DOI:
10.1039/D1RA02629A
(Paper)
RSC Adv., 2021,
11, 22461-22466
In situ gelation of aqueous sulfuric acid solution for fuel cells
Received
3rd April 2021
, Accepted 18th June 2021
First published on 24th June 2021
Abstract
Aqueous sulfuric acid solution is a versatile liquid electrolyte for electrochemical applications and gelation of it has the advantages of easy shaping and reduced leaking. Herein, aqueous sulfuric acid solutions with concentrations of 1–4 mol L−1 are fabricated into gel membranes by in situ polymerization of acrylamide as a monomer and divilynbenzene as a crosslinker for fuel cell applications. The gel membrane with an acid concentration of 3.5 mol L−1 exhibited the maximum proton conductivity of 184 mS cm−1 at 30 °C. Tensile fracture strength of the gel membrane reached 53 kPa with a tensile strain of 14. Thermogravimetric analysis reveals that the gel membranes are thermally stable at temperatures up to 231 °C. The gel membranes are successfully assembled into fuel cells and a peak power density of 74 mW cm−2 is achieved. The fuel cell maintains steady operation over 200 h. In situ gelation of aqueous sulfuric acid solution offers an efficient strategy to prepare gel electrolytes for electrochemical devices.
1. Introduction
Hydrogels consisting of hydrophilic polymeric networks and a high-volume ratio of water, have been regarded as promising electrolyte candidates owing to their flexibility and electrochemical activity.1 After doping with various additives, hydrogels become applicable in a variety of electrochemical devices.2,3 Polyacrylamide hydrogels containing NaCl could serve as ionic conductors operating at high frequencies and voltages to actuate producing sound over the entire audible frequency range.4 Long et al. imbedded hydroxypropyl cellulose into a polyvinyl alcohol (PVA) matrix to prepare conductive hydrogels containing NaCl for artificial flexible electronics.5 Liu et al. prepared an asymmetric polyacrylic acid hydrogel containing Ag0 for highly sensitive tactile sensors.6 In addition, LiNO3,7 Li2SO4,8 and ZnCl2 (ref. 9) have ever been added to hydrogels to prepare electrolytes for aqueous rechargeable metal-ion battery. Although the doped hydrogels were electro-active, limited electrochemically stable potential window and side effect of polymeric networks to the electrochemical reaction prevented them from being extensively applied in rechargeable batteries.
Acid hydrogels were found to be useful for constructing supercapacitors. A few PVA hydrogels doped with H2SO4 and H3PO4 have been prepared to fabricate flexible supercapacitors.10,11 However, the presence of acids would break intermolecular hydrogen bonds between PVA chains and water molecules to result in dehydration and aggregation of PVA chains.12–15 Accordingly, dimensional stability and mechanical performance are still challenging for the acid PVA hydrogel electrolytes. In situ polymerization of monomers in acid solution represented an alternative route to obtain hydrogel electrolytes. Wieczorek et al. used acrylamide (AM) as a monomer and N,N′-methylenebisacrylamide (MBA) as a crosslinker to prepare acid hydrogel electrolytes by copolymerization in H2SO4 solution.16 The resultant polyacrylamide (PAM) hydrogels exhibited a proton conductivity of 17 mS cm−1 at room temperature when molar ratio of H2SO4/AM was greater than 1.6. Nevertheless, the hydrogel exhibited limited thermal stability and began to decompose at 70 °C owing probably to the hydrolysis of MBA.17
In this work, stable acid hydrogels were prepared from aqueous H2SO4 solution by in situ polymerization for fuel cell applications. Owing to the resistance of the aromatic skeleton to acids and temperature,18 divinylbenzene (DVB) was employed as a cross-linker to synthesize the gels. Fourier transform infrared (FT-IR) spectroscopy and scanning electron microscopy (SEM) observations were employed to characterize structures of the acid hydrogels. Thermal stability of the hydrogels was evaluated by thermogravimetric (TG) and differential scanning calorimetric (DSC) analyses. Mechanical properties of the hydrogels were determined by uniaxial tensile and compression tests. Conductivities of the hydrogels were obtained by electrochemical impedance spectroscopy (EIS) measurements. Finally, the acid hydrogels were assembled into hydrogen oxygen fuel cells to evaluate performance under simulated working conditions.
