
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCyanuric chloride as the basis for compositionally diverse lipids†
David Nardoa,
Caleb M. Akersa,
Nicholas E. Cheunga,
Cierra M. Isoma,
Jason T. Spaudea,
Daniel W. Packab and
Vincent J. Venditto *a
*a
aDepartment of Pharmaceutical Sciences, University of Kentucky College of Pharmacy, Lexington, KY 40536, USA. E-mail: vincent.venditto@uky.edu
bDepartment of Chemical and Materials Engineering, University of Kentucky College of Engineering, Lexington, KY 40536, USA
First published on 15th July 2021
Abstract
Cyanuric chloride has been utilized in the development of new synthetic lipid compounds using two differing schemes. The resulting lipids, presented in this manuscript, were characterized and evaluated for their ability to form nanoparticles and subsequently tested for their utility in various biological applications, including gene delivery and immunization. Of the 12 lipids synthesized, 8 formed nanoparticles that remained stable, based on dynamic light scattering, for at least one month. The compounds were then assessed for their toxicity, and subsequently tested for their ability to encapsulate drugs, genes and peptides. While the compounds did not seem to encapsulate carboxyfluorescein, we demonstrate that these lipids are capable of plasmid delivery in vitro, and inducing antibody profiles similar to other hydrophobic anchors in liposomal peptide vaccines. This strategy for accessing diverse lipid compounds offers a way to easily optimize lipid-based therapeutics for research in an expedited manner.
Introduction
Liposomes provide an optimal vehicle for pharmaceutical delivery due to their versatility as amphipathic vectors, which allows for delivery of hydrophobic and hydrophilic agents.1,2 By altering the lipid composition in these nanoparticles, a multitude of properties can be honed to optimize their functionality. In the last few decades, liposome research has fuelled the development of synthetic lipids to improve therapeutic delivery, particularly nucleic acids.3 However, the complexity and cost of novel lipids limits liposome research.1,4–6 To overcome this, various research groups have developed synthetic, cationic lipid libraries with the goal of improving siRNA and mRNA delivery using cost effective and high-throughput schemes, taking advantage of specific chemical structures that allow for rapid headgroup diversification.5–7In addition to their utility as gene delivery vectors, liposomes have been investigated extensively for vaccine development using nucleic acids or proteins8,9 both as adjuvants,10 and as anchors of antigens on liposomal surfaces.11 Incorporating adjuvants and antigens in the same formulation has also improved antigen exposure to immune cells and enhanced the efficacy of liposomal vaccines.12,13 However, the need for synthetic lipids that serve as a platform to generate structure immunogenicity relationships are critical to advance the field of liposomal vaccine design.
Chemical entities that can facilitate efficient and cost-effective lipid synthesis provide opportunities to access diverse compositional space for use in gene, drug, and vaccine delivery. Cyanuric chloride is a heteronuclear aromatic molecule used as a chemoselective linker due to the thermally controlled reactivity of its three electrophilic carbons.14 Previous studies have evaluated the utility of cyanuric chloride for synthesis of a variety of molecules including dendrimers and ionizable lipids.15,16 In 2007 Candiani et al. reported a series of cationic, reducible lipids using cyanuric chloride as a linker.15 These were made from two single tailed triazine molecules, with a cationic diaminopropane headgroup, joined via a disulphide linker. The Candiani lipids resulted in successful plasmid delivery into cells and exhibited limited toxicity, suggesting that cationic TZ lipids capable of forming stable nanoparticles could be employed in this manner.15 The lipids described by Candiani et al. evaluate the usefulness of cyanuric chloride within the scope of single tailed dimerizable redox-sensitive lipids, while establishing a groundwork for continued evaluation of alternative cyanuric chloride based architectures for gene delivery and other applications. It was hypothesized that by altering the functionality of the headgroup structure, cyanuric chloride could provide a simple and cost-effective strategy to synthesize variable compounds with lipid-like properties that could be optimized for liposomal delivery across different areas of research, beyond gene delivery.
In this manuscript, a library of triazine (TZ) based lipids was synthesized using cyanuric chloride as a linker, dialkylamines as lipid tails and various small molecules to generate diverse head groups. The headgroups were chosen due to their cost effectiveness, commercial availability, and diversity in functional moieties, which provide a platform for future evaluation of an expanded library of triazine based lipids. Here we discuss the synthetic pathways used to produce these compounds, compare some of the properties conferred by different headgroups and evaluate the biological utility of some of the molecules generated through this process.
