DOI:
10.1039/D1RA02928B
(Paper)
RSC Adv., 2021,
11, 16173-16178
Chemical dynamics simulations of energy transfer in CH4 and N2 collisions
Received
15th April 2021
, Accepted 15th April 2021
First published on 30th April 2021
Abstract
Chemical dynamics simulations have been performed to study the energy transfer from a hot N2 bath at 1000 K to CH4 fuel at 300 K at different bath densities ranging from 1000 kg m−3 to 30 kg m−3. At higher bath densities, the energy transfer from the bath to the fuel was rapid and as the density was decreased, the energy transfer rate constant decreased. The results show that in combustion systems with CH4 as a prototype fuel, the super pressure regimes control the fuel heating and combustion processes.
Introduction
Combustion is an important process which involves synchronized multiphase fluid dynamics, chemical kinetics, heat transfer and the chaotic mixing of different species.1 Among these processes, heat transfer is very crucial because of its strong relation to the energy utilization efficiency of the system during combustion. On a microscopic scale, energy transfer between a molecule and its surroundings takes place depending on the degrees of freedom of the molecule and the nature of the surroundings.2,3 In order to simulate the energy transfer processes, N2 gas is used as a surrounding gas, since the contribution of N2 is large in the ambient air. Further, N2 has been shown to be a dominant collision partner in lean combustion flames.4–6 Since, practical fuels are complex mixtures of hydrocarbons, understanding the energy transfer between the hydrocarbons and the surrounding gas is vital to evaluate the energy utilization efficiency of fuels. Further, energy transfer efficiency is measured in terms of the degree of vibrational excitation and the nature of the deactivating collision partner.7,8 The combustion of fuel in a dense supercritical fluid at high pressures9–11 has potential applications in jet propulsion, tertiary oil and gas recovery, toxic waste treatment etc. In high-pressure combustion devices such as propellant rocket motors, the comportment of fuel in dense fluids and the associated chemical kinetics play a vital role in determining the efficacy of combustion processes.12
To develop a high-performance aircraft, fuel is the main heat sink and prior to combustion, the temperature of jet fuel rises. The most heat generating combustion reaction is with CH4, the most abundant natural gas with reaction enthalpy of −890.7 ± 0.4 kJ mol−1.13 As such, heating of CH4 by the surrounding gas to increase combustion efficiency will be of interest in the context of energy utilization. In the present work, CH4 is used as a model fuel and is heated from 300 to 1000 K in N2 bath at pressures from the binary collision regime and the super pressure regime by means of chemical dynamics simulations. The energy transfer process between thermalized N2 bath (1000 K) and CH4 (300 K) is determined by crucial factors such as energy and temperature difference between CH4 and N2 bath as well as the energy and temperature difference of CH4 before and after complete equilibration with the bath. A unified protocol for simulating liquid and gas phase intermolecular energy transfer was reported in our earlier work7 and applied for energy transfer from a hot N2 bath to cold C6F6 molecule.14 This simulation procedure will be followed in the present work to study the intermolecular energy transfer of the system of CH4 in the thermalized N2 bath at 1000 K.
Computational methodology
The chemical dynamics simulations can be accurately performed by the use of proper potential energy function. In previous works,7,8 it was shown that there is an overall very good agreement of theoretical studies of intermolecular energy transfer by chemical dynamics simulation with the accurate intermolecular potentials, compared with experimental studies. The potential energy function for the CH4 + N2 system is expressed as a sum of intramolecular and intermolecular potentials. In CH4–N2 bath, the intermolecular pair potentials such as CH4–CH4, N2–N2 and CH4–N2 determine the properties of the system. The intramolecular potential for CH4 is obtained in terms of stretching and bending coordinates. The intermolecular potential for N2–N2 was presented and described in previous publications7,8 and is used in the current simulations. The CH4–N2 intermolecular potential is calculated by performing electronic structure calculations at CCSD(T)/aug-cc-pVTZ level of theory. The ab initio potential energy curves along with the analytic potential fitted using two body potentials according to Buckingham potential using genetic algorithm15 are presented in Fig. 1. In order to obtain the intermolecular potential of CH4–N2, the electronic structure calculations for four orientations were performed, as shown in Fig. 1. In the panel (a), N2 molecule approaches along C–H bond axis. In the panel (b), the N2 molecule approaches bisecting the H–C–H angle of CH4. In panels (c) and (d), the midpoint of N2 molecule approaches the carbon atom directly and along the C–H bond axis perpendicularly, respectively. The V0 and R0 by ab initio calculation for a–d are 0.02 kcal mol−1 and 4.1 Å, −0.02 kcal mol−1 and 3.9 Å, −0.01 kcal mol−1 and 3.5 Å, and −0.01 kcal mol−1 and 3.2 Å. The parameters obtained from potential energy function were used for chemical dynamics simulations.