2. Experimental
2.1. Materials
AM, DVB, 1-hydroxycyclohexylphenylketone (HCHPK) and sulfuric acid (>98%) were obtained from TCI and Acros. Hispec 3000 Pt/C catalysts (contain 20 wt% and 40 wt% Pt, respectively) were purchased from Johnson Mattey. Nafion ionomer solution that with 5 wt% Nafion was obtained from DuPont. Gas diffusion layers (thickness = 270 μm; resistivity ≤ 13 mΩ cm2, pressure drop ≥ 20 mmH2O) were received from Suzhou Yilongsheng Energy Technology Co., Ltd.
2.2. Synthesis of acid hydrogels
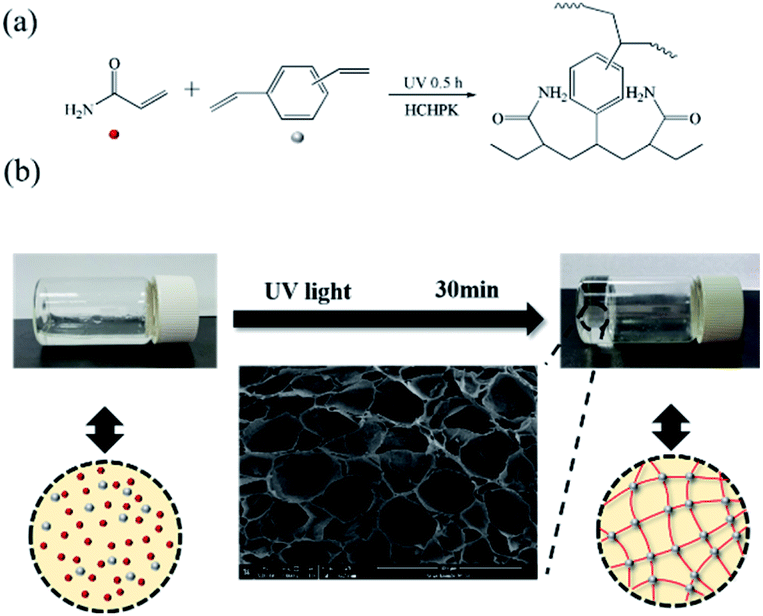
The acid hydrogels (GSA-n, which n represented molar concentration of H2SO4 in the resultant hydrogel) were prepared by in situ polymerization of AM and DVB in H2SO4 aqueous solution. The crosslinker concentration was 1.8 mol% and the synthetic route is shown in Fig. 1a. Typically, AM (30 wt%) and DVB (1 wt%) were introduced to H2SO4 aqueous solution at room temperature and then mixed thoroughly by ultrasonic vibration for 2 h. Then the mixture was poured into a silicon mould and sealed with a piece of glass. After irradiation (6 mW cm−2) under a 365 nm ultraviolet lamp (GY500) for 0.5 h, the mixture changed into a transparent gel as shown in Fig. 1b.
 |
| | Fig. 1 (a) Synthetic route and (b) schematical illustration of the gelation. | |
2.3. Instrumentation
FT-IR spectra were measured on a PerkinElmer Spectrum II spectrometer at room temperature within 4000 to 600 cm−1 region with a wavenumber resolution of 1 cm−1. GSA-n xerogels were obtained by lyophilization on a Scientz-10N freeze dryer. SEM observations were performed on a Quanta 250 environmental scanning electron microscope. TG analysis was implemented at a heating rate of 10 °C min−1 over the temperature range of 35–800 °C under air atmosphere using a Netzsch TG209F1 thermogravimetry. The sample was dried in a vacuum oven for 72 h before the TG analysis to reduce the impact of water. DSC measurements were carried out on a TA Q200 DSC instrument. Rheological behavior was recorded on a MCR302 rheometer. The frequency scanning was tested from 0.01–100 Hz in the region of elastic deformation. Tensile and compressive measurements of GSA-n were carried out on an Instron 5967 electronic universal test machine. Specimens for the tensile test were dumbbell-shaped with the cross section of 10 × 2 mm. The stretching rate was 20 mm min−1. For compressive test, specimens were cylinder-shaped with the size of Ø 25 × 30 mm and the compressing rate was 5 mm min−1. EIS measurements were carried out on a Princeton VersaSTAT 3 electrochemical workstation over a frequency range from 1 MHz to 1000 Hz under an AC amplitude of 10 mV. A sample was held in a perforated PVC spacer and then sandwiched between two pieces of platinum coated electrodes to form a test cell. In order to prevent water from evaporating, the test cell was sealed with parafilm. During the test, a WT-3000 hostage was used to control the sample temperature.
