
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis, structures, and catalytic efficiency in ring opening polymerization of rac-lactide with tridentate vs. bidentate cobalt(II), zinc(II), and cadmium(II) complexes containing N-substituted N,N-bis((3,5-dimethyl-1H-pyrazol-1-yl)methyl)amine ligands†
Solhye Choe a,
Hyosun Lee*a and
Saira Nayab*b
a,
Hyosun Lee*a and
Saira Nayab*b
aDepartment of Chemistry and Green-Nano Materials Research Center, Kyungpook National University, 80 Daehakro, Bukgu, Daegu, 41566, Republic of Korea. E-mail: hyosunlee@knu.ac.kr; Fax: +82-53-950-6330; Tel: +82-53-950-5337
bDepartment of Chemistry, Shaheed Benazir Bhutto University, Sheringal Dir (U), Khyber Pakhtunkhwa, Islamic Republic of Pakistan. E-mail: drnayab@sbbu.edu.pk; sairanayab07@yahoo.com
First published on 25th May 2021
Abstract
A series of Co(II), Zn(II), and Cd(II) complexes supported by 1-(3,5-dimethyl-1H-pyrazol-1-yl)-N-((3,5-dimethyl-1H-pyrazol-1-yl)methyl)-N-(furan-2-ylmethyl)methanamine (LA) and N,N-bis((3,5-dimethyl-1H-pyrazol-1-yl)methyl)-4-isopropylaniline (LB) were synthesized. The direct chelation of CoCl2·6H2O, ZnCl2, and CdBr2·4H2O by the ligands produced [LnMX2] (Ln = LA or LB; M = Zn or Co, with X = Cl; M = Cd, with X = Br) complexes in high yields. Structural studies revealed that [LBCoCl2] and [LBZnCl2] adopted distorted tetrahedral geometries, as LB coordinated the metal centers in a bidentate fashion, while LA coordinated the metal centers in a tridentate fashion through the nitrogen atoms of the pyrazole and amine moieties, so that [LACoCl2] and [LAZnCl2] exhibited trigonal bipyramidal geometries and [LACdBr2] a square pyramidal geometry. [LBCdBr2] has two Cd-containing structures per unit cell, whereby one Cd center adopted a distorted tetrahedral geometry and the other exhibited square bipyramidal geometry. The in situ-generated alkyl derivatives of the synthesized complexes were assessed in the ring-opening polymerization of rac-lactide. Heterotactic polylactides (PLAs) were furnished with all complexes. The [LBZnCl2]/MeLi system produced PLA with a superior heterotactic bias (Pr up to 0.94) at −25 °C. PLAs with wide-ranging polydispersity indices (1.16–2.23) and low molecular weights were produced in all cases, irrespective of the specific M(II) center and ancillary ligand utilized.
1. Introduction
Biodegradable polymers have been developed as excellent substitutes for conventional petroleum-based polymers.1,2 Aliphatic polyesters like polylactide (PLA) are regarded as the most promising candidates to fulfil the described role.3,4 PLA has garnered particular interest owing to its biocompatibility and biodegradability, and the possibility of producing its component monomers starting from renewable resources.5,6 Well-defined PLAs with controlled physical and mechanical properties have been employed in a vast range of applications, including packaging, medical implants, tissue engineering, and pharmacology.7–10 In the recent past, numerous metal complexes, including complexes of Al,11–15 Zn,16,17 Ga,18 In,19 Cu,20,21 Co,22,23 and trivalent rare earths,24 have been utilized as catalysts for the ring-opening polymerization (ROP) of lactide (LA); notably, such catalysts afforded excellent control over reaction initiation and propagation, as well as the stereochemical arrangement of the polymer chains.25The architecture of the ligand coordinating the metal center is one of the key factors determining the catalyst's ability to control the stereochemistry and properties of the resultant polymer,26,27 alongside the identity of the metal center and the temperature at which the polymerization reaction is conducted. The prevalent ligand architectures, including diketiminate,28,29 Schiff bases,30,31 bis(phenolate),32,33 salen,34–36 iminomethylpyridines,37 (benzoimidazolylmethyl)amine,38 and (pyrazolylmethyl)pyridine/amine,39–41 have been widely modified to control their stereo-electronic properties, which in turn affect the activity and stereoselectivity of ROP initiators. Thus, choosing carefully the architecture of the ligand is a N-substituted bis(pyrazolyl)amine have been attracting particular interest as nitrogen donor ligands, because the pyrazole nitrogen is a weaker δ-donor than imine and pyridine nitrogens, and it affords complexes comprising highly electrophilic M(II) centers.42 Additionally, the fact that it is possible to modify the linker units comprising two or three pyrazole moieties43,44 renders various bidentate, tridentate or/and polydentate coordination geometries and nuclearities achievable,45 and it has been shown to allow the overall adjustment of the steric and electrophilic properties of the resultant metal complexes.46,47 For instance, the Yu group reported the synthesis of pyrazole-based Zn(II) complexes that proved to be effective ROP catalysts; in fact, they afforded the polymerization of 100 equivalents of rac-LA within 1–3 min in the presence of [BeOH] at 25 °C; nevertheless, their use resulted in a negligible stereoregularity of the resultant PLA.42 Similarly, Kang studied the ability of in situ-generated (bdmppea)ZnEt2 (bdmppea: N,N-bis[(3,5-dimethyl-1H-pyrazol-1-yl)methyl]-1-phenylethylamine; Et: ethyl) to catalyze the polymerization of rac-LA; the relevant reaction afforded 91% rac-LA conversion within 2 h, and it led to the production of moderately heterotactic PLA.48 In a recently published study conducted by our group, we focused on developing late transition metal complexes based on N,N-bis((1H-pyrazol-1-yl)methyl)amine derivatives with various amine substituents, and we studied their catalytic performances in the ROP of rac-LA.49 The catalytic capabilities of these complexes and the stereoselectivity they afforded were promising, and the relevant reactions yielded PLAs of moderate-to-high molecular weight.50,51 In the present contribution, we intended to study the catalytic properties of N-substituted N,N-bis((3,5-dimethyl-1H-pyrazol-1-yl)methyl)amine-based Zn(II), Co(II), and Cd(II) complexes in the ROP of rac-LA.