Experimental
Materials and instrumentation
Beta-alanine-tert-butyl ester, cyanuric chloride, didodecylamine, diisopropylethylamine (DIPEA), 2-mercaptoethylamine HCl, morpholine, ninhydrin, N,N-dimethyl diaminopropane and trityl chloride were purchased from TCI America (Portland, OR). Dioctadecylamine was purchased from Sigma-Aldrich (Milwaukee, WI). N-Boc-1,3-diaminopropane was purchased from Matrix Scientific (Columbia, SC). 1,2-Dimyristoyl-sn-glycero-3-phosphocholine (DMPC), 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC), 1,2-dimyristoyl-sn-glycero-3-phospho-(1′-rac-glycerol) (DMPG), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), 1,2-di-O-octadecenyl-3-trimethylammonium propane (DOTMA) and cholesterol were purchased from Avanti Polar Lipids, Inc. (Alabaster, AL). Solvents for reactions were purchased from various suppliers through VWR (Radnor, PA). Thin layer chromatography (TLC; Millipore Sigma, Silica gel 60 F254) was visualized under UV light or with 2% ninhydrin in DMSO. Final compound purity was assessed via a Waters 2707 Autosampler, Waters 2545 Quaternary Gradient Module pump and Waters 2998 Photodiode Array Detector following injection into a Waters XBridge C18 3.5 μm column (part no. 186003034) using a water, acetonitrile and methanol mixture as described in the figures below and detected at 205 and 254 nm. 1H and 13C NMR spectra were recorded in deuterated chloroform using a Varian 400 MHz or Varian 500 MHz spectrometer equipped with a 5 mm OneProbe (Cambridge Isotope Laboratories, Inc.; Tewksbury, MA). HR-MS was performed on an Agilent 6230B TOF LC/MS instrument in positive ion by direct injection of the compounds. Lipopeptide purification was performed using the Waters system described above.Synthesis
A full description of the synthesis used to generate each compound is provided in the ESI.† Briefly, two approaches were taken for the synthesis of the TZ lipids: a convergent and a divergent route. In the convergent approach, two small molecule nucleophiles with protected, ionizable moieties were reacted with cyanuric chloride through nucleophilic aromatic substitution (NAS). The resulting monochlorotriazine was then reacted with a long-chained secondary amine lipid tail (dioctadecylamine or didodecylamine) to yield the final protected lipid. In the divergent approach, the lipid tail was reacted first to form a dichloro-triazine, followed by headgroup diversification through addition of various nucleophilic small molecule moieties as headgroups. In both approaches, the first NAS was initiated on ice and allowed to stir at room temperature in chloroform for at least 4 hours. The second substituent was added at room temperature in chloroform and heated to 50 °C for at least 24 hours. The final NAS reaction was performed in xylenes or dioxane and heated from room temperature to 80 °C for at least 72 hours. In each reaction, excess nucleophile or DIPEA served as base. The reactions were monitored at each step via thin layer chromatography and characterized by nuclear magnetic resonance and mass spectrometry. Small molecule nucleophiles with reactant pendant moieties were protected with acid labile protecting groups and deprotected as the final step in the lipid synthesis with trifluoroacetic acid in dichloromethane.Biophysical characterization of lipids and nanoparticles
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ethanol/water ratio and crystalized at −20 °C. The crystalized CF was filtered and washed multiple times with distilled water and dried overnight. Solid CF was then dissolved in water and 5 M NaOH to a concentration of 250 mM and passed over an LH-20 Sephadex column. Five mL were purified on a 10 × 2 cm column by elution at room temperature with distilled water. CF eluted as a dark orange-red band that was quantified via absorbance at 492 nm using of coefficient of 6-CF (76900 M cm−1) as described by Weinstein et al.18 For the encapsulation assay, thin lipid films of CC lipids were prepared as described above. After evaporating remaining organic solvent overnight, the lipids were resuspended in a solution of 200 mM CF. Control phosphatidylcholine liposomes were then purified using a PD10 desalting column (GE Life Sciences).
:2 ethanol/water ratio and crystalized at −20 °C. The crystalized CF was filtered and washed multiple times with distilled water and dried overnight. Solid CF was then dissolved in water and 5 M NaOH to a concentration of 250 mM and passed over an LH-20 Sephadex column. Five mL were purified on a 10 × 2 cm column by elution at room temperature with distilled water. CF eluted as a dark orange-red band that was quantified via absorbance at 492 nm using of coefficient of 6-CF (76900 M cm−1) as described by Weinstein et al.18 For the encapsulation assay, thin lipid films of CC lipids were prepared as described above. After evaporating remaining organic solvent overnight, the lipids were resuspended in a solution of 200 mM CF. Control phosphatidylcholine liposomes were then purified using a PD10 desalting column (GE Life Sciences).Biological application of triazine lipids
000 in 200 μL of medium and allowed to settle overnight for subsequent assays.:1 molar ratio of cationic lipid/DOPE were rehydrated in a 20 mM HEPES solution at pH 4. The nanoparticles were mixed at equal volumes (5 μL) with plasmid DNA (5 μL) at the amine to phosphate (N:P) ratios indicated in the figure legends and incubated at room temperature for 10 minutes. For the triazine lipids the amine quantity per lipid was assumed to be 2 (one per headgroup), while DOTMA was considered to have 1 amine per lipid. After 10 minutes, 10 μL of the lipoplex was mixed with 2 μL of 6× loading dye (Boston BioProducts) and loaded onto a 1% agarose gel containing 0.5 μg mL−1 of ethidium bromide and run at 100 mV for 60 minutes. The gels were visualized and photographed using a Bio-Rad ChemiDoc XR system using the manufacturer's software.000 cells per well and incubated for 24 hours at 37 °C in 5% CO2. Liposomes made with a 1:1 ratio of DOPE and TZ lipid were added to 200 ng pGL3 Luciferase Reporter Vector (Promega) at N:P ratios of 2.5, 5 and 10, and incubated at 37 °C for 10 minutes before being diluted in 100 μL of non-supplemented EMEM and added to the cells. Following a four-hour incubation at 37 °C, the media was changed, and the cells were incubated for another twenty hours, at which point the cells were lysed with