
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCopolyesters of ε-caprolactone and L-lactide catalyzed by a tetrabutylammonium phthalimide-N-oxyl organocatalyst†
Zhiheng Fenga,
Li Wua,
Huan Donga,
Boping Liub and
Ruihua Cheng *a
*a
aSchool of Chemical Engineering, East China University of Science and Technology, Meilong Road 130, Shanghai, 200237, China. E-mail: rhcheng@ecust.edu.cn
bCollege of Materials and Energy, South China Agricultural University, Guangzhou, 510642, China
First published on 26th May 2021
Abstract
Aliphatic polyesters are biocompatible materials that can be used in biomedical applications. We report here the use of tetrabutylammonium phthalimide-N-oxyl catalyst (TBAPINO), as a thermally stable organocatalyst for the ring-opening polymerization (ROP) of cyclic esters under mild conditions. In the solution ROP of ε-caprolactone (ε-CL), quantitative conversion and Mn of ∼20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 are achieved in a wide temperature range from −15 to 60 °C. Under bulk condition, the conversion of ε-CL reaches over 85% at 120 °C within 2 h. The living ROP character of L-lactide (L-LA) catalyzed over TBAPINO is proved by multiple additions of monomer in the bulk polymerization. The catalyst shows comparable selectivity towards the ring-opening polymerization of L-LA and ε-CL. Their copolymerization over TBAPINO is carried out in one-pot bulk condition in terms of the reaction time, monomer feed ratio, and sequence of addition. The colorless poly(ε-caprolactone-co-lactide) (PCLA) is obtained with considerable conversion of both monomers with the Mn over 22000 g mol−1.
000 g mol−1 are achieved in a wide temperature range from −15 to 60 °C. Under bulk condition, the conversion of ε-CL reaches over 85% at 120 °C within 2 h. The living ROP character of L-lactide (L-LA) catalyzed over TBAPINO is proved by multiple additions of monomer in the bulk polymerization. The catalyst shows comparable selectivity towards the ring-opening polymerization of L-LA and ε-CL. Their copolymerization over TBAPINO is carried out in one-pot bulk condition in terms of the reaction time, monomer feed ratio, and sequence of addition. The colorless poly(ε-caprolactone-co-lactide) (PCLA) is obtained with considerable conversion of both monomers with the Mn over 22000 g mol−1.
Introduction
Aliphatic polyesters, such as poly(lactide) (PLA), poly(ε-caprolactone) (PCL), poly(glycolide) (PGA), etc. and their copolymers are found to be excellent biodegradable and biocompatible materials, with applications in drug delivery,1–3 tissue engineering,4 orthopedic stents5 and agricultural production.6 As they are gaining more awareness in daily production and medical fields, higher requirements are put forward for the performance of biodegradability and biocompatibility etc. To improve the properties, copolymerization of two or more monomers provides an alternative way to create materials with complementary properties. For example, poly(lactide-co-glycolide) (PLGA) is extensively used in biomedical applications for its hydrolysis-mediated degradation.7 Poly(caprolactone-co-lactide) (PCLA) is more permeable and has a tunable degradation and hydrolysis rate.8,9 Thus, more compatible catalysts to activate diverse monomers are crucial to the copolymerization system.Up to now, diverse and versatile catalysts were applied in the ring-opening polymerization (ROP) of cycloesters.10 Stannous octoate is the commercial metal catalysts for the homo- and copolymerization of various cyclic esters.11 However, the tin residue should be removed due to its toxicity, especially in biomedical application.8 Alternatively, Al, Mg, and Zn with various anions have been studied for ROP of polyesters.12 Aluminum complexes modified by varying the ligand were utilized to produce well-defined polymers with high catalytic efficiency and selectivity to ROP of lactones.13,14 Zinc alkoxides were friendly substitution of Tin analogue due to their bioresorbable characteristic15 and proved to be high effective for ROP of L-LA at room temperature with good control of polymer structure.16
In recent years, the application of metal-free strategies to ROP reactions has been developed into an important aspect of the field. These systems offer advantages including the absence of metal contamination, simple synthesis, wide scope of monomers and enhanced stability to trace impurities such as water and oxygen.17
Hedrick and coworkers18 first reported 4-(dimethylamino) pyridine (DMAP) as a simple organocatalyst for the ROP of L-LA in solution at room temperature, while several days were needed to reach considerable conversion of lactones (ε-CL and δ-valerolactone, δ-VL). 1, 5, 7-Triazabicyclo[4.4.0]dec-5-ene (TBD) could present the comparative catalytic efficiency as metal catalysts.19 Up to now, various types and chemical structures catalysts, including N-heterocyles, N-heterocyclic carbenes (NHCs), (thio)ureas, acids, phosphazene bases etc. have been reported.20
Beyond that, the organocatalysts derived from multi-component catalysts, as the combinations of the acidic/basic catalysis modes have been proven useful for the ROP reactions, demonstrating advantages of efficiency, stableness, and mildness. In addition to the DMAP·HX complex,21,22 the DBU· BA (benzoic acid) conjugate catalyst could also catalyze the ROP of L-LA, demonstrating a more controlled manner on polymerization.23 The combination of N-methyl-TBD (MTBD) with trifluoro-methanesulfonic acid (TFA) was proved to be efficient for ROP in synthesis of block copolymer poly(trimethylene carbonate)-b-poly(L-lactide) (PTMC-b-PLA).24 Some of these organocatalysts demonstrated bifunctional ability, which could activate the monomer and initiator at the same time.25,26
Commercially, the copolymer PCLA was prepared by the organometallic promoters at temperatures ranged from 100 to 180 °C. Bulk copolymerization could be carried out with high conversion and without disposing of tons of toxic solvent and trouble of separation. For example, using Sn(Oct)2 catalyst, Kim et al. synthesized block PCLA copolymers using the hydroxyl-terminated PCL macroinitiator, which was previously synthesized at 140 °C for 24 h, followed by addition of L-LA at 110 °C for 48 h.27