2. Experimental
2.1. General considerations
The syntheses of ligands 1-(3,5-dimethyl-1H-pyrazol-1-yl)-N-((3,5-dimethyl-1H-pyrazol-1-yl)methyl)-N-(furan-2-ylmethyl)methanamine (LA), and N,N-bis((3,5-dimethyl-1H-pyrazol-1-yl)methyl)-4-isopropylaniline (LB) and their corresponding M(II) (M = Co, Zn, and Cd) complexes were carried out employing benchtop techniques. The ROP reactions were conducted using a high-vacuum Schlenk line and glove box. The starting materials, including CoCl2·6H2O, ZnCl2, CdBr2·4H2O, furfurylamine, 4-isopropylanilie, magnesium sulfate (MgSO4), and molecular sieves (0.4 nm), were purchased from Aldrich. Anhydrous solvents, such as ethanol (EtOH), n-hexane, and diethyl ether (Et2O), were purchased from Merck and used without further purification. THF was dried over benzophenone ketyl radical whereas CH2Cl2 was dried over CaH2, and these solvents were deoxygenated by distillation under argon before use. 3,5-Dimethyl-1H-pyrazolyl-1-methanol was prepared as reported previously.52 Notably, the ligands employed in the current study, LA53 and LB,54 and complex [LACdBr2]51 were synthesized according to previously reported methods. It is important to note that safety protocol should be followed using cadmium bromide salt (CdBr2·4H2O) for synthesis of Cd-complexes as they are highly toxic and mishandling can cause skin and respiratory irritation.The 1H NMR (500 MHz) and 13C NMR (125 MHz) spectra of LA and LB (Fig. S1 and S2,† respectively) and their Zn(II) and Cd(II) complexes (Fig. S3–S9†) were recorded on a Bruker Avance Digital NMR spectrometer. Chemical shifts are reported in ppm relative to tetramethylsilane (SiMe4) used as internal standard. Coupling constants are reported in Hertz (Hz). Resonance peaks are reported as m = multiplet, br = broad, s = singlet, d = doublet, t = triplet, and q = quartet. The infrared (IR) spectra of ligands and complexes were recorded on a Bruker FT/IR-Alpha (neat) instrument, and data were reported in wavenumbers (cm−1) (Fig. S10–S17†). Elemental analyses of the synthesized complexes were performed on an elemental analyzer (EA 1108; Carlo-Erba, Milan, Italy) (Fig. S18†). The monomer conversion to PLA was determined by integrating monomer versus polymer methine resonances in 1H NMR spectrum of PLAs (Fig. S19–S30†). The microstructure analysis of PLAs was performed by inspecting the methine proton region of homodecoupled 1H NMR spectra of the purified polymers (Fig. S31–S44†).55–58 The number average molecular weight (Mn) and molecular weight (Mw) values for the purified PLA samples were determined using a Waters Alliance e2695 instrument possessing differential refractive index detectors; this instrument was calibrated against a polystyrene standard. The Water Styragel columns; HR3, HR4, and HR5E, were employed, and THF was utilized as the eluting solvent at a flow rate of 1.0 mL min−1 at 35 °C. The polydispersity index (PDI) and Mn values of the polymers were reported with respect to the polystyrene standard (Fig. S45–S58†).
2.2. Synthetic procedures
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) 1549 s; ν(CC) + ν(CN)/pz ring, 1493 s, 1463 m; νbend(C–H sp3) 1426 m; ν(N–C) 1248 s; ν(C–O) 1151 (s); νbend(C–H sp2) 834 w; ν(N–M) 599 w.CH–CHC–), 6.41 (dd, 1H, J = 1.84 Hz, 1.83 Hz, –O–CHCH–CHC–), 6.30 (d, 1H, J = 3.81 Hz, –O–CHCH–CHC–), 6.04 (s, 2H, –N–C(CH3)CH–C(CH3) N–), 5.02 (s, 4H, –N–CH2–N–), 3.72 (s, 2H, –C–CH2–N–), 2.66 (s, 6H, –N–C(CH3)CH–C(CH3)N–), 2.20 (s, 6H, –N–C(CH3)CH–C(CH3)N–). 13C NMR (DMSO-d6, 125 MHz): d 150.61 (s, 1C, –O–CHCH–CHC–), 147.83 (s, 2C, –N–C(CH3)CH–C(CH3)N–), 143.95 (d, 1C, J = 203.4 Hz, –O–CHCH–CHC–), 140.94 (s, 2C, –N–C(CH3)CH–C(CH3)N–), 111.46 (d, 1C, J = 192.6 Hz, –O–CHCH–CHC–), 111.33 (d, 1C, J = 174.4 Hz, –O–CHCH–CHC–), 107.18 (d, 2C, J = 174.4 Hz, –N–C(CH3)CH–C(CH3)N–), 64.06 (t, 2C, J = 301.5 Hz, –N–CH2–N–), 46.70 (t, 1C, J = 278.8 Hz, –C–CH2–N–), 15.08 (q, 2C, J = 381.47 Hz, –N–C(CH3)CH–C(CH3)N–), 12.04 (q, 2C, J = 386.0 Hz, –N–C(CH3)CH–C(CH3)N–). Analysis calculated for C17H23Cl2N5OZn (%): C, 45.63, H, 5.19, N, 15.66. Found: C, 45.63, H, 5.24, N, 15.92. FTIR (solid neat; cm−1): ν(C–H) 2956 w; ν(CC) 1610 s; ν(CC) + ν(CN)/pz ring, 1514 s, 1458 s; νbend(C–H sp3) 1417 m; ν(N–C) 1214 s; ν(C–O) 1120 (s); νbend(C–H sp2) 832 w; ν(N–M) 571 w.C) 1608 s; ν(CC) + ν(CN)/pz ring, 1512 s, 1460 s; νbend(–C–H sp3) 1414 m; ν(N–C) 1210 s; νbend(C–H sp2) 831 w; ν(N–M) 570 w.CH–C(CH3)N–), 5.35 (s, 4H, –N–CH2–N–), 2.95–2.90 (m, 1H, J = 27.62 Hz, iso-C3H7–), 2.72 (s, 6H, –N–C(CH3))CH–C(CH3)N–), 1.83 (s, 6H, –N–C(CH3))CH–C(CH3)N–), 1.27 (d, 6H, J = 6.78 Hz, iso-C3H7–). 13C NMR (CDCl3, 125 MHz): δ 151.77 (s, 2C, –N–C(CH3)CH–C(CH3)N–), 147.07 (s, 1C, –C6H4–), 143.22 (s, 1C, –C6H4–), 143.18 (s, 2C, –N–C(CH3)CH–C(CH3)N–), 128.28 (d, 2C, J = 159.85 Hz, –C6H4–), 124.41 (d, 2C, J = 163.49 Hz, –C6H4–) 108.91 (d, 2C, J = 178.02 Hz, –N–C(CH3)CH–C(CH3)N–), 62.92 (t, 2C, J = 305.18 Hz, –N–CH2–N–), 33.88 (d, 1C, J = 127.16 Hz, iso-C3H7), 25.23 (q, 2C, J = 377.84 Hz, iso-C3H7), 15.36 (q, 2C, J = 386.92 Hz, –N–C(CH3))CH–C(CH3)N–), 11.96 (q, 2C, J = 388.74 Hz, –N–C(CH3))CH–C(CH3)N–). FTIR (solid neat; cm−1): ν(C–H) 2956 w; ν(CC) 1610 s; ν(CC) + ν(CN)/pz ring, 1514 s, 1458 s; νbend(C–H sp3) 1417 m; ν(N–C) 1214 s; νbend(C–H sp2) 832 w; ν(N–M) 571 w.CH–C(CH3)N–), 5.88 (s, 4H, –N–CH2–N–), 2.79–2.73 (m, 1H, J = 27.62 Hz, iso-C3H7–), 2.13 (s, 6H, N–C(CH3)CH–C(CH3)N–), 2.08 (s, 6H, –N–C(CH3)CH–C(CH3)N–), 1.14 (d, 6H, J = 6.87 Hz, iso-C3H7–). 13C NMR (CDCl3, 125 MHz): δ 147.56 (s, 2C, –N–C(CH3))CH–C(CH3)N–), 143.78 (s, 1C, –C6H4–), 141.98 (s, 1C, –C6H4–), 140.60 (s, 2C, –N–C(CH3))CH–C(CH3)N–), 127.55 (d, 2C, J = 156.22 Hz, –C6H4–), 119.76 (d, 2C, J = 157.13 Hz, –C6H4–) 160.97 (d, 2C, J = 174.39 Hz, –N–C(CH3))CH–C(CH3)N–), 64.12 (t, 2C, J = 304.27 Hz, –N–CH2–N–), 33.18 (d, 1C, J = 131.70 Hz, iso-C3H7), 25.52 (q, 2C, J = 386.92 Hz, iso-C3H7), 15.00 (q, 2C, J = 365.12 Hz, –N–C(CH3))CH–C(CH3)N–), 11.97 (q, 2C, J = 370.57 Hz, –N–C(CH3))CH–C(CH3)N–). FTIR (solid neat; cm−1): ν(C–H) 2960 w; ν(CC) 1553 s; ν(CC) + ν(CN)/pz ring, 1510 s, 1464 s; νbend(C–H sp3) 1418 m; ν(N–C) 1219 s; νbend(C–H sp2) 802 w; ν(N–M) 586 w.