a cell culture lysis reagent at pH 7.8 composed of 25 mM Tris–phosphate buffer, 0.7 g L−1 1,2-diaminocyclohexane, 10% glycerol, 1% Triton X-100, and 1% protease inhibitor cocktail (Millipore). Total protein content was determined with a bicinchoninic acid assay (G-Biosciences) and luciferase protein expression was quantified by a luciferase assay (Promega). Cell viability was assessed using a Cell Titer Blue assay kit (Promega) based on the manufacturer's instructions. In each of the three independent experiments performed, transfection was compared with cells treated with Lipofectamine 3000 (Thermo), following the manufacturer's instructions, and with DNA treated cells.000 cells per well using D-MEM containing 10% fetal bovine serum (Gemini), 100 U mL−1 penicillin, 0.1 mg mL−1 streptomycin and 500 μg mL−1 geneticin (VWR) and incubated until they reached 70–90% confluency. Lipoplexes were formed by combining TZ lipid liposomes made with a 1:1 ratio of DOPE and TZ lipid in Opti-MEM (Thermo) with 400 ng of human alpha-1 antitrypsin (hAAT) plasmid DNA (Addgene no. 126704) and incubating for 12 minutes in Opti-MEM, before being added to cells. After 24 hours the media was removed for evaluation of viability and replaced with fresh media. The cells were then incubated for another 72 hours and then transferred to 1.5 mL microcentrifuge tubes and centrifuged at 400 rpm for 5 minutes. The media was removed and assessed for hAAT via ELISA and the cells were lysed using RIPA buffer (Thermo) for determination of total protein concentration (Thermo). In each of the three independent experiments performed, transfection was compared with cells treated with Lipofectamine 3000 (Thermo), following the manufacturer's instructions, and with DNA treated cells.:5000 dilution and incubated for 30 minutes at 37 °C. The plate was then washed six times and binding was quantified by incubating the samples with 100 μL of tetramethylbenzidine (Rockland) for 30 minutes at room temperature, followed by quenching with 100 μL of 0.5 M H2SO4. Absorbance at 450 nm was recorded using a BioTek Synergy H1 microplate reader. After quantifying hAAT using the standard curve, hAAT in each well was normalized to total cell protein in respective plate, which was quantified using a Pierce BCA Assay Kit (Thermo) using the manufacturer's instructions.:2:3:0.3 molar ratio and 0.5 mg mL−1 of lipid-conjugated peptide. The peptide used for these experiments was the lecithin–cholesterol acyltransferase domain of apolipoprotein A-I (sequence βAGGLSPVAEEFRDRMRTHVDSLRTQLAPHSEQMRESLAQRLAELKSN). As a control, the original peptide anchor (cholesteryl hemisuccinate) was used to immunize one group of mice, while two other groups were immunized with the peptide was conjugated to intermediate D and the third group was immunized with peptide free liposomes. To assess the efficacy of immunizations, blood was collected by superficial temporal vein puncture using a small animal lancet (Medipoint) into a microcentrifuge tube and centrifuged for 10 min at 21000 × g after standing at room temperature for 2 h. Serum was stored at −80 °C for later antibody detection. The mice were sedated during any procedures using isoflurane gas. All animal procedures were performed in accordance with the United States Department of Health and Human Services, Office of Laboratory Animal Welfare, Public Health Service Policy on Humane Care and Use of Laboratory Animals and approved by the University of Kentucky Institutional Animal Care and Use Committee.:200 and incubated for 30 minutes at 37 °C. The wells were then washed six times and treated with 100 μL of goat anti-mouse IgG–HRP (Invitrogen no. 16066) diluted 1:2000 in PBS–C and incubated for 30 minutes at 37 °C before being washed again. Binding was quantified by incubating the samples with 100 μL of tetramethylbenzidine (Rockland) for 30 minutes at room temperature, followed by quenching with 0.5 M H2SO4. Absorbance at 450 nm was recorded using a BioTek Synergy H1 microplate reader. Reciprocal endpoint titers were then calculated by plotting the absorbance vs. serum dilution and dividing the slope of the curve by two times the average of the blank (PBS–C only) wells.Results and discussion
The thermally controlled, chemo-selective reactivity of cyanuric chloride provides a platform to add a multitude of functional headgroups and develop a wide array of synthetic lipids.14 In general, cyanuric chloride undergoes nucleophilic aromatic substitution at 0 °C for the first substitution, 25 °C for the second, and 70 °C for the third but the reactions are influenced by the nucleophilicity and steric hinderance of the reactants. Using this framework, two dichlorotriazine molecules were generated as the basis for lipids and several small molecules were tested as headgroups (Fig. 1 and S1†). The relative scarcity of commercially available long-chain secondary amines (tails) as compared to the abundance of potential head groups, results in a system in which compositional diversity is introduced in the headgroups rather than the tails. Therefore, a divergent approach was initially utilized for the synthesis of these compounds by adding the lipid tails to prepare a dichlorotriazine that was further diversified with various headgroups. This strategy, however, was not viable for all headgroups used, particularly those with sterically hindered moieties. Therefore, a convergent strategy was attempted by initiating the synthesis with the addition of headgroups to the cyanuric chloride ring to form a monochlorotriazine to which tails were then added (Fig. 1).14 | ||
| Fig. 1 Synthetic schemes for TZ lipids. | ||
Synthesis of all lipids (excluding those containing morpholine) was attempted using both routes for comparison (Table 1) and the resulting products were characterized by NMR and HRMS. Lipids 1–4 proceeded well under both routes with similar yields for the convergent and divergent route using the beta-alanine headgroups (lipid 2: 20% and 21%) and the diaminopropane headgroups (lipid 4: 48% and 53%). This was not the case for lipids 5 and 6, which employed trityl-protected cysteamine (Trt-Cys). Divergent synthesis of lipidated dichlorotriazine molecules with Trt-Cys resulted in an insoluble compound with exceedingly low yield and could only be successfully synthesized using the convergent route with protected beta-alanine as the first substitution on cyanuric chloride.