2.4. Fuel cell performance
Pt/C particles and Nafion ionomers (20 wt%) were added to solvent consisting of equivalent deionized water and isopropanol. After mixing thoroughly by ultrasonic vibration, the ink was sprayed onto the gas diffusion layers to fabricate the gas diffusion electrodes (GDE) with a catalyst load of 0.5 mg Pt cm−2. A membrane electrode assembly (MEA) was constructed by sandwiching a GSA-n membrane with a thickness of 200 μm between two identical GDEs. A pair of graphite plates with serpentine gas flow channels was used to clamp the MEA to form a single cell with an active electrode area of 1 cm2. Fully humidified H2 and O2 were continuously fed to the anode and cathode at flow rates of 120 and 60 mL min−1, respectively. The single cell was connected to a programmable electronic load DC (IT8500+, ITECH) to record the polarization curve immediately after assembling. Independent measurements of 3 samples were taken for each GSA-n.
3. Results and discussion
3.1. FT-IR characterization
The FT-IR spectra of AM and GSA-n xerogels were checked to confirm the polymerization and reveal interaction between H2SO4 and the polymeric skeleton. As shown in Fig. 2, the bands centered at 989 and 962 cm−1 in the spectrum of AM due to the bending vibrations of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C group disappeared in the spectra of GSA-n xerogels, indicating the successful polymerization of AM. The vibrations centered at 1180 and 1121 cm−1 in the spectra of GSA-n xerogels suggested the presence of HSO4− and SO42−, respectively.16 Accordingly, H2SO4 should involve in the protonation of the NH2 amide groups of PAM. The characteristic peaks at 3347 cm−1, 3192 cm−1 and 1617 cm−1 were ascribed to the hydrogen-bonded stretching, free stretching and bending vibration of N–H, respectively. These bands weakened significantly in the spectra of GSA-n xerogels, indicating possible protonation of the NH2 amide groups by H2SO4. In addition, area ratio between the peak at 3192 cm−1 (N–H free stretching) and that at 1659 cm−1 (CO stretching) decreased with the increasing H2SO4 concentration, revealing the increasing degree of protonation of PAM chains.
C group disappeared in the spectra of GSA-n xerogels, indicating the successful polymerization of AM. The vibrations centered at 1180 and 1121 cm−1 in the spectra of GSA-n xerogels suggested the presence of HSO4− and SO42−, respectively.16 Accordingly, H2SO4 should involve in the protonation of the NH2 amide groups of PAM. The characteristic peaks at 3347 cm−1, 3192 cm−1 and 1617 cm−1 were ascribed to the hydrogen-bonded stretching, free stretching and bending vibration of N–H, respectively. These bands weakened significantly in the spectra of GSA-n xerogels, indicating possible protonation of the NH2 amide groups by H2SO4. In addition, area ratio between the peak at 3192 cm−1 (N–H free stretching) and that at 1659 cm−1 (CO stretching) decreased with the increasing H2SO4 concentration, revealing the increasing degree of protonation of PAM chains.
 |
| | Fig. 2 FT-IR spectra of AM and GSA-n xerogels. | |
3.2. Thermal stability and mechanical properties
DSC and TG measurements were performed to determine thermal stability of GSA-n. As shown in Fig. 3a, no endotherm peak appeared in the DSC curve of GSA-3.5 during the heating process until water significantly vaporize around 100 °C, suggesting that GSA-n did not decompose before boiling of water. As shown in Fig. 3b, GSA-n xerogels with different H2SO4 concentrations exhibited similar trend in weight loss. The weight loss at temperatures below 150 °C was due to the water retained in the xerogels. As GSA-4 xerogel lost the largest amount of retained water, GSA-4 was the best in water retention. GSA-4 had the least stability and began to decompose at 231 °C, suggesting that the protonation of PAM chains reduced thermal stability of the GSA-n skeleton. Owing to the adoption of DVB, GSA-n exhibited much superior thermal stability than the MBA crosslinked PAM/H2SO4 hydrogels,17 guaranteeing stable operation of GSA-n in fuel cells.
