
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn investigation of two copper(II) complexes with a triazole derivative as a ligand: magnetic and catalytic properties†
Yuliia P. Petrenko a,
Karolina Piastad,
Dmytro M. Khomenko*a,
Roman O. Doroshchuka,
Sergiu Shovab,
Ghénadie Novitchic,
Yuliya Toporivskad,
Elzbieta Gumienna-Kontecka*d,
Luísa M. D. R. S. Martins*e and
Rostyslav D. Lampekaa
a,
Karolina Piastad,
Dmytro M. Khomenko*a,
Roman O. Doroshchuka,
Sergiu Shovab,
Ghénadie Novitchic,
Yuliya Toporivskad,
Elzbieta Gumienna-Kontecka*d,
Luísa M. D. R. S. Martins*e and
Rostyslav D. Lampekaa
aDepartment of Chemistry, Taras Shevchenko National University of Kyiv, Volodymyrska Street, 64/13, Kyiv 01601, Ukraine. E-mail: dkhomenko@ukr.net
b“Petru Poni” Institute of Macromolecular Chemistry, Laboratory of Inorganic Polymers, Aleea Grigore Ghica Voda No. 41A, RO-700487 Iasi, Romania
cLaboratoire National des Champs Magnétiques Intenses, UPR CNRS 3228, Université Grenoble-Alpes, 25 rue des Martyrs, B.P. 166, 38042 Grenoble Cedex 9, France
dFaculty of Chemistry, University of Wroclaw, F. Joliot-Curie 14, 50-383 Wroclaw, Poland. E-mail: elzbieta.gumienna-kontecka@chem.uni.wroc.pl
eCentro de Química Estrutural, Departamento de Engenharia Química, Instituto Superior Técnico, Universidade de Lisboa, Av. Rovisco Pais, 1049-001 Lisboa, Portugal. E-mail: luisammartins@tecnico.ulisboa.pt
First published on 2nd July 2021
Abstract
Two new copper(II) complexes [Cu2(L)2(OAc)2(H2O)2] (1) (L = 3-methyl-5-pyridin-2-yl-1,2,4-triazole) and [CuL2] (2) were prerared and thoroughly studied. The complexes are able to selectively catalyze the oxidation of styrene towards benzaldehyde and of cyclohexane to KA oil. The 2D coordination polymer 1 showed an antiferromagnetic behaviour attributed to the intrachain magnetic coupling.
Introduction
The coordination chemistry of 5-pyridin-2-yl-1,2,4-triazoles has been investigated from the mid-1970s. The first structural characterization of a binuclear triazole copper(II) compound appeared in 1985 in a publication dedicated to the investigation of its magnetic properties.1 Since that time most studies of triazole copper complexes have focused on this topic. However, the possibilities of magnetostructural exchange in this type of compound are rather limited, as most of the described dimeric triazole complexes have almost planar structures of the inner Cu2N4 core.2,3 The equatorial coordination consists of two deprotonated ligands to which copper(II) atoms are linked by two triazole nuclei via their N(1)–N(2) bridging mode.4,5 Additional ligands coordinate copper(II) atoms axially, and the coordination is completed by a water molecule and anions as nitrate, perchlorate etc., that influence on the process of crystal packing.6 Thus, the coordination polymers constructed from different paramagnetic transition metal ions and triazole ligands have recently attracted intense interest due to their intriguing inherent magnetostructural relationships, as well as the promising applications in memory storage and spintronic structural architecture,7 important for the understanding of devices.8,9 On the other hand, the use of such metal complexes as catalysts of some industrial processes, like an oxidation of alkanes, remains unexplored.10In the present work, we have obtained binuclear (1) and mononuclear (2) complexes based on copper(II) acetate and 3-methyl-5-pyridin-2-yl-1,2,4-triazole. The structural characterization of the mononuclear complex reveals planarly located ligands. Surprisingly, the crystals of the binuclear complex contain non-planar central motif featuring combination between rotational and reflection symmetry of elements. This fact enforced the investigation of magnetic properties of the title compound. Moreover, in pursuit of our interest in the use of Cu-based complexes as catalysts for improving industrial oxidation reactions,11–15 herein we have tested the catalytic activity of complexes 1 and 2 for the selective oxidation of styrene to benzaldehyde and for the far more challenging oxidation of cyclohexane to KA oil (cyclohexanol and cyclohexanone mixture). These reactions were chosen due to the significant industrial importance of their products. Traditionally benzaldehyde is produced by catalytic partial oxidation of toluene or by hydrolysis of benzylidene chloride.16 Both processes require harsh conditions, present low yields, and a huge generation of waste. Therefore, there is a need to find more environmentally friendly processes using green oxidation agents (e.g., hydrogen peroxide) and heterogeneous catalytic systems. Despite the advances that have been already achieved,17–19 the selective oxidation of styrene still remains a challenge. Further, the even more challenging oxidation of saturated hydrocarbons bonds was attempted, using cyclohexane as a model substrate. In fact, copper complexes bearing various nitrogen-containing ligands are among the most active catalysts known20,21 for reactions occurring with functionalization of C–H bonds by hydrogen peroxide and/or organic hydroperoxides. These reactions lead to the production of extremely valuable chemical substances, in particular in the case of cyclohexane, as KA oil that is essential to the production of Nylon 6,6.22 The liquid-phase oxidation of cyclohexane is conducted at temperatures up to 165 °C and high O2 pressure (8–15 bar). These conditions are applied to shorten retention time and to avoid oxidative side reactions. For the same reason, cyclohexane conversion is limited to 10–12% per cycle.23 Therefore, efficient selective oxidation of cyclohexane also remains a challenge that is addressed herein.
It is also worth to highlight that, to the best of our knowledge, to date (search in WOS), there are no alkane oxidation catalysts involving 1,2,4-triazoles, whereas for the oxidation of styrene, only one report was found.24 A thio-Schiff base derived from salicylaldehyde and 4-amino-2,4-dihydro-1,2,4-triazole-5-thione encapsulated in the nanopores of zeolite-Y acted as heterogeneous catalysts for styrene oxidation, using H2O2 as an oxidant. Under the optimized conditions, these catalysts led to styrene oxide, benzaldehyde, 1-phenylethane-1,2-diol and phenylacetaldehyde, thus exhibiting poor selectivity.