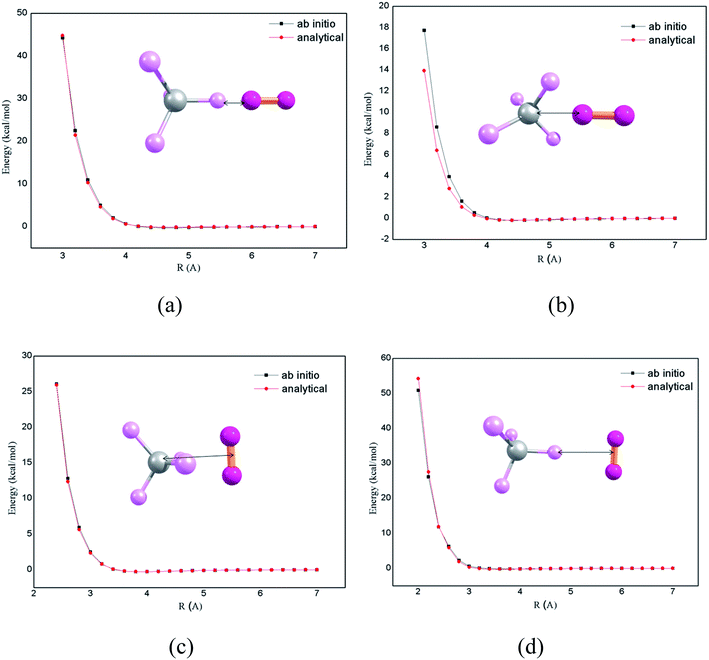 |
| | Fig. 1 CH4–N2 interaction potential energy for four different orientations. The ab initio curves are calculated at CCSD(T)/aug-cc-pVTZ level. The analytical curves are generated by fitting the ab initio potential to the two body potentials according to Buckingham potential. | |
The simulations were performed by considering CH4 at a temperature of 300 K and placing the N2 bath with 1000 N2 molecules at 1000 K temperature and collisional energy transfer takes place from N2 bath to CH4, thereby heating the “cold” CH4. The simulations were studied at different densities ranging from 1000 kg m−3 to 30 kg m−3. Classical microcanonical sampling is used for the simulations accounting for the initial vibrational energy of 5.3 kcal mol−1 for CH4 at 300 K as implemented in the condensed phase version of VENUS chemical dynamics program.7,16,17 The initial translational and rotational energies of CH4 are chosen from their thermal distributions at 300 K. Also, the initial translational, vibrational and rotational energy of N2 molecules were prepared by their thermal distribution at 1000 K. The procedure for choosing the initial conditions for the molecule and the bath are described in our earlier studies.14,18,19 Collisional activation of CH4 in N2 bath was simulated by placing CH4 at the center of a cubical box and CH4 was surrounded by thermally equilibrated N2 bath at 1000 K. The simulations were performed using periodic boundary conditions (PBCs) and a neighbour list algorithm with a cut-off distance of 15 Å. A set of 100 trajectories with random initial conditions for both CH4 and N2 bath were calculated. For the densities, 800 and 1000 kg m−3, the trajectories were integrated upto 6000 ps and for the remaining densities, the trajectories were integrated upto 8000 ps, with 2 fs of integration step size.
Results and discussion
The average total energy of CH4, 〈E(t)〉 versus time for N2 bath densities of 30, 60, 120, 300, 500, 800 and 1000 kg m−3 averaged over 100 trajectories is shown in Fig. 2. The 〈E(t)〉 are fit by a biexponential function given in eqn (1) and the fitting parameters are tabulated in Table 1.| |
〈E(t)〉 = [E(∞) − E(0)](1 − f1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(−k1t) − f2exp(−k2t)) + E(0) exp(−k1t) − f2exp(−k2t)) + E(0)
| (1) |
where f1 + f2 = 1, E(0) and E(∞) are the initial and final energies of CH4, k1 and k2 are the rate constants.