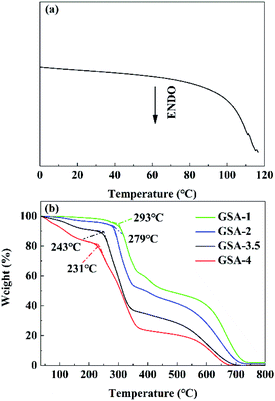 |
| | Fig. 3 (a) DSC curve of GSA-3.5 during the heating and (b) TG curves of GSA-n in air. | |
Rheologic behavior of GSA-n was characterized by controlling the shear strain at 1% to maintain the elastic deformation. As shown in Fig. 4, the storage moduli (G′) changed little with H2SO4 concentration, indicating that H2SO4 had little impact on the crosslinking of polyacrylamide chains. However, the loss moduli (G′′) was quite affected by H2SO4 concentration. GSA-1 exhibited higher G′′ probably because less interaction between H2SO4 and the polymeric chains increased the friction loss of chains. At higher H2SO4 concentration, effect of H2SO4 on polymeric deformation became complicated and no monotonous tendency of G′′ was observed. For all the GSA-n, G′ was always higher than G′′ over the frequency region of 0.01–10 Hz. When the deformation frequency was higher than 40 Hz, G′′ increased rapidly and tended to approach G′. The fact revealed that GSA-n was more elastic at low deformation frequency, which favored the application as electrolytes in fuel cells.
 |
| | Fig. 4 Changes of G′ and G′′ as a function of frequency at 25 °C for GSA-n. | |
GSA-n exhibited considerable mechanical strength by virtue of the DVB crosslinking. It was demonstrated that H2SO4 could inhibit the ionization of the carboxyl groups on the PAM chains and thus to reinforce the mechanical properties of hydrogels by providing high density of the hydrogen bonds.19 As shown in Fig. 5a, tensile fracture strength of GSA-n increased with the increase in H2SO4 concentration and reached 62.8 kPa for GSA-4. Meanwhile, GSA-3.5 showed the greatest stretchability instead of GSA-4. The stretchability of GSA-4 was inhibited owing likely to the higher rigidity produced by the higher H2SO4 concentration. GSA-n showed a Young's modulus of ∼0.04 MPa during the elastic deformation. As shown in Fig. 5b, all the GSA-n could resist compressive strain up to 0.9 without collapse.
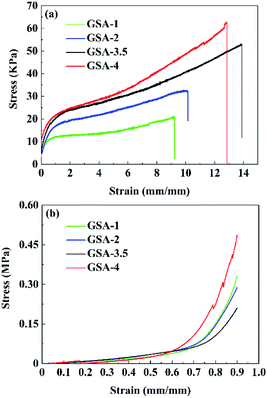 |
| | Fig. 5 (a) Tensile and (b) compressive stress–strain curves of GSA-n. | |
GSA-4 exhibited the maximum compressive stress of 0.47 MPa for the rich hydrogen bonds available. The satisfactory compressive strength of GSA-n could reduce the chance of short circuit when it is used as a proton conductive electrolyte in fuel cell.20.
3.3. Conductivity
Ionic conductivities of aqueous H2SO4 solutions and GSA-n were characterized by EIS measurements. For all the species, conductivities increased with the increase in temperature. However, conductivities changed non-monotonously with acid concentration over the measure concentration range. As shown in Fig. 6, 3.5 M concentration resulted in the peak conductivities for both H2SO4 solution and GSA-n. A decrease in conductivity was observed when H2SO4 concentration increased from 3.5 to 4 M, in consistence with the previous observations.21 The observed peak conductivities should arise from the compromise between acid dissociation and protonation of waters.22–24 At low concentrations (<3.5 M), acid dissociated thoroughly and hydronium number increased with the increase in acid concentration. At high concentrations (>3.5 M), insufficient water inhibited H2SO4 dissociation and the hydronium number decreased with the increase in acid concentration. As a result, conductivities of 3.5 M H2SO4 solution and GSA-3.5 at 30 °C reached the peak values of 307 and 184 mS cm−1, respectively. After the gelation, conductivity decreased by only 40% due likely to contribution of the amide moieties of PAM to conduction as discussed later.