Although some organocatalysts have showed excellent catalytic activity towards certain kinds of cyclic esters, the relative reactivity of different monomers, e.g. L-LA and ε-CL, varies greatly on the same catalyst. For the ROP using (thio)urea systems catalyst, thiourea catalysts were more active than their urea analogues in the bulk ROP of L-LA conducted at 100 °C, which was contrary to the case of ROP of ε-CL with better performance by urea catalyst.28 Using TBD catalyst, the polymerization of ε-CL in benzene-d6 was less active than that of L-LA in CH2Cl2, the reaction time being 5 h vs. 15 s.19 MTBD and 1, 8-diazabicyclo[5.4.0]-undec-7-ene (DBU) could catalyze the ROP of L-LA within 1 h, but 120 h was required to obtain PCL with Mn of 8000 g mol−1. DMAP·HCl showed excellent catalytic activity towards L-LA, achieving conversion of 95% within 1.5 h at 140 °C, but failed to activate ε-CL or VL at 90 °C.21
The attempt to copolymerize L-LA and ε-CL simultaneously using TBD catalyst achieved limited success, only PLA homopolymer was observed.19 Alternatively, by the sequential reaction of ε-CL by TBD in toluene for 7 h, and DL-Lactide in CH2Cl2 solution for 30 s, the block copolymer PCLA was obtained. Applying a similar methodology, block PCLA copolymers were prepared by sequential reaction by DBU/phenol dual catalytic system in 6 days.29 Based on the H-bonding activated Brønsted acid, Guo and coworkers30 reported the block copolymerization of ε-CL and L-LA in CH2Cl2 by the combination of thiophosphoric triamide and methanesulfonic acid. The two monomers were sequentially added and stirred for 45 h and 20 h respectively to obtain the block PCLA with 95% conversion. The pentablock terpolymers were synthesized by one-pot sequential polymerization of ethylene oxide (EO), ε-CL, and L-LA by mild phosphazene base (t-BuP2).31
The bulk ROP of L-LA is still identified as a challenge for organocatalysts,32 not to mention the copolymerization with other monomers. Due to the selectivity of organocatalysts towards ROP of L-LA and ε-CL, it is difficult to find a compatible organocatalyst to obtain PCLA copolymer in bulk condition. Apart from the selectivity, the elevated reaction temperature can also be problematic. At least 100 °C is required for L-LA to reach its melting point. However, the organocatalysts might decompose,33 and some binary organocatalysts could be deactivated due to the weakened H-bonding at high temperature.
To date, only few organocatalysts have been reported in the copolymerization in bulk manner. Benzoic acid accompanied with the alcohol initiator was proved to be active for copolymerization of ε-CL and L-LA mixture to obtain the PCLA at 155 °C.34 More than 24 h was required to reach reasonable conversion, apart from which, another equivalent catalyst was added along with the other monomer to obtain block copolymers. The dibenzoylmethane (DBM) was studied to catalyze the PCLA at 155 °C, Mn of which ranged from 1000 to 4100 g mol−1, while long reaction time over 48 h was also inevitable.35
The air/moisture-tolerant tetrabutylammonium phthalimide-N-oxyl (TBAPINO), synthesized with cheap raw materials, have found applications in cyanosilylation of carbonyl compounds, cyclotrimerization of aryl and alkyl isocyanates, etc.36–38 Our previous work has proved its excellent catalytic efficiency towards ROP of L-LA both in bulk and solution polymerization.39 Therefore, in present work, we expanded the application of TBAPINO and further described the catalytic activity towards ROP of ε-CL (Fig. 1) and L-LA to obtain the optimal condition. Furthermore, their copolymerization in bulk condition was studied in terms of feed ratios and addition sequences, reaction times and the structure of product was characterized by nuclear magnetic resonance (NMR), size exclusion chromatography (SEC) and differential scanning calorimetry (DSC) analysis.
 | ||
| Fig. 1 ROP of ε-CL catalyzed by TBAPINO. | ||
Experimental
Materials and methods
ε-Caprolactone (ε-CL, 99%) was dried over CaH2 for 48 h prior to distillation under reduced pressure. L-lactide (L-LA, 98%) was dissolved in ethyl acetate, recrystallized after reflux for 4 h, and dried under vacuum for 24 h. Toluene (99.5%, Sinopharm Chemical Reagent Co.) using benzophenone as indicator and nitrogen as protective gas, was purified by refluxing until benzophenone turned dark purple. Sodium tert-butoxide (tBuONa, 99%), benzoic acid (BA, 99.5%), absolute ethyl alcohol (EtOH, AR), phthalimide (98%), tetrabutylammonium hydroxide (25 wt% in water) were used as received. Compounds were stored in a glove box (O2 ≤1 ppm, H2O ≤0.1 ppm). All reagents and chemicals, except for those specified, were purchased from Shanghai Titan Scientific Co.1H NMR spectra was recorded on a AVANCE 500 spectrometer at 25 °C in CDCl3 to determine the monomer conversion, using tetramethylsilane (TMS) as an internal standard. 13C NMR spectra was recorded on an ASCEND 600 spectrometer with 1024 scans in CDCl3 to investigate the copolymer composition, using tetramethylsilane (TMS) as an internal standard. The molecular weights (Mn) of products were measured using a size exclusion chromatography (SEC) system (Waters-1515 gel permeation chromatograph). The SEC system was calibrated by a series of polystyrene standards with polydispersities of 1.02, and tetrahydrofuran (THF) was used as the mobile phase at an elution rate of 1 mL min−1 Differential scanning calorimetry (DSC) measurements were carried out with a TA Q200 DSC apparatus under nitrogen flow. Thermal procedure of polymers is showed as follow. The PCL/PLA/PCLA samples were melted at 100/200/200 °C for 3 min to eliminate the thermal history. Then they were cooled to −80/0/−80 °C at the cooling rate of −10 °C min−1, after which the samples were reheated to 100/200/200 °C at the heating rate of 10 °C min−1, respectively.