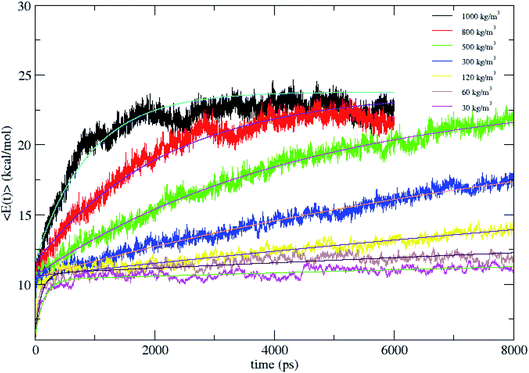 |
| | Fig. 2 Average energy of CH4 versus time for N2 bath densities of 1000, 800, 500, 300, 120, 60, and 30 kg m−3 and this was fit to biexponential equation. The energies were averaged over 100 trajectories. | |
Table 1 Parameters for fits to 〈E(t)〉 for various bath densities
| ρ (kg m−3) |
E(∞) (kcal mol−1) |
f1 |
f2 |
k1 (s−1) |
k2 (s−1) |
C1 |
C2 |
| 1000 |
23.8 |
0.3185 |
0.6815 |
1.42559 |
0.00103835 |
0.001426 |
1.04 × 10−6 |
| 800 |
23.8 |
0.3095 |
0.6904 |
1.22559 |
0.00049534 |
0.001532 |
6.19 × 10−7 |
| 500 |
23.8 |
0.3090 |
0.6910 |
0.9315 |
0.0002331 |
0.001863 |
4.66 × 10−7 |
| 300 |
23.8 |
0.3078 |
0.6921 |
0.7315 |
7.13296 × 10−5 |
0.002438 |
2.38 × 10−7 |
| 120 |
23.8 |
0.2965 |
0.7034 |
0.010821 |
0.109 × 10−5 |
9.02 × 10−5 |
9.08 × 10−9 |
| 60 |
23.8 |
0.2987 |
0.7012 |
0.005499 |
0.57 × 10−6 |
9.17 × 10−5 |
9.5 × 10−9 |
| 30 |
23.8 |
0.2930 |
0.7069 |
0.002939 |
2.80 × 10−7 |
9.8 × 10−5 |
9.33 × 10−9 |
Since from statistical mechanics, the final energy of fully equilibrated CH4 at 1000 K which includes translational, rotational and vibrational energy should be 23.8 kcal mol−1, all the energy curves in Fig. 2 are fit to this energy value. E(∞) is fixed with the value for the fitting. 〈E(t)〉 is highly non-exponential and the fluctuations in the energy curve are due to the relatively small size of the molecule and there is a strong oscillation between the kinetic and potential energy. As given in Table 1, as the density decreases, the weight of the larger rate constant component (k1) decreases. That is, at high pressures frequent collisions in the bath populate molecular vibrational states and lead to intramolecular processes in nearly equilibrium ensembles which are rate determining.20 As the solvent density is lowered, the energy transfer dynamics enters the independent, single collision limit.14,18,19 Here, the rate constants k1 and k2 are proportional to the bath density, ρ with proportionality constants C1 and C2 defined by k1 = C1 × ρ and k2 = C2 × ρ, with the criterion for single collision