 |
| | Fig. 6 Conductivities as a function of temperature for (a) H2SO4 solution and (b) GSA-n. | |
Vogel–Tammann–Fulcher (VTF) equation was fit the temperature dependent conductivities of H2SO4 solution and GSA-n well (R2 > 0.999), indicating that proton conduction was dominated by the vehicle mechanism.25
| |
σ = σ0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp[−Ea/R(T − T0)] exp[−Ea/R(T − T0)]
| (1) |
where
σ0,
Ea,
R, and
T0 are prefactor (S cm
−1), activation energy (kJ mol
−1), the ideal gas constant (8.314 J mol
−1 K
−1), and the Vogel temperature (K), respectively. The derived
Ea values of 1, 2, 3.5, and 4 M H
2SO
4 solutions for the proton conduction were 0.34, 0.21, 0.12 and 0.16 kJ mol
−1, respectively. In contrast,
Ea values of GSA-1, GSA-2, GSA-3.5, and GSA-4 were derived as 1.95, 1.58, 0.43 and 1.26 kJ mol
−1, respectively. Obviously,
Ea of GSA-
n was always higher than that of the corresponding H
2SO
4 solution because the polymeric skeletons in the gels had negative effect on proton conduction. Interestingly, the species with 3.5 M concentration exhibited the lowest
Ea. In this case, the maximum hydronium promoted proton conduction efficiently
via the vehicle mechanism.
According to the FT-IR spectra and temperature dependent conductivities, we proposed proton conduction in GSA-n occurred as follows. Dissociated protons from H2SO4 combined with H2O to form hydronium ions, whose movement made a major contribution to the measured conductivity. Meanwhile, the protonated amide groups of PAM could involve in the conduction by relaying protons across the polymeric skeletons via the hopping mechaism.16,26,27 The proposed proton conduction process is schematically illustrated in Fig. 7.
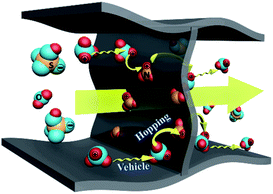 |
| | Fig. 7 Schematic illustration of the proposed conduction pathways in GSA-n. | |
3.4. Fuel cell performance
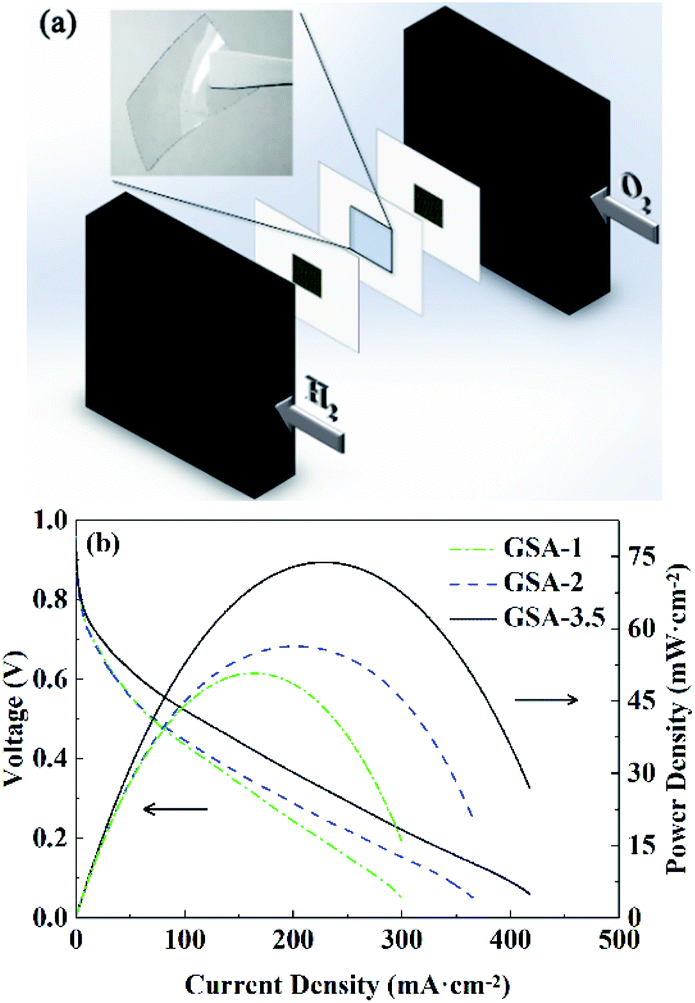
GSA-1, GSA-2, and GSA-3.5 hydrogel membranes were prepared for fuel cell characterization, as shown in Fig. 8a. The resultant polarization curves of the fuel cells at 30 °C are shown in Fig. 8b. The open-circuit voltage (OCV) was 0.953 V for the GSA-1 cell and decreased slightly with the increasing H2SO4 concentration. The slight decrease in OCV may be ascribed to the decreased catalytic activity of platinum at higher H2SO4 concnetrations.28,29 In contrast, the short-circuit current (ISC) increased with the increase in H2SO4 concentration by virtue of the enhanced proton conductivity and reached 437 mA cm−2 for the GSA-3.5 cell. A peak power density of 74 mW cm−2 was achieved for the GSA-3.5 cell. The VOCs, ISCs and peak power densities of the fuel cells constructed from GSA-n are summarized in Table 1.
 |
| | Fig. 8 (a) Diagram of the fuel cell assembly and (b) polarization curves of the fuel cells prepared from GSA-n. | |
Table 1 OCVs, ISCs and peak power densities of the fuel cells constructed form GSA-n
| Fuel cell |
OCV/V |
ISC/mA cm−2 |
Peak power density/mW cm−2 |
| GSA-1 |
0.953 ± 0.005 |
300 ± 2 |
52 ± 2 |
| GSA-2 |
0.950 ± 0.005 |
375 ± 10 |
58 ± 2 |
| GSA-3.5 |
0.940 ± 0.010 |
437 ± 20 |
74 ± 1 |
Performance stability of the fuel cell fabricated from GSA-3.5 was evaluated by recording OCV at 30 °C with certain intervals.30 As shown in Fig. 9, OCV kept relative constant (0.934–0.988 V) for more than 200 h. The result suggested that GSA-n was stable under actual operating conductions, guaranteeing long-term operation of fuel cells.
 |
| | Fig. 9 The OCV of GSA-4 membrane as a function of time at 30 °C. | |
4. Conclusions
Acid hydrogel electrolytes were successfully synthesized by in situ polymerization of AM and DVB in H2SO4 solution. Considerable mechanical strength arose from polymeric networks formed by the crosslinking. The amide moieties of PAM chains participated in proton conduction to promote electrochemical performance of the hydrogels. The acid hydrogel fuel cell maintained a long-term steady operation by virtue of the stable mechanical and electrochemical properties. In situ gelation of aqueous sulfuric acid solution offers an efficient strategy to construct gel electrolytes for diverse electrochemical applications.
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Notes and references
- Y. S. Zhang and A. Khademhosseini, Science, 2017, 356, 3627–3636 CrossRef PubMed.
- Z. F. Wang, H. F. Li, Z. J. Tang, Z. X. Liu, Z. H. Ruan, L. T. Ma, Q. Yang, D. H. Wang and C. Y. Zhi, Adv. Funct. Mater., 2018, 28, 1804560–1804589 CrossRef.
- C. Y. Chan, Z. Q. Wang, H. Jia, P. F. Ng, L. Chow and B. Fei, J. Mater. Chem. A, 2021, 9, 2043–2069 RSC.
- C. Keplinger, J. Y. Sun, C. C. Foo, P. Rothemund, G. M. Whitesides and Z. Suo, Science, 2013, 341, 984–987 CrossRef CAS PubMed.
- Y. Zhou, C. J. Wan, Y. S. Yang, H. Yang, S. C. Wang, Z. D. Dai, K. J. Ji, H. Jiang, X. D. Chen and Y. Long, Adv. Funct. Mater., 2019, 29, 1806220–1806227 CrossRef.
- H. Y. Ding, Z. Q. Xin, Y. Y. Yang, Y. F. Luo, K. L. Xia, B. L. Wang, Y. F. Sun, J. P. Wang, Y. Y. Zhang, H. Wu, S. S. Fan, L. Zhang and K. Liu, Adv. Funct. Mater., 2020, 30, 1909616–1909625 CrossRef CAS.
- Z. Liu, H. Li, M. Zhu, Y. Huang, Z. Tang, Z. Pei, Z. Wang, Z. Shi, J. Liu, Y. Huang and C. Zhi, Nano Energy, 2018, 44, 164–173 CrossRef CAS.