| Lipid structures | # | Tail | Conv. | Div. | Tm (°C) | Size (nm) | PDI | Charge (mV) | LD50 (μM) |
|---|---|---|---|---|---|---|---|---|---|
| a Yield (%) is calculated based on all synthetic steps, including first divergent step to prepare dichlorotriazine intermediates D and F, which are used for all subsequent headgroup reactions. ND = not determined. | |||||||||
 |
1 | C12 | 22 | 49 | <10 | 124 ± 2 | 0.24 ± 0.02 | −63 ± 4 | 1098 |
| 2 | C18 | 20 | 21 | 58 | 131 ± 0 | 0.24 ± 0.00 | −59 ± 5 | 894 | |
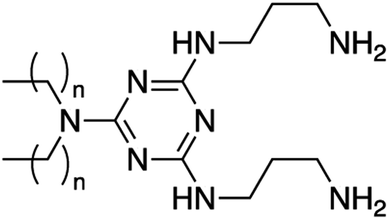 |
3 | C12 | 28 | 43 | <10 | 130 ± 2 | 0.44 ± 0.02 | 52 ± 3 | 133 |
| 4 | C18 | 48 | 53 | 33 | 126 ± 1 | 0.28 ± 0.00 | 63 ± 3 | 180 | |
 |
5 | C12 | 90 | ND | <10 | 87 ± 2 | 0.40 ± 0.00 | −75 ± 6 | 679 |
| 6 | C18 | 72 | ND | 40 | 93 ± 2 | 0.27 ± 0.01 | −71 ± 5 | 988 | |
 |
7 | C18 | ND | 91 | 64 | ND | ND | ND | ND |
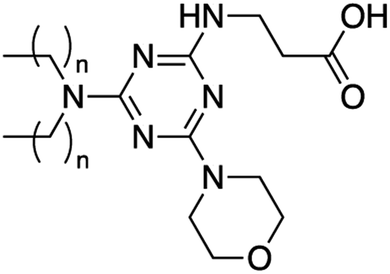 |
8 | C18 | ND | 80 | 51 | ND | ND | ND | ND |
 |
9 | C12 | ND | 88 | <10 | 159 ± 1 | 0.24 ± 0.00 | 45 ± 2 | 337 |
| 10 | C18 | ND | 89 | 28 | 186 ± 1 | 0.28 ± 0.01 | 47 ± 1 | 261 | |
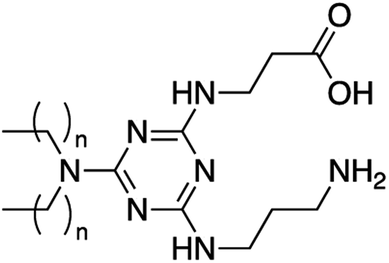 |
11 | C12 | ND | 46 | <10 | Unstable | Unstable | 42 ± 5 | 277 |
| 12 | C18 | ND | 23 | 42 | Unstable | Unstable | 70 ± 5 | 227 | |
Since the third addition to the TZ ring is generally more difficult to achieve, and morpholine is a strong nucleophile,14 lipids 7 and 8 were only synthesized using the divergent route with overall yields of 91% and 80%, respectively. The divergent route was also used to synthesize lipids 9 and 10 containing N,N-dimethyldiaminopropane, as well as lipids 11 and 12 using both beta-alanine and diaminopropane in the headgroup. When using the convergent approach to synthesize these lipids, the purification of the monochlorotriazine headgroup molecules in the absence of the lipid tails was problematic requiring a slow and lengthy purification by column chromatography (>12 hours). Conversely, the divergent route facilitated synthesis and purification of the final lipids.
Using these two routes, the divergent synthetic route reduces the total number of reactions needed to prepare a library of molecules by 25–33% depending on the final composition of the lipids. While some lipids result in similar overall yield between convergent and divergent routes, the challenges with nucleophilicity, steric hinderance and purification of intermediate molecules resulted in synthetic preference for one route over the other for certain headgroups. The divergent route results in increased compositional diversity with fewer steps, while the convergent route serves as an important complementary role for the synthesis of certain lipids.