C) 1549 s; ν(CC) + ν(CN)/pz ring, 1493 s, 1463 m; νbend(C–H sp3) 1426 m; ν(N–C) 1248 s; ν(C–O) 1151 (s); νbend(C–H sp2) 834 w; ν(N–M) 599 w.CH–CHC–), 6.41 (dd, 1H, J = 1.84 Hz, 1.83 Hz, –O–CHCH–CHC–), 6.30 (d, 1H, J = 3.81 Hz, –O–CHCH–CHC–), 6.04 (s, 2H, –N–C(CH3)CH–C(CH3) N–), 5.02 (s, 4H, –N–CH2–N–), 3.72 (s, 2H, –C–CH2–N–), 2.66 (s, 6H, –N–C(CH3)CH–C(CH3)N–), 2.20 (s, 6H, –N–C(CH3)CH–C(CH3)N–). 13C NMR (DMSO-d6, 125 MHz): d 150.61 (s, 1C, –O–CHCH–CHC–), 147.83 (s, 2C, –N–C(CH3)CH–C(CH3)N–), 143.95 (d, 1C, J = 203.4 Hz, –O–CHCH–CHC–), 140.94 (s, 2C, –N–C(CH3)CH–C(CH3)N–), 111.46 (d, 1C, J = 192.6 Hz, –O–CHCH–CHC–), 111.33 (d, 1C, J = 174.4 Hz, –O–CHCH–CHC–), 107.18 (d, 2C, J = 174.4 Hz, –N–C(CH3)CH–C(CH3)N–), 64.06 (t, 2C, J = 301.5 Hz, –N–CH2–N–), 46.70 (t, 1C, J = 278.8 Hz, –C–CH2–N–), 15.08 (q, 2C, J = 381.47 Hz, –N–C(CH3)CH–C(CH3)N–), 12.04 (q, 2C, J = 386.0 Hz, –N–C(CH3)CH–C(CH3)N–). Analysis calculated for C17H23Cl2N5OZn (%): C, 45.63, H, 5.19, N, 15.66. Found: C, 45.63, H, 5.24, N, 15.92. FTIR (solid neat; cm−1): ν(C–H) 2956 w; ν(CC) 1610 s; ν(CC) + ν(CN)/pz ring, 1514 s, 1458 s; νbend(C–H sp3) 1417 m; ν(N–C) 1214 s; ν(C–O) 1120 (s); νbend(C–H sp2) 832 w; ν(N–M) 571 w.C) 1608 s; ν(CC) + ν(CN)/pz ring, 1512 s, 1460 s; νbend(–C–H sp3) 1414 m; ν(N–C) 1210 s; νbend(C–H sp2) 831 w; ν(N–M) 570 w.CH–C(CH3)N–), 5.35 (s, 4H, –N–CH2–N–), 2.95–2.90 (m, 1H, J = 27.62 Hz, iso-C3H7–), 2.72 (s, 6H, –N–C(CH3))CH–C(CH3)N–), 1.83 (s, 6H, –N–C(CH3))CH–C(CH3)N–), 1.27 (d, 6H, J = 6.78 Hz, iso-C3H7–). 13C NMR (CDCl3, 125 MHz): δ 151.77 (s, 2C, –N–C(CH3)CH–C(CH3)N–), 147.07 (s, 1C, –C6H4–), 143.22 (s, 1C, –C6H4–), 143.18 (s, 2C, –N–C(CH3)CH–C(CH3)N–), 128.28 (d, 2C, J = 159.85 Hz, –C6H4–), 124.41 (d, 2C, J = 163.49 Hz, –C6H4–) 108.91 (d, 2C, J = 178.02 Hz, –N–C(CH3)CH–C(CH3)N–), 62.92 (t, 2C, J = 305.18 Hz, –N–CH2–N–), 33.88 (d, 1C, J = 127.16 Hz, iso-C3H7), 25.23 (q, 2C, J = 377.84 Hz, iso-C3H7), 15.36 (q, 2C, J = 386.92 Hz, –N–C(CH3))CH–C(CH3)N–), 11.96 (q, 2C, J = 388.74 Hz, –N–C(CH3))CH–C(CH3)N–). FTIR (solid neat; cm−1): ν(C–H) 2956 w; ν(CC) 1610 s; ν(CC) + ν(CN)/pz ring, 1514 s, 1458 s; νbend(C–H sp3) 1417 m; ν(N–C) 1214 s; νbend(C–H sp2) 832 w; ν(N–M) 571 w.CH–C(CH3)N–), 5.88 (s, 4H, –N–CH2–N–), 2.79–2.73 (m, 1H, J = 27.62 Hz, iso-C3H7–), 2.13 (s, 6H, N–C(CH3)CH–C(CH3)N–), 2.08 (s, 6H, –N–C(CH3)CH–C(CH3)N–), 1.14 (d, 6H, J = 6.87 Hz, iso-C3H7–). 13C NMR (CDCl3, 125 MHz): δ 147.56 (s, 2C, –N–C(CH3))CH–C(CH3)N–), 143.78 (s, 1C, –C6H4–), 141.98 (s, 1C, –C6H4–), 140.60 (s, 2C, –N–C(CH3))CH–C(CH3)N–), 127.55 (d, 2C, J = 156.22 Hz, –C6H4–), 119.76 (d, 2C, J = 157.13 Hz, –C6H4–) 160.97 (d, 2C, J = 174.39 Hz, –N–C(CH3))CH–C(CH3)N–), 64.12 (t, 2C, J = 304.27 Hz, –N–CH2–N–), 33.18 (d, 1C, J = 131.70 Hz, iso-C3H7), 25.52 (q, 2C, J = 386.92 Hz, iso-C3H7), 15.00 (q, 2C, J = 365.12 Hz, –N–C(CH3))CH–C(CH3)N–), 11.97 (q, 2C, J = 370.57 Hz, –N–C(CH3))CH–C(CH3)N–). FTIR (solid neat; cm−1): ν(C–H) 2960 w; ν(CC) 1553 s; ν(CC) + ν(CN)/pz ring, 1510 s, 1464 s; νbend(C–H sp3) 1418 m; ν(N–C) 1219 s; νbend(C–H sp2) 802 w; ν(N–M) 586 w.2.3. X-ray crystallographic studies
The X-ray-quality single crystal was coated with paratone-N oil, and the relevant diffraction data were collected at different levels of synchrotron radiation and at different temperatures: for the blue crystal, [LACoCl2], λ = 0.700 Å at 100(2) K; for the colorless crystal, [LAZnCl2], λ = 0.700 Å at 98(2) K; for the blue crystal, [LBCoCl2], λ = 0.630 Å at 100(2) K; for the colorless crystal, [LBZnCl2], λ = 0.610 Å at 100(2) K; for the colorless crystal, [LBCdBr2], λ = 0.700 Å at 293(2) K. These data were obtained on an ADSC Quantum-210 detector at 2D SMC with a silicon (111) double crystal monochromator (DCM) at the Pohang Accelerator Laboratory, South Korea. The PAL BL2D-SMDC program59 was used for data collection (detector distance was 63 mm, omega scan; Δω = 3°, exposure time was 1 s per frame), and HKL3000sm (Ver. 703r)60 was used for cell refinement, reduction, and absorption correction. Structures were solved by direct methods and refined by full-matrix least-squares refinement using the SHELXL-2018 (ref. 61) computer program. The positions of all non-hydrogen atoms were refined with anisotropic displacement factors. All hydrogen atoms were placed using a riding model, and their positions were constrained relative to their parent atoms using the appropriate HFIX command in SHELXL-2014. X-ray crystallography with PLS-II 2D-SMC beamline was supported by MSIP and POSTECH.Crystals of [LBCdBr2] were picked up with paratone-N oil and mounted on a Bruker SMART CCD diffractometer equipped with a graphite-monochromated Mo Kα (λ = 0.71073 Å) radiation source under a nitrogen gas cold stream (223(2) K). Data collection and integration were performed using SMART and SAINT-Plus software packages.62 Semi-empirical absorption corrections based on equivalent reflections were applied by SADABS.63 The structures were solved employing direct methods and refined using a full-matrix least-squares method on F2 using SHELXTL.64 All non-hydrogen atoms were refined anisotropically. Hydrogen atoms were added to their geometrically ideal positions. A summary of the crystallographic refinement data is presented in Table S1.†
2.4. rac-Lactide polymerization
The active species for the catalysis of rac-LA polymerization were prepared by dissolving 0.50 mmol of the synthesized Co(II), Zn(II), and Cd(II) complexes (i.e., 0.2216 g of [LACoCl2], 0.2248 g of [LAZnCl2], 0.2928 g of [LACdBr2], 0.2407 g of [LBCoCl2], 0.2439 g of [LBZnCl2], and 0.3119 g of [LBCdBr2]) in 7.35 mL of THF under inert atmosphere. To these solutions was the added methyllithium (MeLi, 1.00 mmol, 0.63 mL of 1.6 M solution in Et2O) dropwise to generate in situ the active catalytic species. The resulting mixture was then stirred for 2 h at room temperature, until a solution was obtained; a 1.00 mL aliquot of the said THF solution of the dimethyl Co(II), Zn(II), and Cd(II) complexes (0.0625 mmol) was then employed as initiator of the rac-LA ROP. [LnMX2] (Ln = LA or LB; M = Co or Zn, with X = Cl; M = Cd, with X = Br) itself does not exhibit any activity for ROP of rac-LA without MeLi, thus active species for initiation of polymerization are the combination of [LnMX2] and MeLi to give alkyl complex “[LMR1 or 2]” (R = Me, H,CH2 etc.). The characterization of actual catalytic species is not confirmed yet, and this pending issue will be emphasized in future work.
