
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolydopamine coated hypodermic needles as a microextraction device for the determination of tricyclic antidepressants in oral fluid by direct infusion MS/MS†
Carmina Vejar-Vivar ab,
María Teresa García-Valverdea,
Claudia Mardonesb,
Rafael Lucena*a and
Soledad Cárdenasa
ab,
María Teresa García-Valverdea,
Claudia Mardonesb,
Rafael Lucena*a and
Soledad Cárdenasa
aAffordable and Sustainable Sample Preparation (AS2P) Research Group, Departamento de Química Analítica, Instituto Universitario de Investigación en Química Fina y Nanoquímica IUNAN, Universidad de Córdoba, Campus de Rabanales, Edificio Marie Curie, E-14071 Córdoba, Spain. E-mail: rafael.lucena@uco.es
bDepartamento de Análisis Instrumental, Facultad de Farmacia, Universidad de Concepción, Casilla 237, Correo 3, Concepción, Chile
First published on 28th June 2021
Abstract
In-needle microextraction consists of the confinement of the sorbent, by coating or packing, inside a metallic needle. The size of the needles reduces the eluent requirements providing an efficient preconcentration of the analytes. In this work, hypodermic needles coated with polydopamine (PDA) are presented as microextraction devices to isolate six tricyclic antidepressants from oral fluid samples. The coating consists of the in-surface polymerization of dopamine at pH 8.5 and mild conditions (room temperature and water as solvent). The PDA coating over the stainless-steel surface confers the needles with a high extraction ability towards the target analytes. After the extraction, the eluates were analyzed by direct infusion MS spectrometry, working in multiple reaction monitoring (MRM) mode, which provided a sample throughput of 30 samples per hour. The variables affecting the synthesis (number of coating cycles, the concentration of dopamine, and needle surface pre-treatment) and the extraction (sample salinity, sample loading cycles, and the number of elution strokes) were studied in depth. Under the optimum conditions, a matrix-matched calibration model was built. The limits of quantification are between 2 and 5 ng mL−1 with linear ranges up to 1000 ng mL−1 for all analytes. The precision, expressed as relative standard deviation (RSD), is better than 10% for all analytes. Accuracy was calculated as recovery, and the obtained values are between 84% and 107%. A single-blind assay was also performed to evaluate the suitability of the method for real application.
1. Introduction
Tricyclic antidepressants (TCAs) are psychoactive drugs used to treat depression and other psychiatric disorders such as anxiety and phobias.1 Lately, they have also been used to treat other conditions like chronic pain and migraines.2 The determination of TCAs in biological samples is of great importance in clinical and forensic toxicology.3 The high variability of the treatment response makes it important to control drug levels for each patient. Therapeutic drug monitoring of TCAs is highly recommended because of TCAs' narrow therapeutic window, given the risk of cardiac and central nervous system toxicity.4 TCAs should also be controlled since they may cause sedation and could affect cognitive and psychomotor functioning. In fact, they are considered driving-impairing medicines.5Although blood, plasma, or urine are the common bioanalytical specimens for determining antidepressants and other pharmaceuticals,6–11 oral fluid (OF) has demonstrated many advantages as an alternative matrix. OF is easy to be collected and presents high stability if properly stored. Human OF can be produced at a rate up to 6 mL min−1,12 making multisampling possible for repetitive or periodical analysis, such as pharmacodynamics studies. Besides, the concentration of drugs in OF and plasma are usually correlated,13,14 which is of paramount importance as the plasma concentration defines the actual therapeutic or toxic effects. However, OF analysis faces the same challenges observed for conventional biosamples, namely: the low concentration of the analytes and the high presence of interferents.15 The extraction of TCAs from OF by solid-phase extraction16 and liquid–liquid extraction17 has been reported as suitable approaches to overcome both issues.
In-needle microextraction techniques consist of the confinement of the sorbent inside a metallic needle. The sorbent can be coated as a thin film on the inner surface of the needle18–20 or packed (as particles or monolith) inside it.21–23 In both cases, the technique allows the sample to flow through the needle boosting the interaction with the target analytes. The miniaturized size of the needles requires little solvent for the elution, even when high sample volumes are processed, which is particularly interesting for an efficient preconcentration. In-needle microextraction, often named as needle trap, has been extensively used in biological,20,24–26 food,27,28 and environmental analysis.29 The technique is commonly combined with a previous headspace extraction, as a clean-up step, when complex samples are processed.25–28 Abdel-Rehim et al. reported using molecularly imprinted polymers as a strategy to overcome the sample complexity, thus allowing the direct extraction of urine samples.24 The search for new materials is of great interest to improve the performance and expand the application scope of the technique.30
Hypodermic needles (HN) are cheap and disposable materials extensively used in medicine. HNs have been mainly used to house the extraction probes for in vivo approaches in the sample preparation context.31–33 In this article, the use of HNs as the actual microextraction device is presented. The surface of a stainless-steel needle was coated with polydopamine (PDA), a very promising sorbent.34 PDA is a highly adhesive polymer that can be easily obtained by the in situ oxidative polymerization of dopamine, a small catecholamine.35 Inspired by mussel adhesive proteins, Lee et al. developed a single-step surface modification method based on dip-coating in an alkaline dopamine solution.36 The self-polymerization of dopamine can be used to deposit a thin film of PDA on a wide variety of organic and inorganic materials. The chemical structure of PDA is still under discussion, but the adhesion properties are related to the presence of many catechols and amine groups.37 Although the synthetic mechanism is not fully described, this method is a versatile and straightforward coating procedure.38 These advantages, added to its unique chemical and physical properties, allow its application in various fields, such as chemical, biological, medical, and materials sciences.39 In the microextraction field, PDA has been used to coat nickel foam40 and cellulose substrates.41,42 Also, it has been combined with nanomaterials like graphene43 or magnetic nanoparticles.44,45
In this article, PDA coated HNs are proposed as microextraction devices to isolate six TCAs from oral fluid samples. The low price and availability of the HNs and the simple coating process allow to develop individual extraction units for each sample. This disposability results essential in bioanalysis to avoid cross contamination and reduce the risks associated to the continuous exposure to the analysts to biosamples. The needles were attached to a plastic syringe or a micropipette, depending on the specific step of the extraction protocol, to handle the different solutions. The analytes were determined by direct infusion mass spectrometry (MS), which increased the sample throughput up to 30 samples per hour, guaranteeing a good selectivity level.