limit being C1 and C2 becoming independent. The proportionality constant C1 is same for the densities 120, 60 and 30 kg m−3, with C2 also showing the same trend. Thus C1 and C2 become independent of density between 120 and 30 kg m−3, suggesting that the simulation in the 120 kg m−3 is in the single collision limit as predicted in earlier studies.14,18,19
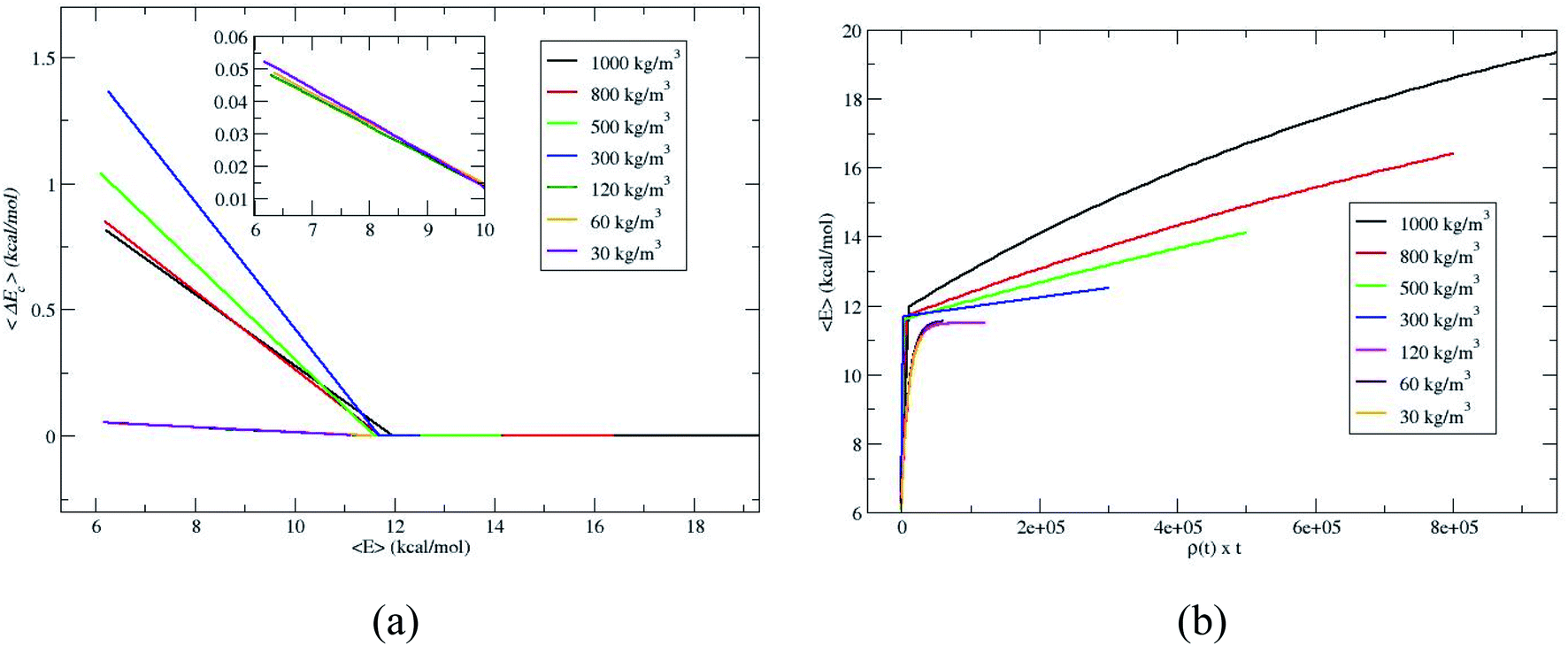
To further verify whether the C1 and C2 are identical for the densities 120, 60 and 30 kg m−3, the average energy transfer per collision is calculated.14,18,19 The average energy transfer per unit time for the value of 〈E(t)〉 in eqn (1) is obtained by differentiating with respect to time, d〈E(t)〉/dt. In the single collision limit, the average energy transfer per collision is obtained by dividing d〈E(t)〉/dt by the collision frequency, ω, i.e.,
| | |
〈ΔEc〉 = [d〈E(t)〉/dt]/ω
| (2) |
The collision frequency of ω = 2.8 × 108 s−1 is used in the calculation of 〈ΔEc〉. Thus using eqn (2) and 〈E(t)〉 in Fig. 2 and the collision frequency, the average energy transferred per collision, 〈ΔEc〉 as a function of 〈E〉 for the different densities is shown in Fig. 3. 〈ΔEc〉 is a composite of energy transfer from and to CH4, i.e. 〈ΔEupc〉 and 〈ΔEdownc〉, respectively. At large 〈E〉,〈ΔEupc〉 dominates and at long times, when equilibrium is attained, 〈ΔEupc〉 and 〈ΔEdownc〉 are same and 〈ΔEc〉 equals zero. The curves for 120, 60 and 30 kg m−3 are nearly identical, coinciding with the identical proportionality constants for 120, 60 and 30 kg m−3. The slopes of 〈ΔEc〉 versus 〈E〉 plots become identical at or below ρ for single collision limit as shown in the inset of Fig. 3a. These discussions reveal that ρ = 30 kg m−3 is the good depiction of single collision limit and the collisional activation rate constant is directly proportional to the density. A plot of 〈E〉 versus ρ × t is shown in Fig. 3b to illustrate the single collision regime more clearly. The curves at densities 120, 60 and 30 kg m−3 coincide depicting that the system is in or near the single collision regime for these densities.
