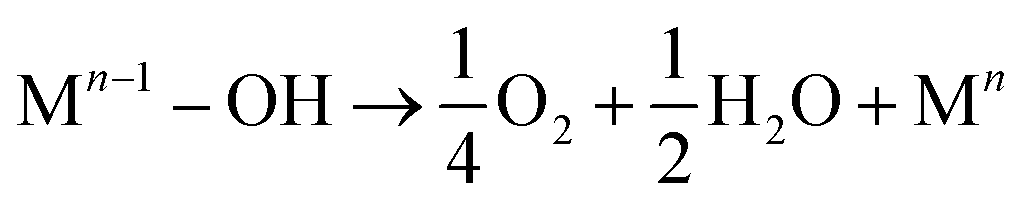DOI:
10.1039/D1RA02788C
(Paper)
RSC Adv., 2021,
11, 19943-19955
Study on the mechanism of synthetic (Ce,La)CO3F sulfuric acid acidification and NH3-SCR loaded with Mn and Fe
Received
10th April 2021
, Accepted 18th May 2021
First published on 3rd June 2021
Abstract
A hydrothermal method was used to synthesise (Ce,La)CO3F grain simulated minerals, in accordance with the Ce–La ratio of bastnaesite in the mineralogy of the Bayan Ebo process. The NH3-SCR catalytic activity of the synthesised (Ce,La)CO3F was improved by loading transition metals Mn and Fe and sulphuric acid acidification treatments. The activity test results showed that the catalysts which were simultaneously acidified with sulphuric acid and loaded with transition metals Mn and Fe had a NOx conversion of 92% at 250 °C. XRD, SEM, XPS and in situ Fourier transform infrared spectroscopy (FTIR) were used to investigate the physical phase structure, surface morphology, reaction performance and mechanism of the catalysts, to provide theoretical guidance for the specific reaction path of cerium fluorocarbon ore in the NH3-SCR reaction. The results showed that the introduction of transition metals and sulphuric acid greatly increases the proportion of adsorbed oxygen (Oα) and facilitates the adsorption of NH3 and NO. The catalyst surface metal sulphate and metal oxide species act as the main active components on the catalyst surface to promoted the reaction, and cracks and pores appear on the surface to facilitate the adsorption of reactive gases. The reaction mechanism of the SO42−–Mn–Fe/(Ce,La)CO3F catalyst, and characterisation of the adsorption and conversion behaviour of the reactive species on the catalyst surface, were investigated by Fourier transform infrared spectroscopy (FTIR). The results showed that the catalyst follows the E–R and L–H mechanisms throughout the reaction, with the E–R mechanism being the main reaction. The reaction species were NH4+/NH3 species in the adsorbed state and NO. The NH3(ad) species on the Lewis acidic site is the main NH3(g) adsorbed species for the reaction, bonded to Ce4+ in the carrier (Ce,La)CO3F to participate in the acid cycle reaction, and undergo a redox reaction on the catalyst surface to produce N2 and H2O. The SO42− present on the catalyst surface can also act as an acidic site for the adsorption of NH3. The above results indicated the excellent performance of the SO42−–Mn–Fe/(Ce,La)CO3F catalyst, which provided a theoretical basis for the high value utilization of bastnaesite.
1 Introduction
In recent years, domestic coal consumption has been increasing. The air pollution problem caused by coal combustion is becoming more and more serious. The NOx produced from coal, diesel and gasoline will cause a series of environmental problems,1 so nitrogen oxides emission reduction is particularly important. The existing denitration catalyst V2O5–WO3(MoO3)/TiO2 still has some problems such as toxicity and narrow active temperature window (320–400 °C), so it is necessary to develop new catalysts. The Bayan Obo Mine is a super large-scale polymetallic symbiosis deposit dominated by iron, rare earths and niobium, with the largest reserve of rare earth resources in the world.2,3 The composition of rare earth minerals is complex and there are many kinds of rare earth elements. Our research group has shown that rare earth minerals as denitration catalysts have better catalytic activity, better sulfur resistance, and can realize the reuse of resources. Meng et al.4 took the rare earth concentrate rich in Ce oxide from Bayan Obo as the carrier for catalytic materials, and obtained a series of mineral catalytic materials by an impregnation method of iron nitrate solution and microwave roasting. The results of denitration activity show that when the solution of iron nitrate is 0.5 mol L−1, the denitration efficiency of rare earth concentrate loaded with Fe2O3 active powder is the best, and the denitration rate can reach 80.6% when the reaction temperature is 350 °C. Zhang et al.5,6 obtained the active component of denitration catalyst by used the roasting weak acid–weak base leaching method to remove impurities and modify the rare earth concentrate powder. The activity test results showed that the denitration efficiency of the catalyst sample was 92.8% at 400 °C. Jiao et al.7 obtained rare earth concentrate catalyst by roasting Bayan Obo rare earth concentrate. It was found that SO2 significantly promoted the denitration performance of rare earth concentrate catalyst. The NO conversion rate increased from 28% to 50% at 300 °C, and from 42% to 75% at 350 °C. FT-IR results showed that the adsorption of SO2 increased the Brønsted acid sites on the surface of the rare earth concentrate catalyst, and the adsorption capacity of NH3 on the catalyst is enhanced, so it is beneficial to improve the activity of the catalyst.
Rare earth mineral catalysts have excellent NH3-SCR denitration performance, but the mechanism is still unclear due to the complex internal composition of the mineral. Currently, reports on the reaction mechanism of NH3-SCR are mainly about pure substances. Wu Xiaomin8 et al. enhanced the oxygen vacancy defect by using Ce-doped manganese oxide octahedral molecular sieve (Ce-K-OMS-2), and proved by density functional theory (DFT+U) calculation that NH3 tends to adsorb on the Ce part of Ce-K-OMS-2, and follows Langmuir–Hinshelwood mechanism, NO is more likely to adsorb at the O site. F. Eigenmann et al.9 studied the FT-IR spectra of Mn–Ce mixtures. The results showed that NOx and NH3 were adsorbed at different locations. Successive adsorption measurements of the reactants showed similar absorptions to those measured individually, indicating no interference between the adsorbed reactants. A study by varying the order of absorption of the reactants (NOx, NH3) revealed that at 100–150 °C the catalyst followed the Eley–Rideal mechanism, where the adsorbed ammonia reacts with NOx in the gas phase, while the adsorbed NOx does not change significantly. Ge et al.10 investigated the denitrification reaction mechanism by loading Mn onto CeO2 microspheres. The results showed that the Mn/CeO2 catalyst was pre-sorbed with NH3 1 h at 90 °C, followed by the addition of NO + O2. The change of catalyst surface species was observed, and the catalyst surface followed the E–R mechanism. NO + O2 was preadsorbed at 90 °C, then NH3 was added. It was found that the surface followed L–H mechanism. Finally, it is concluded that the surface of the Mn/CeO2 catalyst follows both the E–R and L–H mechanism.
The complex mineral intergrowth relationship create difficulties in defining the catalytic mechanism and conditions for catalytic reactions. Existing characterisation has no way of to determine the specific reaction changes of a multi-element doped catalyst, so the role and mechanism played by each mineral phase in the concentrate needs to be investigated on a case-by-case basis. As the main mineral phase of rare earth minerals, bastnaesite plays a key role in the catalytic reaction. Since pure bastnaesite cannot be extracted by existing technical means. I have synthesised (Ce,La)CO3F by hydrothermal methods using analytical purity, the XRD and cell parameters show that the synthesised (Ce,La)CO3F is in good agreement with the higher pure bastnaesite from the Bayan Obo mine in terms of diffraction peaks, crystal plane spacing and prism length. The catalytic performance of (Ce,La)CO3F was improved by loading transition metals Mn, Fe and sulphate acidification, and to investigate the physical properties, reaction performance, reaction mechanism and reaction pathway of NH3-SCR by XRD, SEM, XPS and Fourier infrared spectroscopy. The specific mechanism within cerium fluorocarbon is clarified to provide theoretical guidance significance for the reaction mechanism of rare earth mineral catalysts. The specific mechanism within bastnaesite is clarified to provide theoretical guidance significance for the reaction mechanism of rare earth mineral catalysts.
2 Experimental method
2.1 Materials and methods
Reagents used in the experiment: Ce(NO3)3·6H2O (mass fraction), analytically pure, Tianjin Komiou Chemical Reagent Co; La(NO3)3·6H2O (mass fraction), analytically pure, Tianjin Komiou Chemical Reagent Co; NaHCO3, analytical purity, Tianjin Fengchuan Chemical Reagent Technology Co; NaF, analytical purity, Tianjin Fengchuan Chemical Reagent Technology Co; 50% Mn(NO3)2, analytical purity, Tianjin Fengchuan Chemical Reagent Technology Co; 98% sulfuric acid, AR, Sinopharm Chemical Reagent Co.
2.2 Preparation of catalyst
Synthesis of (Ce,La)CO3F by hydrothermal method. A certain amount of Ce(NO3)3·6H2O, La(NO3)3·6H2O, NaF and NaHCO3 was placed in 100 ml of PTFE liner at room temperature, 80 ml of distilled water was poured into the PTFE liner with constant stirring. Put the PTFE liner into an autoclave at atmospheric pressure and 120 °C for 2 h with stirring and heating for hydrothermal reaction. After cooling, the mixture was filtered and dried at 50 °C to obtain the synthesized (Ce,La)CO3F catalyst, which was treated at different calcination temperatures. It was found that among the catalysts prepared by calcination at 200–600 °C, the synthesis of (Ce,La)CO3F by calcination at 500 °C for 2 h had the best catalytic performance. Synthetic (Ce,La)CO3F was used as a carrier to load the transition metals Mn and Fe by impregnation. Mn(NO3)2 solutions with different mass fractions were calculated and added to the synthetic (Ce,La)CO3F, with distilled water to 50 ml. The mixture was ultrasonically impregnated at room temperature and pressure for 2 h, then filtered and dried. Roasted at 500 °C for 2 h in a muffle furnace to produce Mn/(Ce,La)CO3F. The Fe/(Ce,La)CO3F catalysts were produced by adding different proportions of Fe2O3 powder according to the molar ratio of Ce to Fe in (Ce,La)CO3F, following the same method. The optimum loading amounts of Mn and Fe were loaded simultaneously by ultrasonic impregnation to produce the Mn + Fe/(Ce,La)CO3F catalyst. Acidification of synthetic (Ce,La)CO3F. Different concentrations of sulphuric acid were added to the synthetic (Ce,La)CO3F and impregnated for 2 h, dried at 50 °C and roasted in a microwave heating oven at 350 °C for 25 min to obtain the SO42−/(Ce,La)CO3F catalyst. Finally the optimum loading of transition metal was loaded onto synthetic (Ce,La)CO3F treated with the optimum acid concentration, impregnated at room temperature, dried at 50 °C and roasted for 2 h in a muffle furnace at 400 °C to obtain the SO42−–Mn–Fe/(Ce,La)CO3F catalyst.