2. Experimental
2.1 Reagents and materials
All reagents were of analytical grade and purchased from Sigma-Aldrich (Madrid, Spain), unless otherwise indicated. Stock standard solutions of the tricyclic antidepressants (clomipramine, trimipramine, imipramine, amitriptyline, desipramine, and nortriptyline) were prepared in methanol at a concentration of 5000 mg L−1 and stored at 4 °C. Working standard solutions were prepared by diluting the stock solutions in Milli-Q water (Millipore Corp., Madrid, Spain) or oral fluid samples. Deuterated clomipramine-d3, desipramine-d3, and nortriptyline-d3 were used as the internal standards (IS) for MS measurements. A stock solution containing the three IS was prepared in methanol at a concentration of 10 mg L−1 and stored at −20 °C.Ammonia and sodium sulphate solutions were used for pH and ionic strength adjustment of aqueous solutions (standards and samples), respectively.
Trizma® buffer, dopamine, and sodium hydroxide solutions were employed to coat hypodermic needles of 40 mm in length and 0.8 mm in diameter (Becton Dickinson, Huesca, Spain) with a PDA layer.
Acetonitrile, Milli-Q water, and acetic acid were used as solvents for direct infusion MS analysis.
2.2 Oral fluid sample collection
For the optimization, blank oral fluid samples were collected using the Salivette® sampler (Sarstedt, Nümbrecht, Germany), which consists of a cotton roll that is introduced into the patient's mouth for at least 2 min without chewing. The rolls were then squeezed with a disposable 5 mL plastic syringe, and the recovered oral fluid was stored in glass vials at 4 °C until analysis. Because of the current world's sanitary situation (SARS-CoV-2 pandemic), oral fluid samples were donated only by the first author of this publication. For real samples analysis, including the matrix-matched calibration, using a cotton pad for sampling is not recommended for compounds with amine groups since a strong interaction between the analytes and the cellulose substrate has been found.46 For this purpose, the spitting method is proposed for sample acquisition.47Blank oral fluid samples spiked with the analytes were finally used for matrix-matched calibration. Before analysis, oral fluid was diluted at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio with a solution of 2% Na2SO4 to adjust the salinity to approximately 12 mmho cm−1. The pH was also adjusted to 10 with dilute ammonia solution.
:1 ratio with a solution of 2% Na2SO4 to adjust the salinity to approximately 12 mmho cm−1. The pH was also adjusted to 10 with dilute ammonia solution.
2.3 Preparation of PDA-coated hypodermic needles
Stainless-steel HNs were immersed at a depth of approximately 2.5 cm into a solution of dopamine (2 mg mL−1) freshly prepared in 10 mM Tris–HCl buffer (pH 8.5). The solution was magnetically stirred for 24 h at room temperature to allow the deposition of a PDA layer on the SS surface. The HNs were washed with Milli-Q water and subjected to ultrasound sonication for 5 min to remove the non-bonded PDA. After washing, the modified HNs were dried in an oven for 5 min. They were then left at room temperature until being used.2.4 Microextraction procedure
The design of the PDA-coated needles (PDA-HNs) permits the solutions to flow through their inner volume. For sample processing, the PDA-HN was directly attached to a 2 mL plastic syringe by the conventional Luer connector. For needle conditioning and elution, when a lower volume of solutions is required, the needle was attached to a 10–100 μL Labmate-Pro micropipette. The upper part (approximately 2.5 cm in length) of a plastic 200 μL pipette tip was used as a connector, as shown in Fig. 1.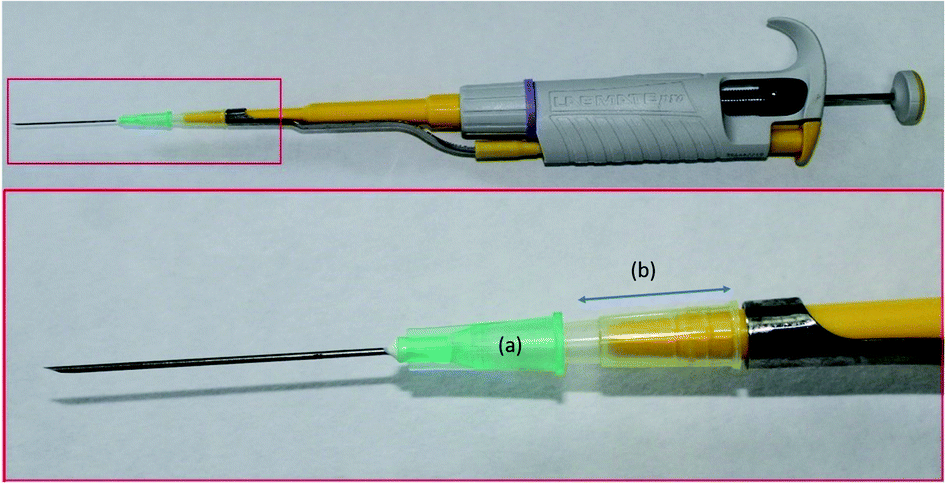 | ||
| Fig. 1 Image of the attachment of the PDA-coated hypodermic needle to the micropipette. The Luer connector of the hypodermic needle (a) is connected to the micropipette using a section of a 100 μL plastic tip (b). This device is used for needle conditioning and elution when a lower volume of solutions is required. For sample processing, however, the needle is attached to a 2 mL disposable plastic syringe. | ||
The sample preparation was achieved as follows: first, the PDA-HN was conditioned by the aspiration and ejection of 100 μL of methanol followed by 100 μL of an alkaline aqueous solution (pH = 10) using the micropipette. A volume of 2 mL of diluted oral fluid, which contained the deuterated standards at a final concentration of 50 ng mL−1 each, was placed into a centrifuge tube. Then, the sample was drawn and ejected 5 times (each time is named extraction cycle), using the 2 mL plastic syringe. The PDA-HN was then attached to the pipette again, and two washing steps using 100 μL of the alkaline aqueous solution (pH 10) were performed. Finally, the needle was eluted in a single stroke with 200 μL of methanol, collected in an HPLC vial. A volume of 5 μL of the extract was analyzed by direct infusion MS for identification and quantification of the analytes.