Results and discussion
Synthesis of complexes
Complexes were synthesized from a DMF (N,N-dimethylformamide) solution of copper(II) acetate and 3-methyl-5-pyridin-2-yl-1,2,4-triazole (Scheme 1). Detailed preparation and elementary characterization see in ESI.† The ligand was synthesized by following standard procedures.25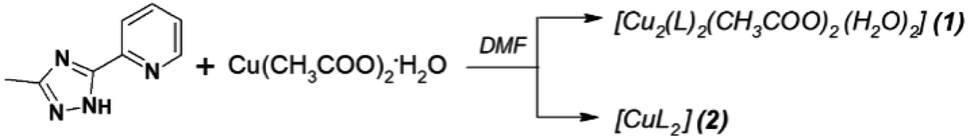 | ||
Scheme 1 Synthesis of complexes 1, 2: (1) with a metal-to-ligand molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1; (2) with a metal-to-ligand molar ratio of 1:2. :1; (2) with a metal-to-ligand molar ratio of 1:2. | ||
The reaction of HL with Cu(OAc)2·H2O in a 1:1 molar ratio gives, on slow crystallization of the reaction mixture, green crystals of [Cu2(L)2(OAc)2(H2O)2] (1) in 80% yield. In order to obtain mononuclear complex, the reaction in a 1:2 molar ratio under analogous conditions was examined. The slow crystallization of reaction mixture produced black prisms of [CuL2] (2) in 95% yield. Crystal structures for both complexes were obtained and confirmed the existence of binuclear species [(CuL(H2O))2]2+ for 1 and mononuclear CuL2 for 2, that was also examined in solution studies.
Solution studies
K1 = 10.67 describes the protonation of triazole N−, acidic logK2 = 2.83 and logK3 = 2.12 correspond to the protonation of pyridine N and triazole N atoms. This is consistent with the literature data for 1,2,4-triazole ligands26,27 and pyridine derivatives28 and is in line with the described trend: triazole N− > pyridine ≈ triazole N29 (Table 1). A significant increase in the acidity of pyridine nitrogen results from the “electron withdrawing” effect of the triazole ring. It is worth mentioning that the obtained constants are higher than for the analogous ligand described earlier, which results from the electron donating effect of the methyl substituent on the triazole ring.30
| UV-Vis | EPR | |||||
|---|---|---|---|---|---|---|
| Species | logβ |
logK |
λ (nm) | ε (M−1 cm−1) | AII (G) | gII |
| a Solvent: MeOH/H2O 80:20 w/w, I = 0.1 M (NaNO3), T = (25.0 ± 0.2) °C. |
||||||
| Protonation of L | ||||||
| HL | 10.67(1) | |||||
| H2L+ | 13.50(1) | 2.83 | ||||
| H3L2+ | 15.62(1) | 2.12 | ||||
|
||||||
| Cu(II):L = 1:1 |
||||||
| [CuHL]2+ | 14.64(3) | 730 | 40 | 142.5 | 2.34 | |
| [CuL]+ | 10.35(3) | 4.24 | 715 | 60 | 142.5 | 2.34 |
| [Cu2L2(OH)]+ | 17.87(9) | 695 | 150 | Not determined | ||
| [Cu2L2(OH)2] | 9.2(1) | 8.67 | 680 | 175 | Not determined | |
|
||||||
| Cu(II):L = 1:3 |
||||||
| [CuHL]2+ | 14.64 | 730 | 40 | 142.5 | 2.34 | |
| [CuHL2]+ | 23.46(3) | Not determined | Not determined | |||
| [CuL2] | 17.82(2) | 5.64 | 690 | 125 | 154.8 | 2.29 |
| [CuL2(OH)]− | 7.69(4) | 10.13 | 670 | 115 | 154.8 | 2.27 |
:HL, with a metal to ligand molar ratio of both 1:1 and 1:3, showed the formation of monomeric species successfully attributed to {[CuL]+}+ m/z = 223.00, {[CuL]+ + HClO4}+ m/z = 321.95, {[CuL2] + H+}+ m/z = 382.07 and {[CuL2] + H+ + HClO4}+ m/z = 482.03 complexes (Fig. S4†). All peak assignments were based on the comparison between the experimental and calculated isotope patterns. | ||
| Fig. 1 Species distribution profiles for Cu(II) complexes of HL in molar ratio (A): Cu(II):HL 1:1 and (B): Cu(II):HL 1:3. | ||
With an increase of pH, the UV-Vis spectra show a hypsochromic shift from 730 nm (ε = 40 M−1 cm−1) to 680 nm (ε = 175 M−1 cm−1), which is associated with an increase in the number of nitrogen donor atoms from 2N to 3N in the coordination sphere of Cu(II) ions (Table 1 and Fig. S5†). EPR parameters, represented by AII = 142.5 G and gII = 2.34 at pH 2.64 also support the model based on the formation of monomeric [CuHL]2+ and [CuL]+ with two N atoms in Cu(II) coordination sphere. The vanishing of the EPR signal above pH 8 confirms the disappearance of the monomeric species (Table 1 and Fig. S6†).31
The solution studies carried out for 1:3 Cu(II):HL molar ratio suggest the formation of mononuclear [CuHL2]+, [CuL2] and [CuL2(OH)]− complexes, preceded by the presence of the [CuHL]2+ species (Table 1 and Fig. 1B) for which the logβCuHL value was entered in the calculations as constant. The maximum of absorption spectra moves from 730 nm (ε = 40 M−1 cm−1) at acidic pH, up to 670 nm (ε = 115 M−1 cm−1) under basic pH conditions (Table 1 and Fig. S5†) which is due to the growing number of coordinated nitrogen atoms in Cu(II) coordination sphere. No signal silencing occurs on recorded EPR spectra, which is in line with the expectations for mononuclear complexes (Table 1 and Fig. S6†). The increase in AII parameters and lowering gII parameters in solutions containing an excess of ligand relative to Cu(II) ions, compared to the values simulated for equimolar systems, indicates a higher number of nitrogen atoms in the coordination sphere of Cu(II) ions (Table 1). This is expected behavior, since the two ligands coordinating to one of the central ion requires the involvement of four nitrogen atoms, and not, as in the dimeric complexes, three only.