2.3 Catalytic performance test
The experiments were carried out in a reaction apparatus with quartz tubes for testing the activity of the catalyst NH3-SCR. The reaction apparatus consists of a gas mixing tank-flow meter, standpipe furnace, quartz tube, Fourier infrared spectroscopy flue gas analyser and computer data acquisition system. The standpipe furnace is from Nanjing Boynton Instrument Technology Co. It is heated using a silicon–molybdenum rod model 1800 with a rated temperature of 1600 °C and an inner diameter of 20 mm and a length of 1.2 m. The Fourier infrared spectroscopy (FTIR) flue gas analyser and data acquisition system were produced in Finland and the model number was GASMET-DX4000. The simulated gas components were NH3 500 ppm, NO 500 ppm, O2 at 3% of the total volume fraction and N2 as the equilibrium gas, with a total gas flow of 100 ml min−1, an air velocity of approximately 8000 h−1 g−1 and a catalyst dosage of 0.6 g for each test.
2.4 In situ infrared microscopic experiment design
(1) Change with time at the optimal denitration temperature. The catalyst was first placed in a reaction cell and dehydrated at 200 °C, using N2 as the carrier gas. When the temperature was raised to 250 °C, data was collected as a background, followed by the introduction of 5% NH3/5% NO + 3% O2 and the 5–30 min curve was recorded. After recording, the NH3 was turned off and purged with N2 for 30 min in order to test the stability of the generated species, and finally the recorded data were used to subtract the background.
(2) Thermal stability experiments. The catalyst was first dehydrated at a constant temperature of 400 °C for 30 min, with N2 as the carrier gas. As the temperature was dropped from 400–50 °C, a curve was recorded at 50 °C intervals as background. NH3/NO + O2 was adsorbed at 50 °C for 1 h and then N2 was introduced into the reaction cell. A curve was recorded at 50 degree intervals from 50–400 °C, and finally the recorded curves were subtracted from the background.
(3) Transient pre-sorption experiments. This experiment investigates the mechanism of NH3-SCR reaction at the optimum denitrification temperature. Firstly, dehydration was carried out at a constant temperature of 200 °C for 30 min, and the background was recorded when the temperature was raised to 250 °C. Then NH3/NO + O2 was pre-sorbed for 1 hour and data was recorded. N2 purge was used for 10–20 min and the background was recorded. Then NO + O2 was inserted, the data were recorded within 2–30 min, and the background was subtracted from the recorded curve.
3 Results and discussion
3.1 Activity test
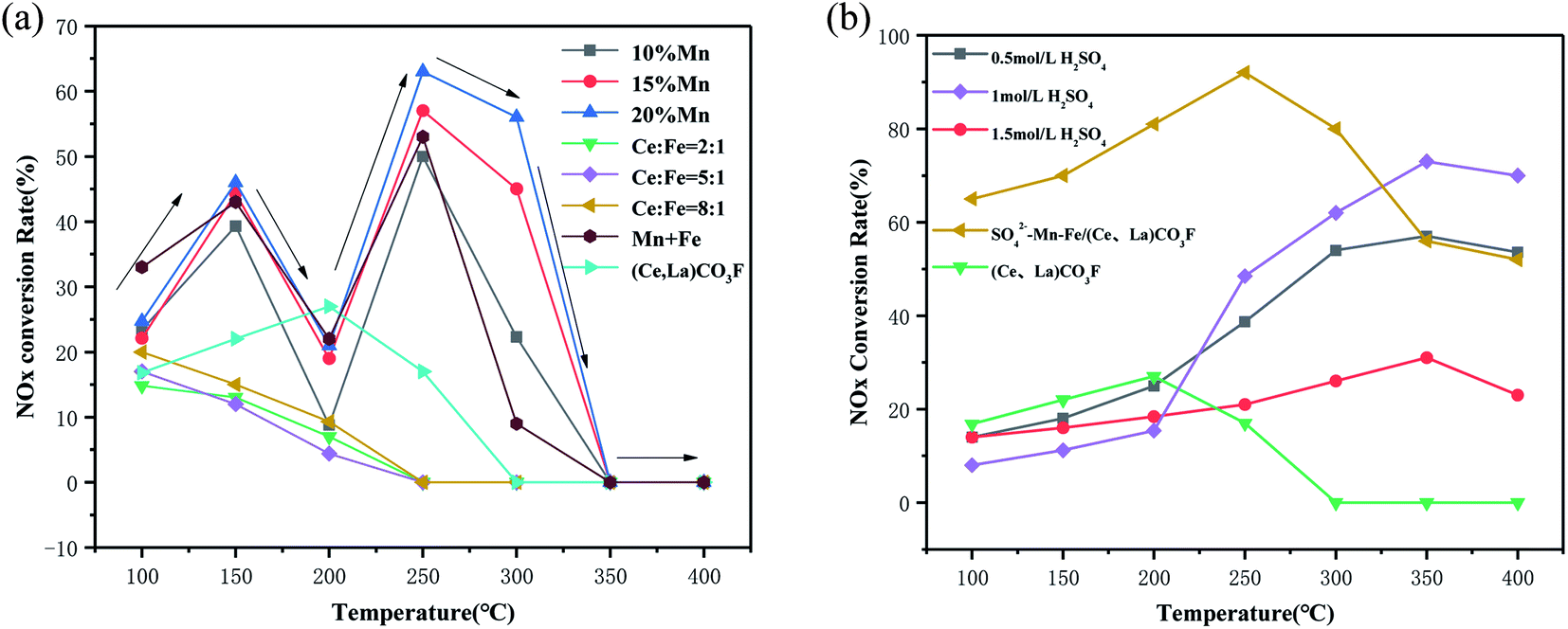
Fig. 1 shows the catalytic activity of the synthetic (Ce,La)CO3F catalyst after roasting, loading with transition metals and acidification with sulphuric acid. Fig. 1(a) shows the NOx conversion of the synthetic (Ce,La)CO3F catalysts loaded with different contents of transition metals. From Fig. 1(a) it can be seen that the NOx conversion of the Mn/(Ce,La)CO3F catalysts with different loadings all increased with temperature from 100–150 °C, but as the temperature continued to increase, the NOx conversion started to decrease at 200 °C, and then rose again at 250 °C to reach the optimum denitrification efficiency. This may occur due to the large amount of NO adsorbed on the catalyst surface in the 100–150 °C range, resulting in a reduced amount of NOx, but NOx is not reacting on the catalyst surface at this point and only occupies the active site. When the temperature reaches 200 °C, NO cannot continue to be adsorbed due to the absence of active sites on the catalyst surface. It was not until 250 °C that the NH3-SCR reaction on the surface of the catalyst began to occur, allowing the catalyst to achieve maximum denitration efficiency. It is also possible that the poor stability of the activated species (NH3/NH4+ and NO2, nitrate, nitrite, etc.) by adsorption on the catalyst surface of NH3 and NO, and are particularly susceptible to temperature effects, with a tendency to decompose at 200 °C, leading to a decrease in activity. The results showed that the catalytic activity was best with 20% Mn loading, and the activity of the Fe loaded catalyst was inhibited compared to (Ce,La)CO3F. This may be due to the fact that the introduction of Fe greatly increases the redox capacity of the catalyst, while the acidic sites on the catalyst surface do not increase, making the adsorption of NH3 less favourable and thus leading to a decrease in activity.
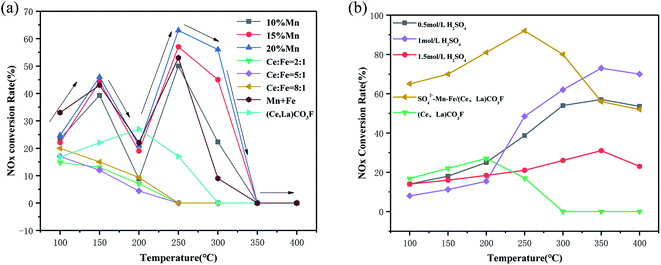 |
| | Fig. 1 NH3-SCR activity of (a) loaded transition metal and (b) sulphate acidification catalysts (reaction conditions: 500 ppm NO, 500 ppm NH3, 3% O2, N2 as balance gas). | |
The synthetic (Ce,La)CO3F was acidified with different concentrations of sulfuric acid to enhance its catalytic activity. Fig. 1(b) shows that (Ce,La)CO3F acidified with 1 mol L−1 sulphuric acid exhibited the highest NOx conversion, which reached 75% at 350 °C. The activity of the (Ce,La)CO3F catalyst was increased due to the introduction of sulphuric acid, which increases the acidic sites on the catalyst surface, the denitrification temperature window is widened compared to untreated (Ce,La)CO3F. Although the activity of the acid-treated (Ce,La)CO3F was improved in the high temperature section, but the activity in the low temperature section was poor. Therefore, in order to improve the activity of the low temperature section, the acid-treated (Ce,La)CO3F was used as a carrier loaded with transition metals Mn–Fe, and roasted at 400 °C to obtain the SO42−–Mn–Fe/(Ce,La)CO3F catalyst. The results showed that the catalytic activity was greatly improved, and the NOx conversion rate was 92% at 250 °C. Comparing the catalysts in Table 1 for the rare earth minerals after roasting, loading and sulphuric acidification, the NOx conversions were all higher than for the synthetic (Ce,La)CO3F, but the differences were not significant. This indicates that (Ce,La)CO3F contributes significantly to the catalytic activity of the rare-earth concentrate, and occupies the main active component of the rare-earth concentrate. Not only that, compared to CeO2 after acidification and doping with K, (Ce,La)CO3F after loading and acidification, the difference in NOx conversion is not significant. This indicates that (Ce,La)CO3F alone has very good catalytic performance as a denitrification catalyst.