2.5 Direct infusion MS analysis
All analyses were performed into an Agilent 6420 Triple Quadrupole MS with an electrospray source working in the positive ionization mode. A carrier phase (90% ACN with 0.1% acetic acid) at a 2 mL min−1 rate was used for driving the analytes to the spectrometer providing a sample throughput of 30 samples per hour. The general MS operating conditions were set as follows: the capillary voltage was established at 2000 V, the flow rate of the drying gas (N2) was 9 mL min−1, its temperature was set at 300 °C, and the nebulizer pressure was 40 psi. Agilent MassHunter Software (version B.06.00, Santa Clara, CA, USA) was used for data analysis. The detection was achieved by Multiple Reaction Monitoring (MRM) transitions, and the specified parameters for each analyte and internal standards are presented in Table S1 (ESI†).The quantitative results were expressed as the ratio between the analyte and the internal standard areas. Clomipramine, desipramine, and nortriptyline were corrected by their corresponding deuterated labeled compounds, while clomipramine-d3 was also used for correcting the signals from trimipramine, imipramine, and amitriptyline.
3. Results and discussion
3.1 Optimization of the coating procedure
The optimization of the PDA coating process considered three different variables, namely: the number of coating cycles, the concentration of dopamine, and HN surface pre-treatment. All these variables were evaluated following a simple extraction procedure that is described in detail in the ESI.†The number of coating cycles needed to obtain an efficient coverage of the needle with PDA was initially evaluated. Each coating cycle consisted of immersing the HN to a depth of approximately 2.5 cm into a freshly prepared dopamine solution (2 g L−1) for 24 h under magnetic stirring. This variable was evaluated at four different levels, namely: 0 (uncoated needles), 1, 2, and 3 cycles. The results, shown in Fig. 2, indicate the negligible capacity of uncoated needles to extract the analytes and the pivotal role of PDA for the efficient isolation of the compounds. Although for some analytes (clomipramine and imipramine) a better extraction can be achieved for larger coating cycles, one coating cycle was selected as the optimum value to safe resources (dopamine, energy) and make the synthesis faster.
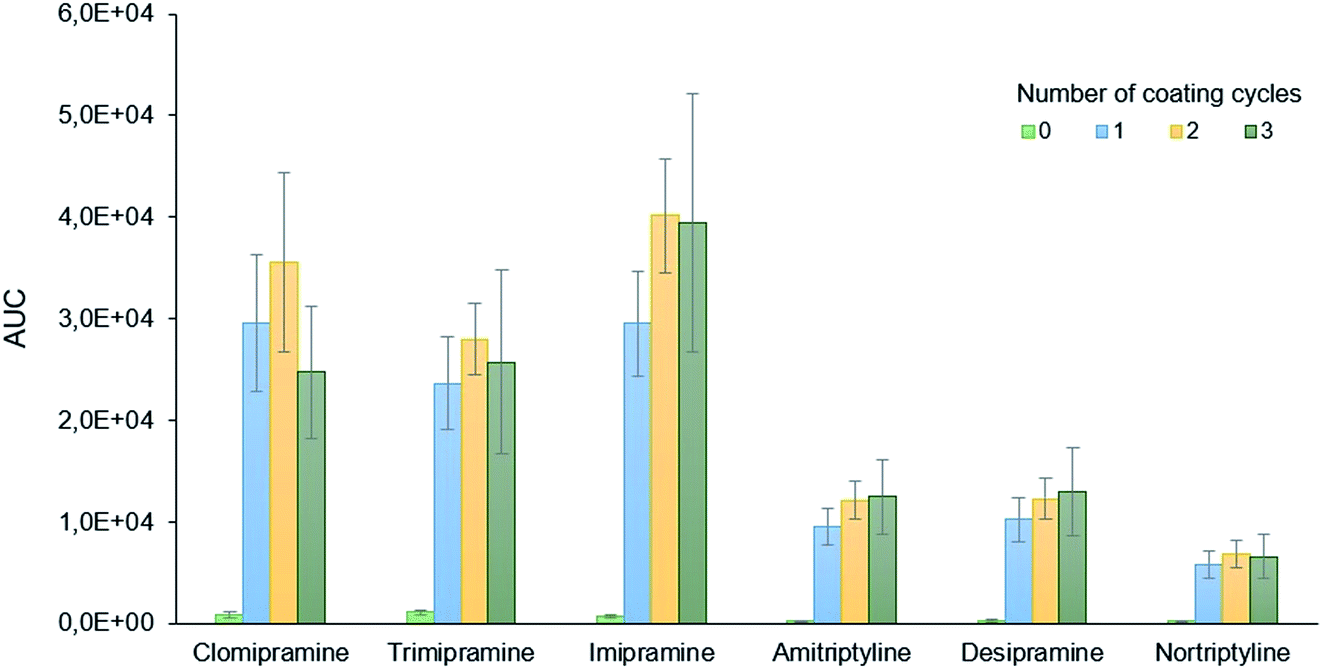 | ||
| Fig. 2 Effect of the PDA-coating cycles on the extraction of the target analytes. The results are presented as the area under the curve (AUC) values once the MRM transition for each analyte is isolated. | ||
The concentration of dopamine in the precursor solution may impact the HN coating, and it was studied at three different levels (2, 4, and 6 g L−1). As it is shown in Fig. S1,† the extraction capability of the PDA-HN decreases as the concentration of dopamine increased. This result could be explained since a higher concentration of dopamine in solution may favor the bulk polymerization over the PDA deposition on the stainless-steel needle, resulting in a more ineffective coating. In fact, the more concentrated solutions (4 and 6 g L−1) becomes darker during the synthesis due to the aggregation of PDA in the solution.
Commercial HNs are coated with a thin layer of silicone oil to reduce friction during skin penetration. Although the experiments mentioned above have demonstrated the capability of PDA-coated needles for the extraction of TCAs, two different surface treatments previous to the HNs coating were evaluated to ensure optimal functionalization. The first one consisted of the sonication of the needles in an aqueous solution of HNO3 (32.5% v/v) for 15 minutes to obtain a surface rich in hydroxyl groups.48 The second pre-treatment involved the sonication of the HN with toluene for 15 minutes to eliminate the silicone layer. As observed in Fig. S2,† the best results are obtained for the needles without any pre-treatment before being coated with PDA. For some analytes, such as clomipramine or nortriptyline, a slight improvement in the signal may be noticed for the extraction using needles treated with HNO3. These results indicate that the potential presence of silicon oil does not interfere in the deposition of PDA over the needle surface. For simplicity, no pre-treatment was applied to the HNs in further studies.
3.2 Optimization of the extraction method
The extraction performance was optimized considering the effect of three variables, namely: ionic strength, sample loading cycles, and the number of elution strokes. The pH was fixed at 10 to assess that the analytes are mainly in their non-protonated form. The sample volume was established at 2 mL, an affordable volume for oral fluid analysis, especially if the oral fluid needs to be diluted before its processing. Aqueous standards containing the analytes at a final concentration of 50 ng mL−1 were processed following the general workflow presented in Section 2.4.The ionic strength of the sample may have a thermodynamic (salting-out effect) and kinetic (reducing the diffusion rates of the analytes) impact on the extraction. The effect of the salinity was evaluated by extracting 5 standard solutions with increasing concentrations of Na2SO4, selected as the model electrolyte, in the range from 0 to 2%. These Na2SO4 concentrations were transformed into salinity units (in the 0.03–19.18 mmho cm−1 interval) using a conductometer. As it is shown in Fig. S3,† the presence of salt negatively affects the extraction of the analytes. However, the extraction performance remains almost constant for concentrations higher than 1% Na2SO4 (salinity over 10.8 mmho cm−1). Oral fluid samples have their own salinity, which is ca. 2.13 mmho cm−1 (average value obtained after measuring different oral fluid pools) when oral fluid is 1:1 diluted in alkaline water. This 1:1 dilution has been found to be an adequate ratio in our previous works to reduce the oral fluid viscosity for an appropriate extraction. To minimize the salinity effect on the extraction, it should be adjusted to a value between 10 and 15 mmho cm−1 when analyzing real samples.
The extraction cycles, defined as the times the sample is drawn and ejected from the 2 mL syringe, were evaluated from 1 to 10. As is shown in Fig. 3, the extraction efficiency increases with the number of cycles. For clomipramine, trimipramine, and amitriptyline, a decrease is observed from 5 to 10 cycles, while a slight increase is observed for the rest of the analytes. The precision values and the sample throughput, however, are better for 5 cycles which were finally selected as the optimum conditions.
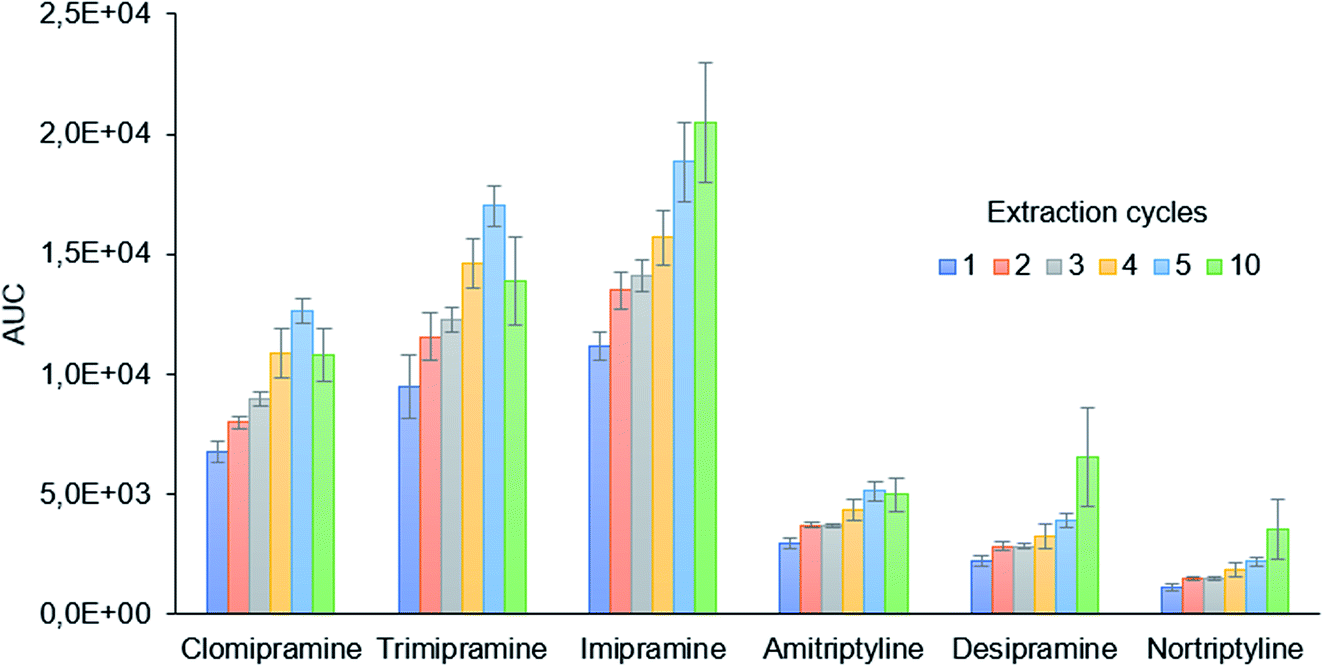 | ||
| Fig. 3 Study of the effect of the sample extraction cycles on extraction efficiency. | ||
Finally, the number of elution strokes was evaluated. The results show (Fig. S4†) that a single stroke is enough to obtain an efficient elution of the TCAs for MS analysis.
3.3 Method validation
The developed analytical method was validated in terms of linearity, sensitivity, precision, and accuracy using matrix-matched calibration curves for each antidepressant. Oral fluid samples were initially analysed to confirm the absence of the target analytes. Blank oral fluid samples were initially 1:1 v/v diluted with a solution of 2% Na2SO4 to adjust the salinity to approximately 12 mmho cm−1. The pH was also adjusted to 10 with dilute ammonia solution. These standards were spiked with the target analytes at seven concentration levels up to 1000 ng mL−1, maintaining the concentration of the three IS at 50 ng mL−1. Each standard was processed following the optimized microextraction workflow, and the final extracts were analyzed by direct infusion mass spectrometry. The calibration curves obtained by representing the analyte/IS area ratio versus the concentration are shown in Fig. 4, while the slope, intercept, and R2 values are summarized in Table S2.†
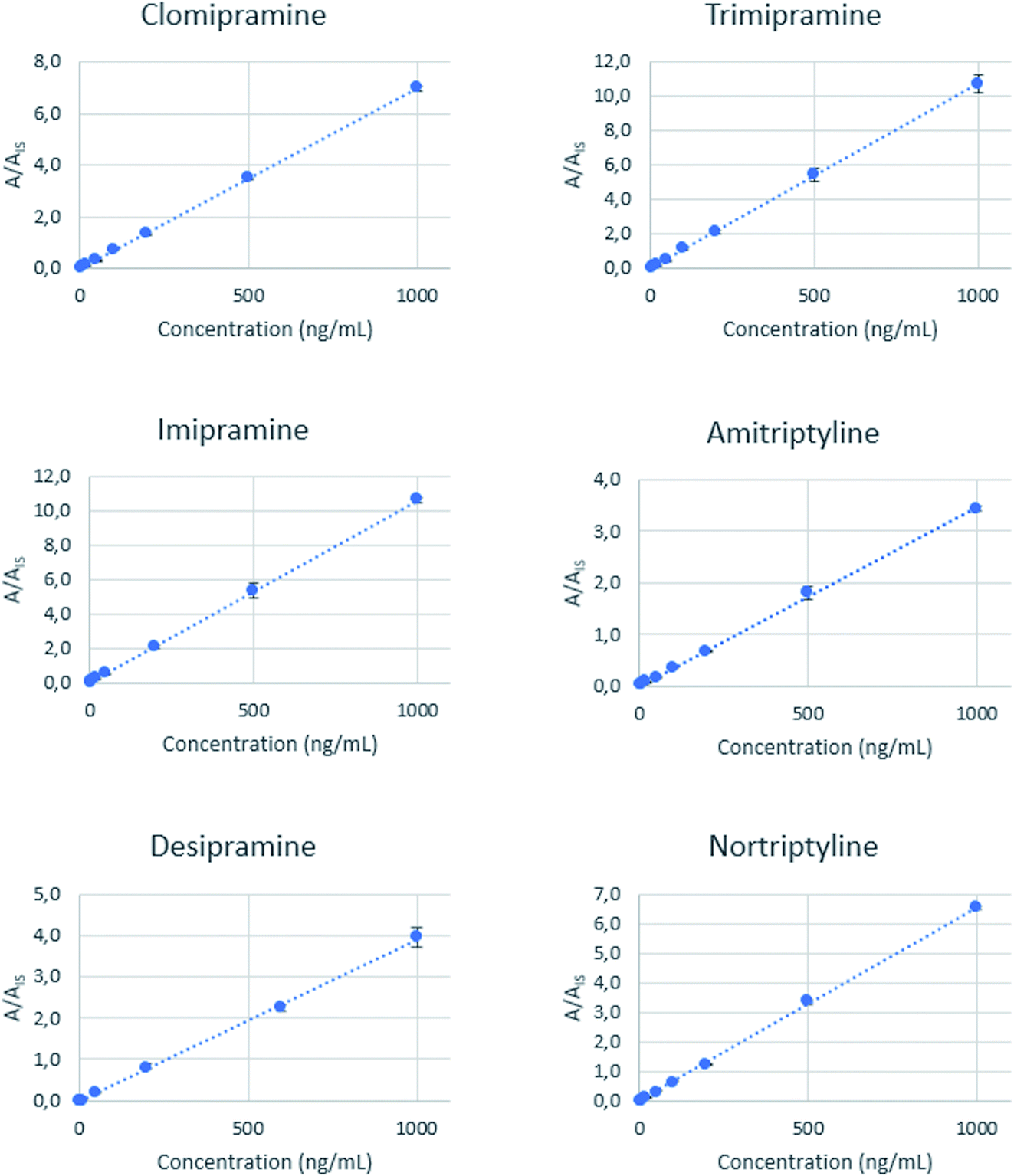 | ||
| Fig. 4 Calibration curves obtained for each analyte by representing the analyte/IS area ratio against the concentration of the analytes. Each point represents the average of three independent measurements, and error bars illustrate the standard deviation of the corresponding mean value. | ||
Good linearity values (R2 > 0.99) are achieved for all the analytes. Limits of quantification (LOQ) are defined as the lower concentration that could be determined with a relative standard deviation (RSD) less than 20%, presenting a signal-to-noise ratio higher than 10. These values are between 2 and 5 ng mL−1 for all analytes. The precision of the method was expressed as the relative standard deviation (RSD) and was calculated from oral fluid samples spiked at three different concentration levels (6, 60, and 600 ng mL−1) and analyzed in triplicate. The obtained RSD values range from 2.1% to 9.6%. A recovery study was done using an independent oral fluid pool, and the results are found to be between 84% and 107%, showing a good accuracy level. Each extraction was performed using independent PDA-coated needles, so batch-to-batch reproducibility could also be evaluated. Table 1 summarizes the figures of merit of the method. Table 2 compares the sensitivity of the proposed method with other alternatives reported for the determination of tricyclic antidepressants in biofluids, including saliva, urine, plasma and blood.1,6,7,9,12,16,49–53 Our method provides a wider linear range, allowing the determination of the target compounds at the therapeutic level. In the oral fluid applications, the sensitivity of the new method is intermediate and only surpassed by the method based on high-resolution MS, although the latter provided a narrower linearity range. The reported methods made use of a previous separation (by gas or liquid chromatography) of the analytes, thus providing an enhanced selectivity. However, this enhancement is done at the expense of the analysis time. The use of the direct infusion MS provides a high sample throughput, and the chromatographic selectivity is compensated by an efficient extraction and the use of MS as the instrumental technique.
| Analyte | LOD (ng mL−1) | LOQ (ng mL−1) | R2 | Linear range (ng mL−1) | RSD (%, n = 3) | Recovery (% n = 3) | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| 6 ng L−1 | 60 ng L−1 | 600 ng L−1 | 6 ng L−1 | 60 ng L−1 | 600 ng L−1 | |||||
| a LOD, limit of detection; LOQ, limit of quantification; RSD, relative standard deviation. | ||||||||||
| Clomipramine | 1.5 | 5 | 0.9997 | 5–1000 | 2.2 | 2.1 | 4.7 | 95 ± 2 | 99 ± 2 | 92 ± 4 |
| Trimipramine | 1.5 | 5 | 0.9969 | 5–1000 | 7.2 | 6.0 | 7.1 | 101 ± 6 | 87 ± 5 | 95 ± 7 |
| Imipramine | 0.6 | 2 | 0.9983 | 2–1000 | 6.6 | 3.2 | 2.1 | 100 ± 8 | 106 ± 3 | 98 ± 2 |
| Amitriptyline | 1.5 | 5 | 0.9984 | 5–1000 | 9.6 | 2.8 | 2.1 | 96 ± 11 | 94 ± 3 | 95 ± 2 |
| Desipramine | 0.6 | 2 | 0.9966 | 2–1000 | 9.6 | 2.8 | 2.1 | 90 ± 10 | 84 ± 2 | 84 ± 2 |
| Nortriptyline | 1.5 | 5 | 0.9994 | 5–1000 | 3.0 | 4.6 | 3.6 | 92 ± 3 | 107 ± 5 | 96 ± 3 |
| Matrix | Sample preparation | Instrumental technique | Analytes | LOD (ng mL−1) | LOQ (ng mL−1) | Linear range | Ref. |
|---|---|---|---|---|---|---|---|
| a PDA-Ag-PPy, polydopamine, silver nanoparticles, and polypyrrole composite; MEPS, microextraction in packed syringe; poly-(GMA-co-EDMA-MWCNTs), poly-(methacrylate-co-ethylene glycol dimethacrylate) containing multiwalled carbon nanotubes; SPE, solid phase extraction; LC, liquid chromatography; UHPLC, ultrahigh pressure liquid chromatography; GC, gas chromatography; MS, mass spectrometry; QTOF, quadrupole time-of-flight; LOD, limit of detection; LOQ, limit of quantification. | |||||||
| Oral fluid | PDA coated in-needle microextraction | LC-MS/MS | Clomipramine | 1.5 | 5 | 5–1000 | This paper |
| Trimipramine | 1.5 | 5 | 5–1000 | ||||
| Imipramine | 0.6 | 2 | 2–1000 | ||||
| Amitriptyline | 1.5 | 5 | 5–1000 | ||||
| Desipramine | 0.6 | 2 | 2–1000 | ||||
| Nortriptyline | 1.5 | 5 | 5–1000 | ||||
| Urine | MEPS (PDA-Ag-PPY) | GC-MS | Amitriptyline | 0.03 | 0.10 | 0.03–100 | 1 |
| Imipramine | 0.05 | 0.20 | 0.05–100 | ||||
| Whole blood | Protein precipitation | LC-QTOF-MS | Amitriptyline | n.a. | 25 | 25–600 | 6 |
| Clomipramine | 20 | 20–900 | |||||
| Desipramine | 50 | 50–600 | |||||
| Imipramine | 20 | 20–600 | |||||
| Trimipramine | 10 | 10–600 | |||||
| Urine | Poly-(GMA-co-EDMA-MWCNTs) monolith pipette tip | LC-UV | Desipramine | 9 | 14 | 14–1000 | 7 |
| Amitriptyline | 15 | 30 | |||||
| Trimipramine | 15 | 29 | |||||
| Plasma | Cloud-point extraction | LC-MS/MS | Amitriptyline | n.a. | 10 | 10–750 | 9 |
| Clomipramine | |||||||
| Desipramine | |||||||
| Imipramine | |||||||
| Nortriptyline | |||||||
| Trimipramine | |||||||
| Oral fluid | MEPS (C8/SCX) | UHPLC-TOF-MS | Desipramine | 0.04 | 0.14 | 0.1–10 | 12 |
| Nortriptyline | 0.01 | 0.03 | |||||
| Imipramine | 0.03 | 0.09 | |||||
| Amitriptyline | 0.02 | 0.08 | |||||
| Oral fluid | SPE (mixed mode Cerex® Trace-B cartridges) | LC-MS/MS | Amitriptyline | — | 10 | 10–1000 | 16 |
| Clomipramine | |||||||
| Desipramine | |||||||
| Imipramine | |||||||
| Nortriptyline | |||||||
| Trimipramine | |||||||
| Urine | MWCNTs SPE | LC-UV | Desipramine | 40.6 | 135 | 49 | |
| Imipramine | 50.0 | 166 | |||||
| Nortriptyline | 35.9 | 119 | |||||
| Amitriptyline | 20.1 | 67 | |||||
| Trimipramine | 30.0 | 97 | |||||
| Clomipramine | 18.1 | 59 | |||||
| Plasma | Liquid–liquid extraction | UHPLC-MS/MS | Amitriptyline | 0.2 | 10 | 10–1000 | 50 |
| Clomipramine | 0.4 | 10 | |||||
| Desipramine | 2.0 | 10 | |||||
| Imipramine | 2.0 | 10 | |||||
| Nortriptyline | 1.0 | 10 | |||||
| Postmortem blood | Salting-out assisted liquid–liquid extraction | UPLC-QTOF-MS | Amitriptyline | 0.010 | 0.05 | 0.05–2 | 51 |
| Clomipramine | 0.005 | 0.05 | 0.05–2 | ||||
| Desipramine | 0.010 | 0.05 | 0.05–2 | ||||
| Imipramine | 0.005 | 0.05 | 0.05–2 | ||||
| Nortriptyline | 0.010 | 0.05 | 0.05–2 | ||||
| Trimipramine | 0.005 | 0.05 | 0.05–2 | ||||
| Postmortem blood | Mini-QuEChERS | UHPLC-MS/MS | Amitriptyline | 0.0003 | 0.001 | 0.001–0.500 | 52 |
| Nortriptyline | 0.0003 | 0.001 | 0.001–0.500 | ||||
| Whole blood | Supported liquid extraction | UPLC-MS/MS | Imipramine | 0.0030 | 0.010 | 0.001–200 | 53 |
| Desipramine | 0.0003 | 0.001 | |||||
| Clomipramine | 0.0003 | 0.001 | |||||
| Amitriptyline | 0.0003 | 0.001 | |||||
3.4 Application of the developed method for TCA analysis in oral fluid
Since no positive samples for these analytes were available to evaluate the validated method, a “single-blind” analysis was performed. For this purpose, an analyst prepared six “blind” samples by spiking blank oral fluid with different concentrations of the target analytes, while a different analyst for whom the composition and concentration of each sample were unknown, performed the extraction and analysis of the samples. The method has allowed the correct identification of all analytes in all samples, while the recovery values are also calculated (Table 3) with achieved values ranging between 77 ± 1 for clomipramine in sample 6 and 119 ± 8 for imipramine in sample 3.| Spiked sample | Identified analyte | Spiked concentration (ng mL−1) | Recovery (%) |
|---|---|---|---|
| 1 | Clomipramine | 100 | 104 ± 7 |
| Amitriptyline | 50 | 89 ± 9 | |
| 2 | Trimipramine | 60 | 97 ± 6 |
| Nortriptyline | 40 | 99 ± 4 | |
| 3 | Imipramine | 250 | 119 ± 8 |
| Desipramine | 150 | 106 ± 5 | |
| 4 | Clomipramine | 50 | 102 ± 2 |
| Imipramine | 370 | 117 ± 12 | |
| Desipramine | 350 | 89 ± 1 | |
| 5 | Trimipramine | 100 | 93 ± 6 |
| Nortriptyline | 80 | 103 ± 1 | |
| 6 | Clomipramine | 500 | 77 ± 1 |
| Amitriptyline | 300 | 96 ± 5 |
4. Conclusion
A microextraction device consisting of the coating of stainless-steel hypodermic needles with polydopamine is described in the present article. The extraction capability of the needles towards six TCA drugs in oral fluid is illustrated, demonstrating the pivotal role of the PDA coating in the analyte's isolation.The coating of the HN is straightforward and reproducible, and it allows to obtain inexpensive and disposable extraction devices, which makes them highly suitable for bioanalysis. In fact, these features are relevant to avoid cross-contamination and unnecessary handling of biological materials.
PDA-HN were employed to extract clomipramine, trimipramine, imipramine, amitriptyline, desipramine, and nortriptyline from oral fluid samples, allowing the rapid and successful direct MS analysis of up to 30 samples per hour. The precision and accuracy of the measurements fulfill the quality criteria in bioanalysis.
The use of oral fluid as the sample presents several advantages compared to other matrixes like blood or urine, since its collection is non-invasive and uncomplicated, and it does not require an extensive sample treatment.
Although it has not been evaluated in this article, the biocompatibility of polydopamine opens the door to be in vivo applied to make simultaneous sampling and extraction possible.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Spanish Ministry of Economy and Competitiveness (CTQ2017-83175R) is gratefully acknowledged. C. V.-V. expresses her gratitude to the Gobierno de Chile for the grant “Beca ANID Doctorado Nacional Folio No. 2117044”.References
- H. Bagheri, S. Banihashemi and F. K. Zandian, Microchim. Acta, 2016, 183, 195–202 CrossRef CAS.
- C. Coulter, M. Taruc, J. Tuyay and C. Moore, J. Anal. Toxicol., 2010, 34, 64–72 CrossRef CAS PubMed.
- N. Manousi and V. F. Samanidou, Mini-Rev. Med. Chem., 2019, 20, 24–38 CrossRef PubMed.
- A. de Castro, M. Concheiro, O. Quintela, A. Cruz and M. López-Rivadulla, J. Pharm. Biomed. Anal., 2008, 48, 183–193 CrossRef CAS PubMed.
- E. Gutiérrez-Abejón, F. Herrera-Gómez, P. Criado-Espegel and F. J. Álvarez, Pharmaceuticals, 2020, 13(4), 61 CrossRef PubMed.
- A. T. Roemmelt, A. E. Steuer and T. Kraemer, Anal. Chem., 2015, 87, 9294–9301 CrossRef CAS PubMed.
- B. Fresco-Cala, Ó. Mompó-Roselló, E. F. Simó-Alfonso, S. Cárdenas and J. M. Herrero-Martínez, Microchim. Acta, 2018, 185, 127 CrossRef PubMed.
- V. Alves, J. Gonçalves, C. Conceição, H. M. T. Câmara and J. S. Câmara, J. Chromatogr. A, 2015, 1408, 30–40 CrossRef CAS PubMed.
- E. Gniazdowska, N. Korytowska, G. Kłudka and J. Giebułtowicz, Pharmaceuticals, 2020, 13, 1–25 CrossRef PubMed.
- A. Luiz Oenning, L. Birk, S. Eller, T. Franco de Oliveira, J. Merib and E. Carasek, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2020, 1143, 122069 CrossRef CAS PubMed.
- L. Mifsud Buhagiar, C. Sammut, Y. Chircop, K. Axisa, N. Sammut Bartolo, J. Vella Szijj, A. Serracino Inglott and G. LaFerla, Biomed. Chromatogr., 2019, 33, 1–9 CrossRef PubMed.
- M. Woźniakiewicz, R. Wietecha-Posłuszny, A. Moos, M. Wieczorek, P. Knihnicki and P. Kościelniak, J. Chromatogr. A, 2014, 1337, 9–16 CrossRef PubMed.
- S. Chojnowska, T. Baran, I. Wilińska, P. Sienicka, I. Cabaj-Wiater and M. Knaś, Adv. Med. Sci., 2018, 63, 185–191 CrossRef PubMed.
- E. Bassotti, G. M. Merone, A. D’Urso, F. Savini, M. Locatelli, A. Tartaglia, P. Dossetto, C. D’Ovidio and U. de Grazia, Forensic Sci. Int., 2020, 312, 110330 CrossRef CAS PubMed.
- R. Lucena, J. Appl. Bioanal., 2015, 1, 72–75 CrossRef CAS.
- S. S. Shin, D. Borg and R. Stripp, J. Anal. Toxicol., 2020, 44, 610–617 CrossRef CAS PubMed.
- B. Desharnais, M. J. Lajoie, J. Laquerre, P. Mireault and C. D. Skinner, Forensic Sci. Int., 2020, 317, 110506 CrossRef CAS PubMed.
- H. Bagheri, E. Babanezhad and F. Khalilian, Anal. Chim. Acta, 2009, 634, 209–214 CrossRef CAS PubMed.
- D. Djozan, M. A. Farajzadeh, S. M. Sorouraddin, T. Baheri and J. Norouzi, Chromatographia, 2012, 75, 139–148 CrossRef CAS.
- Y. Li, J. Li and H. Xu, RSC Adv., 2017, 7, 11959–11968 RSC.
- H. Bagheri, Z. Ayazi and A. Aghakhani, Anal. Chim. Acta, 2011, 683, 212–220 CrossRef CAS PubMed.
- Y. Yang, W. Mai, J. Gao, Z. Hu, J. Xu and S. Zou, J. Sep. Sci., 2019, 42, 1750–1756 CrossRef CAS PubMed.
- X. Zhang, J. Chen, L. Lian, X. Wang, X. Guo, H. Chen, B. Zhu, S. Hou and D. Lou, Chromatographia, 2019, 82, 953–960 CrossRef CAS.
- M. M. Moein, D. Jabbar, A. Colmsjö and M. Abdel-Rehim, J. Chromatogr. A, 2014, 1366, 15–23 CrossRef CAS PubMed.
- P. Porto-Figueira, J. A. M. Pereira and J. S. Câmara, Anal. Chim. Acta, 2018, 1023, 53–63 CrossRef CAS PubMed.
- J. Ghafari, M. Vahabi, S. F. Dehghan and R. Zendehdel, Biomed. Chromatogr., 2020, 34, e4924 CrossRef CAS PubMed.
- J. M. Warren, D.-R. Parkinson and J. Pawliszyn, J. Agric. Food Chem., 2013, 61, 492–500 CrossRef CAS PubMed.
- J. Chen, B. Zhang, D. Zheng, X. Dang, Y. Ai and H. Chen, Anal. Methods, 2018, 10, 5783–5789 RSC.
- H. Li, C. Bi, X. Li and Y. Xu, Chemosphere, 2020, 250, 126284 CrossRef CAS PubMed.
- K. Kędziora and W. Wasiak, J. Chromatogr. A, 2017, 1505, 1–17 CrossRef PubMed.
- D. Vuckovic, I. de Lannoy, B. Gien, R. E. Shirey, L. M. Sidisky, S. Dutta and J. Pawliszyn, Angew. Chem., Int. Ed., 2011, 50, 5344–5348 CrossRef CAS PubMed.
- P. Donabella, N. Rogers, R. Levin and F. M. Musteata, Bioanalysis, 2015, 7, 661–670 CrossRef CAS PubMed.
- B. Bojko, N. Looby, M. Olkowicz, A. Roszkowska, B. Kupcewicz, P. Reck dos Santos, K. Ramadan, S. Keshavjee, T. K. Waddell, G. Gómez-Ríos, M. Tascon, K. Goryński, M. Cypel and J. Pawliszyn, J. Pharm. Anal., 2021, 11, 37–47 CrossRef PubMed.
- D. Che, J. Cheng, Z. Ji, S. Zhang, G. Li, Z. Sun and J. You, TrAC, Trends Anal. Chem., 2017, 97, 1–14 CrossRef CAS.
- V. Ball, D. Del Frari, V. Toniazzo and D. Ruch, J. Colloid Interface Sci., 2012, 386, 366–372 CrossRef CAS PubMed.
- H. Lee, S. M. Dellatore, W. M. Miller and P. B. Messersmith, Science, 2007, 318, 426–430 CrossRef CAS PubMed.
- J. Liebscher, Eur. J. Org. Chem., 2019, 2019, 4976–4994 CrossRef CAS.
- J. H. Ryu, P. B. Messersmith and H. Lee, ACS Appl. Mater. Interfaces, 2018, 10, 7523–7540 CrossRef CAS PubMed.
- Y. Liu, K. Ai and L. Lu, Chem. Rev., 2014, 114, 5057–5115 CrossRef CAS PubMed.
- T. Sun, M. Wang, D. Wang and Z. Du, Talanta, 2020, 207, 120244 CrossRef CAS PubMed.
- J. Feng, M. Sun, S. Han, X. Ji, C. Li, X. Wang and Y. Tian, J. Sep. Sci., 2019, 42, 2163–2170 CrossRef CAS PubMed.
- C. Ye, Y. Wu and Z. Wang, RSC Adv., 2016, 6, 9066–9071 RSC.
- Q. Xu, K. Qiao, C. Yan, Z. Liu, R. Lu and W. Zhou, Anal. Methods, 2020, 12, 3115–3122 RSC.
- W. Fan, D. Yang, N. Ding, P. Chen, L. Wang, G. Tao, F. Zheng and S. Ji, Anal. Methods, 2021, 13, 1412–1421 RSC.
- J. Grau, J. L. Benedé and A. Chisvert, Talanta, 2021, 122375 CrossRef CAS PubMed.
- Ł. Sobczak and K. Goryński, Analyst, 2020, 145, 7279–7288 RSC.
- F. G. Bellagambi, T. Lomonaco, P. Salvo, F. Vivaldi, M. Hangouët, S. Ghimenti, D. Biagini, F. Di Francesco, R. Fuoco and A. Errachid, TrAC, Trends Anal. Chem., 2020, 124, 115781 CrossRef CAS.
- A. Bekmurzayeva, W. J. Duncanson, H. S. Azevedo and D. Kanayeva, Mater. Sci. Eng., C, 2018, 93, 1073–1089 CrossRef CAS PubMed.
- M. Cruz-Vera, R. Lucena, S. Cárdenas and M. Valcárcel, Anal. Bioanal. Chem., 2008, 391, 1139–1145 CrossRef CAS PubMed.
- M. del Mar Ramírez Fernández, S. M. R. Wille and N. Samyn, Ther. Drug Monit., 2012, 34, 11–24 CrossRef PubMed.
- S. Bidny, K. Gago, P. Chung, D. Albertyn and D. Pasin, J. Anal. Toxicol., 2017, 41, 181–195 CrossRef CAS PubMed.
- A. Pouliopoulos, E. Tsakelidou, A. Krokos, H. G. Gika, G. Theodoridis and N. Raikos, J. Anal. Toxicol., 2018, 42, 337–345 CrossRef CAS PubMed.
- W. Ma, X. Gao, H. Guo and W. Chen, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2021, 1171, 122608 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra02721b |
| This journal is © The Royal Society of Chemistry 2021 |