The utility of the divergent route was then explored further by reacting the C18 dichloro-triazine compound (intermediate D) with the N-terminal amine of a protected peptide on rink amide resin, to provide an alternative synthetic route to present lipid-anchored peptides in a liposomal bilayer for vaccination. Using the 44 amino acid sequence from apolipoprotein A-I (ApoA-I) that our group has previously investigated,22 we achieved 95% lipopeptide purity which is improved as compared to previously described lipopeptide synthetic strategies using cholesteryl hemisuccinate and other lipid anchors.23 In addition to the improved overall yield, the ease of lipopeptide synthesis using intermediate D provides a convenient platform for continued vaccination studies with minimal downstream purification, as evidenced by the HPLC trace of the lipopeptide (Fig. S45†).
Next, we sought to prepare liposomal formulations using each lipid. First, the transition temperature (Tm) of each compound was determined by forming nanoparticles of pure lipid via rehydration of thin lipid films in 20 mM HEPES and sonicated at high temperature. The resultant formulations were then transferred into Hastelloy ampules to assess the transition temperature by differential scanning calorimetry. All lipids made with a didodecylamine tail yielded a Tm below 10 °C, while those containing a dioctadecylamine tail ranged in Tm from 28–64 °C (Fig. 2A and Table 1).
 | ||
| Fig. 2 (A) Transition temperature of TZ lipids determined by DSC. (B) In vitro toxicity of triazine lipids. Toxicity of TZ lipids on bone marrow derived macrophages as compared to commercially available cationic (DOTMA) and zwitterionic (DMPC) lipids using the lactate dehydrogenase assay. Liposomes were made by thin film hydration followed by sonication and used immediately to treat cells for 24 hours, prior to testing LDH release in cell media. Representative data from one of three independent experiments is shown; bars indicate mean values for three technical replicates of duplicate experiments ± SEM. | ||
All lipids were initially formulated at pH 7, but failed to properly hydrate. Therefore, hydration of lipids 1 and 2 were tested at increasing pH and found that pH 10 was ideal for hydration. All other lipids hydrated well under acidic conditions (pH 4). Lipids 7 and 8, which contained morpholine in the headgroup, failed to form liposomes, alone or in combination with distearoyl phosphatidylcholine (DSPC) or DSPC and cholesterol from 5 to 90 mol% TZ lipids. Other lipids made with isonipecotic acid also failed to form liposomes (data not shown) indicating that steric hinderance of the headgroups may preclude liposome formation. Additionally, while lipids 11 and 12 initially formed nanoparticles, the structures were unstable past 24 hours as determined by dynamic light scattering. Aside from these, all other lipids formed nanoparticles that appear stable at one month after preparation when stored in the refrigerator, based on dynamic light scattering.
For lipids that formed nanoparticles, the size of the nanoparticles ranged from 87 to 186 nm in diameter (Table 1), with no clear trend between diameter and structural characteristics, such as lipid tail and charge. Lipids containing cysteamine in the headgroup achieved the smallest size, while lipids 9 and 10, exhibited the largest initial diameter. Lipids 11 and 12 also formed nanoparticles >300 nm in diameter, but are unstable as denoted in Table 1 with PDI of 0.98 and 0.51, respectively. The charges of each formulation also aligned with the headgroup used and ranged from −75 to 70 mV for anionic and cationic headgroups, respectively. Lipids 11 and 12, which contained beta-alanine and 1,3-diaminopropane in the headgroup, were hydrated in acidic conditions yielding a positive charge for this formulation. Attempts were made to formulate these in both acidic (pH 4) and basic (pH 10) solutions, but the lipids only hydrated well in acidic conditions.
Prior to testing the nanoparticles in further applications, the toxicity of the compounds was assessed in vitro. The primary mechanisms of toxicity associated with lipid nanoparticles, particularly cationic ones, are cell lysis and activation of immune responses.24,25 Therefore, macrophages were chosen to test this aspect of TZ nanoparticles as these are among the primary cells responsible for the uptake of nanoparticles from circulation and are associated with the immune responses observed following in vivo nanoparticle administration.26 To assess the toxicity of the nanoparticles in vitro, the lipids were tested for induction of lactate dehydrogenase (LDH) release from bone marrow derived macrophages (BMDMs) from C57BL/6J mice, which has been demonstrated to be a more sensitive method of early liposomal toxicity.25 BMDMs were treated with TZ lipids at concentrations ranging from 31.25 to 250 nmol mL−1. As can be seen in Fig. 2B and Table 1, the toxicity of the nanoparticles ranged between that of the synthetic, cationic lipid DOTMA, and the natural zwitterionic phospholipid DMPC (Table 1). The LD50 values of the cationic lipids was considerably higher than that of other lipids (133 and 180 mM for lipids 3 and 4, respectively), approximating the toxicity of DOTMA (LD50 = 78 mM). Lipids 9 and 10 also had higher toxicity than other TZ lipids (LD50 = 337 and 261 mM, respectively), which did not differ significantly from DMPC (LD50 = 969 mM).
The first assessment of the utility of the nanoparticles was performed by testing whether those TZ lipid nanoparticles that remained stable for several weeks they could retain therapeutics in their aqueous core, and carboxyfluorescein (CF) encapsulation was used to test this.18 Intriguingly, when pure TZ lipids were used to encapsulate CF, they formed a gel indicating that they may be unable to encapsulate aromatic molecules even when mixed with DSPC at ratios as low as 10% TZ lipid. Additional experiments are needed to determine the utility of these lipids for delivery of various aromatic and non-aromatic small molecules alone or as part of more complex nanoparticles.