Table 1 Comparison of catalytic denitration activities
| Catalyst |
Preparation method |
Roasting temperature |
Reaction conditions |
Optimum NOx conversion |
Literature cited |
| Rare earth concentrate |
Roasted |
500 °C – 4 h |
500 ppm NO, 500 ppm NH3, 4% O2 |
32.2% (500 °C) |
6 |
| Mn/fluorocarbon cerium concentrate |
Impregnation method |
500 °C – 2 h |
500 ppm NO, 500 ppm NH3, 3% O2 |
85% (150 °C) |
11 |
| Fe/rare earth concentrate |
Impregnation method |
350 °C – 2 h |
500 ppm NO, 500 ppm NH3, 3% O2 |
82% (350 °C) |
12 |
| SO42−/rare earth tailings |
Impregnation method |
Microwave 250 °C – 25 min |
500 ppm NO, 500 ppm NH3, 6% O2 |
>80% (350–400 °C) |
13 |
| K/CeO2(SO42−) |
Impregnation method |
500 °C – 4 h |
500 ppm NH3, 500 ppm NO, 5% O2, |
>90% (300–400 °C) |
14 |
3.2 Physical structure and surface morphology analysis
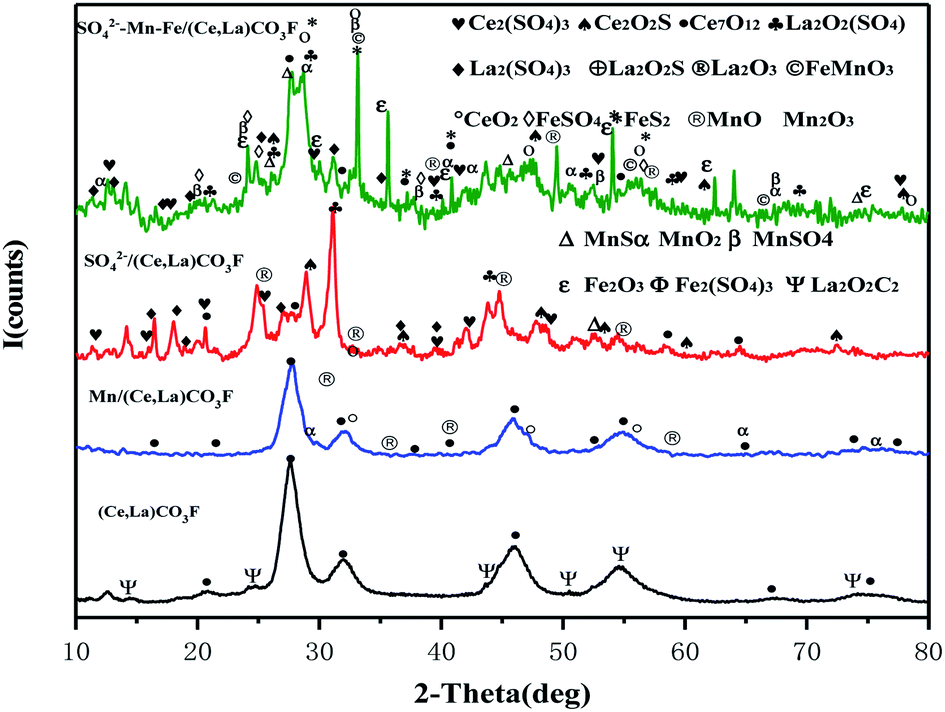
Fig. 2 shows the XRD spectrum of the synthetic (Ce,La)CO3F after loaded transition metals and acidification with sulfuric acid.
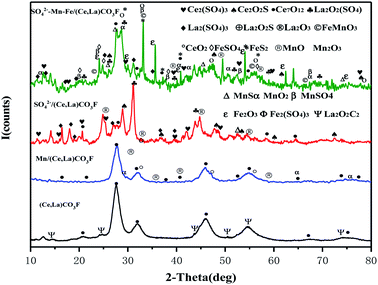 |
| | Fig. 2 XRD diagram of the catalyst. | |
It can be seen from the figure that after roasting of (Ce,La)CO3F, the main species on the catalyst surface are Ce7O12 and La2O2C2. After loaded with Mn, the diffraction peaks of Ce7O12 species on the catalyst surface were reduced, and MnOx species distributed in an amorphous state were detected on the catalyst surface. The amorphous state was favourable for the catalytic activity.15 After acidification with sulphuric acid, Ce2(SO4)3 and La2(SO4)3 species appeared on the catalyst surface. Related studies have shown that denitrification catalysts with metal sulphate species as the active component have higher denitrification activity compared to metal oxides.16 Many species were detected on the surface of the catalysts loaded with Mn, Fe and sulphate acidification at the same time, with characteristic peaks of oxides of different composite metal species appearing. This indicated that the S on the carrier would prevent the metal from dispersing, causing the metal to aggregate to form a solid solution synergistically.
Fig. 3 shows SEM images of the synthetic (Ce,La)CO3F after loaded with transition metals and acidification with sulphuric acid. Scanning electron microscopy was used to observe the surface structure of the catalyst. Fig. 3(a) shows that the surface structure of the synthetic (Ce,La)CO3F catalyst is dense with few pores and gaps, which is not conducive to mass transfer and diffusion during the gas–solid catalytic reaction. Fig. 3(b) shows the surface morphology of the Mn/(Ce,La)CO3F catalyst. It can be observed that the active component is distributed in very fine particles on the surface of the carrier, and that the surface structure is flat, with slightly larger pores and a few cracks on the surface, which are favourable for the adsorption of gases. Fig. 3(c) shows the surface morphology of the catalyst after sulphuric acidification. It can be observed that the morphology of (Ce,La)CO3F was greatly changed by sulphuric acidification, most of them are blocky, with a large number of cracks, which increased the contact surface area. Fig. 3(d) shows the surface morphology of the catalyst after both loaded and acidified. It can be observed that on the basis of the acidification forming a blocky structure, the loaded active components are present on the catalyst surface in granular form, and with a more developed pore structure, which facilitates full contact between the reacting gas and the catalyst. This is probably due to the interaction of Mn and Fe on the surface of the carrier, which induces the conversion of large particles into smaller ones, and increases the active sites. These properties favour the adsorption–desorption behaviour of the gas on the catalyst surface.
 |
| | Fig. 3 From left to right: (Ce,La)CO3F, Mn/(Ce,La)CO3F, SO42−/(Ce,La)CO3F, and SO42−–Mn–Fe/(Ce,La)CO3F. | |
3.3 XPS analysis
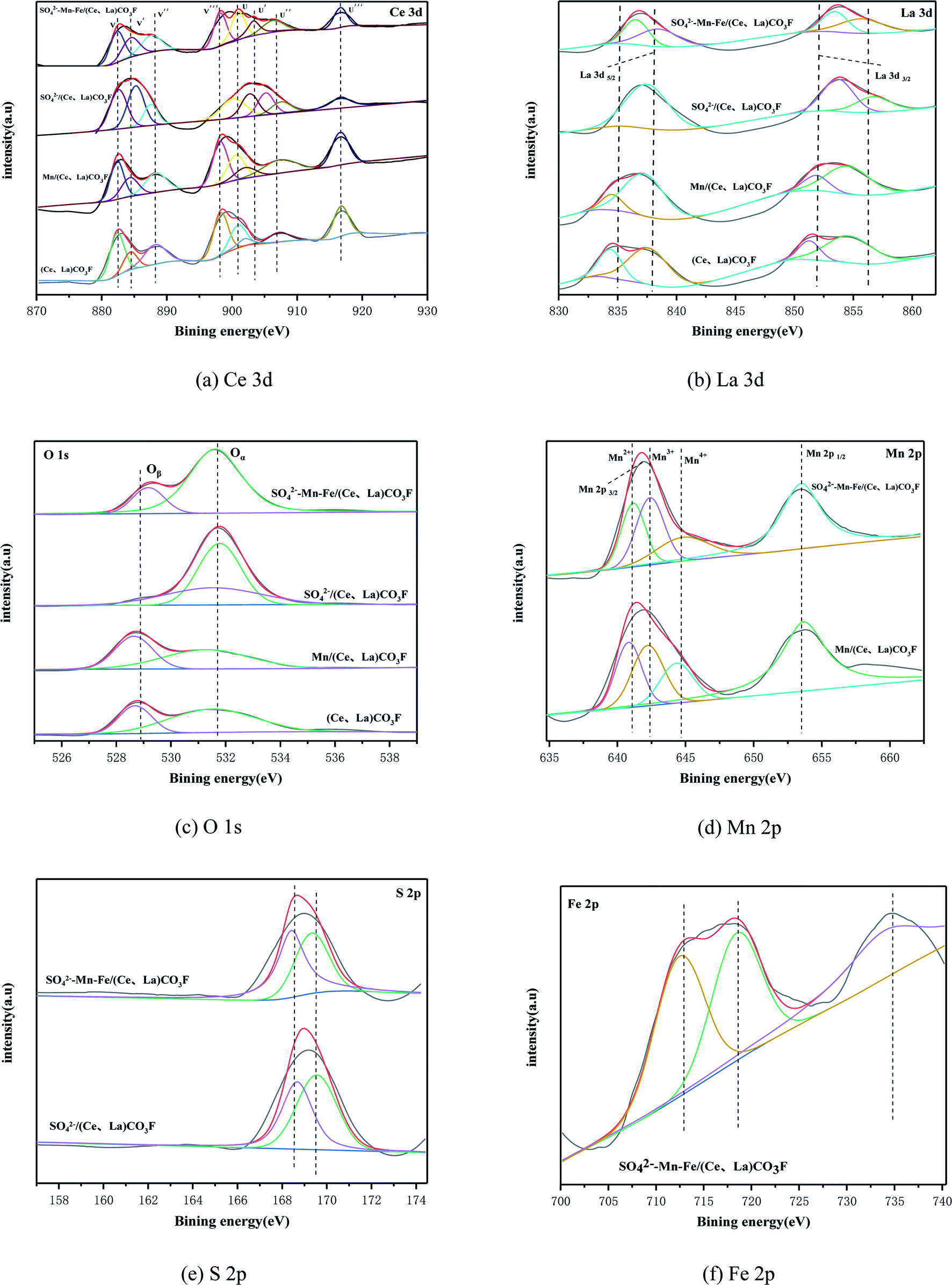
In order to determine the surface chemical state and surface composition of catalysts, the XPS analysis was carried out on various types of catalysts. Fig. 4(a) shows the Ce 3d spectra, which contains eight characteristic peaks. These are u (900.8 eV), u′′ (907.5 eV), u′′′ (916.5 eV), v (882.3 eV), v′′ (888.9 eV), v′′′ (898.4 eV) are attributed to Ce4+ and the characteristic peaks of u′ (903.8 eV), v′ (884.7 eV) are attributed to Ce3+.17,18 In general the higher the Ce3+/(Ce4+ + Ce3+) ratio, it indicates that this catalyst exhibits unsaturated chemical energy bands and more oxygen vacancies, which will promote the adsorption of NH3 from the reactants, and Ce3+ can promote the oxidation of NO to NO2, thus improving the low temperature activity of the catalyst.19–21 The calculated proportions of each element are shown in Table 2, from which it can be seen that the Ce3+ content is increased compared to the original carrier when loaded alone, acidified alone and when loaded by simultaneous acidification. The acidified (Ce,La)CO3F catalysts contain the highest amount of Ce3+., For cerium-based catalysts, the performance was improved after sulphuric acid treatment, it is due to the fact that SO42− can promotes the interconversion of Ce4+ and Ce3+, enhancing the oxygen storage and release ability of the cerium-based catalysts, promotin the increase of active sites, and improved the activity.22 However, based on activity tests, the catalytic activity of the catalyst with simultaneous acidification and loaded was the best. The possible reason for this is the introduction of Mn–Fe, after acidification. Sulphuric acid simultaneously promotes redox properties between the polymetallic ions, which together participate in the reaction, At this time, Ce3+ is no longer the only active ion.