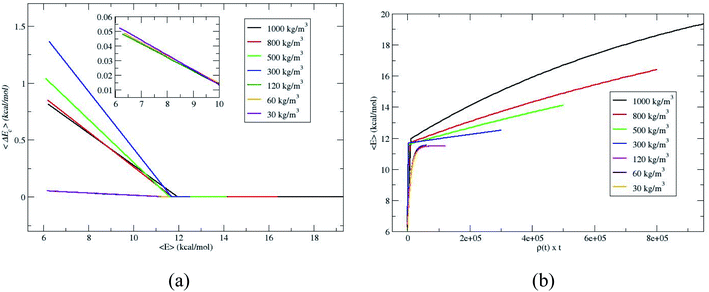 |
| | Fig. 3 Average energy transferred per collision, 〈ΔEc〉, for the simulations. | |
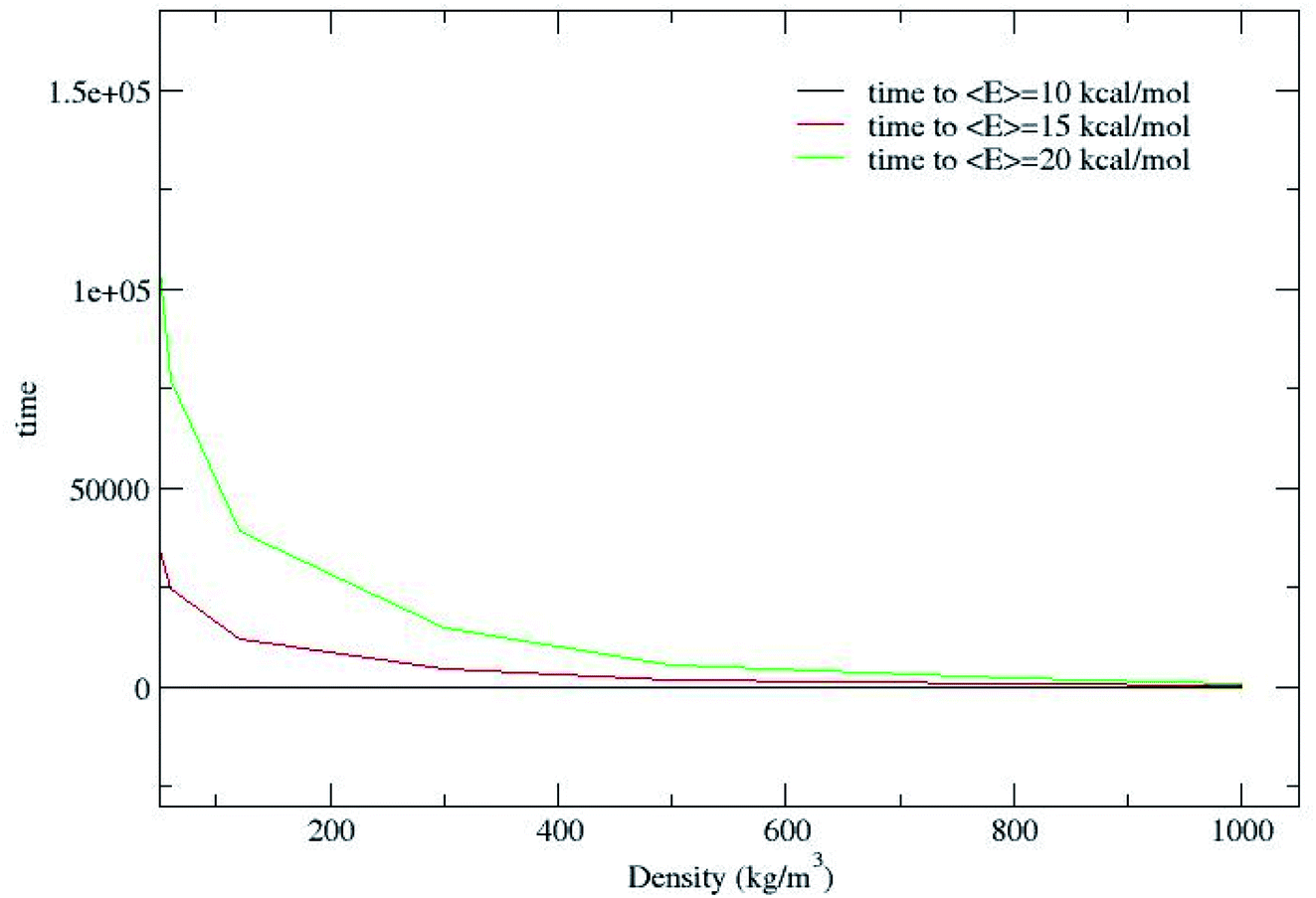
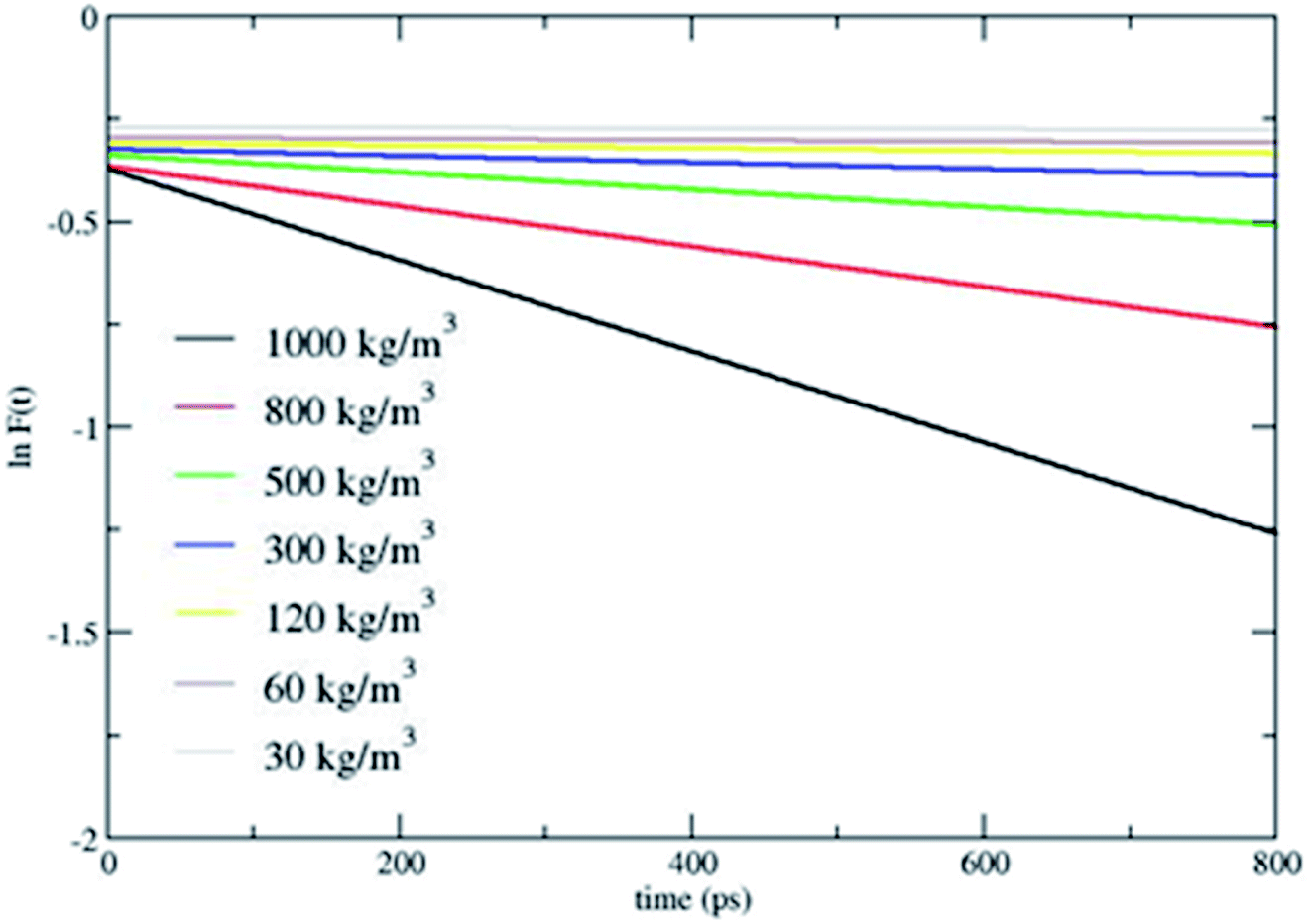
Fig. 2 shows that CH4 is heated at relatively lesser time at higher densities 1000 and 800 kg m−3 and as the density is decreased, energy transfer from the bath to CH4 is very moderate. A more precise plot illustrating the time required for each density to reach average energies of 10, 15 and 20 kcal mol−1 is presented in Fig. 4. The energy transfer of 10 kcal mol−1 from N2 bath to CH4 is instantaneous independent of the density of the bath. The time required to transfer 15 and 20 kcal mol−1 energy from the bath to CH4 is relatively shorter for densities 500, 800 and 1000 kg m−3 densities, whereas at low densities relatively longer time is required to transfer energy from N2 bath to heat CH4 and equilibration of the system. The plot shown in Fig. 4 is tantamount to assume that the fuels are heated rapidly at higher bath pressures. The dynamics of energy transfer from the bath to CH4 can thus be expressed as lnF(t) versus time for each density for clarity purpose, where lnF(t) = ln[(f1) − k1T] or ln[(f2) − k2T] as shown in Fig. 5. The energy transfer rate constant plots show linear behaviour and at the single collision regime, the magnitude of the collisional activation rate constant, k is similar and the collisional activation is remarkably sensitive to the high pressures.