 | ||
| Fig. 2 Competition plot between studied ligand, its corresponding analogues and Cu(II), describes complex formation at different pH values in a hypothetical situation in which equimolar amounts of the three reagents are mixed. Calculations are based on binding constants from Table S3.† Conditions: [Cu(II)] = [A] = [B] = [C] = [HL] = 1 mM. | ||
The obtained results were compared with the data for 1,2,4-triazole containing an aminomethyl substituent 3-(aminomethyl)-5-methyl-4H-1,2,4-triazol-4-amine (A),32 which possesses similar distribution of donor atoms as in the HL ligand, as well as ligands 6-methyl-3-(pyridin-2-yl)-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazine (B)33 and 3-(ethylthio)-5-(pyridin-2-yl)-4H-1,2,4-triazol-4-amine (C),34 with analogous structure to the studied HL ligand, but containing substituents on the nitrogen atom N(4) (Table S3†).
For A and C ligands (Table S3†), the hypsochromic shift of the d–d transition band can be observed from a maximum at over 700 nm to 594 nm and 660 nm, respectively,32,34 as a result of the increase in the number of nitrogen atoms in the Cu(II) coordination environment, which is similar to studied Cu(II):HL complexes. The stronger band shift for ligand A is due to the presence of an amino group which is a stronger donor than pyridine.
All described ligands tend to form CuL2 complexes, and the determined logβ values are in a similar range. It should be mentioned that ligand B is able to form Cu2L2 dimeric complexes in solid state, however, such behaviour was not observed in aqueous solutions, most likely due to the participation of water molecules in the coordination of Cu(II) ions.33 Similarly, dimer formation was not observed for ligands A and C.32,34 Further on to compare the ability of these ligands to chelate Cu(II), a competition plot was made (Fig. 2).
The presented competition diagram clearly indicates that the tested HL ligand shows the highest affinity for Cu(II) ions in relation to A, B and C ligands in solutions with a pH above 8.5. This may be due to the presence of additional groups that may constitute alternative donor atoms in ligands B and C, as well as the higher acidity of ligands A, B and C alone. In addition, it should be noted that the free aminomethyl group present in ligand A is a stronger donor than nitrogen of the pyridyl ring (Table S3†).
X-ray crystallography
Crystal data and some further details concerning X-ray analysis are given in Table 3. The bonds lengths and angles are listed in Tables S4–S6, CCDC-2008470, 2008469.† The results of X-ray diffraction study for [Cu2(L)2(OAc)2(H2O)2] (1) are depicted in Fig. 3. The crystal structure consists of discrete dinuclear entities, assembled around the inversion center located in the middle of the Cu1⋯Cu1 separation equal to 3.976(9) Å. Each Cu(II) atom is five-coordinated in slightly distorted N3O2 square-pyramidal geometry provided by three nitrogen atoms of organic ligands and two oxygen atoms from water molecule and monodentate acetate anion. As mentioned above, for earlier described dimeric triazole complexes, the inner Cu2N4 core have almost planar conformation.2,3 By contrary, in compound 1, the coordination of the acetate and water oxygens leads to the formation of non-planar six-membered metal cycle (Cu1N1N2Cu1iN1iN2i), in a “twist-boat” conformation. Nevertheless, the bond lengths of [Cu1–O3w, 2.289(2) Å; Cu1–O1, 1.966(2) Å] is typical for the Cu–O complexes with water molecule, and acetate anion35,36 while those of Cu–N [Cu1–N1, 1.999(2) Å; Cu1–N4, 2.046(2) Å; Cu1– N21, 1.989(2)] are comparable to the lengths found for mononuclear copper(II) compounds.37| Structure | gx | gy | gz | Ax | Ay | Az | R | τ |
|---|---|---|---|---|---|---|---|---|
| 1 | 1.832 | 2.099 | 2.249 | 10 | 20 | 10 | 0.56 | 0.21 |
| 2 | 2.031 | 2.073 | 2.12 | 1 | 9 | 1 | 1.12 | 0 |
| Empirical formula | C20H24Cu2N8O6 | C16H14CuN8 |
|---|---|---|
| a R1 = Σ‖Fo| − |Fc‖/Σ|Fo|.b wR2 = {Σ[w(Fo2 − Fc2)2]/Σ[w(Fo2)2]}1/2.c GOF = {Σ[w(Fo2 − Fc2)2]/(n − p)}1/2, where n is the number of reflections and p is the total number of parameters refined. | ||
| Formula weight | 599.55 | 381.89 |
| Temperature/K | 180 | 293 |
| Crystal system | Monoclinic | Monoclinic |
| Space group | C2/c | P21/c |
| a/Å | 22.4328(6) | 5.3383(4) |
| b/Å | 7.23016(16) | 9.0913(6) |
| c/Å | 15.4337(4) | 15.5717(10) |
| α/° | 90 | 90 |
| β/° | 107.996(3) | 95.648(6) |
| γ/° | 90 | 90 |
| V/Å3 | 2380.78(11) | 752.06(9) |
| Z | 4 | 2 |
| Dcalc/mg mm−3 | 1.673 | 1.686 |
| μ/mm−1 | 1.841 | 1.470 |
| Crystal size/mm3 | 0.25 × 0.15 × 0.02 | 0.20 × 0.05 × 0.02 |
| 2θmin, 2θmax (°) | 3.818 to 50.05 | 5.194 to 50.054 |
| Reflections collected | 6663 | 3075 |
| Independent reflections | 2103 [Rint = 0.0362] | 1336 [Rint = 0.0334] |
| Data/restraints/parameters | 2103/0/166 | 1336/0/116 |
| GOFc | 1.066 | 1.041 |
| R1a (I > 2σ(I)) | 0.0312 | 0.0421 |
| wR2b (all data) | 0.0745 | 0.1076 |
| Largest diff. peak/hole/e Å−3 | 0.36/−0.40 | 0.31/−0.32 |
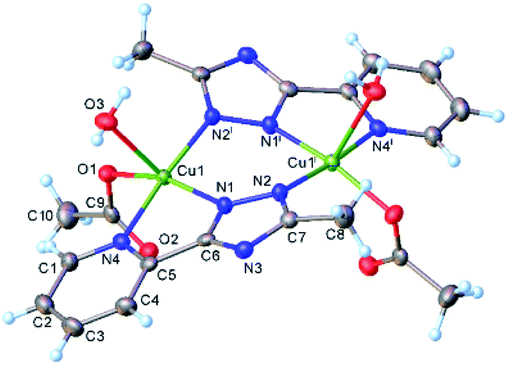 | ||
| Fig. 3 X-ray molecular structure of [Cu2(L)2(OAc)2(H2O)2] (1) with atom labelling and thermal ellipsoids at 50% probability level. Symmetry code: (i) 1 − x, y, 0.5 − z. | ||
The distortion parameter calculated as the sum of deviations from 90° for all 8 cis bond angles is equal 52.0°. The copper atom is displaced from the mean plane formed by the basal atoms at 0.153(1) Å towards axial water molecule. Of importance, the non-coordinated oxygen of acetate anion is involved as acceptor in hydrogen bonding with coordinated water molecule of the adjacent complex. Further analysis of the intermolecular contacts showed the presence of two-dimensional supramolecular layers consolidated by C–H⋯O, O–H⋯O and O–H⋯N intermolecular hydrogen bonds (Fig. S10 and Table S6†). The crystal structure essentially results from the parallel packing of isolated 2D layers (Fig. S11†).