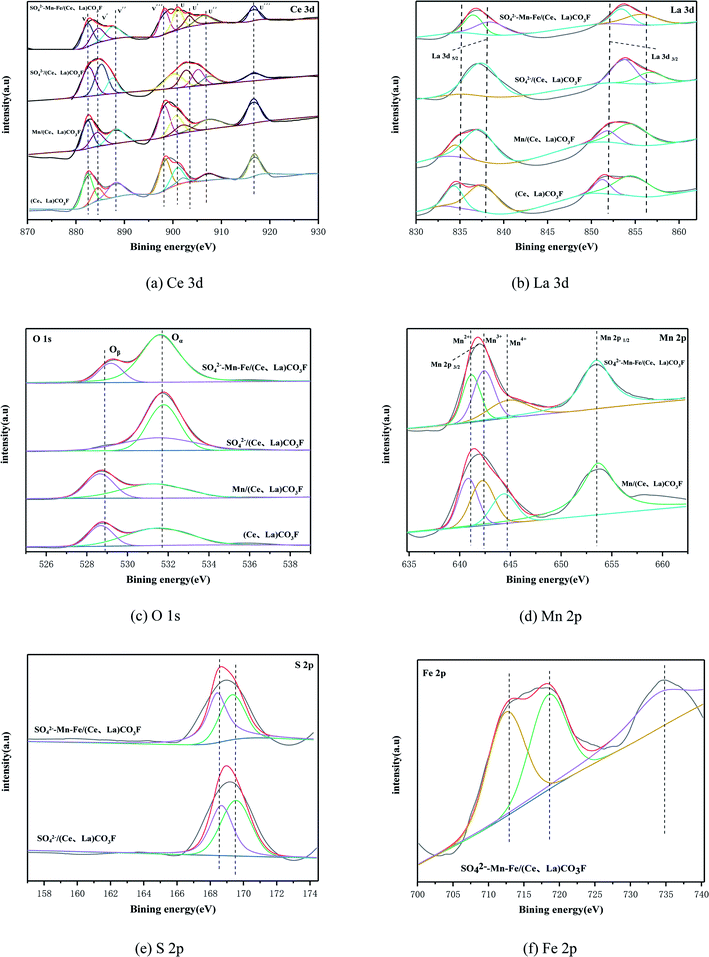 |
| | Fig. 4 XPS of catalysts with different treatment methods: (a) Ce 3d, (b) La 3d, (c) O 1s, (d) Mn 2p, (e) S 2p, and (f) Fe 2p. | |
Table 2 Fitted data of XPS on the catalyst
| Samples |
Ce3+/(Ce3+ + Ce4+) (%) |
Oα/(Oα + Oβ) (%) |
Fe3+/(Fe3+ + Fe2+) (%) |
Mn4+/(Mn4+ + Mn3+ + Mn2+) (%) |
| (Ce,La)CO3F |
10.7 |
71.1 |
— |
— |
| Mn/(Ce,La)CO3F |
13.2 |
58.7 |
— |
14.5 |
| SO42−/(Ce,La)CO3F |
23.6 |
62.8 |
— |
— |
| SO42−–Mn–Fe/(Ce,La)CO3F |
16.9 |
92.0 |
70.8 |
16.1 |
Fig. 4(b) shows the La 3d spectra. The La 3d orbitals in the four catalysts consist of four sub-popular peaks, and the magnitudes of La electron binding energies are 854–855 eV, 850–851 eV, 837–838 eV and 833–834 eV respectively.23 The characteristic peaks of lanthanum metal are bimodal, with electron binding energies of 837.7 eV for La 3d5/2 and 854.3 eV for La 3d3/2.24 The 4f orbital of lanthanum metal is a vacant orbital when it is not affected in any way. When lanthanum metal forms a composite oxide, the La 3d3/2 and La 3d5/2 are both double peaks, which meant that companion peaks appear. This is due to the ionization of electrons in the inner shell layers of La 3d3/2 and La 3d5/2, the transfer of 2p valence electrons from the La-coordinated oxygen to the 4f vacant orbital of La, resulting in the splitting of the La 3d characteristic peak and the resultant vibrational companion peaks of La 3d3/2 and La 3d5/2.25,26 So the change in the intensity of the companion peak can reflect the ability of the 2p electrons of oxygen to give La. The binding energy in the La 3d spectrum of the synthetic (Ce,La)CO3F is low compared to the binding energy of the standard characteristic peak and the accompanying peaks of La. This indicated that there is no transfer of electrons between La,27 where a Ce–La–O solid solution may be formed.28 When loaded with Mn, the electron binding energy of La 3d5/2 is shifted more towards the lower side, probably due to the introduction of a third metal, which enhanced the interaction between the elements and formed a new solid solution (Mn–Ce–La). After acidification with sulphuric acid, the accompanying peaks of La 3d5/2 disappear, indicating that the 2p valence electrons of the ligand oxygen with La are not transferred here. The binding energies of all the accompanying peaks in the La 3d spectrum of the SO42−–Mn–Fe–/(Ce,La)CO3F catalyst are shifted towards higher binding energies. This was probably due to the fact that the introduction of the transition metal made the electron supply weaker, causing a decreased electron cloud density of La, leading to a high binding energy, and the covalency of the La–O bond is decreased.23
Fig. 4(c) shows the O 1s spectra of the four catalysts. The spectral band of O 1s can be obtained from two peak positions based on the split peak fitting. This indicated the presence of two different types of oxygen, corresponding to lattice oxygen and adsorbed oxygen in the catalysts. In Fig. 4(c), the peak at 531.5 eV belongs to the adsorbed oxygen species, denoted as Oα, and the peak at 529.3 eV belongs to the lattice oxygen species, denoted as Oβ.29 Studies have reported that adsorbed oxygen (Oα) is the most active oxygen species, and plays a key role in redox reactions. The formation of adsorbed oxygen (Oα) is due to the presence of oxygen vacancies on the surface, so the proportion of adsorbed oxygen (Oα) is particularly important. The ratio of Oα/(Oα + Oβ) can measure the concentration of oxygen vacancies on the catalyst surface, and a larger ratio means that more active oxygen exists on the catalyst surface, which is conducive to the conversion of NO to NO2 in the SCR reaction, and can promote the SCR reaction at low temperatures.30 From Fig. 4(c), it can be seen that for (Ce,La)CO3F and Mn/(Ce,La)CO3F catalysts, the binding energy of O 1 s is slightly shifted towards the lower occurrence, which indicated that O and metal elements interacted to form the Mn–O–Ce(La) species, and the binding energy was shifted towards the lower due to its shorter bond length.31,32 Table 2 calculates the proportion of Oα. The synthetic (Ce,La)CO3F catalyst itself contains more adsorbed oxygen and after loaded with Mn, the proportion of adsorbed oxygen decreases, which may be that the addition of Mn is not conducive to the increase in the proportion of adsorbed oxygen (Oα). The O 1s binding energy of (Ce,La)CO3F catalysts acidified by sulphuric acid is shifted towards high. This indicated strong interactions between sulfuric acid and the oxides of Ce, La. While accelerating the Cen+ interconversion, metal sulphate species such as Ce2(SO4)3 are produced, which in turn limit the ability to carry oxygen, causing the proportion of adsorbed oxygen to decrease. When simultaneously loaded and acidified, the various species interact to produce metal sulphates that provide more acidic sites for the catalyst, and the addition of Mn, Fe greatly promotes redox, and increases the proportion of oxygen adsorbed by the catalyst, thus facilitating SCR reactivity.
Fig. 4(d) shows the Mn 2p spectra for the Mn/(Ce,La)CO3F, SO42−–Mn–Fe/(Ce,La)CO3F catalysts, from which the Mn 2p can be obtained as two main peaks, Mn 2p1/2 (653.0 eV) and Mn 2p3/2 (641.0 eV) respectively. The Mn 2p spectra of all catalysts can be further decomposed into three peaks, where the binding energy in the range of 641.0–641.3 eV belongs to Mn2+, 642.0–642.6 eV to Mn3+ and 644.0–644.7 eV to Mn4+.33 It is generally believed that the catalytic capacity of MnOx is MnO2 > Mn3O4 > Mn3O4,34–36 and that the catalyst surface produces a large amount of MnO2 to facilitate the SCR reaction.37 The reason why Mn4+ is more favourable than Mn3+ and Mn2+ in the NH3-SCR reaction is that its high redox capacity promotes the conversion of NO to NO2, and enhances the catalytic activity of the low temperature section through the fast SCR reaction NO + NO2 + 2NH3![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) 3N2 + 2H2O, and therefore this reaction is also the main low temperature reaction pathway.38,39 The Mn4+ contents of the Mn/(Ce,La)CO3F, SO42−–Mn–Fe/(Ce,La)CO3F catalysts were calculated and listed in Table 2, from which it can be concluded that the SO42−–Mn–Fe/(Ce,La)CO3F catalyst has the highest content of Mn4+, which is consistent with the denitrification activity of the catalyst. This is probably due to the introduction of sulphuric acid and Fe promoted the dispersion of MnOx, allowing more MnO2 species to be distributed in an amorphous state on the catalyst surface to facilitate the reaction.
3N2 + 2H2O, and therefore this reaction is also the main low temperature reaction pathway.38,39 The Mn4+ contents of the Mn/(Ce,La)CO3F, SO42−–Mn–Fe/(Ce,La)CO3F catalysts were calculated and listed in Table 2, from which it can be concluded that the SO42−–Mn–Fe/(Ce,La)CO3F catalyst has the highest content of Mn4+, which is consistent with the denitrification activity of the catalyst. This is probably due to the introduction of sulphuric acid and Fe promoted the dispersion of MnOx, allowing more MnO2 species to be distributed in an amorphous state on the catalyst surface to facilitate the reaction.
Fig. 4(e) shows the S 2p spectra of SO42−/(Ce,La)CO3F, SO42−–Mn–Fe/(Ce,La)CO3F catalyst. From the figure, the S 2p spectra were divided into two superimposed peaks. The electron binding energies of S 2p are at 169.3–169.9 eV and 168.4–168.8 eV, respectively, both belonging to the S6+ species on the catalyst surface.40,41 This indicates that the presence of SO42− on the catalyst surface, it can provide acidic sites for the catalyst, and SO42− can interact with metal elements to form metal sulphates as active components. Not only that, the peaks of S 2p in both acid-treated catalysts shifted towards lower binding energies, and the SO42−–Mn–Fe/(Ce,La)CO3F catalysts had lower binding energies, which indicated strong interactions between sulphate species and metal species such as Mn, Fe and Ce.