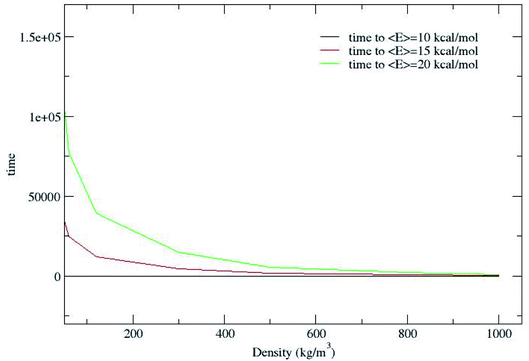 |
| | Fig. 4 Time (ps) to reach average energies of 10, 15 and 20 kcal mol−1 for each density. | |
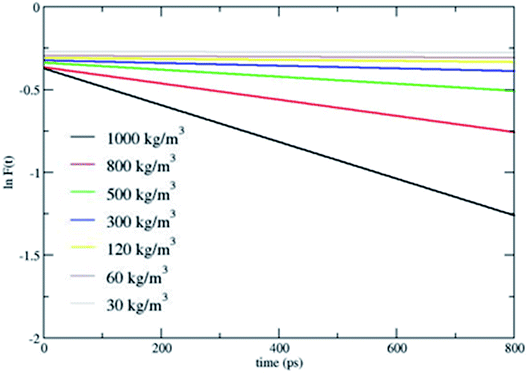 |
| | Fig. 5 lnF(t) versus time. lnF(t) = ln[(f1) − k1T] or ln[(f2) − k2T]. | |
Conclusions
To summarize, in the present study the energy transfer from a thermalized 1000 K N2 bath to CH4 at 300 K is studied using classical chemical dynamics simulations. The simulations were performed for different N2 bath densities ranging from 1000 kg m−3 to 30 kg m−3. The average energy of the ensemble of CH4, 〈E〉 versus time is well fit by the biexponential eqn (1) and the fitting parameters f1 is smaller and f2 is larger and the longer time activation rate constant k2 is smaller than k1. The average energy transfer per collision 〈ΔEc〉, is obtained and 〈ΔEc〉 versus 〈E〉 is plotted. The plots are similar for 120, 60 and 30 kg m−3 densities indicating the single collision regime, whereas at higher densities 〈ΔEc〉 decreases with the increase in density. The average energy transferred from the bath to CH4 per collision is 〈ΔEupc〉 = 0.02 kcal mol−1 at the initial CH4 temperature of 300 K. The energy transfer from the bath to CH4 is comparatively faster at higher bath densities than at the lower densities. The collisional activation rate constant decreases as the density is decreased. Thus, the heating of fuels is largely determined by high pressures and the combustion processes is controlled at these high pressures. The density dependence reveals that over wide density ranges from low pressure gases to highly compressed gases, various factors influence the intermolecular energy transfer and the diffusion control induced by the surrounding medium at high densities is one of the major factors. At supercritical pressures, the combustion mechanism is diffusion controlled and the rate will increase with pressure. Incorporation of diffusion controlled rate in the intramolecular dynamics of N2 bath has more impact at gas/liquid phase transition.20,21 In the diffusion controlled range, bimolecular reactions pre-dominate. Hence, the diffusion control is expected not to have much influence on the collisional activation rate constant for intermolecular energy transfer process in the current study. To conclude, in combustion systems the fuel is heated rapidly at super pressure regimes than at binary collision regimes and the energy transfer from the bath at pressures higher than the supercritical pressures to the fuels will be of future interest.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The research reported here is based upon work supported by the Air Force Office of Scientific Research (AFOSR) under Grant No. FA9550-17-1-0119 and the Robert A. Welch Foundation under Grant No. D-0005. The simulations were performed on the Quanah computer cluster of the High Performance Computing Center (HPCC) of Texas Tech University and the Chemdynm computer cluster of the Hase Research Group.
References
- M. Janbozorgi, K. E. Far and H. Metghalchi, Combustion Fundamentals in Handbook of Combustion, Fundamentals and Safety, ed. M. Lackner, F. Winter, and A. K. Agarwal, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2010, vol. 1 Search PubMed.
- D. C. Tardy and B. S. Rabinovitch, Intermolecular vibrational energy transfer in thermal unimolecular systems, Chem. Rev., 1977, 77, 369–408 CrossRef CAS.
- I. Oref and D. C. Tardy, Energy transfer in highly excited large polyatomic molecules, Chem. Rev., 1990, 90, 1407–1445 CrossRef CAS.