Compound 2 has a molecular crystal structure built up from the centrosymmetric neutral entities [CuL2] (Fig. 4), where copper atom is located in the inversion center. The presence of two intramolecular C–H⋯N hydrogen bonds, which stabilizes the planar conformation is also to be mentioned. The copper(II) atom exhibits a square-planar coordination with Cu1–N1 2.036(3) Å and Cu1–N2 1.946(3) Å distances, bond angles N2–Cu1–N1 and N2i–Cu1–N1 correspond 81.33(11) and 98.67(11), which is in the normal range.38 The main crystal structure motif can be considered as 1D ladder-like supramolecular array of mononuclear [CuL2] units sustained by a weak intermolecular contacts Cu1⋯N4(1 + x, y, z) of 3.106(3) Å as shown in Fig. S12.† The crystal structure of 2 is determined by the parallel packing of discrete 1D architecture along a crystallographic axis (Fig. S13†).
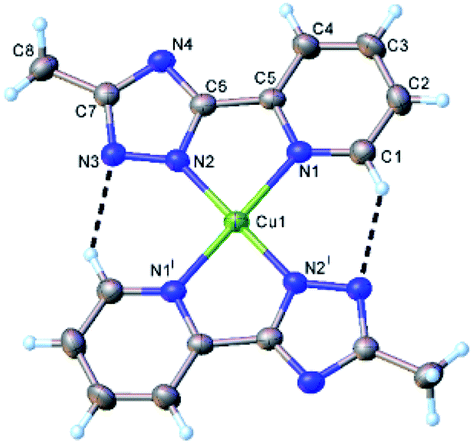 | ||
| Fig. 4 X-ray molecular structure of [CuL2] (2) with atom labelling and thermal ellipsoids at 50% probability level. Symmetry code: (i) 2 − x, 1 − y, 1 − z. H-bond parameters: C1–H⋯N3 [C1–H 0.93 Å, H⋯N3(2 − x, 1 − y, 1 − z) 2.36 Å, C1⋯N3 3.148(5) Å, ∠C1HN3 141.9°]. | ||
Thermogravimetric measurements
The thermograms (TG, DTG and DTA) of complexes were carried out in the temperature range 30–900 °C under an air atmosphere with a heating rate of 10 °C min−1. Complex 1 shows a weight loss of 6% at 120 °C, attributed to the depletion of water (Fig. S8†). The second weight loss occurs between 220 and 260 °C, corresponding to the removal of remaining acetate anions that have some weak coordination interaction with the Cu2 center (calcd: 19.6%). Further heating corresponds to the decomposition of ligand molecules and affords loss of crystallinity. The compound 2 gradually decomposes under heating in the range 300–450 °C which could be ascribed to a collapse of the coordination framework (Fig. S9†).Magnetic properties
Before proceeding to the magnetic and catalytic characterization, the powder products of compounds have been characterized by X-ray powder diffraction (PXRD) at room temperature. The observed XRD patterns are in good agreement with the results simulated from the single crystal data, indicating the purity of the synthesized samples. The differences in intensity may be due to the preferred orientation of the crystalline powder samples. The most informative part of XRD patterns of complexes (2Θ range is 5–50°) is shown in Fig. S3.† The thermal dependence of magnetic susceptibility for compound 1 is shown in Fig. 5. At room temperature, the χMT product has a value of 0.74 cm3 K mol−1 which is close to the expected χMT value (0.75 cm3 K mol−1) for two non-interacting S = 1/2 ions (g = 2.0). While decreasing the temperature, the χMT product continuously decreases and finally reaches a value close to zero at 20 K. This behavior suggests the presence of important antiferromagnetic interactions in 1. According to the X-ray investigation, the complex has a binuclear structure in which the magnetic interaction is carried out via two bridged triazole ligands. In this context the magnetic interaction in 1 can be estimated using classical Bleaney–Bowers coupling scheme (H = −2JS1S2 with S1 = S2 = 1/2)39,40where N is the Avogadro number, β is the magnetic moment and kB is the Boltzmann constant, g is the Landé factor of Cu(II) in the complex, and J is the magnetic exchange constant between paramagnetic ions in the dimer. The fitting procedure leads to a good agreement between the experimental points and the theoretical curve (Fig. 5). The parameters extracted from the fit are J = −46.88(6) cm−1, ρ = 0.003(1), g = 2.12(1). The obtained values of J and g are close to those obtained from EPR analysis (assuming giso = (gz + gy + gx)/3) and consistent with previously reported compounds in the square-pyramidal coordination environment.41,42 The EPR spectra of both 1 and 2 are rhombic (Table 2). The parameter R = (gy − gx)/(gz − gy) with gz > gy > gx (Table 2) indicates that the ground state is dx2–y2 for 1 and dz2 for 2.43,44 For a geometry intermediate between the idealised square-pyramidal and trigonal-bipyramidal extremes, the structural index parameter τ5 = (β − α)/60 can be used as an index of the degree of trigonality (β is the greatest of the basal angles, α is the smallest one). For a perfectly tetragonal geometry τ5 = 0, while it becomes 1 for a perfectly trigonal-bipyramidal geometry.45 For compound 1 τ5 is 0.21 which indicates 79% share of the square pyramidal geometry. For compound 2, where Cu(II) is tetra-coordinated, τ4 calculated as [360 − (β + α)]/141 (ref. 46) is equal to zero. Accordingly, the coordination environment of the central atom exhibits a square-planar geometry.
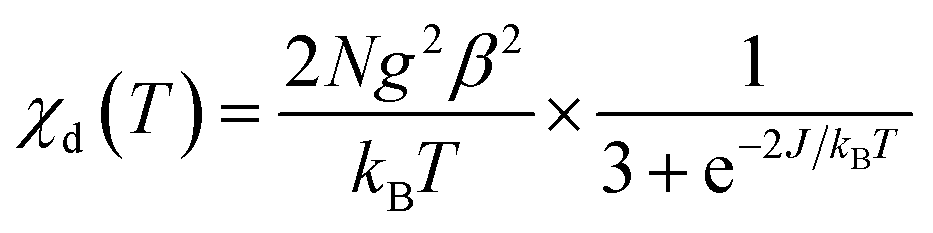
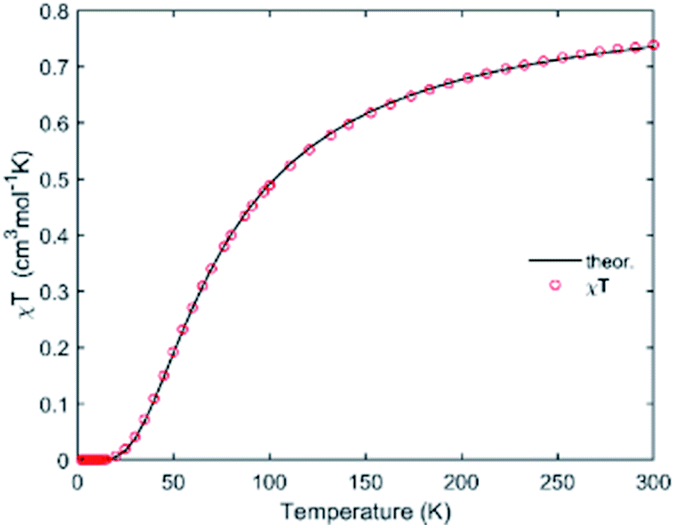 | ||
| Fig. 5 Thermal variations of χMT for 1. Solid line represents the best fit. | ||
Magnitude of magnetic interaction in the case of bis-triazole bridge is determined by distortion of the coordination geometry around Cu(II) centers (parameter τ in the case of trigonal-bipyramidal geometry) and symmetry of the Cu–(N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N)2–Cu units.45,47–52 These two factors impact orientation of the resulting magnetic orbital and overlapping of metal orbitals with orbital of bridged ligands. The literature reports a large numbers of triazole bridged compounds.2,53–59 We found only one that have the similar donor atoms set [N3O2] in trigonal-bipyramidal environment [Cu2(pt)2(SO4)(H2O)3](H2O)3 (were pt = 3-pyridin-2-yl-1,2,4-triazolato ligand).59 The magnitude of magnetic interaction in this complex is −49 cm−1 and based of EPR measurement g = 2.14 which is consistent with the results obtained for compound 1 reported in this work. It should also be noted that the structural parameters of the Cu–(NN)2–Cu units are also similar.
N)2–Cu units.45,47–52 These two factors impact orientation of the resulting magnetic orbital and overlapping of metal orbitals with orbital of bridged ligands. The literature reports a large numbers of triazole bridged compounds.2,53–59 We found only one that have the similar donor atoms set [N3O2] in trigonal-bipyramidal environment [Cu2(pt)2(SO4)(H2O)3](H2O)3 (were pt = 3-pyridin-2-yl-1,2,4-triazolato ligand).59 The magnitude of magnetic interaction in this complex is −49 cm−1 and based of EPR measurement g = 2.14 which is consistent with the results obtained for compound 1 reported in this work. It should also be noted that the structural parameters of the Cu–(NN)2–Cu units are also similar.
Catalytic properties
Both copper complexes 1 and 2 were successfully tested as heterogeneous catalysts for the microwave-assisted peroxidative oxidations of styrene and cyclohexane (Scheme 2) by aqueous hydrogen peroxide, in acetonitrile, under mild conditions. They yield selectively (>99%) benzaldehyde (Scheme 2a) under optimized reaction conditions (microwave irradiation, 90 °C, 30 min, 600 rpm, 15 W, nperoxide/nstyrene = 2; 1 mol% vs. styrene; see Table S1 of ESI†) up to 60% of conversion and a turnover frequency (moles of product per mole of catalyst per unit of time, TOF) of 120 h−1. They are also able to catalyse the selective oxidation of inert cyclohexane to KA oil (Scheme 2b) yielding under optimized (additive-free) reaction conditions (microwave irradiation, 60 °C, 2 h, 600 rpm, 10 W, nperoxide/ncyclohexane = 2; 0.2 mol% vs. cyclohexane; see Table S2 of ESI†) 37 and 23% (for complex 1 and 2, respectively) of KA oil. Cyclohexyl hydroperoxide is formed as a primary product and evolves to a mixture of the final products, cyclohexanol and cyclohexanone (KA oil). MW irradiation was used as an alternative energy source over the conventional (oil bath) heating method. This source of energy has already proved to remarkably enhance the activity of catalytic systems and promote product yields.11,21,60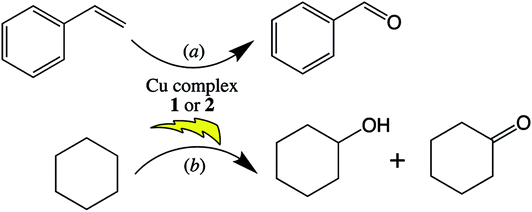 | ||
| Scheme 2 Microwave-assisted peroxidative oxidation of (a) styrene to benzaldehyde or (b) cyclohexane to KA oil, catalysed by complexes 1 and 2. | ||
This was also observed in the present study. For comparative purposes, an exhaustive study was carried out using complex 1 as catalyst under similar conditions to those described above but using conventional thermal heating instead of MW irradiation (Fig. 6). In fact, for both substrates, the selective conversion into the desired product is attained much quickly under MW irradiation compared with the thermal heating. Concerning one of the goals of this catalytic study, the search for a greener benzaldehyde production method, the optimized conditions provided benzaldehyde as the only product, which is a great result taking into account the usual selectivity problems associated to this oxidation reaction.16–19 The selectivity towards benzaldehyde suffers a significant decrease for higher quantities of catalyst or oxidant. The temperature has also shown an important role in the selectivity, and for lower temperatures, the oxidation process is more selective towards the formation of 1-phenylethane-1,2-diol (up to 13% was detected at 60 °C, maintaining the other optimized conditions). Temperatures higher than 90 °C were not tested due to safety reasons. The mechanism for styrene oxidation is believed to be initiated by the Cu-assisted formation of oxygen-centred radicals (e.g., hydroxyl) which oxidise styrene, what is also suggested by the reaction inhibition upon addition of the radical scavenger TEMPO to the reaction mixture under the optimized conditions. In addition, the possible epoxide product was not detected, neither acetophenone nor further epoxidation products such as phenylacetaldehyde (from isomerization of the epoxide). An increase in the microwave irradiation time above 30 minutes led to the formation of benzoic acid by oxidation of benzaldehyde. The recycling of complexes 1 and 2 was tried by recovering them by centrifugation (see Experimental, ESI†) and use the recovered solids in a further catalytic cycle by addition of new typical portions of all other reagents. Unfortunately, the catalytic activity of recovered 1 and 2 for the second cycle was significantly lower (up to 17% conversion) what prevented their reuse.
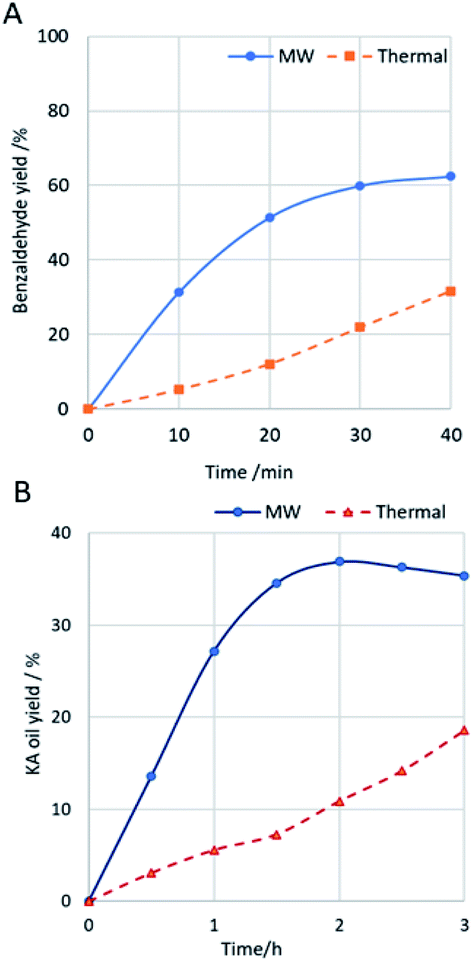 | ||
Fig. 6 (A) Benzaldehyde yield from MW-assisted ( ) or thermal ( ) or thermal ( ) oxidation of styrene, catalysed by 1. (B) KA oil yield from MW-assisted ( ) oxidation of styrene, catalysed by 1. (B) KA oil yield from MW-assisted ( ) or thermal ( ) or thermal ( ) oxidation of cyclohexane, catalysed by 1. ) oxidation of cyclohexane, catalysed by 1. | ||
The obtained yields for KA oil are ca. four times higher than those reported for the industrial process (8%) to assure a suitable selectivity.23 Moreover, the use of copper complexes 1 or 2 led to a high selectivity towards the formation of KA oil, as cyclohexanol and cyclohexanone were the only products detected by GC-MS analysis. Complex 2 exhibits a slightly higher activity in comparison with complex 1, on account of the nuclearity of both complexes (19% yield of KA oil per Cu atom for 1 compared to 23% for 2). Under the tested conditions, the peroxidative oxidation cyclohexane is believed to proceed mainly through a radical mechanism, similar to that proposed in other cases that are also of a radical type11–15,61 and as also suggested by the reaction inhibition upon addition of the radical scavenger diphenylamine to the reaction mixture under the best conditions (a maximum yield of 8% was obtained for complex 1). The recyclability of complexes 1 and 2 was examined by recovering them by centrifugation (see Experimental, ESI†). Although a slight colour change was detected for the recovered 2, both complexes were used in a new cycle. Unfortunately, their catalytic activity for the second cycle was significantly lower (up to 7% yield of KA oil) than the exhibited for the first cycle, what prevented their reuse.
Experimental part
General procedure
All solvents used were laboratory reagent grade. All reagents used in this work were of analytical grade and used without further purification. The characterization and purity monitoring of the obtained compounds has been done by IR, 1H NMR spectroscopy, elemental and thermogravimetric analyses, and X-ray crystallography. Coordination compounds were also investigated by means of magnetochemistry. Elemental analysis was carried out with Perkin-Elmer 2400 CHN Analyzer. Melting point was measured with OptiMelt Automated Melting Point System (MPA 100). 1H NMR spectrum in CDCl3 solution was performed on a Varian 400 spectrometer at room temperature (Fig. S1†). The IR spectra (KBr, pellet) were recorded with Spectrum BX Perkin Elmer spectrometer (Fig. S2†). The phase composition of prepared samples was determined by X-ray powder diffraction (XRD) using SHIMADZU XRD-6000 diffractometer with a linear detector and Cu Kα radiation (λ = 1.5418 Å).Preparation and elementary characterization of complexes
Conclusions
Two new copper(II) complexes have been successfully synthesized from 3-methyl-5-pyridin-2-yl-1,2,4-triazole. Despite different crystal structure and magnetic properties, they both represent first examples of catalyst with a high selectivity towards the formation of KA oil, as cyclohexanol and cyclohexanone. Complex 2 exhibits a bit higher activity in comparison with of complex 1, on account on the nuclearity of both complexes. The presence of binuclear species and mononuclear bis complexes has been confirmed in MeOH/H2O (80/20 w/w) solution, both stoichiometries predominating the speciation above pH 6 for metal to molar ligand ratio of 1:1 and 1:3, respectively. It has to be underlined that so far, the formation of dimers by pyridin-2-yl-1,2,4-triazole compounds has been observed only in solid state, which distinguishes the ligands studied in this work.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been partially supported by the Fundação para a Ciência e a Tecnologia (FCT), Portugal, through project UIDB/00100/2020 of Centro de Química Estrutural, and by the National Science Centre Poland, UMO-2015/19/B/ST5/00413.Notes and references
- R. Prins, P. J. M. W. L. Birker, J. G. Haasnoot, G. C. Verschoor and J. Reedijk, Inorg. Chem., 1985, 24, 4128–4133 CrossRef CAS.
- P. M. Slangen, P. J. van Koningsbruggen, J. G. Haasnoot, J. Jansen, S. Gorter, J. Reedijk, H. Kooijman, W. J. J. Smeets and A. L. Spek, Inorg. Chim. Acta, 1993, 212, 289–301 CrossRef CAS.
- Y.-H. Zhou, W.-Q. Wan, D.-L. Sun, J. Tao, L. Zhang and X.-W. Wei, Z. Anorg. Allg. Chem, 2014, 640, 249–253 CrossRef CAS.
- R. Doroschuk, Acta Crystallogr., Sect. E: Crystallogr. Commun., 2016, 72, 486–488 CrossRef CAS PubMed.
- A. N. Gusev, V. F. Shul’gin, E. A. Ugolkova, N. N. Efimov, G. G. Aleksandrov, V. V. Minin and I. L. Eremenko, Russ. J. Inorg. Chem., 2014, 59, 699–705 CrossRef CAS.
- W. Li, J. Zhang, C. Li and Y. Yang, Zeitschrift für Kristallographie – New Crystal Structures, 2010, 225, 181–182 CAS.
- J. P. Zhang, Y. B. Zhang, J. Bin Lin and X. M. Chen, Chem. Rev., 2012, 112, 1001–1033 CrossRef CAS PubMed.
- M. Kurmoo, Chem. Soc. Rev., 2009, 38, 1353–1379 RSC.
- Y. Y. Zhang, Q. Liu, E. C. Yang and X. J. Zhao, Inorg. Chim. Acta, 2014, 415, 81–87 CrossRef CAS.
- G. Shul’pin, Catalysts, 2016, 6, 50 CrossRef.
- L. M. D. R. S. Martins, Coord. Chem. Rev., 2019, 396, 89–102 CrossRef CAS.
- L. M. D. R. S. Martins and A. J. L. Pombeiro, Eur. J. Inorg. Chem., 2016, 2016, 2236–2252 CrossRef CAS.
- A. P. C. Ribeiro, L. M. D. R. S. Martins, S. Hazra and A. J. L. Pombeiro, C. R. Chim., 2015, 18, 758–765 CrossRef CAS.
- I. Timokhin, C. Pettinari, F. Marchetti, R. Pettinari, F. Condello, S. Galli, E. C. B. A. Alegria, L. M. D. R. S. Martins and A. J. L. Pombeiro, Cryst. Growth Des., 2015, 15, 2303–2317 CrossRef CAS.
- M. Sutradhar, L. M. D. R. S. Martins, M. F. C. Guedes da Silva, K. T. Mahmudov, C.-M. Liu and A. J. L. Pombeiro, Eur. J. Inorg. Chem., 2015, 2015, 3959–3969 CrossRef CAS.
- F. Brühne and E. Wright, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2012, vol. 5, pp. 223–235 Search PubMed.
- N. M. R. Martins, A. J. L. Pombeiro and L. M. D. R. S. Martins, Catal. Commun., 2018, 116, 10–15 CrossRef CAS.
- T. A. G. Duarte, A. P. Carvalho and L. M. D. R. S. Martins, Catal. Sci. Technol., 2018, 8, 2285–2288 RSC.
- T. A. G. Duarte, A. P. Carvalho and L. M. D. R. S. Martins, Catal. Today, 2020, 357, 56–63 CrossRef CAS.
- L. Martins, Catalysts, 2017, 7, 12 CrossRef.
- L. M. D. R. S. Martins and A. J. L. Pombeiro, Coord. Chem. Rev., 2014, 265, 74–88 CrossRef CAS.
- Global Adipic Acid Market Research and Forecast 2018–2023, https://www.researchandmarkets.com/reports/3821034/global-adipic-acid-market-research-and-forecast, accessed 29 July 2020 Search PubMed.
- Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, Germany, 6th edn, 2002 Search PubMed.
- H. S. Abbo and S. J. J. Titinchi, in Topics in Catalysis, Springer, 2010, vol. 53, pp. 1401–1410 Search PubMed.
- D. M. Khomenko, R. O. Doroshchuk, O. V. Vashchenko and R. D. Lampeka, Chem. Heterocycl. Compd., 2016, 52, 402–408 CrossRef CAS.
- P. Lumme, I. Pitkänen, N. Nigri, A. Kjekshus, B. Klewe and D. L. Powell, Acta Chem. Scand., Ser. A, 1974, 28, 1106–1118 CrossRef.
- R. Konášová, J. J. Dytrtová and V. Kašička, J. Chromatogr. A, 2015, 1408, 243–249 CrossRef PubMed.
- K. Zdyb, M. O. Plutenko, R. D. Lampeka, M. Haukka, M. Ostrowska, I. O. Fritsky and E. Gumienna-Kontecka, Polyhedron, 2017, 137, 60–71 CrossRef CAS.
- J. I. Lachowicz, V. M. Nurchi, G. Crisponi, M. D. G. Jaraquemada-Pelaez, M. Ostrowska, J. Jezierska, E. Gumienna-Kontecka, M. Peana, M. A. Zoroddu, D. Choquesillo-Lazarte, J. Niclós-Gutiérrez and J. M. González-Pérez, J. Inorg. Biochem., 2015, 151, 94–106 CrossRef CAS PubMed.
- Y. P. Petrenko, D. M. Khomenko, R. O. Doroshchuk, S. Shova, G. Novitchi, K. Piasta, E. Gumienna-Kontecka and R. D. Lampeka, Inorg. Chim. Acta, 2020, 500, 119216 CrossRef CAS.
- Y. S. Moroz, K. Kulon, M. Haukka, E. Gumienna-Kontecka, H. Kozłowski, F. Meyer and I. O. Fritsky, Inorg. Chem., 2008, 47, 5656–5665 CrossRef CAS PubMed.
- F. Gaccioli, R. Franchi-Gazzola, M. Lanfranchi, L. Marchiò, G. Metta, M. A. Pellinghelli, S. Tardito and M. Tegoni, J. Inorg. Biochem., 2005, 99, 1573–1584 CrossRef CAS PubMed.
- S. Tardito, O. Bussolati, M. Maffini, M. Tegoni, M. Giannetto, V. Dall'Asta, R. Franchi-Gazzola, M. Lanfranchi, M. A. Pellinghelli, C. Mucchino, G. Mori and L. Marchiò, J. Med. Chem., 2007, 50, 1916–1924 CrossRef CAS PubMed.
- F. Dallavalle, F. Gaccioli, R. Franchi-Gazzola, M. Lanfranchi, L. Marchiò, M. A. Pellinghelli and M. Tegoni, J. Inorg. Biochem., 2002, 92, 95–104 CrossRef CAS.
- S. Zhang, S. Zhang and H. Zhang, CN106432286A20170222, 2017, p. 7.
- F. L. Yang, G. Z. Zhu, B. B. Liang, Y. H. Shi and X. L. Li, Polyhedron, 2017, 128, 104–111 CrossRef CAS.
- J. Pons, A. Chadghan, A. Alvarez-Larena, J. F. Piniella and J. Ros, Inorg. Chim. Acta, 2001, 324, 342–346 CrossRef CAS.
- M. Du, S. T. Chen, Y. M. Guo, X. H. Bu and J. Ribas, J. Mol. Struct., 2005, 737, 17–21 CrossRef CAS.
- B. Bleaney, Rev. Mod. Phys., 1953, 25, 161–162 CrossRef.
- O. Kahn, Molecular magnetism, VCH Publishers, Inc., USA, 1993 Search PubMed.
- N. Ritterskamp, K. Sharples, E. Richards, A. Folli, M. Chiesa, J. A. Platts and D. M. Murphy, Inorg. Chem., 2017, 56, 11862–11875 CrossRef CAS PubMed.
- B. M. Kozyrev and S. A. Al’tshuler, Electron Paramagnetic Resonance in Compounds of Transition Elements, Wiley, 2nd edn, 1974 Search PubMed.
- E. Garribba, G. Micera, D. Sanna and L. Strinna-Erre, Inorg. Chim. Acta, 2000, 299, 253–261 CrossRef CAS.
- E. Garribba and G. Micera, J. Chem. Educ., 2006, 83, 1229–1232 CrossRef CAS.
- A. W. Addison, T. N. Rao, J. Reedijk, J. Van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- L. Yang, D. R. Powell and R. P. Houser, J. Chem. Soc., Dalton Trans., 2007, 955–964 RSC.
- S. Tanase, I. A. Koval, E. Bouwman, R. De Gelder and J. Reedijk, Inorg. Chem., 2005, 44, 7860–7865 CrossRef CAS PubMed.
- V. P. Hanoi, T. D. Robert, J. Kolnaar, J. G. Haasnoot, J. Reedijk, H. Kooijman and A. L. Spek, J. Chem. Soc., Dalton Trans., 1996, 4275–4281 Search PubMed.
- A. N. Gusev, I. Nemec, R. Herchel, E. Bayjyyev, G. A. Nyshchimenko, G. G. Alexandrov, I. L. Eremenko, Z. Trávníček, M. Hasegawa and W. Linert, Dalton Trans., 2014, 43, 7153–7165 RSC.
- M. H. Klingele, P. D. W. Boyd, B. Moubaraki, K. S. Murray and S. Brooker, Eur. J. Inorg. Chem., 2005, 2005, 910–918 CrossRef.
- S. Ferrer, J. Hernández-Gil, F. J. Valverde-Muñoz, F. Lloret and A. Castiñeiras, RSC Adv., 2019, 9, 29357–29367 RSC.
- D. Kaase, C. Gotzmann, S. Rein, Y. Lan, S. Kacprzak and J. Klingele, Inorg. Chem., 2014, 53, 5546–5555 CrossRef CAS PubMed.
- J. Klingele, A. I. Prikhod’ko, G. Leibeling, S. Demeshko, S. Dechert and F. Meyer, Dalton Trans., 2007, 2003–2013 RSC.
- R. Prins, P. J. M. W. L. Birker, J. G. Haasnoot, G. C. Verschoor and J. Reedijk, Inorg. Chem., 1985, 24, 4128–4133 CrossRef CAS.
- H. Matsushima, H. Hamada, K. Watanabe, M. Koikawa and T. Tokii, J. Chem. Soc., Dalton Trans., 1999, 971–977 RSC.
- A. W. Addison and T. N. Rao, J. Chem. Soc., Dalton Trans., 1984, 1349 RSC.
- P. Martín-Ramos, M. R. Silva, J. d. A. e Silva, N. D. Martins, C. Yuste-Vivas, P. S. Pereira da Silva, A. J. F. N. Sobral and L. C. J. Pereira, J. Mol. Struct., 2016, 1108, 278–287 CrossRef.
- E. Sugawara and H. Nikaido, Antimicrob. Agents Chemother., 2014, 58, 7250–7257 CrossRef PubMed.
- P. M. Slangen, P. J. van Koningsbruggen, K. Goubitz, J. G. Haasnoot and J. Reedijk, Inorg. Chem., 1994, 33, 1121–1126 CrossRef CAS.
- M. Sutradhar, L. M. D. R. S. Martins, M. F. C. Guedes da Silva and A. J. L. Pombeiro, Appl. Catal., A, 2015, 493, 50–57 CrossRef CAS.
- T. F. S. Silva and L. M. D. R. S. Martins, Molecules, 2020, 25, 748 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. Experimental section, spectroscopic characterisation, crystal structures, solution studies, and other related materials. CCDC 2008470 and 2008469. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra03107d |
| This journal is © The Royal Society of Chemistry 2021 |