Fig. 4(f) shows the Fe 2p spectra for the SO42−–Mn–Fe/(Ce,La)CO3F catalyst. The Fe 2p spectra is divided into three peaks, where the binding energy belongs to Fe3+ at 711.9 eV and 733.6 eV, Fe2+ at 716.4 eV,42 which indicated that the iron species on the catalyst surface existed in Fe3+ and Fe2+ states. Fe3+ is more favourable to the SCR reaction at low temperatures, and the appropriate proportion of Fe3+ can promote NOx conversion and improve catalytic performance.43 From the Fig. 4, it can be seen that the Fen+ is shifted towards higher binding energies compared to the standard spectrum. This may be due to two reasons, on the one hand, the strong interaction of the carrier with the Fe species as the active component, and the electron transfer between the active component and the carrier can promote the redox cycle in the SCR process, thus increasing the activity.44 On the other hand, the strong interaction forces between S and Fe, Mn, due to sulphuric acidification. This results in a strong attraction of electrons to the sulphur centre, causing an increase in the density of negative charges surrounding the sulphur centre, producing charge-rich and charge-deficient sites centred on Fe, Mn and S atoms, respectively.45,46
3.4 In situ infrared analysis and reaction mechanism research
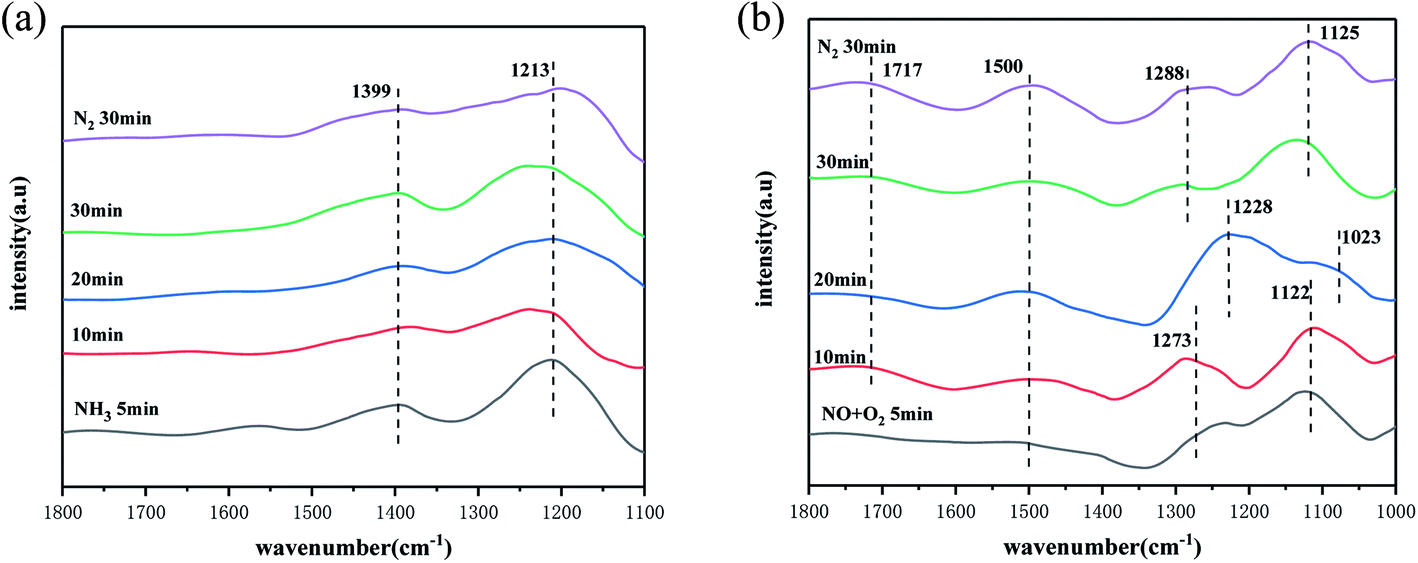
Infrared reaction diagram of SO42−–Mn–Fe/(Ce,La)CO3F catalyst. From the activity test results, it can be concluded that the SO42−–Mn–Fe/(Ce,La)CO3F catalyst reaches its optimum activity at 250 °C. In order to investigated the effect of the SO42−–Mn–Fe/(Ce,La)CO3F catalyst on the activation performance of NH3 adsorption at optimum denitration temperature conditions. This experiment investigated its NH3 adsorption at 250 °C with time. The results are shown in Fig. 5(a), where two distinct infrared absorption peaks at 1399 cm−1 and 1213 cm−1 appeared on the surface of the SO42−–Mn–Fe/(Ce,La)CO3F catalyst after 5 min of NH3 passaged. The peak at 1399 cm−1 is attributed to the deformation vibration of the N–H bond in the NH4+ species at the Brønsted acidic site.47 The peak at 1213 cm−1 is attributed to NH3 species in the ligand state on the Lewis acidic site.48,49 This indicated that the SO42−–Mn–Fe/(Ce,La)CO3F catalyst has an abundance of acidic sites on its surface. The stronger infrared absorption peak of the Lewis acidic site can be seen in Fig. 5(a), which indicates that the Lewis acidic site is the main NH3 adsorption acid site on the catalyst surface. The NH4+/NH3 species adsorbed at the Brønsted/Lewis acidic site were both present in a very stable state as the time of NH3 gas introduction increased, and their absorption peaks did not change significantly even after 30 min of N2 purging, which indicated that the SO42−–Mn–Fe/(Ce,La)CO3F catalyst had a very good ability to adsorb NH3, which facilitated the SCR reaction.
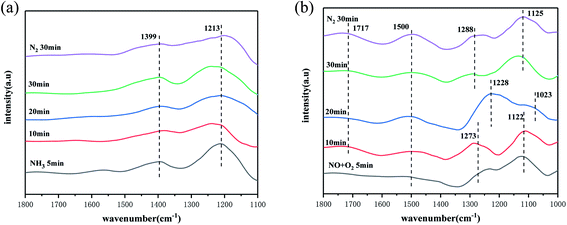 |
| | Fig. 5 In situ infrared spectra of (a) NH3 and (b) NO + O2 adsorption of the catalyst at the optimal denitration temperature over time. | |
To further investigated the effect of the catalyst on the adsorption and activation performance of NO. The in situ infrared spectra of the SO42−–Mn–Fe/(Ce,La)CO3F catalyst at 250 °C for NO + O2 adsorption with time were examined. As shown in Fig. 5(b), when NO + O2 was passed through, distinct infrared absorption peaks soon appeared on the catalyst surface, with the peak at 1717 cm−1 attributed to the bidentate nitrate species,50 and the peak at 1500 cm−1 attributed to the monodentate species.51 With the increase of NO + O2 pass time, it can be found that the monodentate and bidentate nitrate species all existed in a stable state and did not decompose even after N2 purging, so the stability was good. It can also be found that at 5 min, the catalyst surface shows infrared absorption peaks at 1273 cm−1 and 1122 cm−1, with the peak at 1273 cm−1 attributed to the monodentate nitrate species, and the peak at 1122 cm−1 attributed to the secondary nitrate species [–(N2O2)2−],47 which starts to decompose when the pass time is 20 min, and new adsorption peaks appeared on the catalyst surface at 1228 cm−1 and 1023 cm−1. The peaks at 1228 cm−1 belonged to the bridged nitrate species, and the peaks at 1023 cm−1 belonged to the secondary nitrate species,52,53 but with the increase of the passage time, the adsorption peaks returned to their initial positions and remained relatively stable after the N2 purging. The shift at 20 min is probably due to the formation of bridging nitrates from the metal ions bonded to O ions in the catalyst, with the increase in reaction time, the redox capacity weakens and the M–O bond breaks, reverting back to monodentate nitrate species. This showed that the abundance of nitrate species on the catalyst surface, and indicated that catalyst has better NO adsorption and activation ability, which can effectively improve the SCR activity of the catalyst. Based on the number of nitrate species and the peak intensity, it is clear that the main nitrate species bonded to the metal ions on the catalyst surface are monodentate species, and accompanied by bidentate and bridged nitrate species to promote the reaction.
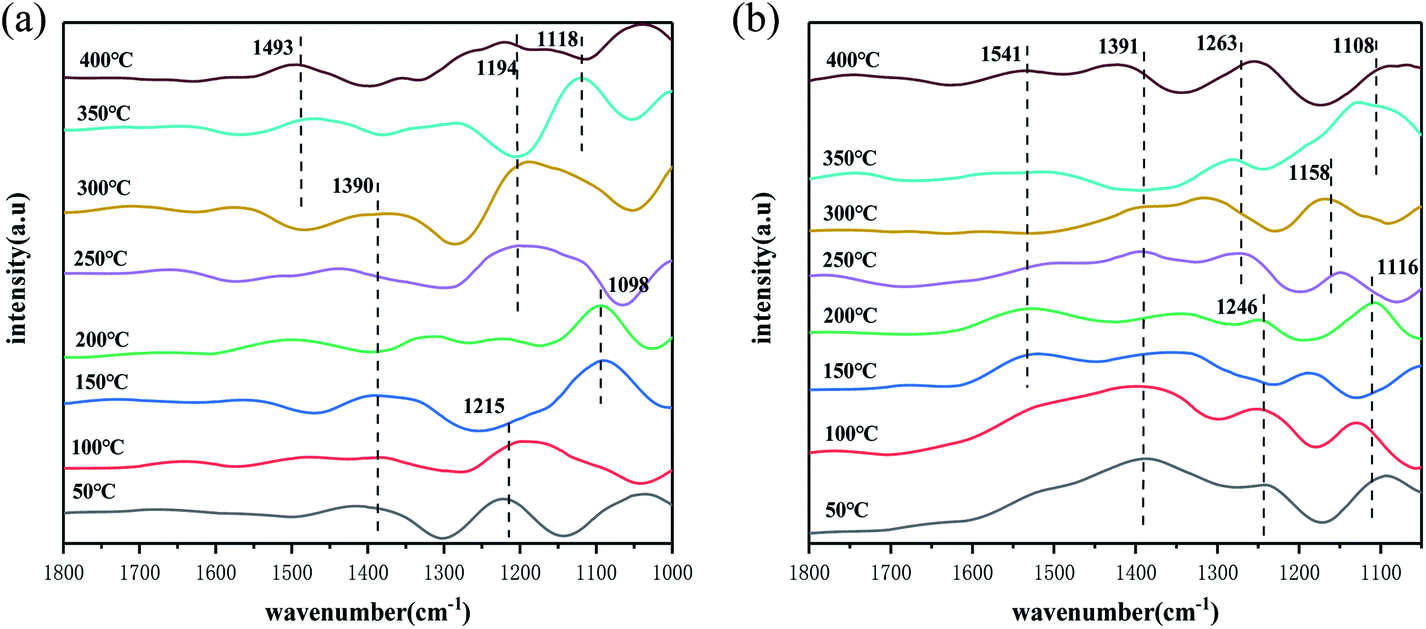
In this experiment, the adsorption and activation of NH3 on the catalyst surface under different temperature conditions and the presence of NH3 species on the catalyst surface under different temperature conditions were investigated by in situ infrared spectroscopy. The results are shown in Fig. 6(a). Firstly, it can be found that in the low temperature range of 50–100 °C, the peaks of NH3 species attributed to the ligand state on the Lewis acidic site appear at 1215 cm−1 on the catalyst surface.49 As the temperature increases, a peak for NH4+ species in the Brønsted acidic site appears at 1098 cm−1,54 indicating that both the Brønsted/Lewis acidic site play a role throughout the reaction temperature interval. It is noteworthy that in the interval from 50–300 °C, a weaker absorption peak appeared at 1390 cm−1, where the peak belongs to the deformation vibration of the N–H bond in the NH4+ species on the Brønsted acidic site,47 which shifts with increasing temperature at 200 °C, which may be an intermediate product generated by the dehydrogenation reaction of the NH4+ species on the Brønsted acidic site, and gradually recovered at 250 °C. In the temperature interval of 250–300 °C, the peak of NH3 species belonging to the ligand state on Lewis acid site appeared at 1194 cm−1 on the catalyst surface,55 which is consistent with the results in Fig. 5(a). According to the activity test results, the Lewis acidic site is more favourable than the Brønsted acidic site on the SO42−–Mn–Fe/(Ce,La)CO3F catalyst surface. In the high temperature interval, it can be found that the peak at 1493 cm−1 belongs to the amide(–NH2) species, and the peak at 1118 cm−1 belongs to the NH4+ species on the Brønsted acidic site,56 indicating that the Brønsted acidic site occupies the main active site in the high temperature section of the catalyst surface.