Having shown great success in preclinical studies, many synthetic lipids with cationic headgroups are used in gene transfection as commercial reagents for laboratory use.3 More recently, the first siRNA therapeutic, patisiran, was approved for clinical use by the United States FDA and two lipid-based mRNA vaccines were approved under emergency use authorization for prevention of COVID-19.27,28
To determine whether TZ lipids with cationic headgroups that form stable formulations (3, 4, 9 and 10) could complex nucleic acids, lipid nanoparticles made from a 1:1 molar ratio of cationic lipids and DOPE were incubated with plasmid DNA at increasing ratios of cationic amine (N) to anionic nucleic acid phosphate (P) and assessed for migration in an agarose gel. Formulations with a 1:1 molar ratio of DOPE and cationic lipid have been extensively reported in the literature and provides a simple starting point for assessing the potential of cationic lipid formulations.29,30 Of note, the N content of TZ lipids are based on the distal aliphatic amines of the headgroups, but the other amines in the molecules may contribute to complexation. As shown in Fig. 3A, all four lipids were able to complex RNA at an N:P ratio of 5 or above. By comparison, DOTMA/DOPE nanoparticles inhibited RNA migration at an N:P ratio of 10 while pure DOPE lipids were unable to prevent migration. The TZ/DOPE lipoplexes were also evaluated by DLS at N:P ratios of 1 and 5 and their size and charge was determined by DLS. As evidenced by Table S1,† the lipoplexes increased in size, compared with the free nanoparticles, but they increased slightly in size as more DNA was added, suggesting complexation.
 | ||
| Fig. 3 Efficacy of TZ lipids in gene transfection. (A) Gel shift assay of plasmid DNA complexed with TZ lipids. (B) pKa assessment of cationic lipids measured by TNS fluorescence at pH range 2.5 to 10. Plots represent the sigmoidal, best fit analysis of one of three independent experiments. (C–E) Transfection of HeLa cells with luciferase reporter gene using Lipofectamine 3000 or TZ lipids at an N:P ratio of 10, 5 and 2.5 (left to right). Bars represent the mean values from one of three representative experiments, except for the LDH assay which was performed twice. (C) Luciferase expression in transfected HeLa cells. (D) LDH release from HeLa cells transfected with luciferase plasmid 4 hours after transfection. (E) Viability of cells treated with plasmid and lipids 24 hours after transfection. (F–H) Transfection of HEK293-T cells with hAAT using Lipofectamine 3000 or TZ lipids at N:P ratios of 6, 3 and 1.5 (left to right). Bars represent the mean values from one of three representative experiments, except for the viability assay which was performed twice. (F) hAAT expression 72 hours after transfection based on ELISA and normalized to total cell protein. (G) LDH release from cells transfected with hAAT plasmid 24 hours after transfection. (H) Viability of cells treated with plasmid and lipids 48 hours after transfection. In both experiments, each treatment was compared to the Lipofectamine control using the Kruskal–Wallis non-parametric test. Bars indicate mean values for triplicates ± SEM and p = <0.05. | ||
In addition to size, charge and complexation, pKa is another crucial aspect of cationic lipids that has been directly correlated to the success of lipid nanoparticles in gene delivery.19,31,32 Particularly, ionizable lipids with a pKa ranging from 6.2 to 6.4, have been shown to achieve a high degree of efficacy when used to deliver siRNA.3 To assess the pKa of the cationic TZ lipids, liposomes made from these lipids were rehydrated in buffered solutions ranging from pH 2.5 to 12 and mixed with TNS, as described in the methods section. Interestingly, the pKa of the TZ lipids differed in both cases due to the tail length (Fig. 3B), despite having the same headgroup, with lipids 3 and 4 varying by almost two units (7.37 for lipid 3 and 5.59 for lipid 4).
The mixture of cationic TZ lipids with DOPE was then used to deliver plasmid DNA into HeLa cells using a luciferase reporter vector, comparing their efficacy with free DNA and Lipofectamine 3000.30 As shown in Fig. 3C, all four lipids improved plasmid transfection compared with naked plasmid, with the shorter tailed lipids (3 and 9) demonstrating better efficacy than the lipids with C18 tails (4 and 10), which concurs with the findings of Candiani et al. who reported improved transfection with shorter length tails.15 Overall, TZ lipid transfection was only modest compared to Lipofectamine, with optimal luciferase expression reaching an average of 462 RLU per mg for lipid 9 at an N:P ratio of 5 (vs. 7937 RLU per mg for Lipofectamine), and LDH release and cell viability approximating that of Lipofectamine (Fig. 3D and E).
To confirm these findings in a more clinically relevant context, HEK293-T cells were transfected with a plasmid encoding human alpha-1 antitrypsin (hAAT) using the same lipid mixtures, and hAAT expression was assessed by ELISA. As evidenced in Fig. 3F–H, the cationic TZ lipids significantly improved pDNA transfection, except for lipid 10, and exhibited a similar toxicity profile to that of Lipofectamine. The data from these experiments suggests that TZ lipids may have utility in the delivery of nucleic acids and warrants further exploration of their capabilities in vivo, which are currently underway. The efficacy of the nanoparticles did not seem to correlate with a lower pKa of the nanoparticles, which was unexpected, based on previous reports.3 However, the influence of the lipid architecture on complexation with DNA as compared to siRNA may explain the deviation from the optimally reported pKa of 6.2–6.5.3 This finding highlights the need for studying the behaviour of aromatic lipids, as suggested by Martinez-Negro, et al.33 and more specifically the behaviour of cationic TZ lipids, which may also serve a role as helper lipids in more complex gene delivery platforms.