- R. A. Copeland, M. L. Wise and D. R. Crosley, Vibrational energy transfer and quenching of OH (A2∑+, v' = 1), J. Phys. Chem., 1988, 92, 5710–5715 CrossRef CAS.
- P. H. Paul, A model for temperature-dependent collisional quenching of OH A2∑+, J. Quant. Spectrosc. Radiat. Transfer, 1994, 51, 511–524 CrossRef CAS.
- P. H. Paul, Vibrational energy transfer and quenching of OH A2∑+ (v' = 1) measured at high temperatures in a shock tube, J. Phys. Chem., 1995, 99, 8472–8476 CrossRef CAS.
- A. K. Paul, S. C. Kohale, S. Pratihar, R. Sun, S. W. North and W. L. Hase, A unified model for simulating liquid and gas phase, intermolecular energy transfer: N2 + C6F6 collisions, J. Chem. Phys., 2014, 140, 194103 CrossRef PubMed.
- A. K. Paul, S. C. Kohale and W. L. Hase, Bath model for N2 + C6F6 gas-phase collisions. details of the intermolecular energy transfer dynamics, J. Phys. Chem. C, 2015, 119, 14683–14691 CrossRef CAS.
- W. Schilling and E. U. Franck, Combustion and diffusion flames at high pressures to 2000 bar, Ber. Bunsen-Ges. Phys. Chem, 1988, 92, 631 CrossRef CAS.
- E. U. Franck, High pressure combustion and flames in supercritical water, Proc. Znt. Symp. Supercritical Fluids, 1991, p. 91 Search PubMed.
- J. Zhang, S. Wang, M. Ren, J. Lu, S. Chen and H. Zhang, Effect mechanism of auxiliary fuel in supercritical water: a review, Ind. Eng. Chem. Res., 2019, 58(4), 1480–1494 CrossRef CAS.
- P. E. Savage, S. Gopalan, T. I. Mizan, C. J. Martino and E. E. Brock, Reactions at supercritical conditions: applications and fundamentals, AIChE J., 1995, 41, 1723–1778 CrossRef CAS.
- D. A. Pittam and G. Pilcher, Measurements of heats of combustion by flame calorimetry. Part 8.-methane, ethane, propane, n-butane and 2-methylpropane, J. Chem. Soc., Faraday Trans., 1972, 68, 2224–2229 RSC.
- A. K. Paul, D. Donzis and W. L. Hase, Collisional intermolecular energy transfer from a N2 bath at room temperature to a vibrationally “Cold” C6F6 molecule using chemical dynamics simulations, J. Phys. Chem. A, 2017, 121, 4049–4057 CrossRef CAS PubMed.
- J. M. C. Marques, V. F. Prudente, F. B. Pereira, M. M. Almeida, A. M. Maniero and C. E. Fellows, A new genetic algorithm to be used in the direct fit of potential energy curves to ab initio and spectroscopic data, J. Phys. B: At., Mol. Opt. Phys., 2008, 41, 085103 CrossRef.
- W. L. Hase, R. J. Duchovic, X. Hu, A. Komornicki, K. F. Lim, K. D. –H. Lu, G. H. Peslherbe, K. N. Swamy, S. R. Vande Linde, A. Varandas, et al., VENUS96: A General Chemical Dynamics Computer Program, Texas Tech University, Lubbock, TX, 2005 Search PubMed.
- X. Hu, W. L. Hase and T. Pirraglia, Vectorization of the general monte carlo classical trajectory program VENUS, J. Comput. Chem., 1991, 12, 1014–1024 CrossRef CAS.
- H. Kim, A. K. Paul, S. Pratihar and W. L. Hase, Chemical dynamics simulations of intermolecular energy transfer: azulene + N2 collisions, J. Phys. Chem. A, 2016, 120, 5187–5196 CrossRef CAS PubMed.
- H. Kim, H. N. Bhandari, S. Pratihar and W. L. Hase, Chemical dynamics simulation of energy transfer: propylbenzene cation and N2 collisions, J. Phys. Chem. A, 2019, 123, 2301–2309 CrossRef CAS PubMed.
- J. Troe, Elementary reactions in compressed gases and liquids: from collisional energy transfer to diffusion control, J. Phys. Chem., 1986, 90, 357–365 CrossRef CAS.
- H. Hippler, V. Schubert and J. Troe, A low-pressure extension of the Stokes–Einstein relationship, Ber. Bunsen-Ges. Phys. Chem, 1985, 89, 760–763 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab,
Hyunsik Kim*a and
William L. Hase†
a
*ab,
Hyunsik Kim*a and
William L. Hase†
a