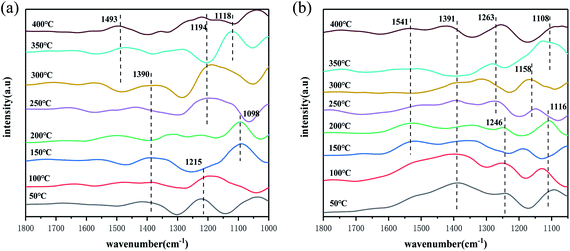 |
| | Fig. 6 In situ infrared spectra of (a) NH3 and (b) NO + O2 adsorbed species on the catalyst surface under different temperature conditions. | |
The adsorption and activation processes of NO + O2 species on the catalyst surface under different temperature conditions were investigated by in situ infrared spectroscopy, and the forms and changes of the presence of NOx species on the catalyst surface under different temperature conditions were discussed. From Fig. 6(b), it can be obtained that in the temperature interval of 50–100 °C, infrared absorption peaks appear on the catalyst surface at 1391 cm−1, 1246 cm−1 and 1116 cm−1, where the peaks at 1391 cm−1 and 1246 cm−1 belong to the monodentate nitrate species,57 and the peak at 1116 cm−1 belongs to the continuous secondary nitrate species. As the temperature increases, they all decompose, and new nitrate species appear on the catalyst surface at 150 °C, this may be that the action of O2 oxidises the monodentate nitrate adsorbed on the catalyst surface. However, they recover again at 200 °C, indicating that the reaction rate of NO at this point is greater than the oxidation rate. When the temperature reached 250 °C, monodentate nitrate species (1391 cm−1, 1263 cm−1, 1541 cm−1) and secondary nitrate species (1158 cm−1) appeared on the catalyst surface. The absorption peak at 1541 cm−1 almost disappears at 250 °C, indicating that the adsorption is less stable, and is susceptible to pyrolysis and desorption due to temperature. Throughout the reaction temperature range, the monodentate nitrate species occupies the main active site, bonding to the metal ions in the catalyst, and participating in the SCR reaction.
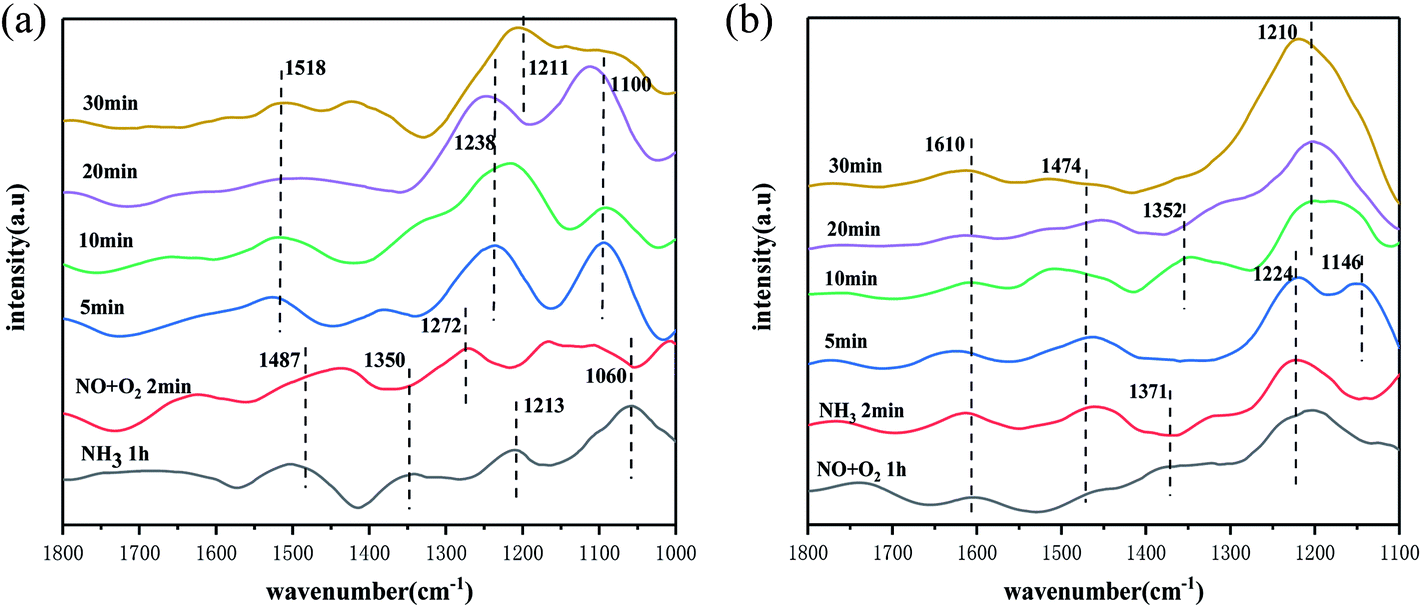
To further investigate the NH3-SCR reaction mechanism of the catalyst, in situ infrared spectroscopy of the catalyst was carried out at 250 °C under different reaction conditions. Firstly, the infrared reaction cell with catalyst was pre-treated with N2 as supplementary gas at 200 °C for 30 min, then NH3 gas was passed to the reaction cell for 60 min, then NH3 was stopped, and purged with N2 for 10 min, after the purging, NO + O2 gas was then passed to the reaction cell. As shown in Fig. 7(a), the changes in the infrared spectrum was observed, firstly after 1 h of adsorption of NH3, NH4+ species belonging to the adsorbed on Brønsted acidic sites (1487 cm−1, 1350 cm−1)47 and NH3 species adsorbed on Lewis acidic sites (1213 cm−1, 1060 cm−1)57 appeared on the catalyst surface, and after 1 h of NH3 adsorption, the NH4+/NH3 species formed were more stable. Turning off NH3 and purging with N2 before passing NO + O2 gas. The peaks of NH3 species adsorbed on the Lewis acidic site and NH4+ species adsorbed on the Brønsted acidic site at 1350 cm−1 disappeared, when the gas was passed for 2 min. This indicated a rapid reaction with the NO species. The peak at 1487 cm−1 was significantly weaker, but did not disappear completely, and only part of the reaction took place. This indicated that the catalyst surface followed the E–R mechanism, and the reacting species were NH4+/NH3 species in the adsorbed state and NO, with the NH3 species on the Lewis acidic site being the main reacting species. As the NO + O2 flux time increased, nitrate species appeared on the catalyst surface, with peaks at 1518 cm−1, 1100 cm−1 and 1211 cm−1 belonged to monodentate nitrate, continuous secondary nitrate and bridged nitrate species, respectively,48,51 and peaks at 1238 cm−1 belonged to nitrate species bound to the Mn–O–Ce site on the catalyst surface,58 These species are more stable, which also indicated the involvement of NH4+/NH3 species in the reaction.
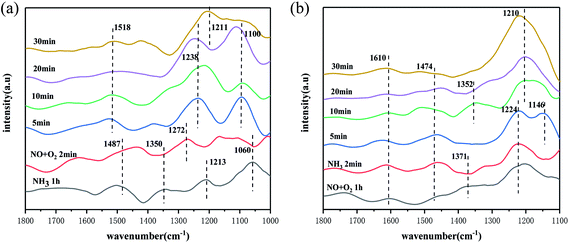 |
| | Fig. 7 In situ infrared spectra of the reaction of (a) NH3 and (b) NO + O2 pre-adsorbed species on the catalyst surface at the optimal reaction temperature. | |
Fig. 7(b) shows the in situ infrared spectrum of the reaction of NH3 with pre-adsorbed NO + O2 species at 250 °C. After passing 60 min of NO + O2 into the reaction cell system, the NO2 species (1610 cm−1),59 monodentate nitrate species (1474 cm−1, 1371 cm−1), and bridged nitrate species (1224 cm−1) can be observed on the catalyst surface. Previous studies60,61 have shown that NO2 is an important intermediate species in the occurrence of the “fast SCR” reaction at low temperatures, and is also an important factor in improved catalytic performance at low temperatures. However, with the passage of NH3, the peaks of the NO2 species on the catalyst surface did not change throughout the reaction, which showed that the NO2 species did not react with NH3, and meant that the chances of “fast SCR” reactions on the catalyst surface were low, which indicated that most of the reactions of this catalyst followed the E–R/L–H mechanism. It was also found that the monodentate nitrate species at 1474 cm−1 and 1371 cm−1 disappeared rapidly when NH3 was introduced, which indicated a reaction with the NH4+/NH3 species. The bridged nitrate species decompose gradually after NH3 was introduced for 5 min, it was not easy to react with NH3 than the monodentate nitrate species, which also suggested that the catalyst surface followed the L–H mechanism, with the NH4+/NH3 species and the monodentate nitrate, bridged nitrate species as the reaction species. However, due to the weak absorption peaks of the nitrate species, this also suggested that the L–H mechanism on the catalyst surface was not the main reaction pathway. When NH3 was passed through, peaks of NH3 adsorbed species appeared on the catalyst surface, with peaks at 1474 cm−1, 1146 cm−1 that belonged to NH4+ species on the Brønsted acidic site, the peak at 1352 cm−1 was associated with the oxidation of NH3 species on the catalyst surface.62 The peak at 1210 cm−1 belonged to NH3 species on the Lewis acidic site, and these species existed in a stable state with increased access time except for the peak at 1146 cm−1 which disappeared.