- Y. Zhao, Y. Zhang, H. Sun, X. Dong, J. Cao, L. Wang, Y. Xu, J. Ren, Y. Hwang, I. H. Son, X. Huang, Y. Wang and H. Peng, Angew. Chem., Int. Ed. Engl., 2016, 55, 14384–14388 CrossRef CAS PubMed.
- Y. X. Zeng, X. Y. Zhang, Y. Meng, M. H. Yu, J. N. Yi, Y. Q. Wu, X. H. Lu and Y. X. Tong, Adv. Mater., 2017, 29, 1700274–1700280 CrossRef PubMed.
- C. Meng, C. Liu, L. Chen, C. Hu and S. Fan, Nano Lett., 2010, 10, 4025–4031 CrossRef CAS PubMed.
- Y. Xie and J. Wang, J. Sol-Gel Sci. Technol., 2018, 86, 760–772 CrossRef CAS.
- N. Wen, B. Jiang, X. Wang, Z. Shang, D. Jiang, L. Zhang, C. Sun, Z. Wu, H. Yan, C. Liu and Z. Guo, Chem. Rec., 2020, 20, 773–792 CrossRef CAS PubMed.
- Z. Wang, F. Tao and Q. Pan, J. Mater. Chem. A, 2016, 4, 17732–17739 RSC.
- Z. Gao, L. Kong, R. Jin, X. Liu, W. Hu and G. Gao, J. Mater. Chem. C, 2020, 8, 11119–11127 RSC.
- S. J. Peng, X. Z. Jiang, X. T. Xiang, K. Chen, G. Q. Chen, X. C. Jiang and L. X. Hou, Electrochim. Acta, 2019, 324, 134874–134884 CrossRef CAS.
- W. Wieczorek and J. R. Stevens, Polymer, 1997, 38, 2057–2065 CrossRef CAS.
- M. J. Caulfield, X. Hao, G. G. Qiao and D. H. Solomon, Polymer, 2003, 44, 3817–3826 CrossRef CAS.
- A. N. Zeldin, L. E. Kukacka, J. Fontana and N. Carciello, Polym. Compos., 1981, 2, 1–7 CrossRef CAS.
- M. X. Wang, Y. M. Chen, Y. Gao, C. Hu, J. Hu, L. Tan and Z. Yang, ACS Appl. Mater. Interfaces, 2018, 10, 26610–26617 CrossRef CAS PubMed.
- L. Zang, Q. Liu, J. Qiu, C. Yang, C. Wei, C. Liu and L. Lao, ACS Appl. Mater. Interfaces, 2017, 9, 33941–33947 CrossRef CAS PubMed.
- H. E. Darling, J. Chem. Eng. Data, 1964, 9, 421–426 CrossRef CAS.
- M. Sampoli, N. C. Marziano and C. Tortato, J. Phys. Chem., 1989, 93, 7252–7257 CrossRef CAS.
- N. Gospodinova, E. Tomšík and O. Omelchenko, Eur. Polym. J., 2016, 74, 130–135 CrossRef CAS.
- H. G. M. Edwards, Spectrochim. Acta, Part A, 1989, 45, 715–719 CrossRef.
- M. B. Karimi, F. Mohammadi and K. Hooshyari, Phys. Chem. Chem. Phys., 2020, 22, 2917–2929 RSC.
- Q. Tang, G. Qian and K. Huang, RSC Adv., 2013, 3, 3520–3525 RSC.
- Q. Qin, Q. Tang, Q. Li, B. He, H. Chen, X. Wang and P. Yang, Int. J. Hydrogen Energy, 2014, 39, 4447–4458 CrossRef CAS.
- E. Peled, V. Livshits, M. Rakhman, A. Aharon, T. Duvdevani, M. Philosoph and T. Feiglin, Electrochem. Solid-State Lett., 2004, 7, A507–A510 CrossRef CAS.
- M. Ahlskog, M. Reghu, T. Noguchi and T. Ohnishi, Synth. Met., 1997, 89, 11–15 CrossRef CAS.
- S. Y. Lee, A. Ogawa, M. Kanno, H. Nakamoto, T. Yasuda and M. Watanabe, J. Am. Chem. Soc., 2010, 132, 9764–9773 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*