The general procedure for the polymerization reaction was as follows: rac-LA (0.901 g, 6.25 mmol) was transferred to a Schlenk flask (100 mL) under argon atmosphere, and 5.00 mL of dried CH2Cl2 were then added to the flask. The polymerization was initiated by the slow addition of the catalyst solution (1.0 mL, 0.0625 mmol) via a syringe under argon atmosphere at 25 °C and −25 °C. The reaction mixture was stirred for the specified time, and the polymerization reactions were quenched via addition of 1.0 mL of H2O followed by the addition of 2.0 mL of n-hexane. The solvent was then evaporated directly to yield a sticky polymeric material, which was subjected to monomer conversion determination, which was monitored by integrating monomer versus polymer methine resonances in 1H NMR spectrum. 1H NMR (CDCl3, 500 MHz) spectrum of the obtained polymer: δ 5.03–5.07 (m, 1H), 1.57–1.62 (m, 3H). The precipitates collected from the bulk mixture were again dissolved with CH2Cl2, and sequentially precipitated into n-hexane. Solvents were decanted, and the white solids thus isolated were first dried in vacuo then in a vacuum oven at 40 °C for 12 h.
Note: Proper precautionary measures need to be taken for handling alkyl-cadmium species, prepared for ROP reaction, owing to their pulmonary toxicity.
3. Results and discussion
3.1. Synthesis and physical parameters
In the current investigation N,N-bis((3,5-dimethyl-1H-pyrazol-1-yl)methyl derivatives were selected to study the effects of the ligand substitution patterns on the polymerization behavior. For instance, the amine moiety possessing an identical N,N-bis((3,5-dimethyl-1H-pyrazol-1-yl)methyl framework was modified/varied from to furanyl-methanamine (LA) and isopropylaniline (LB) to investigate how the steric effects and electronic properties affect the catalytic performance and stereoselectivity of the N,N-bis((3,5-dimethyl-1H-pyrazol-1-yl)methyl-based Zn(II), Co(II), and Cd(II) complexes toward the ROP of rac-LA. The ligands were prepared via a single-step condensation reaction involving furfurylamine or/and 4-isopropylaniline and 3,5-dimethyl-1H-pyrazolyl-1-methanol, as outlined in Scheme S1.† The reaction of ligands LA and LB with CoCl2·6H2O, ZnCl2, and CdBr2·4H2O in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio led to the formation of the desired complexes of [LnMX2] (Ln = LA or LB; M = Co or Zn, with X = Cl; M = Cd, with X = Br) general formula in good yield (Scheme 1).
:1 molar ratio led to the formation of the desired complexes of [LnMX2] (Ln = LA or LB; M = Co or Zn, with X = Cl; M = Cd, with X = Br) general formula in good yield (Scheme 1).
 | ||
| Scheme 1 Schematic representation of the synthesis of Co(II), Zn(II) and Cd(II) complexes. | ||
The FTIR spectra of the synthesized complexes were characterized by the presence of absorption bands corresponding to the ν(CN) and ν(CC) stretching frequencies of the pyrazolyl motif in the 1514–1493 cm−1 and 1464–1458 cm−1 wavenumber ranges, respectively. These values compare favorably to the corresponding ones reported for other complexes in the literature.65 In the spectra of the complexes, the absorption bands due to the stretching of aromatic CC and C–H bonds were observed to shift to higher wavenumbers with respect to the corresponding free ligands. Similarly, the complexes' absorption bands due to the stretching of sp3 and sp2 C–H and CC bonds appeared at the expected wavenumbers, based on literature data.50,51,66 Additionally, the C–O bond stretching appeared in the 1110–1230 cm−1 wavenumber range, in the case of [LBMX2] (M = Zn(II) or Co(II), with X = Cl; M = Cd(II), with X = Br). The absorption bands assigned to the stretching of the (CN)pyrazole bond shifted to higher wavenumbers in the FTIR spectra of [LnMX2] complexes with respect to the corresponding (free) ligands, which suggests the possibility of a back-donation and the involvement of the nitrogen atoms of pyrazolyl or/and amine moieties in metal bonding.67 The newly confirmed bands appearing in the 599–570cm−1 wavenumber range were attributed to the ν(M–N) stretching frequencies (M = Zn(II), Co(II), or Cd(II)).68
Compositional analysis data of the synthesized complexes are consistent with the proposed formulas reported in Scheme 1 and confirmed the purity of the isolated complexes. 1H NMR and 13C NMR spectroscopy data on the Zn(II) and Cd(II) complexes revealed the formation of the expected chemical species and were consistent with the synthesis of mononuclear structures.
3.2. Solid state structures of Co(II), Zn(II) and Cd(II) complexes
The solid-state structures of the synthesized Co(II), Zn(II), and Cd(II) complexes were elucidated by single-crystal X-ray diffraction studies. The geometric parameters defining the coordination environment of the complexes are listed in Table 1; in Fig. 1–6, on the other hand, are reported the ORTEP diagrams of [LACoCl2], [LAZnCl2], [LACdBr2], [LBCoCl2], [LBZnCl2], and [LBCdBr2], respectively. The M(II) center in [LAMX2] (M = Co, Zn, or Cd; X = Cl or Br) was observed to be coordinated in tridentate fashion by the nitrogen atoms of the pyrazolyl and amine moieties of the bis(pyrazolylamine) ligand, thus forming two five-membered chelate rings (Fig. 1–3).51,66,69| [LACoCl2] | [LAZnCl2] | [LACdBr2] | |||
|---|---|---|---|---|---|
| Bond lengths (Å) | |||||
| Co(1)–N(5) | 2.089(2) | Zn(1)–N(1) | 2.118(4) | Cd(1)–N(5) | 2.298(5) |
| Co(1)–N(1) | 2.097(2) | Zn(1)–N(1)#1 | 2.118(4) | Cd(1)–N(1) | 2.298(5) |
| Co(1)–Cl(2) | 2.303(6) | Zn(1)–Cl(1) | 2.264(2) | Cd(1)–Br(2) | 2.562(8) |
| Co(1)–Cl(1) | 2.327(8) | Zn(1)–Cl(2) | 2.288(1) | Cd(1)–Br(1) | 2.597(1) |
| Co(1)–N(3) | 2.334(2) | Zn(1)–N(3) | 2.544(5) | Cd(1)–N(3) | 2.609(5) |
|
|||||
| Bond angles (°) | |||||
| N(5)–Co(1)–N(1) | 110.02(6) | N(1)–Zn(1)–N(1)#1 | 143.4(2) | N(5)–Cd(1)–N(1) | 138.5(2) |
| N(5)–Co(1)–Cl(2) | 101.10(5) | N(1)–Zn(1)–Cl(1) | 100.04(1) | N(5)–Cd(1)–Br(2) | 103.51(1) |
| N(1)–Co(1)–Cl(2) | 103.63(5) | N(1)#1–Zn(1)–Cl(1) | 100.04(1) | N(1)–Cd(1)–Br(2) | 105.92(2) |
| N(5)–Co(1)–Cl(1) | 129.07(5) | N(1)–Zn(1)–Cl(2) | 100.32(1) | N(5)–Cd(1)–Br(1) | 99.09(1) |
| N(1)–Co(1)–Cl(1) | 110.52(5) | N(1)#1–Zn(1)–Cl(2) | 100.32(1) | N(1)–Cd(1)–Br(1) | 97.18(1) |
| Cl(2)–Co(1)–Cl(1) | 97.99(2) | Cl(1)–Zn(1)–Cl(2) | 111.40(6) | Br(2)–Cd(1)–Br(1) | 110.66(3) |
| N(5)–Co(1)–N(3) | 75.04(6) | C(1)–N(1)–Zn(1) | 130.6(3) | N(5)–Cd(1)–N(3) | 69.87(2) |
| N(1)–Co(1)–N(3) | 76.26(6) | N(2)–N(1)–Zn(1) | 117.6(3) | N(1)–Cd(1)–N(3) | 69.50(2) |
| Cl(2)–Co(1)–N(3) | 175.70(4) | N(2)–C(6)–N(3) | 107.1(4) | Br(2)–Cd(1)–N(3) | 147.22(1) |
| Cl(1)–Co(1)–N(3) | 86.01(4) | C(6)#1–N(3)–C(6) | 113.5(5) | Br(1)–Cd(1)–N(3) | 102.12(1) |
| [LBCoCl2] | [LBZnCl2] | [Bi-LBCdBr2] | [Tri-LBCdBr2] | ||||
|---|---|---|---|---|---|---|---|
| Bond lengths (Å) | |||||||
| Co(1)–N(5) | 2.034(2) | Zn(1)–N(1) | 2.049(1) | Cd(1)–N(5) | 2.275(5) | Cd(2)–N(10) | 2.334(5) |
| Co(1)–N(1) | 2.040(2) | Zn(1)–N(5) | 2.058(1) | Cd(1)–N(1) | 2.273(5) | Cd(2)–N(6) | 2.381(5) |
| Co(1)–Cl(2) | 2.235(6) | Zn(1)–Cl(1) | 2.231(5) | Cd(1)–Br(1) | 2.569(1) | Cd(2)–Br(3) | 2.544(1) |
| Co(1)–Cl(1) | 2.241(6) | Zn(1)–Cl(2) | 2.232(5) | Cd(1)–Br(2) | 2.598(1) | Cd(2)–Br(4) | 2.572(1) |
| Co(1)–N(3) | 3.864(2) | Zn(1)–N(3) | 3.886(1) | Cd(1)–N(3) | 2.953(4) | Cd(2)–N(8) | 2.571(5) |
|
|||||||
| Bond angles (°) | |||||||
| N(5)–Co(1)–N(1) | 110.22(6) | N(1)–Zn(1)–N(5) | 109.00(5) | N(5)–Cd(1)–N(1) | 120.80(2) | N(10)–Cd(2)–N(6) | 137.83(2) |
| N(5)–Co(1)–Cl(2) | 102.67(5) | N(1)–Zn(1)–Cl(1) | 112.42(4) | N(5)–Cd(1)–Br(1) | 112.36(1) | Br(3)–Cd(2)–Br(4) | 127.20(4) |
| N(1)–Co(1)–Cl(2) | 110.12(5) | N(5)–Zn(1)–Cl(1) | 105.04(4) | N(1)–Cd(1)–Br(1) | 108.16(1) | N(10)–Cd(2)–Br(3) | 102.05(1) |
| N(5)–Co(1)–Cl(2) | 112.16(4) | N(1)–Zn(1)–Cl(2) | 103.19(4) | N(5)–Cd(1)–Br(2) | 99.27(1) | N(6)–Cd(2)–Br(3) | 99.69(1) |
| N(1)–Co(1)–Cl(2) | 104.64(5) | N(5)–Zn(1)–Cl(2) | 109.02(4) | N(1)–Cd(1)–Br(2) | 108.05(1) | N(10)–Cd(2)–Br(4) | 94.09(1) |
| Cl(2)–Co(1)–Cl(1) | 117.09(2) | Cl(1)–Zn(1)–Cl(2) | 118.01(2) | Br(1)–Cd(1)–Br(2) | 107.02(4) | N(6)–Cd(2)–Br(4) | 100.85(1) |
| C(1)–N(1)–Co(1) | 126.88(1) | C(1)–N(1)–Zn(1) | 130.22(9) | C(1)–N(1)–Cd(1) | 136.1(4) | N(10)–Cd(2)–N(8) | 69.83(2) |
| N(2)–N(1)–Co(1) | 126.81(1) | N(2)–N(1)–Zn(1) | 123.23(8) | N(2)–N(1)–Cd(1) | 118.9(3) | N(6)–Cd(2)–N(8) | 68.79(2) |
| N(3)–C(6)–N(2) | 111.16(1) | N(3)–C(6)–N(2) | 113.53(1) | N(2)–C(6)–N(3) | 108.3(4) | Br(3)–Cd(2)–N(8) | 132.44(1) |
| N(3)–C(7)–N(4) | 113.55(1) | N(3)–C(7)–N(4) | 111.23(1) | N(3)–C(7)–N(4) | 108.3(4) | Br(4)–Cd(2)–N(8) | 100.34(1) |
 | ||
| Fig. 1 An ORTEP drawing of [LACoCl2] with thermal ellipsoids at 50% probability. All hydrogen atoms are omitted for clarity. | ||
 | ||
| Fig. 2 An ORTEP drawing of [LAZnCl2] with thermal ellipsoids at 50% probability. All hydrogen atoms are omitted for clarity. | ||
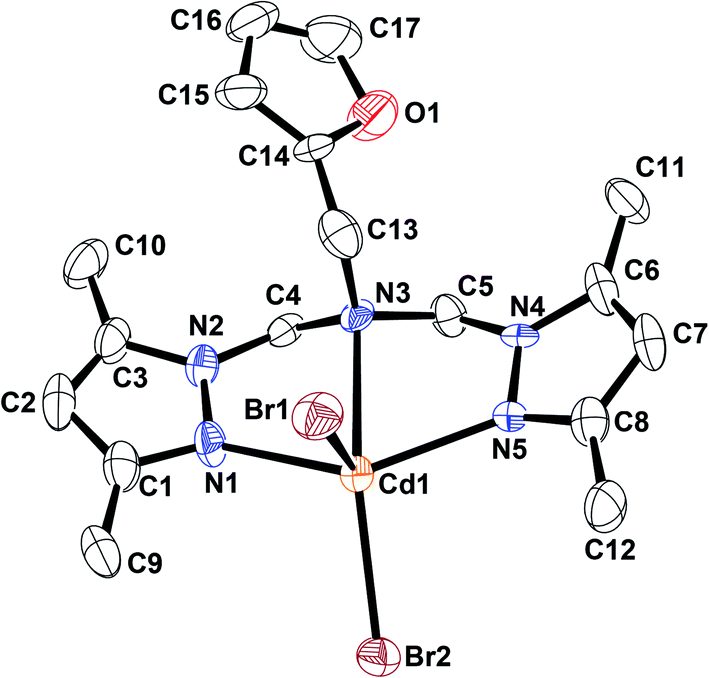 | ||
| Fig. 3 An ORTEP drawing of [LACdBr2] with thermal ellipsoids at 50% probability. All hydrogen atoms are omitted for clarity. | ||
[LACoCl2] and [LAZnCl2] exhibited distorted trigonal bipyramidal geometries, whereas the geometry of [LACdBr2] can be best described as distorted square pyramidal. The M–Npyrazole bond lengths are 2.089(2) and 2.097(2) Å for [LACoCl2], 2.118(4) and 2.118(4) Å for [LAZnCl2], 2.298(5) and 2.298(5) Å for [LACdBr2]. The M–Namine bond distances for these complexes have values within the expected range (2.334(2)–2.609(5) Å).50,66,70 The X-ray diffraction data revealed that the Co–Npyrazole bonds are slightly shorter than the Co–Namine bonds, probably because of the basicity difference between amine and pyrazole nitrogens. The M–Cl and M–Br bond distances had values in the 2.327(8)–2.264(2) Å and 2.597(1)–2.562(8) Å ranges, respectively. Npyrazole–M–Npyrazole bond angles were found to have values of 110.02(6)° [LACoCl2], 143.4(2)° [LAZnCl2], and 138.5(2)° [LACdBr2]. The Clterminal–M–Clterminal bond angle had values of 97.99(2)° and 111.40(6)° for [LACoCl2], and 143.4(2)° for [LAZnCl2], respectively,; the Brterminal–M–Brterminal bond angle of [LACdBr2] was observed to measure 110.66(3)°. X-ray data also revealed that the Npyrazole–M–Npyrazole bond angles had much smaller values than the Clterminal–M–Clterminal or Brterminal–M–Brterminal bite angles (Table 1), a common attribute of [LMX2]-type complexes with tetrahedral geometry.
Interestingly, the potentially tridentate ligand LB did not engage in the expected tridentate coordination of the M(II) centers but displayed a bidentate coordination mode, whereby ligation occurred via the nitrogen atoms of pyrazolyl moieties. [LBCoCl2] and [LBZnCl2] exhibited distorted tetrahedral geometries around the metal centers, as a result of the metal ions being coordinated by the N-substituted bis(pyrazolylamine) ligand in bidentate fashion and by two halide ions (Fig. 4 and 5).68 The M–Npyrazole bond distances in [LBCoCl2] and [LBZnCl2] have values of 2.037 and 2.054 Å, respectively; notably, these geometric parameters are in good agreement with the accepted values.66 Similarly, the M–Cl and M–Br bond distances of these complexes are comparable to the bond lengths reported for structurally related Zn(II) compounds.66 Furthermore, the Npyrazole–M–Npyrazole bond angles had values of 110.22(6)° for [LBCoCl2] and 109.00(5)° for [LBZnCl2], whereas the values for the Clterminal–M–Clterminal bond angles lie in the 102.67(5)°–112.42(4)° range.
 | ||
| Fig. 4 An ORTEP drawing of [LBCoCl2] with thermal ellipsoids at 50% probability. All hydrogen atoms are omitted for clarity. | ||
 | ||
| Fig. 5 An ORTEP drawing of [LBZnCl2] with thermal ellipsoids at 50% probability. All hydrogen atoms are omitted for clarity. | ||
Contrarily, the ORTEP diagram of [LBCdBr2] illustrates that this complex has two crystal structures in one unit cell. These crystal structures are depicted in Fig. 6 and S59,† with values for relevant selected parameters listed in Table 1.
 | ||
| Fig. 6 An ORTEP drawing of tetrahedral [Bi-LBCdBr2] (a) and square pyramidal [Tri-LBCdBr2] (b) with thermal ellipsoids at 50% probability. All hydrogen atoms are omitted for clarity. | ||
In the crystalline phase, this complex has two crystallographically independent Cd(II) sites: one Cd is tetra-coordinated and displays a tetrahedral structure whereas the other is penta-coordinated and displays a square pyramidal geometry. Addison et al.71 have introduced index of trigonality, τ, which provides useful information regarding the geometry of five-coordinate complexes; in particular, a perfect trigonal bipyramidal geometry is characterized by value of τ5 of 1.0 whereas a value for τ5 of 0.0 is indicative of a perfect square pyramidal geometry. Where τ = (β − α)/60, in which α and β are the two largest coordination angles as illustrated in τ5 values (Table S2†).71–74 In the case of the five-coordinate Cd center in [LBCdBr2] in the crystalline phase, t was found to have a value of 0.089; hence this Cd center was concluded to exhibit a distorted square pyramid five-coordinate geometry. Notably, in the mentioned structure, the amine nitrogen occupies the apex of the square pyramid. All the bond lengths and angles around the Cd center in both structures are in agreement with those reported previously for tetra- and/or penta-coordinated Cd(II) complexes.69,75 Additionally, the geometry of [LACdBr2] (τ5 = 0.145) could be described as distorted square pyramidal whereas the geometries of [LACoCl2] (τ5 = 0.777) and [LAZnCl2], (τ5 = 0.533) could be described as distorted trigonal bipyramidal (Table S2†). The angles between the X1–M–X2 (X = Cl or Br) and Npyrazole–M–Npyrazole planes in [LACoCl2], [LAZnCl2], [LACdBr2], and [LBCdBr2] were observed to have values of 95.83(4)°, 90.00(2)°, 91.34(1)°, and 87.12(1)°, respectively.
The geometry of tetra-coordinated complexes can be described by the value of the τ4 parameter. Specifically, in the case of a perfect tetrahedron, τ4 has a value of 1.00 whereas its value is equal to 0.00 in the case of a perfect square planar geometry.76 The values of the τ4 parameters for the tetra-coordinated complexes [LBCoCl2], [LBZnCl2], and [LBCdBr2] are listed in Table S3.†76 Based on these values, the geometry around the M(II) centers of the these complexes could be concluded to be distorted tetrahedral. Additionally, the bond angles around the Co(II), Zn(II), and Cd(II) centers were employed to measure the degree of distortion. The calculated angles between the X1–M–X2 (X = Cl or Br) and Npyrazole–M–Npyrazole planes of [LBCoCl2], [LBZnCl2], and [LBCdBr2] were 95.11(4)°, 94.49(3)°, and 94.69(9)°, respectively.
3.3. rac-Lactide polymerization
The catalytic capabilities of 3,5-dimethylpyrazolylamine based Co(II), Zn(II), and Cd(II) initiators, were tested for rac-LA polymerization at two different temperatures. The catalytic species were generated in situ by treating dichloro Co(II), Zn(II) and dibromo Cd(II) complexes with two equiv. of MeLi in THF. The alkyl derivatives proved significantly active, allowing conversion of rac-LA at 25 °C and −25 °C, inspected by the decrease of monomer signals in 1H NMR spectrum of the resultant PLA. Representative polymerization results are summarized in Tables 2 and 3. The complete conversion was observed within 2 h, where 98% conversion was achieved for LB-containing Zn complex after 5 min of reaction at 25 °C, as revealed by the %-conversion vs. time plot (Fig. S60†).| Entry | Catalysta | Conv.b (%) | Mnc (g mol−1) × 103 (calcd.) | Mnd (g mol−1) × 103 (GPC) | Mwd (g mol−1) × 103 (GPC) | PDId | Pre |
|---|---|---|---|---|---|---|---|
| a Conditions: [initiator] = 0.0625 mmol, [rac-LA]/[initiator] = 100; 5.0 mL of CH2Cl2 as polymerization solvent; polymerization time = 2 h.b Monomer conversion (%) determined by 1H NMR spectroscopy.c Calculated from ([molecular weight of rac-LA] × [mol concentration of used [rac-LA]/[mol concentration of initiator]) × (conversion%).d Determined by gel permeation chromatography (GPC) in THF, relative to polystyrene standard (corrected using the Mark−Houwink factor of 0.58).77e Probability of heterotactic enchainment (Pr) were calculated on the basis of homonuclear decoupled 1H NMR spectra according to literature.55–58 | |||||||
| 1 | MeLi | 99 | 14.27 | 11.44 | 17.21 | 1.50 | 0.47 |
| 2 | [LACoCl2]/MeLi | 98 | 14.12 | 8.57 | 15.57 | 1.82 | 0.53 |
| 3 | [LBCoCl2]/MeLi | 98 | 14.12 | 11.34 | 15.43 | 1.36 | 0.82 |
| 4 | [LAZnCl2]/MeLi | 98 | 14.12 | 8.18 | 18.28 | 2.23 | 0.53 |
| 5 | [LBZnCl2]/MeLi | 97 | 13.98 | 8.02 | 12.16 | 2.02 | 0.68 |
| 6 | [LACdBr2]/MeLi | 98 | 14.12 | 8.04 | 12.72 | 2.11 | 0.51 |
| 7 | [LBCdBr2]/MeLi | 98 | 14.12 | 5.39 | 10.19 | 1.89 | 0.60 |
| Entry | Catalysta | Conv.b (%) | Mnc (g mol−1) × 103 (calcd.) | Mnd (g mol−1) × 103 (GPC) | Mwd (g mol−1) × 103 (GPC) | PDId | Pre |
|---|---|---|---|---|---|---|---|
| a Conditions: [initiator] = 0.0625 mmol, [rac-LA]/[initiator] = 100; 5.0 mL of CH2Cl2 as polymerization solvent; polymerization time = 2 h.b Monomer conversion (%) determined by 1H NMR spectroscopy.c Calculated from ([molecular weight of rac-LA] × [mol concentration of used rac-LA]/[mol concentration of initiator]) × (conversion%).d Determined by gel permeation chromatography (GPC) in THF, relative to polystyrene standard (corrected using the Mark−Houwink factor of 0.58).77e Probability of heterotactic enchainment (Pr) were calculated on the basis of homonuclear decoupled 1H NMR spectra according to literature.55–58 | |||||||
| 1 | MeLi | 99 | 14.27 | 9.945 | 16.49 | 1.66 | 0.78 |
| 2 | [LACoCl2]/MeLi | 97 | 13.98 | 7.77 | 11.80 | 1.52 | 0.69 |
| 3 | [LBCoCl2]/MeLi | 97 | 13.98 | 6.42 | 8.64 | 1.35 | 0.82 |
| 4 | [LAZnCl2]/MeLi | 97 | 13.98 | 6.57 | 8.20 | 1.25 | 0.85 |
| 5 | [LBZnCl2]/MeLi | 97 | 13.98 | 6.52 | 7.56 | 1.16 | 0.94 |
| 6 | [LACdBr2]/MeLi | 97 | 13.98 | 10.12 | 14.03 | 1.39 | 0.80 |
| 7 | [LBCdBr2]/MeLi | 98 | 14.12 | 4.71 | 7.06 | 1.50 | 0.69 |
The experimentally determined Mn (corrected using the Mar–Houwink factor of 0.58)77 had values that were half of those calculated based on the monomer/initiator molar ratio. This observation could be explained by assuming the presence of two growing polymer chains per metal center. Notably, the molecular weights of the resultant PLAs were also influenced by the identity of the M(II) center. In fact, as Cd(II) was replaced by Co(II), the Mn value of the produced PLAs appeared to decrease (Table 2). A general trend was observed toward a linear increase in the Mn value as the polymerization time increased (Fig. S61†). For example, the value of Mn was 6.28 × 103 g mol−1 for 1 min, 8.20 × 103 g mol−1 for 3 min, 8.81 × 103 g mol−1 for 5 min, and 9.64 × 103 g mol−1 for 10 min polymerization time (Fig. S62–S65†). However, the conversion rate did not increase after 5 min; in the case of the LB containing Zn-complex. The plot of the Mn values versus the monomer/catalyst ratio indicated that an increase in monomer/catalyst ratio was associated with a linear increase in the Mn values observed up to rac-LA/catalyst = 400 (Fig. S66†). For example, with an increase in the rac-LA/catalyst ratio from 100 to 400, increase in the Mn values from 9.27 × 103 g mol−1 to 19.26 × 103 g mol−1, respectively, is observed. However, with an increase in the monomer/catalyst ratio from 400 to 500 a decrease in Mn values have been observed (Fig. S67–S71†). Additionally, the % conversion value was not substantially affected by increases in the monomer/catalyst ratio. The Mn of the resultant PLAs was in turn slightly affected by the identity of the amine substituents. For instance, the Mn values of the PLAs obtained via catalysis with the LA-containing complexes were higher than their counterparts measured for the PLAs synthesized using the LB-containing complexes as catalysts, as illustrated by the polymerization data collected (Table 3; entries 2, 4, and 6). This trend contradicted those reported recently for catalysts consisting of Zn(II) and Cu(II) complexes bearing (pyrazol-1-ylmethyl)pyridine ligands, whereby increases in the steric bulk of the amine substituents attached to the ligand framework were associated with increases in the molecular weight of the polymers produced.39 However, in the cases of the hereby studied initiators, the observed Mn values were lower compared to the calculated ones (Tables 2 and 3), probably because of the low initiation efficiencies of these complexes which resulted in a broader range of PDIs (1.36–2.23).78 However, the PDIs values notably decrease (1.16–1.52) as the polymerization temperature decreases from 25 °C to −25 °C (Table 3).
Generally, the steric hindrance around the M(II) center caused by increases in the bulk of ligand substituents/groups reduce the catalytic activity of the complex.79–81 In fact, changing the amine moiety from furanyl-methanamine to isopropylaniline did not significantly affect the catalytic activity of the hereby investigated complexes. This fact is further augmented by the calculation of buried volumes using SambVca program.82 The total steric hindrance provided by ligand with respect to the metal center can be quantitatively calculated by comparing the topographic steric maps of the various M(II) complexes (M = Co, Zn, and Cd). Ball and stick models, space-filling models, and topographic steric maps of these complexes are presented in Fig. S72.† Evidence indicates that, for a particular metal center, five-coordinated complexes are characterized by larger buried volumes than four-coordinated complexes, although these differences did not translate into significant differences in catalytic activity.