Synthesis of TBAPINO
6.8 mmol phthalic diamide and equivalent tetrabutylammonium hydroxide were added to 25 mL flask, and the mixture was stirred for 5 min at room temperature. Then 5 mL deionized water was added to the mixture. The reaction was conducted at 100 °C for 4 h under reflux condition. After the mixture was cooled to room temperature, the solvent was evaporated. The residue was cooled under 0–4 °C for 1 h to afford a white crystal and then dried at vacuum at 60 °C for 12 h.Homopolymerization
Results and discussion
Solution polymerization of ε-caprolactone
First, the ROP of ε-CL were presented in solution in terms of the feed ratio of catalyst to initiator. In Table 1, when the [ε-CL]:[TBAPINO] = 200:2, the conversion was 78% in 5 h, while the similar conversion could be obtained in 4 h at 200:5. And 88.5% conversion was achieved within 2 h when the [ε-CL]:[TBAPINO] ratio increased to 200:10. With the increasing conversion, the molecular weight of PCL changed slightly, but polydispersity increased from 1.49 to 2.78, which was likely due to the intensive transesterification reaction.
| Entry | ε-CL/TBAPINO/tBuONa | t (h) | Conv.b (%) | Mnc (g mol−1) | Đc |
|---|---|---|---|---|---|
| a Polymerization condition: toluene = 5 mL, ε-CL: 300 mg (2.6 mmol) and desired amount of TBAPINO and tBuONa, pressure: 0.1 MPa, 25 °C, inert atmosphere.b Measured by 1H NMR.c Measured by SEC. | |||||
| 1 | 200:2:4 |
5.0 | 78.0 | 26770 |
1.49 |
| 2 | 200:5:4 |
4.0 | 80.4 | 25640 |
1.73 |
| 3 | 200:10:4 |
2.0 | 88.5 | 26030 |
2.78 |
The effect of monomer concentration on polymerization (entry 1–4, Table 2) was studied by varying the volume of toluene. The monomer concentration varied from 0.5 to 0.87 mol L−1 led to an elevated conversion and Mn and broader polydispersity. The highest conversion and Mn were 98.0% and 37490 g mol−1 at 0.87 mol L−1. When the monomer concentration was over 0.87 mol L−1, both the conversion and Mn dropped with the increasing concentration, which might be owing to faster reaction rate of ROP causing partial blockage of catalyst active center and limited contact of ε-CL monomer with catalyst. On the other hand, the increased viscosity of generated PCL during the polymerization process resulted in slower movement of ε-CL monomer and eventually led to the decreased conversion.
| Entry | ε-CL (mol L−1) | t (h) | Conv.b (%) | Mnc (g mol−1) | Đc |
|---|---|---|---|---|---|
| a Polymerization condition: toluene = 2/3/4/5 mL, [ε-CL]:[TBAPINO]:[tBuONa] = 200:2:4, ε-CL: 300 mg (2.6 mmol), pressure: 0.1 MPa, 25 °C, inert atmosphere.b Measured by 1H NMR.c Measured by SEC. |
|||||
| 1 | 0.52 | 2.0 | 78.0 | 26770 |
1.49 |
| 2 | 0.65 | 2.0 | 85.3 | 35250 |
2.55 |
| 3 | 0.87 | 2.0 | 96.9 | 37490 |
2.75 |
| 4 | 1.30 | 2.0 | 87.7 | 36900 |
2.78 |
| 5 | 0.52 | 1.0 | 84.9 | 29110 |
1.57 |
| 6 | 0.52 | 2.0 | 86.8 | 30230 |
1.70 |
| 7 | 0.52 | 3.0 | 92.1 | 30930 |
1.71 |
| 8 | 0.52 | 4.0 | 86.5 | 28540 |
1.67 |
| 9 | 0.52 | 5.0 | 78.0 | 26770 |
1.49 |
In Table 2, the conversion and Mn (entry 5–9) both increased with prolonged reaction time. The highest conversion and Mn of 92.1% and 30930 g mol−1 were obtained when the reaction time reached 3 h, and then decreased dramatically with longer reaction time. The polydispersity remained stable with prolonged time. The correlation between Mn and conversion is plotted in Fig. 2, the Mn increased with conversion with linearly dependence coefficient of R2 ≥0.93, suggesting the living polymerization character of ε-CL solution polymerization over TBAPINO.
 | ||
| Fig. 2 Relationship curve of monomer conversion with molecular weight and Mw/Mn of ε-CL polymerization. | ||
In Table 3, the catalyst showed good activity in a wide temperature range, proved by conversion over 85% from −15 °C to 60 °C and Mn of PCL reaching 10430 g mol−1 at −15 °C and 15170 g mol−1 at 60 °C within 1 h. Higher Mn over 20000 g mol−1 were obtained from 0 °C to 40 °C. When temperature was below 25 °C, the conversion and Mn increased with higher temperature, and Mn reached the highest 33920 g mol−1 at 25 °C, but then, Mn decreased. Higher temperature led to faster reaction rate and consumption of monomer, but when polymerization further proceeded, more intensive transesterification reaction caused broader polydispersity.
