossRef CAS PubMed.
- M. A. Bruckman, K. Jiang, E. J. Simpson, L. N. Randolph, L. G. Luyt, X. Yu and N. F. Steinmetz, Dual-modal magnetic resonance and fluorescence imaging of atherosclerotic plaques in vivo using VCAM-1 targeted tobacco mosaic virus, Nano Lett., 2014, 14(3), 1551–1558 CrossRef CAS PubMed.
- H. Hu, Y. Zhang, S. Shukla, Y. Gu, X. Yu and N. F. Steinmetz, Dysprosium-Modified Tobacco Mosaic Virus Nanoparticles for Ultra-High-Field Magnetic Resonance and Near-Infrared Fluorescence Imaging of Prostate Cancer, ACS Nano, 2017, 11(9), 9249–9258 CrossRef CAS PubMed.
- A. E. Czapar, Y. R. Zheng, I. A. Riddell, S. Shukla, S. G. Awuah, S. J. Lippard and N. F. Steinmetz, Tobacco Mosaic Virus Delivery of Phenanthriplatin for Cancer therapy, ACS Nano, 2016, 10(4), 4119–4126 CrossRef CAS PubMed.
- R. D. Lin and N. F. Steinmetz, Tobacco mosaic virus delivery of mitoxantrone for cancer therapy, Nanoscale, 2018, 10(34), 16307–16313 RSC.
- F. C. Geiger, F. J. Eber, S. Eiben, A. Mueller, H. Jeske, J. P. Spatz and C. Wege, TMV nanorods with programmed longitudinal domains of differently addressable coat proteins, Nanoscale, 2013, 5(9), 3808–3816 RSC.
- K. P. Maresca, S. M. Hillier, F. J. Femia, D. Keith, C. Barone, J. L. Joyal, C. N. Zimmerman, A. P. Kozikowski, J. A. Barrett, W. C. Eckelman and J. W. Babich, A series of halogenated heterodimeric inhibitors of prostate specific membrane antigen (PSMA) as radiolabeled probes for targeting prostate cancer, J. Med. Chem., 2009, 52(2), 347–357 CrossRef CAS PubMed.
- J. Kapuscinski and Z. Darzynkiewicz, Interactions of antitumor agents Ametantrone and Mitoxantrone (Novatrone) with double-stranded DNA, Biochem. Pharmacol., 1985, 34(24), 4203–4213 CrossRef CAS PubMed.
- M. A. Bruckman and N. F. Steinmetz, Chemical modification of the inner and outer surfaces of Tobacco Mosaic Virus (TMV), Methods Mol. Biol., 2014, 1108, 173–185 CrossRef CAS PubMed.
- Y. Lin, Z. Su, G. Xiao, E. Balizan, G. Kaur, Z. Niu and Q. Wang, Self-assembly of virus particles on flat surfaces via controlled evaporation, Langmuir, 2011, 27(4), 1398–1402 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra03166j |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |