
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSupramolecular D⋯A-layered structures based on germanium complexes with 2,3-dihydroxynaphthalene and N,N′-bidentate ligands†
Pavel G. Shangina,
Irina V. Krylovaa,
Andrey V. Lalova,
Anna Y. Kozmenkova a,
Evgeniya A. Saverinaa,
Petr A. Buikinb,
Alexander A. Korlyukovb,
Alyona A. Starikovac,
Elena N. Nikolaevskayaa,
Mikhail P. Egorova and
Mikhail A. Syroeshkin*a
a,
Evgeniya A. Saverinaa,
Petr A. Buikinb,
Alexander A. Korlyukovb,
Alyona A. Starikovac,
Elena N. Nikolaevskayaa,
Mikhail P. Egorova and
Mikhail A. Syroeshkin*a
aN. D. Zelinsky Institute of Organic Chemistry, 119991 Moscow, Russia. E-mail: syroeshkin@ioc.ac.ru
bA. N. Nesmeyanov Institute of Organoelement Compounds, 119991 Moscow, Russia
cInstitute of Physical and Organic Chemistry, Southern Federal University, 344090 Rostov-on-Don, Russia
First published on 17th June 2021
Abstract
The concept of using redox-active ligands, which has become extremely widespread in organometallic chemistry, is often considered from ‘their effect on the metal center properties’ point of view and ‘how to modify the ligands’. In this paper, we present the reverse side of this effective approach – a dramatic change of redox properties of ligands under the influence of a redox-inert metal. Germanium derivatives based on 2,3-dihydroxynaphthalene (1) and N,N′-bidentate ligands, namely 2,2′-bipyridine (2) and 1,10-phenanthroline (3), were obtained and characterized by CV, UV-vis spectroscopy, DFT calculations and in the case of 3 X-ray diffraction. It was shown that the HOMO of the complexes is almost completely located on the naphthalene fragment while the LUMO is on the N,N-ligands. At the same time, there are no boundary molecular orbitals on the germanium atom, but it forms the axial part of the molecule holding two opposite motifs together. Moreover, it sharply affects the level of HOMO and LUMO. Derivatives 2 and 3 are more easily oxidized compared to 2,3-dihydroxynaphthalene by 0.31–0.34 V (7–8 kcal mol−1) and are more easily reduced compared to N,N-donors by 1.08–1.15 V (25–26.5 kcal mol−1). All this together makes it possible to form a system with a narrow HOMO/LUMO gap (∼2 eV). The crystal structure of 3 consists of alternating monomolecular easily oxidizing and easily reducing layers formed due to intermolecular interactions, in particular π-stacking. In addition, in contrast to 1 that starts to decompose noticeably at the temperatures from 200 °C, 2 and 3 have an extremely high thermal stability. They remain stable with no signs of decomposition and melting up to 400 °С. We believe that this approach to the formation of the supramolecular structure may present prospects for obtaining new functional materials.
Introduction
The use of redox-active ligands is an effective approach to control and regulate the electrochemical, photochemical and magnetic properties of organometallic compounds, to manipulate their reactivity in chemical processes.1–5 In this regard, the concept of redox-active ligands has found various applications in the production of new materials with useful properties6 and catalysis7,8 as well as chemistry of organogermanium derivatives9–12 etc.Germanium is a “special” element of the Periodic Table. Its chemical properties are similar to ones of the ancestor of the 14th group – carbon – the basic element of organic chemistry. However, the more complex electron shell makes germanium a typical metalloid.13 Germanium is distinguished from silicon by a relatively higher reactivity of its dioxide, which allows it to enter a variety of reactions under mild conditions, including those close to physiological ones.14 In addition, organic and coordination germanium compounds are significantly more chemically stable, including reactions with water and oxygen.15 In turn, germanium derivatives are distinguished from their heavier analogues in group 14 – tin and, especially, lead – by their significantly lower toxicity.16 This property seems to make germanium unique among organometallic compounds due to, in some cases (for example, derivatives with carboxylic acid17), the toxicity of its salts is comparable to sodium chloride18 and other typical participants of physiological processes. These features led to the using of germanium derivatives as physiologically active compounds19 (in particular, they exhibit antioxidant activity20), as well as in green chemistry21,22 and developing new approaches to functional materials23 such as Li-ion batteries24 etc.
Returning to the concept of using redox-active ligands, it can be noted that it is usually considered as a means of influencing the properties of the metal center, which remains the main participant of the processes. In this paper, we present germanium derivatives containing naphthalene and bipyridine motifs. Germanium in them has no boundary molecular orbitals and plays an axial role, chemically combining the HOMO (dioxonaphthalene) and LUMO (N-donor) centers. At the same time, the level of HOMO increases significantly due to the electronic effects of the germanium atom while level of LUMO decreases; as a result HOMO/LUMO gap becomes more narrow and the crystal is composed of alternating monomolecular easily oxidizing and easily reducing organic layers.
Results and discussion
Synthesis and crystal structure description
Derivative 1 was obtained by reaction of germanium dioxide with double molar excess of 2,3-dihydroxynaphthalene in boiling water. In this case, all reactants do not dissolve in water and product of reaction can be separated by filtration. Further interaction of 1 with 2,2′-bipyridine or 1,10-phenanthroline in boiling either water or acetonitrile or toluene led to formation of complexes 2 and 3, respectively, with high yields (Scheme 1). In case of organic solvents use, the N-donor ligands are in solution while compounds 1, 2 and 3 present as a precipitate; in water solution all the reaction participants do not dissolve.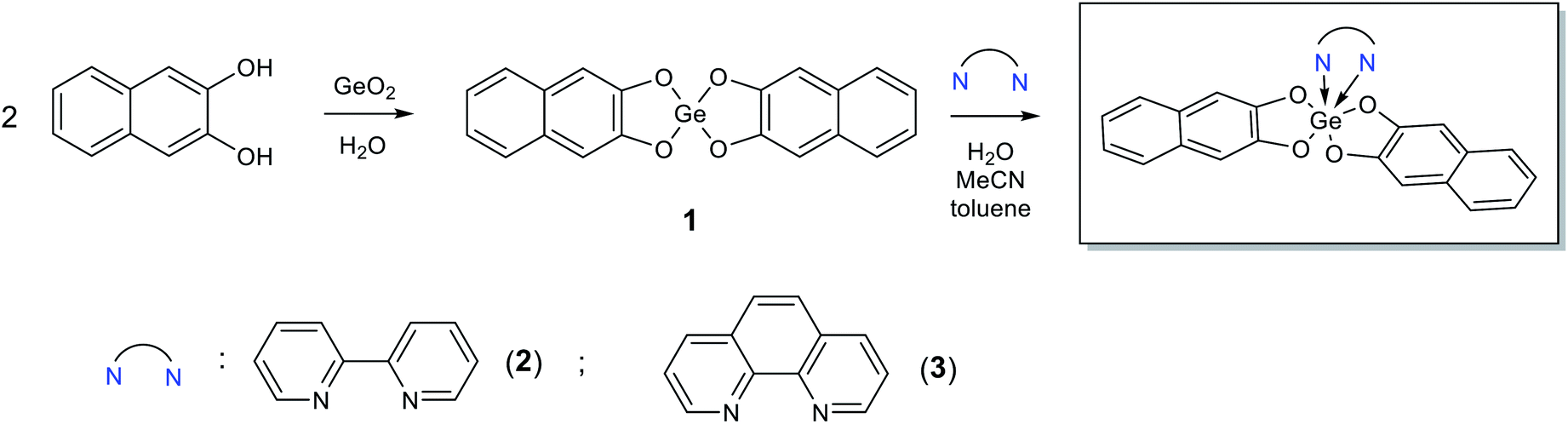 | ||
| Scheme 1 Synthesis of 1–3. | ||
Slow evaporation of ethanol solution of 3 at room temperature resulted in the reproducible formation of its yellow thin plate-shaped single crystals that were further studied by X-ray diffraction. The structure of 3 is one of the few examples of Ge(IV) complexes with phenanthroline ligand. To date the most studied complexes of such type are GeHal4 adducts. The closest structural analog of 3 is the complex of Ge(IV) containing two 3-carboxy-2-oxidopropanoato-O,O ligands and one phenanthroline ligand, the structure of which was published in CSD private communications (refcode JENHOC).25 Similarly to JENHOC, the coordination polyhedron of the germanium atom in 3 can be described as a distorted octahedron (Fig. 1). The lengths of Ge–O bonds in it vary in the narrow range (1.847–1.863 Å, Table 1), while in JENHOC the difference between them is much more pronounced (1.818–1.912 Å). The mean lengths of Ge–O bonds in both structures are almost the same (1.855 and 1.865 Å). The lengths of Ge–N bonds in 3 (2.110(11) and 2.068(11) Å) are somewhat increased as compared to JENHOC (2.057 Å) and complex phenanthroline·GeF4 (2.046 Å).26
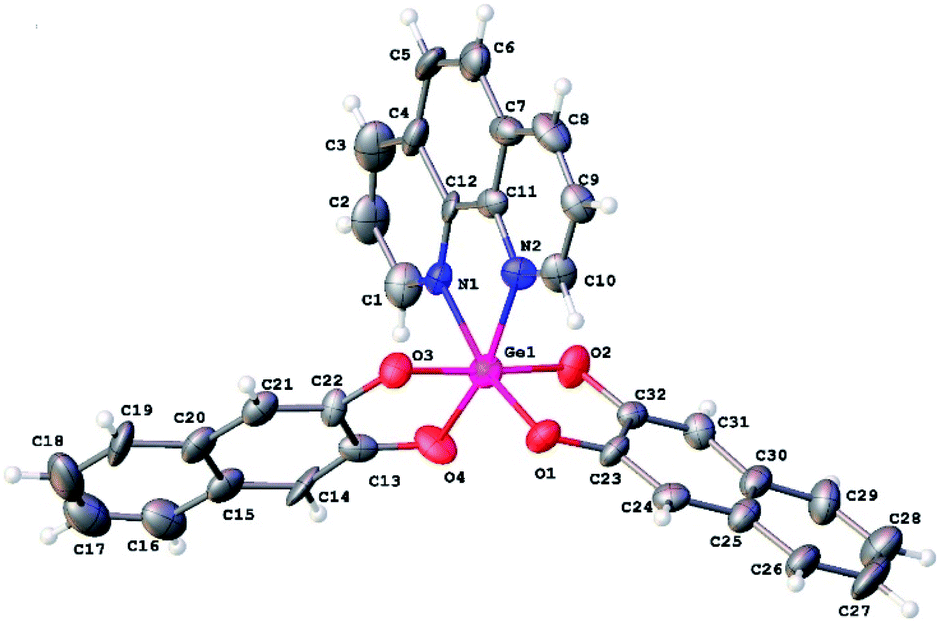 | ||
| Fig. 1 General view of 3. Atoms are presented in ADP ellipsoids drawn at 50% probability. | ||
| Bond | Bond length, Å | |||
|---|---|---|---|---|
| Neutral, crystal, X-ray | Neutral, gas phase, DFT | Radical-anion, gas phase, DFT | Radical-cation, gas phase, DFT | |
| Ge1–N1 | 2.068(11) | 2.177 | 2.053 | 2.104 |
| Ge2–N2 | 2.110(11) | 2.178 | 2.052 | 2.103 |
| Ge1–O1 | 1.862(9) | 1.851 | 1.895 | 1.888 |
| Ge1–O2 | 1.847(9) | 1.856 | 1.889 | 1.856 |
| Ge1–O3 | 1.850(9) | 1.856 | 1.889 | 1.857 |
| Ge1–O4 | 1.863(10) | 1.851 | 1.896 | 1.891 |
In crystal structure of 3 (Fig. 2), aromatic C–H groups form weak C–H⋯O interactions with oxygen atoms of 2,3-dihydroxynaphthalene ligands. At the same time, phenanthroline ligands additionally bind via stacking interactions between pyridine cycles (Fig. 3, the angle between planes of two cycles is 3.8°, centroid–centroid and shift distances are 3.66 and 1.22 Å, respectively). Additionally, a hydrogen atom of the phenanthroline ligand participates in C–H⋯π interaction (Fig. 4, the distance H⋯centroid is equal to 2.56 Å and C–H⋯centroid angle is 136°).
 | ||
| Fig. 2 Crystal packing of 3. | ||
 | ||
| Fig. 3 Stacking interaction in a crystal of 3, (a) top view, (b) side view. Hydrogen atoms are omitted for clarity. Second molecule is generated by x − 1, y, z symmetry operation. | ||
 | ||
| Fig. 4 C–H⋯π interaction in a crystal of 3. The centroid of Ph group is depicted by a black sphere. Second molecule is generated by 1 − x, −1/2 + y, 1 − z symmetry operation. | ||
Cyclic voltammetry
2,3-Dihydroxynaphthalene, 2,2′-bipyridine and 1,10-phenanthroline are capable of both electrochemically reducing and oxidizing (Table 2). However, taking into account that HOMO of complexes 2 and 3 are localized on the catechol motif, and the LUMO present on the N-donor fragments (see the DFT calculations), it would be correct to compare the complexes with substituted naphthalenenaphtalene in terms of their oxidation behavior and with bipyridine and phenanthroline in the context of their reduction properties. The corresponding CV curves are shown in Fig. 5.| Compound | Epred, V | Epox, V | ΔEp, V | λ, nm (eV) | ELUMO − EHOMO, V |
|---|---|---|---|---|---|
| 1 | — | 0.803 | > 4 | 328 (3.78) | 4.739 |
| 2 | −1.302 | 0.810 | 2.112 | 332 (3.74) | 1.917 |
| 3 | −1.282 | 0.841 | 2.123 | 330 (3.76) | 1.985 |
| 2,3-Dihydroxynaphthalene | −2.015 | 1.148 | 3.163 | 325 (3.82) | 4.720 |
| 2,2′-Bipyridine | −2.454 | 2.036 | 4.490 | 281 (4.42) | 4.982 |
| 1,10-Phenanthroline | −2.364 | 2.391 | 4.755 | 263 (4.72) | 4.800 |
 | ||
Fig. 5 CV curves of reduction of 2 (with the reverse of the potential scan after the first and second reduction steps) in comparison with 2,2′-bipyridine (v = 0.1 V s−1, top, left), 3 in comparison with 1,10-phenanthroline (v = 0.1 V s−1, bottom, left; v = 1 V s−1, bottom, right), and oxidation of 2 and 3 in comparison with 2,3-dihydroxynaphthalene (v = 0.1 V s−1, top, right). Concentration of compounds is 1 × 10−3 М, background electrolyte is 0.1 M Bu4NClO4/MeCN (in case of 3, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 MeCN/CH2Cl2 solution was used because of low solubility). Working electrode is glassy carbon (d = 1.7 mm), Т = 298 K. :1 MeCN/CH2Cl2 solution was used because of low solubility). Working electrode is glassy carbon (d = 1.7 mm), Т = 298 K. | ||
In comparison with 2,2′-bipyridine (Epred = −2.454 V), which is chemically reversibly reduced in a sufficiently far range of potentials, complex 2 (Epred = −1.302 V) is reduced by more than 1 V more easily. The potential is close to ones of physiological redox processes, in particular, the reduction of molecular oxygen27 or redox materials С60 or perfluoropentacene.28 In this case, the reduction peak of 2 is completely chemically reversible, which indicates a high stability of the corresponding anion radical. Moreover, the second reversible reduction peak (Epred = −1.862 V) is also present on the CV curve of 2 indicating the formation of its stable dianion. Thus, complex 2 is a reversible acceptor of one or two electrons. As an example of a similar structural motif that reversibly attaches two electrons, we can mention the fluorenone.29
The results obtained can be compared with the electroreduction of chlorogermanes in the presence of 2,2′-bipyridine,10 where the potential shift for various objects was also about 1 V. Comparable values of potential shifts are observed in case of nicotinamide and isoniazid and their complexes with 3,5-di-tert-butyl germanium catecholate.30 In case of nicotinic acid30 and 3- and 4-cyanopyridines, the potential shift is 0.4–0.6 V.31 At the same time, in all cases, complete chemical reversibility of electroreduction is not observed.
The reduction of 1,10-phenanthroline is characterized by a chemically irreversible peak (Epred = −2.364 V) at a potential scan rate of 0.1 V s−1; this peak becomes reversible when the scan rate increases to 1 V s−1 (Fig. 5). In addition, a second reversible peak is present on the CV curve (Epred = −2.530 V). Coordination of phenanthroline, as in the previous case, is accompanied by a strong, more than 1 V, facilitation of reduction. At the same time, the reduction peak of 3 (Epred = −1.282 V) remains irreversible in the studied range of scan rates. It can also be noted that the solubility of 3 in MeCN is acceptable for determining peak potentials and observing UV-vis spectra (Table 2), but it is too low to obtain qualitative CV curves. So, Fig. 5 shows the curve obtained after adding an equivalent volume of CH2Cl2 to the electrolyte. This did not significantly affect the peak potentials.
In contrast to 2 and 3, complex 1 without N-donor ligands does not reduce electrochemically at available potential range, which also confirms conclusion about the key role of these structural motifs in the formation of LUMO of the complexes. The fact that the initial 2,3-dihydroxynaphthalene is reduced in an early region (∼−2 V) looks somewhat anomalous. Indeed, the unsubstituted naphthalene is reduced in a relatively far potential region, and the introduction of donor substituents would further shift the potential to the cathode region. According to,32 the reduction potential of naphthalene in 0.1 M Pr4NClO4/DMF vs. SCE on the mercury dropping electrode is −2.54 V (which correlates, for example, with our data in),33 and for its 2,3-dimethoxy-substituted derivative it shifts to −2.73 V which is close to the background discharge potentials under such conditions. Probably, the relatively easy reduction of the dihydroxy-substituted derivative can be explained by the formation of intermolecular hydrogen bonds, often strongly facilitating the reduction of organic compounds34 and specific for phenols,35 or by the effects of self-protonation.
In comparison with 2,3-dihydroxynaphthalene, the oxidation peaks of 1, 2 and 3 are shifted (Fig. 5) by 0.345, 0.338 and 0.307 V, respectively, to the earlier region (Table 2). In general, it can be seen that 2 and 3 are systems with a relatively small difference in reduction and oxidation potentials (2.11 and 2.12 V, respectively), comparable to ones, for example, of organic semiconductors as pentacene (2.09 V) or perfluoropentacene (1.92 V).28
UV-vis spectra
The UV-vis spectra of compounds 1–3 and N-donor ligands are shown in Fig. 6, and the maxima of the longest-wavelength absorption bands in nm and eV are shown in Table 2. These values for 2,2′-bipyridine and 1,10-phenanthroline correlate well with the potential differences of the reduction and oxidation peaks (4.42 eV vs. 4.49 V and 4.72 eV vs. 4.76 V, respectively). At the same time, the band of 3.82 eV observed in the spectrum of 2,3-dihydroxynaphthalene significantly exceeds the difference in its redox potentials – 3.16 V, which, however, is consistent with the above-mentioned underestimation of its observed reduction potential.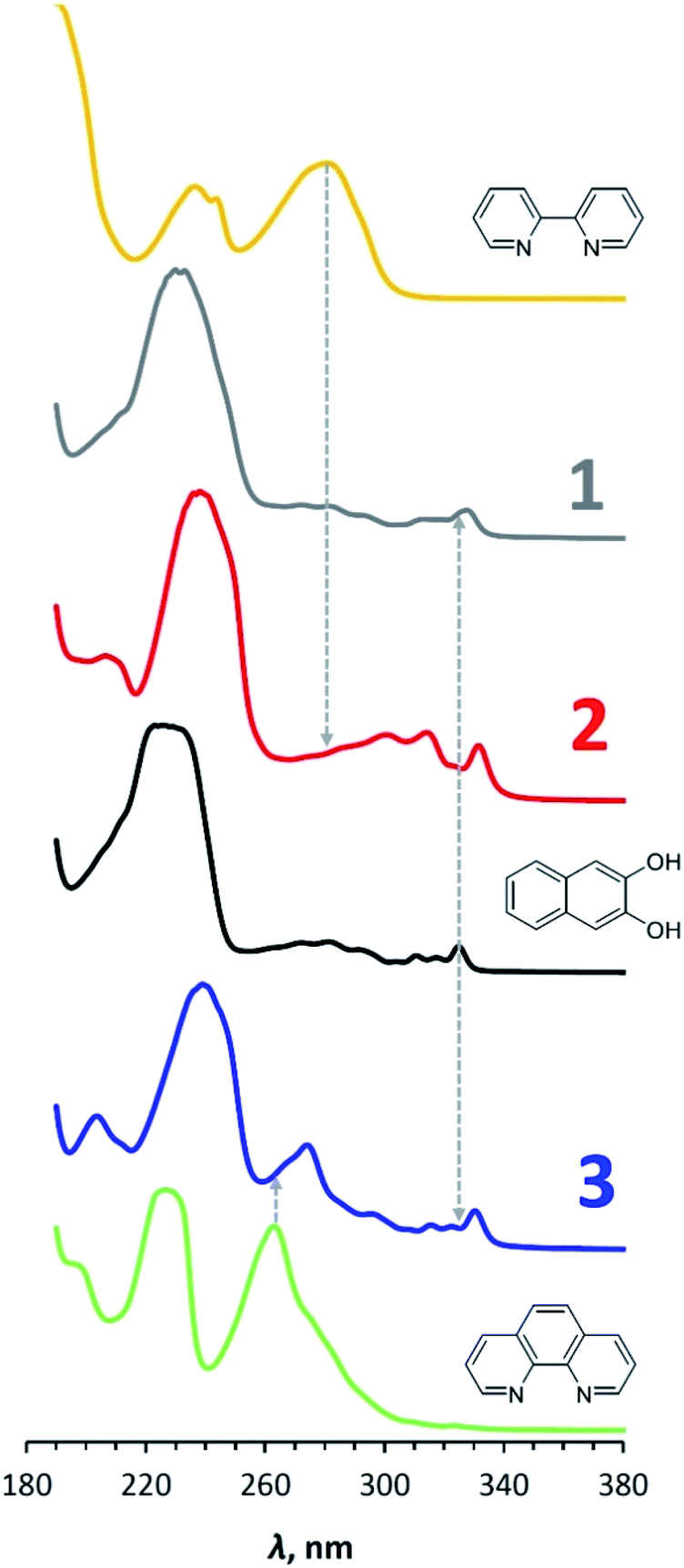 | ||
| Fig. 6 UV-vis spectra of 1–3 in comparison with spectra of 2,2′-bipyridine, 1,10-phenanthroline and 2,3-dihydroxynaphthalene in MeCN. Dashed lines are drawn the longest wave maxima for ligands. Concentration of compounds is 1 × 10−4 М, Т = 298 K. | ||
In the UV-vis spectrum of 2,3-dihydroxynaphthalene, the longest-wavelength band (300–330 nm) is known to correspond π → π* electron transition resulting in the longitudinal polarization of the excited molecule.36 The fine structure of the band can be attributed to vibrational sub-levels. The frequency differences between the corresponding peaks are almost the same with a small decrease in their values with increasing vibrational energy (1223, 1178 and 1114 cm−1 spaces between 291, 281, 272 and 264 nm peaks), which is in accordance with the Franck–Condon principle. A similar picture was earlier observed for anthracene.37 The longest-wavelength band also presents in the spectra for 1, 2 and 3, however, it is red-shifted. The wavelength of its far-right peak increases from 328 to 332 nm in the series 1 – 3 – 2.
Thus, the UV-vis spectra of complexes 1–3 are close to the spectrum of initial 2,3-dihydroxynaphthalene. Transition occurs in the naphthalene part of the compounds under photoexcitation.
DFT calculations
The geometry of the discussed compounds was optimized using DFT calculations (B3LYP/Def2TZVP). The complete results are presented in ESI (Fig. S1†). It should also be noted here that for 2,2′-bipyridine, minima corresponding to the twisted and flat form with an advantage of 6 kcal mol−1 in favor of the flat form were found (Table S1 and Fig. S2†). In turn, 2,3-dihydroxynaphthalene, being an example of aromatic 1,2-diols, can have an intramolecular hydrogen bond, the existence of which is the subject of some discussion in the literature.38,39 Therefore, we have investigated this possibility and found that a structure with an intramolecular hydrogen bond is more profitable than one without it by 3.5 kcal mol−1 (Table S1 and Fig. S2†). In addition, this structure has HOMO−6, in which there is an overlap of the orbitals of H and the neighboring O (Fig. S2†). Thus, in the further discussion, the results of calculations for energetically more favorable structures are used.As can be seen from Table 1, the lengths of the Ge–O bonds in 3 in the gas phase are close to those determined by X-ray and differ by no more than 0.01 Å. In this case, the Ge–N donor–acceptor bonds in the gas phase are longer by 0.07 and 0.11 Å and become almost equal, in contrast to the somewhat asymmetric shape in the crystal. Considering that electron transfer is a destabilizing factor for both covalent and donor–acceptor bonds, we were interested in testing the possibility of eliminating of ion-radical N-donors from electrochemically-generated ion-radical germanium bis-catecholate complexes 2 and 3. Due to potential differences, it would be accompanied by the formation of a stronger reducing agent during reduction, in the case of anion radicals, or a stronger oxidizing agent during oxidation, in the case of cation radicals (redox-upconversion40). However, as can be seen from Table 1 during reduction and oxidation, the lengths of Ge–N bonds in 2 and 3 do not increase, but on the contrary, they slightly decrease.
Fig. 7 shows the distribution of HOMO and LUMO in complexes 2 and 3. In both cases, HOMO is located entirely on the 2,3-dihydroxynaphthalene part without affecting the germanium atom and N-ligand. The LUMO, in contrast, is completely distributed on 2,2′-bipyridine and 1,10-phenanthroline, in turn, without affecting the catecholate fragment and germanium. In this case, the HOMO/LUMO gap correlates well (within 0.1–0.2 eV) with the difference in the oxidation and reduction potentials 2 and 3 (Table 2).
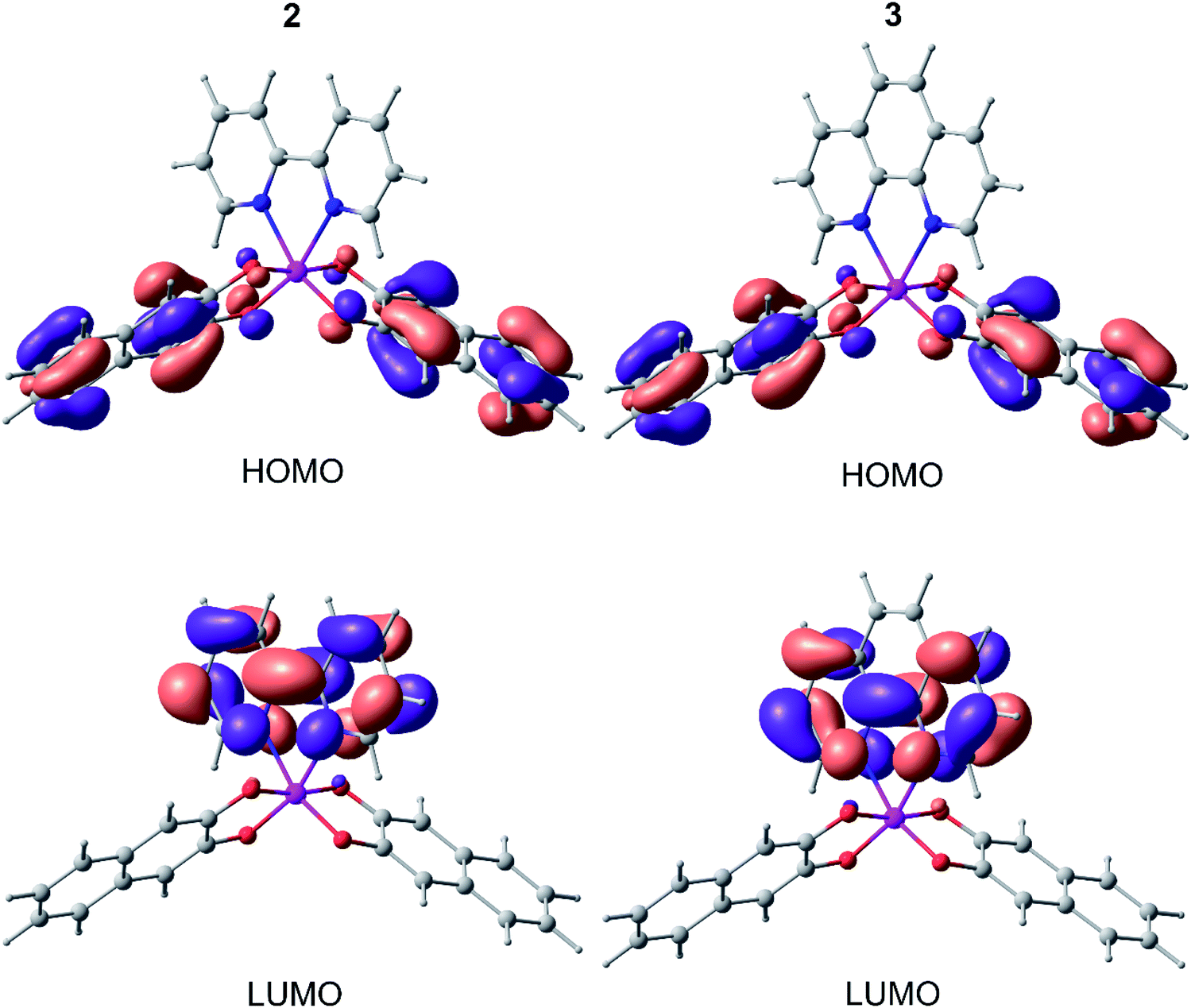 | ||
| Fig. 7 HOMO and LUMO of 2 and 3 (from DFT B3LYP/def2tzvp calculations). | ||
Thermal analysis
A thermal analysis of compounds 1, 2 and 3 was carried out under argon atmosphere at a heating rate of 10 °C min−1 up to 900 °C (Fig. 8). According to obtained thermogravimetric and differential thermal curves, complexes 2 and 3 show a rather high stability up to almost 500 °C. During heating of 3 a sharp endothermic reaction takes place between temperatures of 460 °C and 525 °C with a peak at 500 °C. This process with loss of mass of about 54% corresponds to decomposition of 3. A similar picture is observed for 2 with the relevant temperature values of 430, 500 and 470 °C and loss of mass of 77%, which is consistent with GeO2 formation. A small exothermic peak observed at 325 °C without weight loss implies a possible structural rearrangement of the complex 2. It should be highlighted that the stability of compounds 2 and 3 is notably higher compared to that of 1 that starts to decompose already at 200 °C.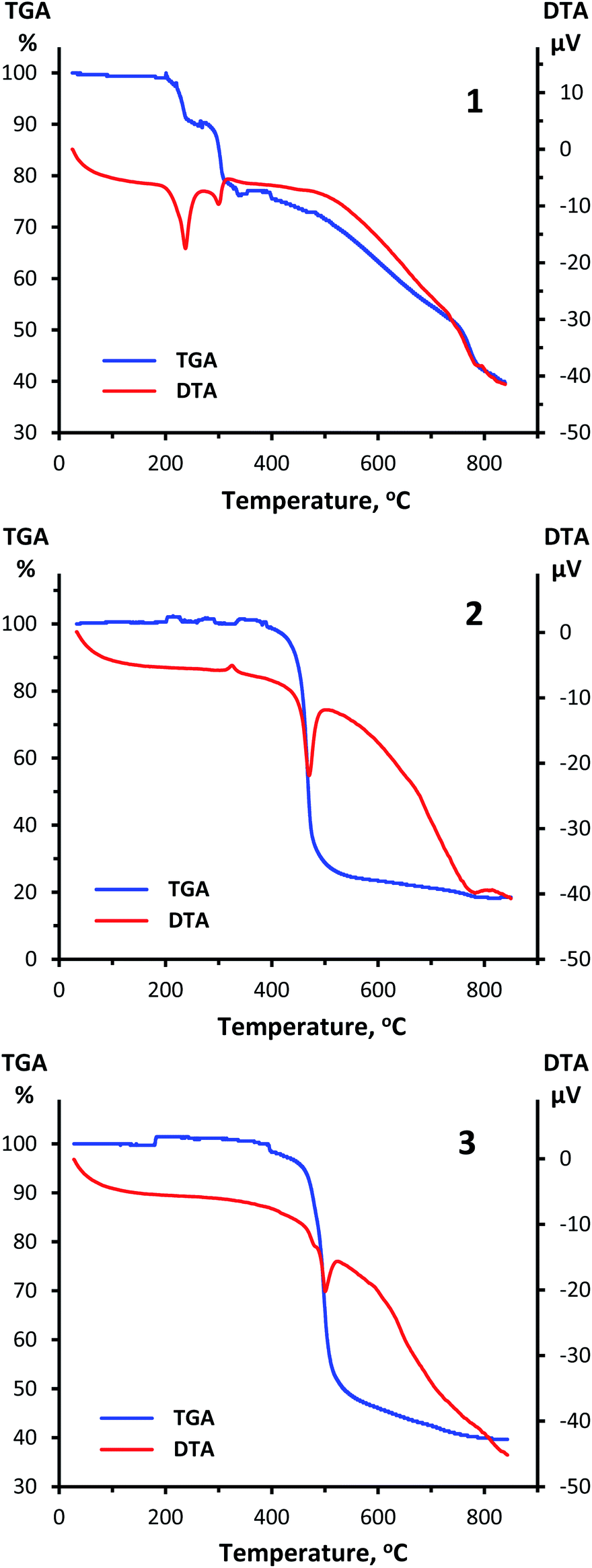 | ||
| Fig. 8 TGA and DTA curves for 1 (top), 2 (center) and 3 (bottom) obtained at a heating rate of 10 °C min−1 in the range from ambient temperature up to 900 °C under argon atmosphere. | ||
Experimental
Materials
Germanium dioxide GeO2 was purchased from “Germanium and Applications Ltd” (DG-B, TY 1774-001-95961127-2010, batch #117), 2,2′-bipyridine, 1,10-phenanthroline, 2,3-dihydroxynaphthalene, tetrabutylammonium perchlorate (electrochemical grade), acetonitrile, dichloromethane and toluene (HPLC grade) were purchased from Sigma Aldrich.Synthesis of 1–3
Instrumentation
Oxidation and reduction behavior of 2,2′-bipyridine, 1,10-phenanthroline, 2,3-dihydroxynaphthalene and complexes 1–3 was analyzed by cyclic voltammetry using a digital potentiostat IPC-Pro-MF (Econix). The compound solutions preparation and all measurements were made in an argon-filled glove box with water and oxygen levels below 1 ppm. Before that, acetonitrile and dichloromethane with initial water content of <100 ppm, were stored over 4 Å molecular sieves preliminarily dried under oil-pump vacuum at 200–250 °С for 4 h. Bu4NClO4 was dried under oil-pump vacuum at 80 °С for 4 h. The water content in the supporting electrolytes, including 0.1 M Bu4NClO4/MeCN and 0.1 М Bu4NClO4/MeCN:CH2Cl2 (1:1, in case of 3), did not exceed 20 ppm, as determined by Karl Fischer titration using a Mettler–Toledo Titrator C10SD. The studied compounds dissolved in the supporting electrolyte with a concentration of 1 × 10−3 M were electrochemically tested in a standard three-electrode glass cell at the potential scan rates of 0.1 and 1 V s−1. The working electrode was a glassy carbon disc electrode with a diameter of 1.7 mm. Before using, it was polished with abrasive paper and then GOI paste until the surface attained a mirror shine. The counter electrode was a Pt wire pre-annealed in a gas burner flame to remove oxides and other possible contaminations. The potentials of the studied processes were measured versus the Ag wire coated with AgCl (prepared by galvanostatic anodizing in 5% HCl solution) separated from the bulk electrolyte solution by an electrolytic bridge filled with the supporting electrolyte. The reference electrode was calibrated with respect to the ferrocene/ferrocenium couple.
UV-vis spectroscopy was performed using an Agilent 8453 instrument. The spectra were registered for 1 × 10−4 М solutions in MeCN in a 10 mm quartz cell with a PTFE stopper. Before taking the cell out of the glove box, the cell-stopper contact was sealed with Parafilm, and then during several minutes the spectrum was recorded. Each CV curve and UV-vis spectrum were reproduced at least three times.
1H NMR (300 MHz) and 13C NMR (75 MHz) were recorded in DMSO-d6 on Bruker AM300 at ambient temperature. NMR spectra were referenced using residual DMSO-d6 (1H NMR δ = 2.5 ppm, 13C NMR δ = 39.5 ppm). High resolution mass spectra (HRMS) were measured on a Bruker micrOTOF II instrument using electrospray ionization (ESI). The measurements were done in a positive ion mode (interface capillary voltage 4.5 kV); mass range from m/z 50 to m/z 1600; external or internal calibration was done with ESI Tuning Mix, Agilent. A syringe injection was used for the solutions in acetonitrile (flow rate 3 μl min−1). Nitrogen was applied as a dry gas (flow rate 4 l min−1); the interface temperature was set at 200 °C.
Elemental analysis was performed using EuroEA-3000 (EuroVector) instrument.
Thermal analysis was performed on DTG-60 Simultaneous Thermogravimetry/Differential Thermal Analyzers, Shimadzu (Japan). All experiments were carried out in alumina crucible under argon flow at a heating rate of 10 °C min−1.
X-ray crystallography
The structure was solved by the direct method and refined in the anisotropic approximation for non-hydrogen atoms. Hydrogens atoms of aromatic fragments were calculated according to those idealized geometry and refined with constraints applied to C–H bond lengths and equivalent displacement parameters (Ueq(H) = 1.2Ueq(C), C – central atom of CH group). All structures were solved with the ShelXT41 program and refined with the ShelXL42 program. Molecular graphics was drawn using OLEX2 (ref. 43) program. The structure‡ of 3 was refined as inversion twin using TWIN and BASF instructions (Flack parameter is equal to 0.09(4)).CCDC 2074622 contains the supplementary crystallographic data for 3.†
Computational details
Density functional theory (DFT) calculations were performed using the Gaussian 16 program package44 at the B3LYP/Def2TZVP level. The applied approximation was recently shown30,31 to give accurate reproduction of the geometry, electronic and energy characteristics of germanium complexes with redox-active ligands. The stationary points on the potential energy surfaces were located by full geometry optimization with the calculation of the force constant matrix and checking for the stabilities of the DFT wave function. Structural visualizations in Fig. 7 were produced with the ChemCraft program suite.45Conclusion
In this paper we present organogermanium derivatives with very available HOMO and LUMO, the gap between which is comparable to many common organic and inorganic semiconductors. Germanium atom, playing a leading role in the setting of boundary molecular orbital levels, does not have them on it; HOMO and LUMO are completely distributed on the organic parts of the metal complexes. Thus, in our opinion, this is an example of how the chemistry of non-transition metals can make a contribution to solving traditional problems of organic chemistry.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the Russian Science Foundation (Project no. 20-73-10234). X-ray study of 3 was performed using the equipment of Center for molecular composition studies of INEOS RAS. High resolution mass spectra were recorded in the Department of Structural Studies of N. D. Zelinsky Institute of Organic Chemistry, Moscow.Notes and references
- J. I. van der Vlugt, Redox-Active Pincer Ligands, in Topics in Organometallic Chemistry, Springer, Berlin, Heidelberg, 2020, p. 1 Search PubMed.
- V. I. Minkin, A. G. Starikov and A. A. Starikova, Pure Appl. Chem., 2018, 90, 811 CrossRef CAS.
- E. N. Nikolaevskaya, N. O. Druzhkov, M. A. Syroeshkin and M. P. Egorov, Coord. Chem. Rev., 2020, 417, 213353 CrossRef CAS.
- J. I. van der Vlugt, Chem.–Eur. J., 2019, 25, 2651 CrossRef CAS PubMed.
- I. V. Ershova, A. V. Piskunov and V. K. Cherkasov, Russ. Chem. Rev., 2020, 89, 1157 CrossRef CAS.
- A. A. Starikova and V. I. Minkin, Russ. Chem. Rev., 2018, 87, 1049 CrossRef CAS.
- O. R. Luca and R. H. Crabtree, Chem. Soc. Rev., 2013, 42, 1440 RSC.
- V. Lyaskovskyy and B. de Bruin, ACS Catal., 2012, 2, 270–279 CrossRef CAS.
- M. G. Chegerev and A. V. Piskunov, Russ. J. Coord. Chem., 2018, 44, 258 CrossRef CAS.
- M. Dieng, D. Gningue-Sall and V. Jouikov, Main Group Met. Chem., 2012, 35, 141 CAS.
- I. V. Krylova, E. A. Saverina, S. S. Rynin, A. V. Lalov, M. E. Minyaev, E. N. Nikolaevskaya, M. A. Syroeshkin and M. P. Egorov, Mendeleev Commun., 2020, 30, 563 CrossRef CAS.
- R. Guilard, J.-M. Barbe, A. Boukhris, C. Lecomte, J. E. Anderson, Q. Y. Xu and K. M. Kadish, J. Chem. Soc., Dalton Trans., 1988, 1109 RSC.
- G. A. Abakumov, A. V. Piskunov, V. K. Cherkasov, I. L. Fedushkin, V. P. Ananikov, D. B. Eremin, E. G. Gordeev, I. P. Beletskaya, A. D. Averin, M. N. Bochkarev, A. A. Trifonov, U. M. Dzhemilev, V. A. Dyakonov, M. P. Egorov, A. N. Vereshchagin, M. A. Syroeshkin, V. V. Zhuikov, A. M. Muzafarov, A. A. Anisimov, A. V. Arzumanyan, Y. N. Kononevich, M. N. Temnikov, O. G. Sinyashin, Y. H. Budnikova, A. R. Burilov, A. A. Karasik, V. F. Mironov, P. A. Storozhenko, G. I. Shcherbakova, B. A. Trofimov, S. V. Amosova, N. K. Gusarova, V. A. Potapov, V. B. Shur, V. V. Burlakov, V. S. Bogdanov and M. V. Andreev, Russ. Chem. Rev., 2018, 87, 393 CrossRef CAS.
- E. N. Nikolaevskaya, A. V. Kansuzyan, G. E. Filonova, V. A. Zelenova, V. M. Pechennikov, I. V. Krylova, M. P. Egorov, V. V. Jouikov and M. A. Syroeshkin, Eur. J. Inorg. Chem., 2019, 676 CrossRef CAS.
- G. E. Filonova, E. N. Nikolaevskaya, A. V. Kansuzyan, I. V. Krylova, M. P. Egorov, V. V. Jouikov and M. A. Syroeshkin, Eur. J. Org. Chem., 2019, 4128 CrossRef CAS.
- K. Yokoi, Germanium, Toxicity, in Encyclopedia of Metalloproteins, ed. R. H. Kretsinger, V. N. Uversky and E. A. Permyakov, Springer, New York, NY, 2013, DOI:10.1007/978-1-4614-1533-6_434.
- H. Kehlbeck and D. Lekim, When intra-abdominal injection to mice, LD50 of germanium citrate is >2500 mg/kg, Pharmaceutically active organic germanium compounds, FRG patent, 3212817, 1983 Search PubMed.
- When intra-abdominal injection to mice, LD50 of sodium chloride is 2602 mg/kg https://www.vaxserve.com/image.cfm?doc_id=11586%26image_type=msds_sheet.
- E. Lukevics and L. Ignatovich, Biological Activity of Organogermanium Compounds, in The Chemistry of Organic Germanium, Tin and Lead Compounds, ed. Z. Rappoport, 2002, vol. 2, ch. 23 Search PubMed.
- A. A. Vishtorskaya, E. A. Saverina, V. M. Pechennikov, I. V. Krylova, A. V. Lalov, M. A. Syroeshkin, M. P. Egorov and V. V. Jouikov, J. Organomet. Chem., 2018, 858, 8 CrossRef CAS.
- M. Glavinovic, M. Krause, L. Yang, J. McLeod, L. Liu, K. M. Baines, T. Friscic and J.-P. Lumb, Sci. Adv., 2017, 3, e1700149 CrossRef PubMed.
- E. A. Saverina, V. Sivasankaran, R. R. Kapaev, A. S. Galushko, V. P. Ananikov, M. P. Egorov, V. V. Jouikov, P. A. Troshin and M. A. Syroeshkin, Green Chem., 2020, 22, 359 RSC.
- D. A. Grishanov, A. V. Churakov, A. G. Medvedev, A. A. Mikhaylov, O. Lev and P. V. Prikhodchenko, Inorg. Chem., 2019, 58, 1905 CrossRef CAS PubMed.
- E. A. Saverina, R. R. Kapaev, P. V. Stishenko, A. S. Galushko, V. A. Balycheva, V. P. Ananikov, M. P. Egorov, V. V. Jouikov, P. A. Troshin and M. A. Syroeshkin, ChemSusChem, 2020, 13, 3137 CrossRef CAS PubMed.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171 CrossRef CAS PubMed.
- F. Cheng, M. F. Davis, A. L. Hector, W. Levason, G. Reid, M. Webster and W. Zhang, Eur. J. Inorg. Chem., 2007, 2007, 4897 CrossRef.
- D. Vasudevan and H. Wendt, J. Electroanal. Chem., 1995, 192, 69 CrossRef.
- Y. Sakamoto, T. Suzuki, M. Kobayashi, Y. Gao, Y. Fukai, Y. Inoue, F. Sato and S. Tokito, J. Am. Chem. Soc., 2004, 126, 8138 CrossRef CAS PubMed.
- A. S. Mendkovich, M. A. Syroeshkin, D. V. Nasybullina, M. N. Mikhailov, V. P. Gultyai, M. N. Elinson and A. I. Rusakov, Electrochim. Acta, 2016, 191, 962 CrossRef CAS.
- E. N. Nikolaevskaya, P. G. Shangin, A. A. Starikova, V. V. Jouikov, M. P. Egorov and M. A. Syroeshkin, Inorg. Chim. Acta, 2019, 495, 119007 CrossRef CAS.
- E. N. Nikolaevskaya, E. A. Saverina, A. A. Starikova, A. Farhati, M. A. Kiskin, M. A. Syroeshkin, M. P. Egorov and V. V. Jouikov, Dalton Trans., 2018, 47, 17127 RSC.
- A. Zweig, A. H. Maurer and B. G. Roberts, J. Org. Chem., 1967, 32, 1322 CrossRef CAS.
- M. A. Syroeshkin, M. N. Mikhailov, A. S. Mendkovich and A. I. Rusakov, Russ. Chem. Bull., Int. Ed., 2009, 58, 41 CrossRef CAS.
- E. Martínez-González, F. J. González, J. R. Ascenso, P. M. Marcos and C. Frontana, J. Org. Chem., 2016, 81, 6329 CrossRef PubMed.
- F. Ramondo, L. Bencivenni, G. Portalone and A. Domenicano, Struct. Chem., 1995, 6, 37 CrossRef CAS.
- D. M. Hercules and L. B. Rogers, Spectrochim. Acta, 1959, 15, 393 CrossRef.
- R. N. Jones, Chem. Rev., 1947, 41, 353 CrossRef CAS PubMed.
- C. Grieco, A. T. Hanes, L. Blancafort and B. Kohler, J. Phys. Chem. A, 2019, 123, 5356 CrossRef CAS PubMed.
- M. Mandado, A. M. Grañaa and R. A. Mosquera, Phys. Chem. Chem. Phys., 2004, 6, 4391 RSC.
- M. A. Syroeshkin, F. Kuriakose, E. A. Saverina, V. A. Timofeeva, M. P. Egorov and I. V. Alabugin, Angew. Chem. Int. Ed., 2019, 58, 5532 (Angew. Chem., 2019, 131, 5588) CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03, Gaussian Inc., Wallingford, CT, 2016 Search PubMed.
- Chemcraft, version 1.8, 2014, http://www.chemcraftprog.com Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Optimized geometries and distribution of HOMO and LUMO for the studied compounds calculated by the DFT B3LYP/Def2TZVP, calculation details and Cartesian coordinates. CCDC 2074622. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra02691g |
| ‡ Crystallographic data for 3: C32H20GeN2O4, formula weight 569.09, diffractometer Bruker D8 Quest, scan mode – ϕ and ω scans, crystal, mm – 0.01 × 0.15 × 0.23, crystal system – monoclinic, a = 6.0837(5) Å, b = 15.6412(17) Å, c = 13.3288(13) Å, β = 98.927(6)°, V = 1253.0(2) Å3, d = 1.508 g cm−3, T = 296.15 K, μ = 1.266 mm−1, space group P21, Z = 2, F(000) = 580, reflections collected 7650, independent reflections 3873, reflections (I > 2σ(I)) 3382, Rint = 0.0667, 2θmin–2θmax 4.044°–52.120°, wR2 (all reflections) 0.1870, R1 (I > σ(I)) 0.0786, GOF 1.077, ρmin/ρmax –0.931/1.616 e Å−3. |
| This journal is © The Royal Society of Chemistry 2021 |