| Entry | T (°C) | Conv.b (%) | Mnc (g mol−1) | Đc |
|---|---|---|---|---|
| a Polymerization condition: solvent: toluene = 3 mL; [ε-CL]:[TBAPINO]:[tBuONa] = 200:2:4; ε-CL: 300 mg (2.6 mmol), t = 1.0 h, pressure: 0.1 MPa, inert atmosphere.b Measured by 1H NMR.c Measured by SEC. |
||||
| 1 | −15 | 84.8 | 10430 |
1.51 |
| 2 | 0 | 86.5 | 22160 |
1.50 |
| 3 | 5 | 87.0 | 26770 |
1.53 |
| 4 | 10 | 87.7 | 27340 |
1.49 |
| 5 | 25 | 88.1 | 33920 |
1.57 |
| 6 | 40 | 94.4 | 24260 |
3.05 |
| 7 | 60 | 84.9 | 15170 |
3.36 |
Fig. 3 shows the monomodal SEC curve of PCL (entry 5, Table 2). The asymmetrical profile with a slight tailing indicated side reactions occurred during the polymerization procedure, resulting in a broader molecular weight distribution. In Fig. 4, a typical melting and crystallization point of PCL sample are at 56.4 and 31.2 °C, respectively.40,41
 | ||
| Fig. 3 SEC curve of PCL (entry 5, Table 2). | ||
 | ||
| Fig. 4 DSC curve of PCL (entry 5, Table 2). | ||
Bulk polymerization of ε-caprolactone
At the same time, the ROP of ε-CL was conducted in bulk condition. The onset decomposition temperature of TBAPINO was nearly 180 °C (ESI, Fig. S1†), suggesting its stableness at the reaction temperature from 100 to 150 °C. In Table 4, 56.1% conversion of ε-CL was observed in 1 h at 120 °C (entry 1), while for the case of DMAP and ionic acid catalyst system, 15 h was required to reach 10% conversion.13| Entry | T (°C) | t (h) | ε-CL/TBAPINO/tBuONa | Conv.b (%) |
|---|---|---|---|---|
| a Polymerization condition: solvent free, ε-CL: 300 mg (2.6 mmol), pressure: 0.1 MPa, inert atmosphere.b Measured by 1H NMR. | ||||
| 1 | 120 | 1.0 | 200/2/1 | 56.1 |
| 2 | 120 | 2.0 | 200/5/1 | 86.8 |
| 3 | 120 | 4.0 | 200/10/1 | 87.1 |
| 4 | 135 | 1.0 | 200/5/1 | 68.9 |
| 5 | 135 | 2.0 | 200/10/1 | 86.5 |
| 6 | 135 | 4.0 | 200/2/1 | 68.4 |
| 7 | 150 | 1.0 | 200/10/1 | 85.7 |
| 8 | 150 | 2.0 | 200/2/1 | 46.4 |
| 9 | 150 | 4.0 | 200/5/1 | 57.7 |
| 10 | 120 | 2.0 | 200/10/1 | 88.3 |
| 11 | 105 | 4.0 | 200/10/1 | 85.4 |
| 12 | 120 | 2.0 | 200/10/4 | 72.3 |
| 13 | 120 | 2.0 | 200/10/2 | 83.5 |
| 14 | 120 | 2.0 | 200/10/0.4 | 89.5 |
According to Table 4, the catalyst loading was the most important to obtain high conversion in the bulk ROP of ε-CL, followed by reaction time and temperature. Within the range of 120 to 150 °C, a decreasing conversion was observed. Another controlled experiment at 105 °C (entry 11, Table 4) suggested the conversion with a tendency of first increasing and then decreasing. And 2 h was selected as the optimal reaction time.
To further investigate the influence of initiator concentration, the initiator feeds of [ε-CL]:[tBuONa] were changed from 50 to 500 (entry 10, 12–14, Table 4). The monomer conversion was as high as 89.5%, then slowed down. The lower initiator concentration meant less activated polymer chains, and therefore more monomers were gathered on each chain. No obvious change in Đ was found with various initiator concentration. The synthesized PCL was confirmed by 1H NMR (Fig. 5) and IR spectrum (ESI Fig. S2†), respectively. The integral value ratio of representative methylene protons (a, b, c and d, Fig. 5) of PCL was approximately the theory value of 1:2:1:1.41 In Fig. S2,† the absorption peak at 1727 cm−1 attributed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching, and the 1260–1140 cm−1 for the C–O stretching and the 1421–1367 cm−1 and 2948–2864 cm−1 for the C–H bending and stretching respectively.42
O stretching, and the 1260–1140 cm−1 for the C–O stretching and the 1421–1367 cm−1 and 2948–2864 cm−1 for the C–H bending and stretching respectively.42
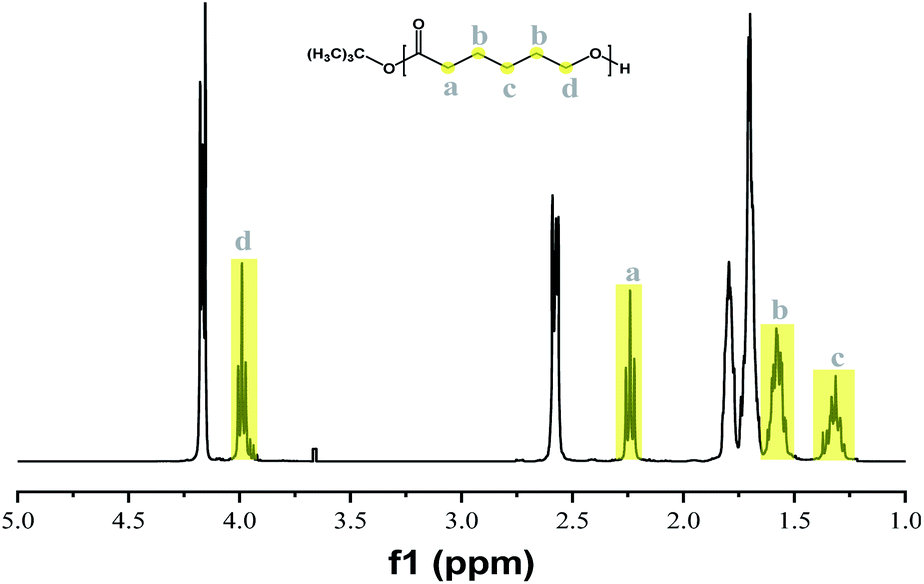 | ||
| Fig. 5 1H NMR spectrum of PCL (entry 10, Table 4). | ||
Bulk polymerization of L-lactide
In our previous work, the catalyst showed strong air/moisture tolerance and thermal stableness. The polymerization of L-LA could be carried out in 120 °C with high conversion in inert atmosphere or even in open air.In Table 5, the influence of reaction time and temperature on bulk polymerization at inert atmosphere was investigated. At 120 °C, the conversion reached 85.7% in 0.5 h and the Mn was 2630 g mol−1, then increased to 3220 g mol−1 with the reaction time prolonged to 2 h. When reaction was conducted for 4 h, the conversion reached 97.2%, but the Mn decreased to 1880 g mol−1 suggested the occurrence of transesterification.
| Entry | T (°C) | t (h) | Conv.b (%) | Mnc (g mol−1) | Đc |
|---|---|---|---|---|---|
| a Polymerization condition: solvent free, [L-LA]:[TBAPINO]:[tBuONa] = 200:10:1, pressure: 0.1 MPa, inert atmosphere.b Measured by 1H NMR.c Measured by SEC. |
|||||
| 1 | 120 | 0.5 | 85.7 | 2630 | 1.27 |
| 2 | 120 | 1.0 | 95.5 | 2930 | 1.23 |
| 3 | 120 | 2.0 | 95.7 | 3220 | 1.19 |
| 4 | 120 | 4.0 | 97.2 | 1880 | 1.28 |
| 5 | 95 | 4.0 | 81.8 | 2330 | 1.20 |
| 6 | 105 | 4.0 | 89.1 | 2350 | 1.32 |
| 7 | 135 | 4.0 | 90.0 | 1570 | 1.68 |
| 8 | 150 | 4.0 | 88.0 | 1430 | 1.72 |
When 4 h for reaction was fixed, the conversion of the monomer increased from 81.8% at 95 °C to 97.2% at 120 °C (entry 5–8, Table 5), and a slight drop of conversion to 88% was observed at 150 °C. With the increased temperature, the Mn of the polymers decreased from 2350 to 1570 g mol−1 (entry 6&7, Table 5), and the polydispersity increased to 1.72. At the higher temperature, the chain initiation and extension were accelerated, shortage of monomer for chain extension led to the slowed down chain growth. Due to the influence of severe transesterification reaction, the reaction suffered from reduced conversion and Mn and broader polydispersity.
The living polymerization experiments were also conducted (Table 6). The concentration of active groups was of vital importance to the living polymerization reaction, and therefore 0.5 h was selected as the interval of monomer feed. Utilizing TBAPINO, the Mn increased from 2310 to 5420 g mol−1 after the addition of L-LA for three times, confirming efficient re-initiation from the PLA chain. Then the Mn decreased to 2330 and 1980 g mol−1 after the following additions. This might be due to the degradation and transesterification during long reaction. It is worth noting that the polydispersity generally decreased upon chain extension with L-LA showing that the ROP was well controlled. At the same time, the living polymerization performed according to the same mechanism suggested the active chain end group could activate monomers when the chain continued to grow, leading to the potential of experiments conducted by sequential polymerization with addition various monomers using a single organocatalyst. And therefore, we explored the copolymerization of L-LA and ε-CL by TBAPINO in bulk condition.
Copolymerization of ε-caprolactone and L-lactide
Based on the above homopolymerization work, the optimized condition of both L-LA and ε-CL was 120 °C. Therefore, the similar reaction conditions were applied in the copolymerization of ε-CL and L-LA.As shown in Table 7, the living character of ROP over TBAPINO was further supported by a second-feed experiment in terms of the feed ratios and reaction times. The ε-CL was first activated and the polymerization was then restarted by subsequent addition of L-LA. The individual reaction of each monomer was carried out for 1 or 2 h, respectively. As the feed ratios of ε-CL to L-LA increased from 1:3 to 3:1, the conversion of ε-CL in 1 h was decreased from 88.1% to 75.6%, while that of L-LA increased from 39.4% to 48.7%. Reasonable conversion and dispersity could be obtained with the equivalent ratio of ε-CL and L-LA. When the individual reaction time was prolonged to 2 h, the conversions of ε-CL and L-LA were greatly improved to 96.5% and 75.2% respectively, and the Mn of the copolymer reached 22100 g mol−1 (entry 4, Table 7). Either increasing the initiator concentration or lower catalyst loading (entry 5&6, Table 7) was not beneficial to copolymerization, leading to reduced Mn and conversion. It is worth noting that the addition sequence of monomers also had influence on the copolymerization (entry 7, Table 7). The reversed addition led to the ε-CL conversion leaped from 96.5% (entry 4, Table 7) to 32.5%, while the L-LA conversion changed slightly. For the sequential polymerization of monolactones, e.g. ε-CL, with L-LA, the latter was usually added as the second monomer.20 Despite faster polymerization rate of ε-CL than that of L-LA, L-LA was usually incorporated first.19,28,35,43 Furthermore, we also synthesized the PCLA by the reaction of equivalent molar feed of ε-CL and L-LA at 120 °C for 2 min. The 1H NMR spectrum in ESI Fig. S3† indicated that both monomers were inserted into the polymer chains and the CL:LA ratio of product was around 1:4 according to the integral value, which was consistent with the preferential insertion of L-LA in the polymer chains.34,43 The strong evidence showed the comparable selectivity towards the two monomers over TBAPINO at high temperature. The copolymerization of L-LA with ε-CL using TBD as the catalyst only resulted in homopolymerization of L-LA and transesterification reaction on PLA. It might be due to the lower reactivity of ε-CL and cannot be initiated by the end group of PLA chain. For the case of PLA-b-PCL-b-PLA triblock copolymer synthesized by sequential addition of ε-CL and L-LA with benzoic acid as catalyst, 75% conversion of ε-CL and Mn of 7730 g mol−1 were achieved after 26 h, while the reverse order resulted in only 27% conversion.41
| Entry | CL/LA | tCL (h) | tLA (h) | Conv. of CLb (%) | Conv. of LAb (%) | Mnc (g mol−1) | Đc |
|---|---|---|---|---|---|---|---|
| a Polymerization condition: solvent free, [Monomer]:[TBAPINO]:[tBuONa] = 200:10:1, 120 °C, pressure: 0.1 MPa, inert atmosphere.b Measured by 1H NMR.c Measured by SEC.d [Monomer]:[TBAPINO]:[tBuONa] = 200:10:4.e [Monomer]:[TBAPINO]:[tBuONa] = 200:2:1.f L-LA was first added. |
|||||||
| 1 | 1:3 |
1 | 1 | 88.1 | 39.4 | 12340 |
1.52 |
| 2 | 1:1 |
1 | 1 | 85.6 | 45.4 | 10500 |
1.26 |
| 3 | 3:1 |
1 | 1 | 75.6 | 48.7 | 10930 |
1.46 |
| 4 | 1:1 |
2 | 2 | 96.5 | 75.2 | 22100 |
1.85 |
| 5d | 1:1 |
2 | 2 | 92.1 | 71.5 | 17330 |
2.21 |
| 6e | 1:1 |
1 | 1 | 71.2 | 41.3 | 8700 | 1.27 |
| 7f | 1:1 |
2 | 2 | 32.5 | 74.4 | 10500 |
1.87 |
The synthesized copolymers were characterized by 1H NMR, 13C NMR, SEC and DSC analysis. In the 1H NMR spectrum (Fig. 6), the formation of block PCLA was confirmed by representative protons of both blocks, 1.58 and 5.17 ppm for the methyl and methine protons of PLA, and 1.30, 1.66, 2.24 and 3.97 ppm for the methylene protons of PCL, which was supported by the hydroxyl-methine PLA end groups at 4.32 ppm.41 Furthermore, analysis by 13C NMR spectroscopy analysis (Fig. 7) also proved the microstructure of the copolymer.34,44 Representative peaks at 173.5 ppm attributed to the CL–CL–CL triads, and those at 169.6 ppm for the LL units sandwiched by LL and CL. Peaks between 169.2 and 169.6 ppm belonged to the LL–LL–LL and CL–LL–LL triads, and the broad ones from 168.8 to 169.2 ppm were the LL–LL–LL in epimerized sequences. The transesterification reactions might be rather negligible due to the absence of various heterotraids.34,45
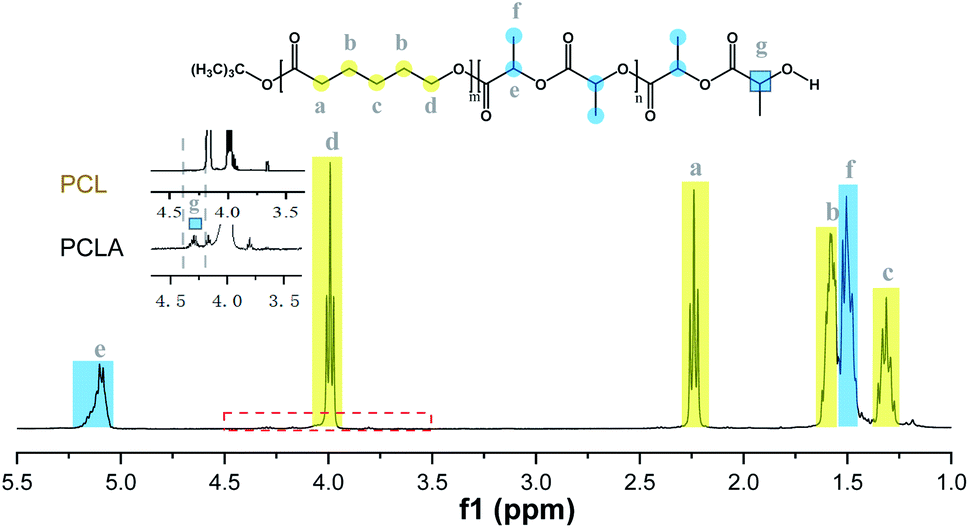 | ||
| Fig. 6 1H NMR spectrum of PCLA (entry 4, Table 7). | ||
 | ||
| Fig. 7 13C NMR spectrum of PCLA (entry 4, Table 7); LL and CL refer to the lactidyl and caproyl units respectively. | ||
In Fig. 8, a monomodal SEC curve of PCLA suggested the synthesis of block PCLA. The slight tailing of SEC curve indicated the occurrence of transesterification reaction. As shown in Fig. 9, a typical glass transition temperature (Tg) of PLA and a melting peak of PCL are at 39.9 °C and 56.4 °C, respectively. The PCLA copolymer showed a combined characteristic of the two homopolymers.
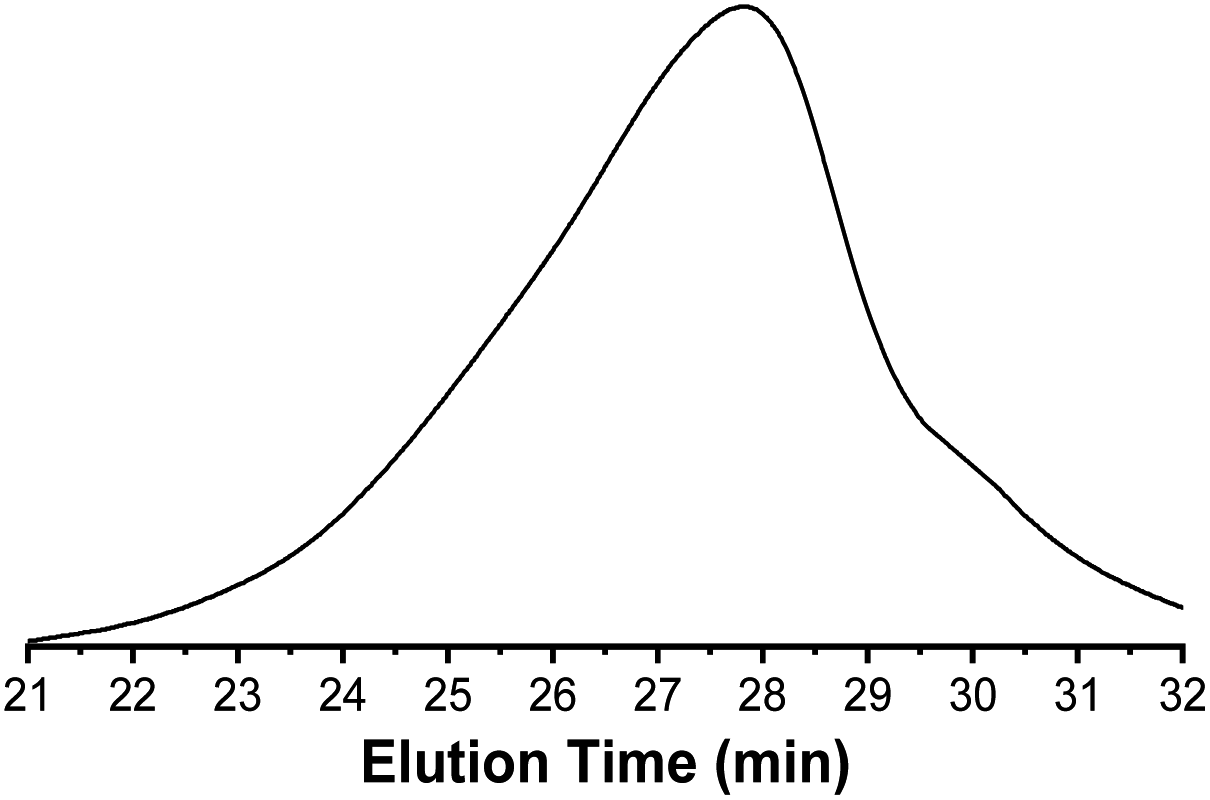 | ||
| Fig. 8 SEC curve of PCLA (entry 4, Table 7). | ||

Conclusion
In summary, this thermally stable catalyst (TBAPINO) could activate the metal-free and bulk ROP of ε-CL and L-LA, and substantial conversion of both monomers was observed within 4 h at 120 °C. The solution ROP of ε-CL could be conducted in a wide temperature range from −15 to 60 °C with conversion over 80%.The catalyst demonstrated stableness at high temperature and similar selectivity towards ε-CL and L-LA in bulk condition, and PCLA copolymer of 22100 g mol−1 was obtained with sequential addition of ε-CL and L-LA. The living character was further proved.
Author contributions
Conceived and designed experiments: R. Cheng, B. Liu. Performed experiments: Z. Feng, L. Wu, H. Dong. Analyzed data: Z. Feng, R. Cheng, B. Liu. Contributed materials/analysis tools: R. Cheng, B. Liu. Wrote the paper: all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Pujiang Talent Projects (16PJD016).References
- M. Qiao, D. Chen, T. Hao, X. Zhao, H. Hu and X. Ma, Int. J. Pharm., 2007, 345, 116–124 CrossRef CAS PubMed.
- M. Qiao, D. Chen, X. Ma and Y. Liu, Int. J. Pharm., 2005, 294, 103–112 CrossRef CAS PubMed.
- H. Chen, F. Jia, C. Zhu, J. Xu, X. Hua, Z. Xi, L. Shen, S. Zhao and L. Cen, Chem. Eng. J., 2018, 339, 346–358 CrossRef CAS.
- H. K. Kim, W. S. Shim, S. E. Kim, K. H. Lee, E. Kang, J. H. Kim, K. Kim, I. C. Kwon and D. S. Lee, Tissue Eng., Part A, 2009, 15, 923–933 CrossRef CAS PubMed.
- M. J. Sandker, A. Petit, E. M. Redout, M. Siebelt, B. Müller, P. Bruin, R. Meyboom, T. Vermonden, W. E. Hennink and H. Weinans, Biomaterials, 2013, 34, 8002–8011 CrossRef CAS PubMed.
- M. Tosin, F. Degli-Innocenti and C. Bastioli, J. Environ. Polym. Degrad., 1998, 6, 79–90 CrossRef CAS.
- D. Pappalardo, T. Mathisen and A. Finne-Wistrand, Biomacromolecules, 2019, 20, 1465–1477 CrossRef CAS PubMed.
- A. Petit, M. Sandker, B. Müller, R. Meyboom, P. Van Midwoud, P. Bruin, E. M. Redout, M. Versluijs-Helder, C. H. A. Van Der Lest, S. J. Buwalda, L. G. J. De Leede, T. Vermonden, R. J. Kok, H. Weinans and W. E. Hennink, Biomaterials, 2014, 35, 7919–7928 CrossRef CAS PubMed.
- X. Leng, Y. Ren, Z. Wei, Y. Bian and Y. Li, Macromol. Chem. Phys., 2017, 218, 1700178 CrossRef.
- C. Jérôme and P. Lecomte, Adv. Drug Delivery Rev., 2008, 60, 1056–1076 CrossRef PubMed.
- M. D. Deokar, S. B. Idage, B. B. Idage and S. Sivaram, J. Appl. Polym. Sci., 2016, 133, 43267 CrossRef.
- E. Stirling, Y. Champouret and M. Visseaux, Polym. Chem., 2018, 9, 2517–2531 RSC.
- H. Du, X. Pang, H. Yu, X. Zhuang, X. Chen, D. Cui, X. Wang and X. Jing, Macromolecules, 2007, 40, 1904–1913 CrossRef CAS.