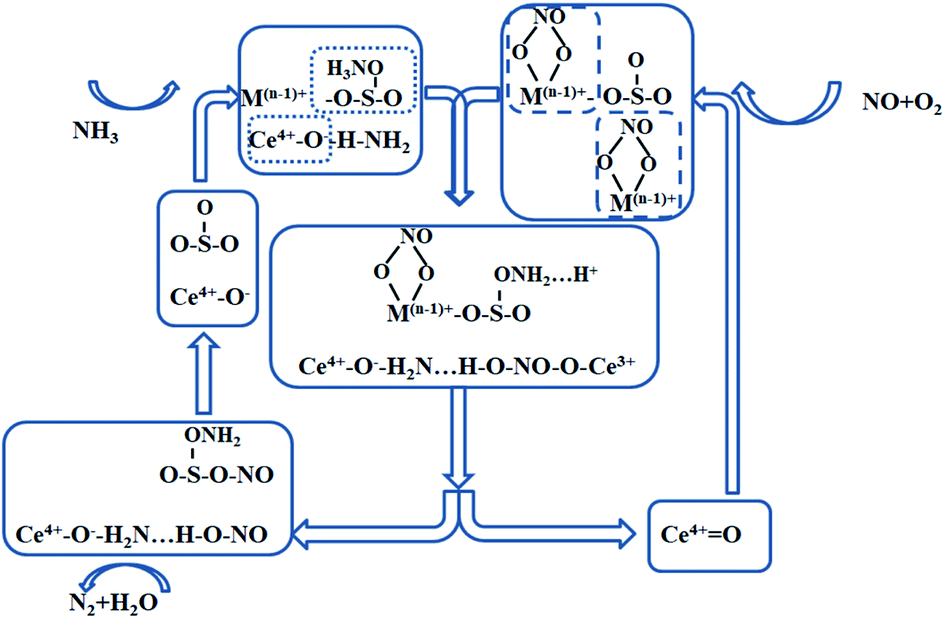
From the above arguments, it can be obtained that the NH3-SCR reaction process occurred on the surface of the SO42−–Mn–Fe/(Ce,La)CO3F catalyst at 250 °C mainly follows the E–R mechanism, with the reaction species being NH4+/NH3 species in the adsorbed state and NO. The NH3(ad) species on the Lewis acidic site is the main reaction NH3(g) adsorbed species, bonded to the Ce4+ in the carrier (Ce,La)CO3F to participate in the acid cycle reaction. While SO42− present on the catalyst surface can also act as an acidic site for NH3 adsorption.63 The E–R mechanism reaction proceeds as follows.
| | |
NH3(g) → NH3(a) (Lewis)
| (1) |
| | |
NH3(g) → NH4+(a) (Brønsted)
| (2) |
| | |
Ce4+–O–NH3 + M(Mn4+,Ce4+,Fe3+,La3+)O → Ce4+–O−–NH2 + M(n−1)+–OH (Lewis)
| (3) |
| | |
SO42−–O–NH3 + M(Mn4+,Ce4+,Fe3+,La3+)O → SO42−–O−–NH2 + M(n−1)+–OH (Lewis)
| (4) |
| | |
□(Ce4+–O,SO42−)–O–NH4+ + MnO → □(Ce4+–O,SO42−)–O−–NH3+ + Mn−1–OH (Brønsted)
| (5) |
| | |
□–O−–NH2–Mn−1–OH + NO → □–O−–NH2–NO–Mn−1–OH (Lewis)
| (6) |
| | |
□–O−–NH3+–Mn−1–OH + NO → □–O−–NH3+–NO–Mn−1–OH (Brønsted)
| (7) |
| | |
□–O−–NH2–NO–Mn−1–OH → □–O−–NH2–NO + Mn−1–OH (Lewis)
| (8) |
| | |
□–O−–NH2–NO → N2 + H2O + □–O− (Lewis)
| (9) |
| | |
□–O−–NH3+–NO–Mn−1–OH → □–O−–NH3+–NO + Mn−1–OH (Brønsted)
| (10) |
| | |
□–O−–NH3+–NO → N2 + H2O + □–O−–H (Brønsted)
| (11) |
| |
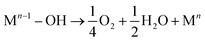 | (12) |
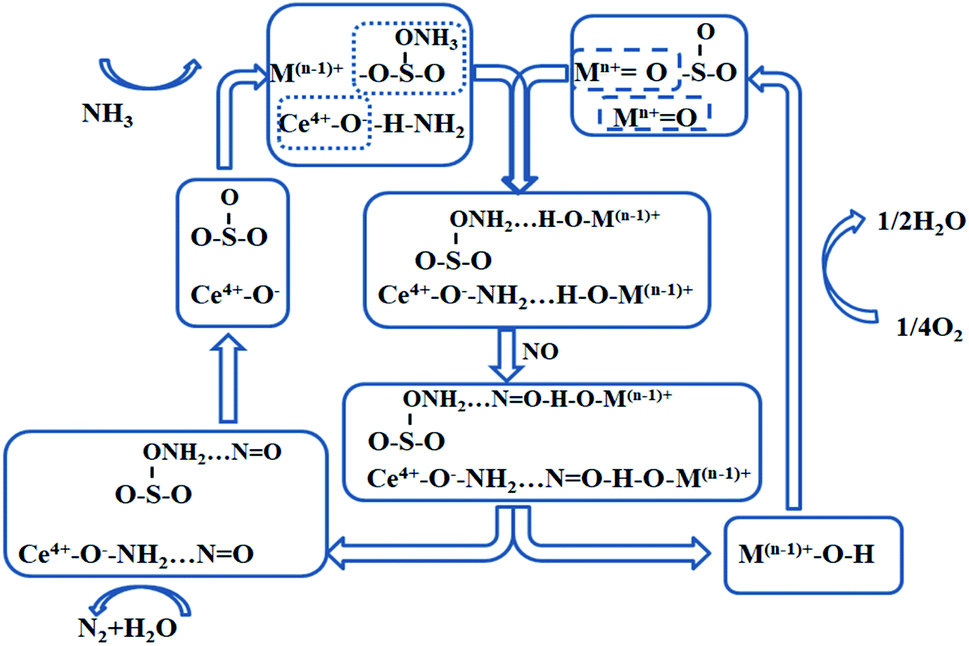
The reaction pathway of the SO42−–Mn–Fe/(Ce,La)CO3F catalyst E–R mechanism is shown in Fig. 8, with metal sulphates and metal oxides participated in the reaction as active components (the acidic sites are dominated by the Lewis acidic sites).
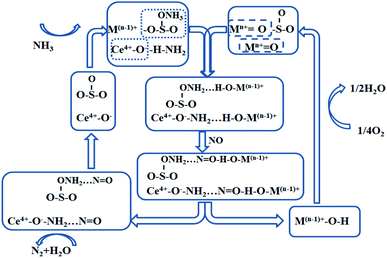 |
| | Fig. 8 E–R mechanism reaction process on the catalyst surface under the optimal denitration temperature. | |
Also at 250 °C the catalyst surface is accompanied by an L–H reaction mechanism. This meant that NO was activated on the catalyst surface mainly into monodentate nitrate species with a small amount of bridging nitrate species, which then react with adsorbed NH3/NH4+ species to form N2 and H2O. The infrared spectrum showed that the main acidic site on the catalyst surface was the Lewis acidic site, and that the monodentate nitrate reacted more readily with NH3 in the adsorbed state than the bridged nitrate species. The L–H reaction proceeded as follows (Fig. 9).
| | |
O2 + 2* → 2O−* (* NO adsorption sites on the catalyst surface)
| (13) |
| | |
NO + O2−* + Mn+ = O* → O–NO–O–M(n−1)+ (monodentate nitrate)
| (14) |
| | |
NO + O2−* + Mn+O* → M(n−1)+–O–NO–O–M(n−1)+ (bridge nitrate)
| (15) |
| | |
□–NH3 + O− → □–O−–NH2 + H+ (Lewis)
| (16) |
| | |
□–NH4+ + O− → □–O−–NH3 + H+ → □–O−–NH2 + 2H+ (Brønsted)
| (17) |
| | |
□–O−–NH2 + O–NO–O–M(n−1)+ → □–O−–NH2–O–NO–O–M(n−1)+
| (18) |
| | |
□–O−–NH2 + M(n−1)+–O–NO–O–M(n−1)+ → □–O−–NH2–M(n−1)+–O–NO–O–M(n−1)+
| (19) |
| | |
□–O−–NH2–O–NO–O–M(n−1)+ → □–O−–NH2–O–NO + OMn+
| (20) |
| | |
□–O−–NH2–M(n−1)+–O–NO–O–M(n−1)+ → □–O−–NH2–O–NO + Mn+–O–Mn+
| (21) |
| | |
□–O−–NH2–O–NO → N2 + H2O + □–O−
| (22) |
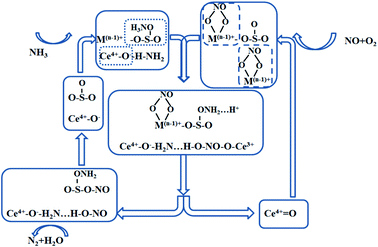 |
| | Fig. 9 L–H mechanism reaction process on the catalyst surface under the optimal denitration temperature. | |
4 Conclusions
In this experiment, the (Ce,La)CO3F grains were synthesised by hydrothermal method to simulate the bastnaesite mineral phase in rare earth minerals, according to the Ce–La ratio of bastnaesite in the mineralogy of the Bayan Obo process. The NH3-SCR catalytic activity of the synthetic (Ce,La)CO3F was enhanced by loaded and acidified treatments. The activity test results showed that the SO42−–Mn–Fe/(Ce,La)CO3F catalyst has a NOx conversion of 92% at 250 °C. The XRD results showed that the diffraction peaks of the composite metal oxides appear on the surface of the catalysts that have been simultaneously loaded with transition metals and acidified with sulphuric acid. This indicated that the S prevents the metals from dispersing, and facilitates the synergistic interaction between the metals. SEM showed that after the treatment, the pores and cracks on the catalyst surface increased, which facilitates the mass transfer diffusion of the reaction gas. XPS results showed that the introduction of transition metals and sulphuric acid greatly increased the proportion of adsorbed oxygen (Oα), and favoured the adsorption of NH3 and NO. After acidification with sulphuric acid, which results in strong interaction forces between S and Fe, Mn. This led to a strong attraction of electrons to the sulphur centre, which results in an increase in the density of negative charges surrounding the sulphur centre, producing charge-rich and charge-deficient sites centred on S and Fe, Mn atoms respectively. After loaded Mn, Fe and sulfuric acid acidification. The metal sulphate and the metal oxide act as active components on the catalyst surface to promote the reaction.
The reaction mechanism was investigated by Fourier transform infrared spectroscopy (FTIR), which characterised the adsorption and conversion behaviour of the reactive species on the catalyst surface. The results showed that the whole reaction process of the SO42−–Mn–Fe/(Ce,La)CO3F catalyst mainly followed the E–R mechanism, and the reaction species were NH4+/NH3 species in the adsorbed state and NO. The NH3(ad) species on the Lewis acidic site is the main reacting NH3(g) adsorbed species, bonded to the Ce4+ in the carrier (Ce,La)CO3F to participate in the acid cycle reaction, where they undergo a redox reaction on the catalyst surface to produce N2 and H2O. The SO42− present on the catalyst surface can also act as an acidic site for the adsorption of NH3. The above results indicated the excellent performance of the SO42−–Mn–Fe/(Ce,La)CO3F catalyst, which provided theoretical guidance on the specific reaction mechanism of bastnaesite in the catalytic reaction.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This study was financially supported by Natural Science Foundation of Inner Mongolia (Grant No. 2018MS02013, 2019ZD13, 2020BS05030), National Natural Science Foundation of China (Grant No. 51866013). Thanks for Start-up Funds for Talent Introduction and Scientific Research of Institutions in Inner Mongolia Autonomous Region.
References
- F. Luck and J. Roiron, Catal. Today, 1989, 4(2), 205–218 CrossRef CAS.
- H. C. Cao, Nonferrous Mining and Metallurgy, New progress in research on copper ore beneficiation technology in my country, 2011, vol. 027(006), pp. 26–28 Search PubMed.
- Y. K. Yang, Nonferrous Metals, Mineral Processing, Experimental study on optimization of flotation process for a low-grade copper ore, 2016, vol. 000(002), pp. 14–17 Search PubMed.