Finally, given the synthetic versatility of the divergent route to append more complex moieties, such as peptides, we examined the immunogenicity of the lipopeptide prepared with C18 TZ linker (intermediate D) as compared to our standard immunization protocol using cholesteryl hemisuccinate as a peptide anchor in the liposomal formulation.
The modular design of liposomes allows for combination of antigens and adjuvants to tailor immune responses for clinically relevant immunization strategies (Fig. 4A).10 Liposomal peptide vaccines increase the bioavailability of antigens by extending their half-life and increasing concentrations in lymphatic tissues.34 Our lab has previously developed a liposomal strategy to induce antiboides toward apolipoprotein A-I (ApoA-I) in mice, as a mechanism to mimic the immunity observed in humans. To achieve this we use a lipid anchored 44 amino acid peptide derived from ApoA-I for liposomal immunizations.22 To determine whether TZ lipids could be used in this setting, formulations were prepared with the respective lipopeptides along with the adjuvant monophosphoryl lipid A (MPLA), which is a toll-like receptor (TLR)-4 agonist. Peptides are formulated in the liposomes (20 mM) at a concentration of 1 mg mL−1 which equates to about 1000 peptides per liposome. The resulting liposomes range in size from ∼110 nm in diameter for the peptide free liposomes to between 150–200 nm for the liposomes containing peptide, which are stable for up to a month under refrigeration.
 | ||
| Fig. 4 TZ lipids as peptide anchors in liposomal vaccines. (A) Liposomal vaccines can include various components, including natural phospholipids and adjuvants, to optimize responses to an immunogen. (B) Lipid linkers anchoring apolipoprotein A-I peptide to the liposomal vaccine, cholesterol hemisuccinate and intermediate D (C18 TZ). (C) Reciprocal endpoint titres 7 days after the second of two immunizations compared with no peptide immunization. Symbols correspond to individual mice and line represents mean ± SEM and p = <0.05. | ||
After liposome preparation, mice were immunized twice with a liposomal vaccine containing one of the lipopeptide conjugates (Fig. 4B) or a control formulation without peptide present and reciprocal endpoint titres (RET) toward the peptide immunogen were assessed seven days after the second immunization. RET from mice immunized with the TZ lipid anchor approximated that of CHEMS (Fig. 4C), which has previously been shown to serve as an optimal peptide anchor for liposomal immunization.23 These data highlight the utility of this synthetic strategy in peptide conjugation and establish a platform for continued studies. Efforts are currently underway to evaluate the secondary structure of peptides anchored into bilayers using different lipid constructs to identify parameters that promote peptide structure and immunogenicity when presented in liposomal formulations.
Conclusions
The present work demonstrates the utility of cyanuric chloride in the development of synthetic lipids with a wide potential for therapeutic delivery, based on the properties of specific headgroups. Furthermore, this strategy provides a simple method to alter the structure of lipids to optimize lipid properties depending on the desired outcome. This work expands on previous research demonstrating the utility of this compound in the development of synthetic structures for drug delivery and provides a novel strategy to access diverse lipids with relative synthetic ease.5,7,14 While continued studies are underway to evaluate the utility of TZ lipids using in vivo models, the present findings suggest that TZ lipids have broad utility in gene delivery and vaccine development.Conflicts of interest
There are no conflicts of interest to declare.Notes and references
- A. G. Kohli, P. H. Kierstead, V. J. Venditto, C. L. Walsh and F. C. Szoka, J. Controlled Release, 2014, 190, 274–287 CrossRef CAS.
- T. M. Allen and P. R. Cullis, Adv. Drug Delivery Rev., 2013, 65, 36–48 CrossRef CAS.
- P. R. Cullis and M. J. Hope, Mol. Ther., 2017, 25, 1467–1475 CrossRef CAS.
- J. Li, X. Wang, T. Zhang, C. Wang, Z. Huang, X. Luo and Y. Deng, Asian J. Pharm. Sci., 2015, 10, 81–98 CrossRef.
- M. R. Molla, A. Böser, A. Rana, K. Schwarz and P. A. Levkin, Bioconjugate Chem., 2018, 29, 992–999 CrossRef CAS.
- C. Y. Zhou, H. Wu and N. K. Devaraj, Chem. Sci., 2015, 6, 4365–4372 RSC.
- C. A. Alabi, K. T. Love, G. Sahay, H. Yin, K. M. Luly, R. Langer and D. G. Anderson, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 12881–12886 CrossRef CAS PubMed.
- L. Miao, L. Li, Y. Huang, D. Delcassian, J. Chahal, J. Han, Y. Shi, K. Sadtler, W. Gao, J. Lin, J. C. Doloff, R. Langer and D. G. Anderson, Nat. Biotechnol., 2019, 37, 1174–1185 CrossRef CAS PubMed.
- J. B. Ulmer and A. J. Geall, Curr. Opin. Immunol., 2016, 41, 18–22 CrossRef CAS.
- C. R. Alving, Z. Beck, G. R. Matyas and M. Rao, Expert Opin. Drug Delivery, 2016, 13, 807–816 CrossRef CAS.
- D. S. Watson, V. M. Platt, L. Cao, V. J. Venditto and F. C. Szoka, Clin. Vaccine Immunol., 2011, 18, 289–297 CrossRef CAS PubMed.
- C. R. Alving, M. Rao, N. J. Steers, G. R. Matyas and A. V. Mayorov, Expert Rev. Vaccines, 2012, 11, 733–744 CrossRef CAS.
- G. Fujii, W. Ernst and J. Adler-Moore, Front. Biosci., 2008, 13, 1968–1980 CrossRef CAS.
- E. E. Simanek, H. Abdou, S. Lalwani, J. Lim, M. Mintzer, V. J. Venditto and B. Vittur, Proc. R. Soc. A, 2010, 466, 1445–1468 CrossRef CAS.
- G. Candiani, M. Frigerio, F. Viani, C. Verpelli, C. Sala, L. Chiamenti, N. Zaffaroni, M. Folini, M. Sani, W. Panzeri and M. Zanda, ChemMedChem, 2007, 2, 292–296 CrossRef CAS.
- O. M. Merkel, M. A. Mintzer, D. Librizzi, O. Samsonova, T. Dicke, B. Sproat, H. Garn, P. J. Barth, E. E. Simanek and T. Kissel, Mol. Pharm., 2010, 7, 969–983 CrossRef CAS.
- E. Ralston, L. M. Hjelmeland, R. D. Klausner, J. N. Weinstein and R. Blumenthal, Biochim. Biophys. Acta, Biomembr., 1981, 649, 133–137 CrossRef CAS.
- J. N. Weinstein, R. Blumenthal and R. D. Klausner, Methods Enzymol., 1986, 128, 657–668 CAS.
- M. Jayaraman, S. M. Ansell, B. L. Mui, Y. K. Tam, J. Chen, X. Du, D. Butler, L. Eltepu, S. Matsuda, J. K. Narayanannair, K. G. Rajeev, I. M. Hafez, A. Akinc, M. A. Maier, M. A. Tracy, P. R. Cullis, T. D. Madden, M. Manoharan and M. J. Hope, Angew. Chem., Int. Ed., 2012, 51, 8529–8533 CrossRef CAS PubMed.
- M. A. Akbar, D. Nardo, M. J. Chen, A. S. Elshikha, R. Ahamed, E. M. Elsayed, C. Bigot, L. S. Holliday and S. Song, Mol. Med., 2017, 23, 57–69 CAS.
- I. Pineda-Torra, M. Gage, A. de Juan and O. M. Pello, Methods Mol. Biol., 2015, 1339, 101–109 CrossRef CAS PubMed.
- M. G. Pitts, D. Nardo, C. M. Isom and V. J. Venditto, ImmunoHorizons, 2020, 4, 455–463 CrossRef CAS PubMed.
- D. S. Watson and F. C. Szoka Jr, Vaccine, 2009, 27, 4672–4683 CrossRef CAS PubMed.
- K. B. Knudsen, H. Northeved, P. Kumar Ek, A. Permin, T. Gjetting, T. L. Andresen, S. Larsen, K. M. Wegener, J. Lykkesfeldt, K. Jantzen, S. Loft, P. Møller and M. Roursgaard, Nanomedicine, 2015, 11, 467–477 CrossRef CAS PubMed.
- K. Lappalainen, I. Jääskeläinen, K. Syrjänen, A. Urtti and S. Syrjänen, Pharm. Res., 1994, 11, 1127–1131 CrossRef CAS.
- H. H. Gustafson, D. Holt-Casper, D. W. Grainger and H. Ghandehari, Nano Today, 2015, 10, 487–510 CrossRef CAS.
- H. Wood, Nat. Rev. Neurol., 2018, 14, 570 Search PubMed.
- K. Rawat, P. Kumari and L. Saha, Eur. J. Pharmacol., 2021, 892, 173751 CrossRef CAS.
- A. Lechanteur, V. Sanna, A. Duchemin, B. Evrard, D. Mottet and G. Piel, Nanomaterials, 2018, 8, 270 CrossRef PubMed.
- Z. Du, M. M. Munye, A. D. Tagalakis, M. D. I. Manunta and S. L. Hart, Sci. Rep., 2014, 4, 7107 CrossRef PubMed.
- C. L. Walsh, J. Nguyen, M. R. Tiffany and F. C. Szoka, Bioconjugate Chem., 2013, 24, 36–43 CrossRef CAS PubMed.
- J. Zhang, H. Fan, D. A. Levorse and L. S. Crocker, Langmuir, 2011, 27, 1907–1914 CrossRef CAS PubMed.
- M. Martinez-Negro, A. L. Barran-Berdon, C. Aicart-Ramos, M. L. Moya, C. T. de Ilarduya, E. Aicart and E. Junquera, Colloids Surf., B, 2018, 161, 519–527 CrossRef CAS PubMed.
- R. Nisini, N. Poerio, S. Mariotti, F. De Santis and M. Fraziano, Front. Immunol., 2018, 9, 155 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra02425f |
| This journal is © The Royal Society of Chemistry 2021 |