- Y. A. Piskun, I. V. Vasilenko, K. V. Zaitsev, Y. F. Oprunenko and S. V. Kostjuk, Macromol. Chem. Phys., 2017, 218, 1600580 CrossRef.
- D. M. Gonzalez, N. B. Cruz, L. A. Hernandez, J. Oyarce, R. Benavente and C. Manzur, J. Polym. Sci., 2020, 58, 557–567 CrossRef CAS.
- B. Gao, R. Duan, X. Pang, X. Li, Z. Qu, H. Shao, X. Wang and X. Chen, Dalton Trans., 2013, 42, 16334–16342 RSC.
- N. E. Kamber, W. Jeong, R. M. Waymouth, R. C. Pratt, B. G. Lohmeijer and J. L. Hedrick, Chem. Rev., 2007, 107, 5813–5840 CrossRef CAS PubMed.
- F. Nederberg, E. F. Connor, M. Möller, T. Glauser and J. L. Hedrick, Angew. Chem., Int. Ed., 2001, 40, 2712–2715 CrossRef CAS PubMed.
- B. G. G. Lohmeijer, R. C. Pratt, F. Leibfarth, J. W. Logan, D. A. Long, A. P. Dove, F. Nederberg, J. Choi, C. Wade, R. M. Waymouth and J. L. Hedrick, Macromolecules, 2006, 39, 8574–8583 CrossRef CAS.
- S. Hu, J. Zhao, G. Zhang and H. Schlaad, Prog. Polym. Sci., 2017, 74, 34–77 CrossRef CAS.
- H. Wang, Z. Yao, Z. Li, Y. Zhu, C. Zhang, Z. Luo, T. Guo, Y. Gao, L. Zhang and K. Guo, Eur. Polym. J., 2020, 127, 109570 CrossRef CAS.
- J. Kadota, D. Pavlovic, H. Hirano, A. Okada, Y. Agari, B. Bibal, A. Deffieux and F. Peruch, RSC Adv., 2014, 4, 14725–14732 RSC.
- D. J. Coady, K. Fukushima, H. W. Horn, J. E. Rice and J. L. Hedrick, Chem. Commun., 2011, 47, 3105–3107 RSC.
- X. Wang, S. Cui, Z. Li, S. Kan, Q. Zhang, C. Zhao, H. Wu, J. Liu, W. Wu and K. Guo, Polym. Chem., 2014, 5, 6051–6059 RSC.
- M. K. Kiesewetter, E. J. Shin, J. L. Hedrick and R. M. Waymouth, Macromolecules, 2010, 43, 2093–2107 CrossRef CAS.
- A. P. Dove, ACS Macro Lett., 2012, 1, 1409–1412 CrossRef CAS.
- J. K. Kim, D. J. Park, M. S. Lee and K. J. Ihn, Polymer, 2001, 42, 7429–7441 CrossRef CAS.
- J. U. Pothupitiya, N. U. Dharmaratne, T. M. M. Jouaneh, K. V. Fastnacht, D. N. Coderre and M. K. Kiesewetter, Macromolecules, 2017, 50, 8948–8954 CrossRef CAS.
- C. Thomas, F. Peruch and B. Bibal, RSC Adv., 2012, 2, 12851–12856 RSC.
- X. Li, Q. Zhang, Z. Li, S. Xu, C. Zhao, C. Chen, X. Zhi, H. Wang, N. Zhu and K. Guo, Polym. Chem., 2016, 7, 1368–1374 RSC.
- J. Zhao, D. Pahovnik, Y. Gnanou and N. Hadjichristidis, Polym. Chem., 2014, 5, 3750–3753 RSC.
- L. Mezzasalma, A. P. Dove and O. Coulembier, Eur. Polym. J., 2017, 95, 628–634 CrossRef CAS.
- D. N. Coderre, K. V. Fastnacht, T. J. Wright, N. U. Dharmaratne and M. K. Kiesewetter, Macromolecules, 2018, 51, 10121–10126 CrossRef CAS.
- L. Mezzasalma, S. Harrisson, S. Saba, P. Loyer, O. Coulembier and D. Taton, Biomacromolecules, 2019, 20, 1965–1974 CrossRef CAS PubMed.
- L. Mezzasalma, J. De Winter, D. Taton and O. Coulembier, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 475–479 CrossRef CAS.
- M. G. Dekamin, S. Sagheb-Asl and M. Reza Naimi-Jamal, Tetrahedron Lett., 2009, 50, 4063–4066 CrossRef CAS.
- M. G. Dekamin, M. Ghanbari, M. R. Moghbeli, M. Barikani and S. Javanshir, Polym.-Plast. Technol. Eng., 2013, 52, 1127–1132 CrossRef CAS.
- M. G. Dekamin, M. Alikhani and S. Javanshir, Green Chem. Lett. Rev., 2016, 9, 96–105 CrossRef CAS.
- H. Li, J. Wang, L. Wu, W. Liu, R. Cheng and B. Liu, Acta Polym. Sin., 2019, 50, 1290–1297 CAS.
- K. Van de Velde and P. Kiekens, Polym. Test., 2002, 21, 433–442 CrossRef CAS.
- L. Mezzasalma, J. De Winter, D. Taton and O. Coulembier, Green Chem., 2018, 20, 5385–5396 RSC.
- N. Bölgen, Y. Z. Menceloğlu, K. Acatay, İ. Vargel and E. Pişkin, J. Biomater. Sci. Polym. Ed., 2005, 16, 1537–1555 CrossRef PubMed.
- M. Baśko and P. Kubisa, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 7071–7081 CrossRef.
- J. Kasperczyk and M. Bero, Makromol. Chem., 1991, 192, 1777–1787 CrossRef CAS.
- J. Kasperczyk and M. Bero, Makromol. Chem., 1993, 194, 913–925 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra02417e |
| This journal is © The Royal Society of Chemistry 2021 |