- Z. L. Meng, Study on NH3-SCR denitration performance of Fe2O3 mineral catalytic materials supported by rare earth concentrates, Inner Mongolia University of Science and Technology, 2020 Search PubMed.
- K. Zhang, J. H. Zhu, B. W. Li, H. J. Luo and W. F. Wu, Minerals, 2019, 9, 246 CrossRef CAS.
- K. Zhang, Y. Ge, J. H. Zhu, N. Li, H. J. Luo, B. W. Li, Z. W. Zhao and W. F. Wu, Mater. Chem. Phys., 2020, 242, 122421 CrossRef CAS.
- K. L. Jiao, Y. G. Zhao, W. F. Wu, Z. F. Wang and Z. J. Gong, J. Chem. Eng., 2019, 70(12), 133–141 Search PubMed.
- X. M. Wu, Y. X. Long, X. Y. He and G. H. Jing, J. Phys. Chem. C, 2019, 123(17), 10981–10990 CrossRef CAS.
- F. Eigenmann, M. Maciejewski and A. Baiker, Appl. Catal., B, 2006, 62(3–4), 311–318 CrossRef CAS.
- G. Ge, J. W. Shi, L. Chang, G. Chen, Z. Fan and C. M. Niu, Appl. Surf. Sci., 2017, 411, 338–346 CrossRef.
- N. Li, Z. D. Chen, H. Li, L. M. Hou, J. J. Wang and K. Zhang, J. Rare Earths, 2021, 1–15 Search PubMed.
- Z. L. Meng, B. W. Li, J. Y. Fu, C. Zhu and W. F. Wu, Process. Eng. J., 2021, 21(03), 363–372 Search PubMed.
- L. M. Hou, X. Yan, C. Y. Qiao, M. K. Cui, W. F. Wu, J. Wang and G. D. Xu, J. Rare Earths, 2021, 1–11 Search PubMed.
- Z. Z. Zhou, J. M. Lan, L. G. Liu and Z. M. Liu, Catal. Commun., 2021, 149, 106230 CrossRef CAS.
- M. Lei, J. Li, K. Rui and L. Fu, J. Phys. Chem. C, 2011, 15, 115 Search PubMed.
- W. Ren, B. Zhao, Y. Q. Zhou and C. H. Chen, J. Chem. Eng., 2011, 62(2), 362–368 CAS.
- L. Chen, X. Yao, J. Cao, F. Yang, C. Tang and L. Dong, Appl. Surf. Sci., 2019, 476, 283–292 CrossRef CAS.
- L. Zhang, W. X. Zou, K. L. Ma, Y. Cao, Y. Xiong, S. G. Wu, C. J. Tang, F. Gao and L. Dong, J. Phys. Chem. C, 2015, 119, 1155–1163 CrossRef CAS.
- T. Boningari, P. R. Ettireddy, A. Somogyvari, Y. Liu, A. Vorontsov, C. A. McDonald and P. G. Smirniotis, Catal, 2015, 325, 145–155 CrossRef CAS.
- Y. Geng, X. L. Chen, S. J. Yang, F. D. Liu and W. P. Shan, ACS Appl. Mater. Interfaces, 2017, 9, 16951–16958 CrossRef CAS PubMed.
- D. W. Kwon, K. B. Nam and S. C. Hong, Appl. Catal., A, 2015, 497, 160–166 CrossRef CAS.
- Q. L. Zhang, J. H. Zhang, P. Ning, Z. X. Song, Y. C. Wang, L. S. Xu and X. S. Tang, J. Kunming Univ. Sci. Technol., Nat. Sci. Ed., 2014, 6, 110–115 Search PubMed.
- H. Q. Ma, X. Tan, H. M. Zhu, J. Y. Zhang and L. Zhang, J. Rare Earths, 2003,(04), 445–448 CAS.
- L. Guo, Preparation, characterization and catalytic performance of core–shell Cu-Ce-La-SSZ-13[D], Nanjing University of Science and Technology, 2018 Search PubMed.
- Y. Jianmei and S. Qiang, Journal of the Chinese Rare Earth Society, 1992, 10(3), 259–261 Search PubMed.
- Y. L. Yu, R. F. Zhang, S. T. Liu and Z. L. Yu, J. Rare Earths, 1992,(02), 134–137 CAS.
- J. S. Wang, M. L. Zhou, J. X. Zhang, Z. R. Nie and T. Y. Zuo, J. Mater. Sci. Eng., 2000, 29(4), 225–227 CAS.
- E. S. Gnanakumar, J. M. Naik, M. Manikandan, T. Raja and C. S. Gopinath, Chemcatchem, 2014, 6(11), 3116–3124 CrossRef CAS.
- K. Zhao, W. L. Han, G. X. Lu, J. Y. Lu, Z. C. Tang and X. P. Zhen, Appl. Surf. Sci., 2016, 379, 316–322 CrossRef CAS.
- H. D. Xu, X. Feng, S. Liu, Y. Wang, M. M. Sun, J. L. Wang and Y. Q. Chen, Appl. Surf. Sci., 2017, 419, 697–707 CrossRef CAS.
- L. H. Chang, N. Sasirekha and Y. W. Chen, Ind. Eng. Chem. Res., 2006, 45(14), 4927–4935 CrossRef CAS.
- J. Fan, X. D. Wu, X. D. Wu, Q. Liang, R. Ran and D. Weng, Appl. Catal., B, 2008, 81(1), 38–48 CrossRef CAS.
- Y. Gao, T. Luan, S. Zhang, W. Jiang, W. Feng and H. Jiang, Catalysts, 2019, 9, 175 CrossRef.
- F. Gao, X. Tang, H. Yi, C. Chu, N. Li, J. Li and S. Zhao, Chem. Eng. J., 2017, 322, 525–537 CrossRef CAS.
- F. Kapteijn, L. Singoredjo, A. Andreini and J. A. Moulijn, Appl. Catal., B, 1994, 3, 173–189 CrossRef CAS.
- S. Luo, W. Zhou, A. Xie, F. Wu, C. Yao, X. Li, S. Zuo and T. Liu, Chem. Eng. J., 2016, 286, 291–299 CrossRef CAS.
- J. Huang, H. Huang, H. T. Jiang and L. C. LIiu, Catal. Today, 2018, 332 Search PubMed.
- P. Sun, S. Huang, R. Guo, M. Li, S. Liu, W. Pan, Z. Fu, S. Liu, X. Sun and J. Liu, Appl. Surf. Sci., 2018, 44, 479–488 CrossRef.
- Q. Xu, R. Su, L. Cao, Y. Li, C. Yang, Y. Luo, J. Street, P. Jiao and L. Cai, RSC Adv., 2017, 7, 48785–48792 RSC.
- S. J. Yang, C. Z. Wang, J. H. Li, N. Q. Yan, L. Ma and H. Z. Chang, Appl. Catal., B, 2011, 110, 71–80 CrossRef CAS.
- Q. Zhang, J. Zhang, Z. Song, P. Ning, H. Li and X. Liu, J. Ind. Eng. Chem., 2015, 34, 165–171 CrossRef.
- A. P. Grosvenor, B. A. Kobe, M. C. Biesinger and N. S. McIntyre, Surf. Interface Anal., 2004, 36, 1564–1574 CrossRef CAS.
- M. Devadas, O. Kröcher, M. Elsener, A. Wokaun, G. Mitrikas, N. Söger, M. Pfeifer, Y. Demel and L. Mussmann, Catal. Today, 2007, 119, 137–144 CrossRef CAS.
- K. Qi, J. L. Xie, D. Fang, F. Li and F. He, Chin. J. Catal., 2017, 38(5), 845–851 CrossRef CAS.
- X. Hou, Y. Qiu, E. Yuan, X. Zhang and G. Liu, Appl. Catal., A, 2017, 537, 12–23 CrossRef CAS.
- D. M. M. Cristian, E. R. S. Alfonso, S. G. Jurado and C. R. Vera, J. Mol. Catal. A: Chem., 2015, 398, 325–335 CrossRef.
- Y. J. Liao, Y. P. Zhang, Y. X. Yu, J. Li and X. Wang, CIESC J., 2016, 67(12), 5033–5037 Search PubMed.
- L. Li, Y. Wu, X. Hou, B. Chu, B. Nan, Q. Qin, M. Fan, C. Sun, B. Li, L. Dong and L. Dong, Ind. Eng. Chem. Res., 2019, 58, 849–862 CrossRef CAS.
- J. Sun, Y. Lu, L. Zhang, C. Ge, C. Tang, H. Wan and L. Dong, Ind. Eng. Chem. Res., 2017, 56, 12101–12110 CrossRef CAS.
- L. Song, R. Zhang, S. Zang, H. He, Y. Su, W. Qiu and X. Sun, Catal. Lett., 2017, 147, 934–945 CrossRef CAS.
- X. Yao, L. Chen, J. Cao, Y. Chen, M. Tian, F. Yang, J. Sun, C. Tang and L. Dong, Chem. Eng. J., 2019, 369, 46–56 CrossRef CAS.
- Z. M. Liu, H. Su, J. H. Li and Y. Li, Catal. Commun., 2015, 65, 51–54 CrossRef CAS.
- M. Y. Mihaylov, E. Z. Ivanova, H. A. Aleksandrov, P. S. Petkov, G. N. Vayssilov and K. I. Hadjiivanov, Appl. Catal., B, 2015, 176–177, 107–119 CrossRef CAS.
- D. M. Meng, Q. Xu, Y. L. Jiao, Y. Guo and Y. L. Wang, Appl. Catal., B, 2018, 221, 652–663 CrossRef CAS.
- D. K. Sun, Q. Y. Liu, Z. Y. Liu and Q. Guo, Appl. Catal., B, 2009, 92(s3–4), 462–467 CrossRef CAS.
- M. A. Larrubia, G. Ramis and G. Busca, J. Mol. Catal. A: Chem., 2001, 30(1), 101–110 CAS.
- N. Z. Yan, R. T. Guo, Q. S. Wang, W. G. Pan and S. X. Wang, RSC Adv., 2016, 6(14), 11226–11232 RSC.
- S. Xie, L. Li, L. Jin, Y. Wu, H. Liu, Q. Qin, X. Wei, J. Liu, L. Dong and B. Li, Appl. Surf. Sci., 2020, 515(15), 146014 CrossRef CAS.
- L. Chen, J. Li and M. Ge, Environ. Sci. Technol., 2010, 44(24), 9590–9596 CrossRef CAS PubMed.
- M. Bendrich, A. Scheuer, R. E. Hayes and M. Votsmeier, Appl. Catal., B, 2018, 222, 76–87 CrossRef CAS.
- Y. Shan, X. Shi and G. He, J. Phys. Chem. C, 2018, 122(45), 25948–25953 CrossRef CAS.
- Y. Liu, T. Gu, X. Weng, Y. Wang, Z. Wu and H. Wang, J. Phys. Chem. C, 2012, 116, 16582–16592 CrossRef CAS.
- W. Ren, Denitration mechanism and preparation of ferrous sulfate SCR catalyst, Tsinghua University, 2010 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence c,
Kunling Jiaoa and
Wenfei Wu
c,
Kunling Jiaoa and
Wenfei Wu