A monomer-activated mechanism can be predicted to take place in the case of the ROP of rac-LA initiated by the types of dimethyl-containing M(II) species investigated in the present study.83 In this mechanistic approach, the monomer would be electronically activated as a result of its coordination to the electrophilic M(II) center. Simultaneously, one of the metal-bound methyl groups nucleophilically attacks the carbonyl carbon of the monomer to initiate the ROP of the lactide by breaking the bond between the endocyclic oxygen and the carbonyl group. This step contrasts with the classical coordination insertion mechanism,84 whereby the opening of the ring occurs via the nucleophilic reactivity of a δ-bonded alkyl/alkoxide group that is bound to the M(II) center. Furthermore, the propagation proceeded by the attack, which contributed to the growth of the polymer chain at the M end, after which the addition of the LA molecule produced the heterotactic PLA. The ROP-initiating solution was predicted to comprise some unreacted MeLi along with the in situ-generated dimethyl M(II) (M = Co, Zn, and Cd) species. Notably, the polymerization reaction conducted in the presence of MeLi only (Table 2; entry 1) furnished PLA with good conversion (99% in 2 h) but with negligible stereoselectivity (Pr = 0.47 at 25 °C). Additionally, the activity using MeLi alone is low as illustrated in Fig. S73;† a negligible amount of PLA has been obtained after 5 min of reaction time comparted to [LBZnCl2]/MeLi system which give 98% conversion within 5 min of reaction time (Fig. S60†). These results highlighted the benefits afforded by the Lewis acidic M(II) center and the bis(pyrazolyl-amine) framework; indeed, use of the resulting complex as catalyst delivered good polymerization control and directed the heterotacticity of the resultant PLA. Moreover, conducting the ROP reaction using [LnMX2] (Ln = LA or LB; M = Co or Zn, with X = Cl; M = Cd, with X = Br) in the absence of MeLi resulted in negligible amounts of PLA.31,85
The stability of the studied systems was determined implementing a sequential five-stage rac-LA polymerization reaction using [LBZnCl2]/MeLi system at 25 °C (Table S4; Fig. S60†). In the first cycle, 98% conversion was achieved within 5 min, in conditions whereby [rac-LA]/[initiator] = 100. Subsequently, another 100 equivalents of monomer were added to the reaction mixture ([rac-LA]/[initiator] = 200), and a conversion value of 93% was measured without the addition of initiator. A slight drop in the activity signifies the stability of the said initiator. Afterwards, sequential additions of 100 equivalents of the monomer were performed at interval of 5 min, and at [rac-LA]/[initiator] = 500, the conversion was observed to have a value of 67%.
The PLAs produced using [LnMCl2]/MeLi (Ln = LA or LB, M = Co or Zn, with X = Cl; M = Cd, with X = Br) as catalysts are predominantly heterotactic. Polymerization results indicated that the preference for heterotactic bias was relatively low when the reaction temperature was 25 °C (Pr = 0.51–0.82 at 25 °C, Table 2), which is consistent with our previously published findings.86,87 However, a drop in reaction temperature from 25 to −25 °C resulted in increased heteroselectivity (Pr = 0.68–0.94). Polymerization data further revealed that the LB-containing complexes bearing the isopropylaniline moiety (i.e., [LBMCl2]/MeLi) afforded superior heteroselectivities at 25 °C to their LA-containing counterparts (Table 2). Thus, enhancing the bulk of the amine moiety was observed to result in an increase of the heterotacticity of the PLAs produced using the studied complexes as catalysts. However, when polymerization was conducted at −25 °C, the opposite trend became apparent (Table 3). The proper orientation of the substituents in the ancillary ligand could thus provide the electronic environment for the effective accommodation of any possible steric clash between the ligand and the propagating PLA chain and the alternative attachment of the incoming monomer molecule. These results indicate that the polymerization of rac-LA monomers catalyzed by the hereby developed catalytic system proceeded by the chain end mechanism. These findings are in good agreement with the results of our previously published studies.50,86,87
The catalytic activity of the Zn(II) system in the context of rac-LA polymerization are comparable with those reported by our group for dimethyl Zn(II) complexes, but use of the hereby developed system yielded PLAs with higher heterotacticity than we reported at 25 °C.87 In comparison with the previously reported Zn(II) system bearing N,N′,N-bis((1H-pyrazol-1-yl)methyl)amines derivatives as ligands (Pr = 0.95 at −50 °C with 90% conversion), the current system exhibited higher activity and comparable heteroselectivity (97% conversion with Pr = 0.94 at −25 °C).49 Moreover, in comparison with the complexes of the tetradentate N,N,N,N-bis(pyrazolyl)methane ligand with Zn(II) and Fe(II) (Pr = 0.79) used as catalysts,88 our system exhibited superior activity and stereoselectivity (Pr = 0.94). Co(II)-based catalysts previously studied by our group, comprising N,N-bis((3,5-dimethyl-1H-pyrazol-1-yl)methyl)-3,5-dimethylaniline and N,N-bis((3,5-dimethyl-1H-pyrazol-1-yl)methyl)-4-methoxyaniline as ligands, afforded PLAs of lower molecular weight and heterotacticity (Pr = 0.81–0.85 at −50 °C) but narrower PDI value range (1.13–1.21).50 Recently, Sutar et al. described the ROP of L-lactide catalyzed by the Co(II)–salen complex, which afforded 79% conversion at 30 °C after 24 h with a narrow range of MWD values (1.12).89 In comparison with the hereby developed Co(II)-based catalytic system, the recently reported co-tripodal complexes/iPrOH catalytic system proved to be effective, affording 81% conversion within 2 min to produce isotactic PLA (Pm = 0.73).90 Chakraborty's group demonstrated that CoCl2·6H2O could effectively catalyze the polymerization of rac-LA, affording 96% conversion within 1 h under melting conditions (145 °C), albeit with negligible stereocontrol.91 Mandal et al. recently reported that Cd(OAc)2 was an effective catalyst in the presence of benzyl alcohol at 140 °C; in fact, use of this catalyst afforded 98% conversion within 18 min (monomer:[Cd]:BnOH ratio = 100:1:2).92 Similarly, the Cd(II) complexes from this recent investigation achieved the complete conversion of the monomer to PLA with stereoselectivities that were equivalent to those achieved using Cd(II) complexes with the N,N′,X-tridentate iminomethylpyridine ligand framework.93 Further research work should focus on the design and optimization of procedures that afford the production of PLAs with improved polymerization control and enhanced stereoregularity.
4. Conclusions
We demonstrated the synthesis of Co(II), Zn(II), and Cd(II) complexes supported with N,N-bis((3,5-dimethyl-1H-pyrazol-1-yl)methyl)amines derivatives and determined their crystal structures by X-ray diffraction. Diverse coordination geometries were afforded through the bi- and/or tri-dentate coordination of the ligands to the M(II) centers via the nitrogen atoms of the ligands' pyrazole and amine moieties. [LnMX2]/MeLi (Ln = LA or LB, M = Co or Zn, with X = Cl; M = Cd, with X = Br) system effectively catalyzed the ROP of rac-LA to yield low-molecular-weight PLAs with a broader range of PDI values (1.36–2.11). The catalytic performance of the initiators was observed to depend to a minor extent on the ligand substitution pattern. Based on the polymerization results, the introduction of substituents that increased steric hindrance around the M(II) center increased the amount of heterotactic enchainment along the growing polymer chain.Author contributions
Solhye Choe: formal analysis-supporting. Hyosun Lee: conceptualization; supervision-lead. Saira Nayab: writing original draft-supporting.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Research Foundation (NRF) of South Korea, funded by the Ministry of the Education, Science, and Technology (MEST) (Grant No. 2019R1A2C1088654). This work was also supported by the Technology Innovation Program (TIP # 20011123, Development of Cyclic Olefin Polymer(COP) with High Heat Resistance and High Transmittance) funded by the Korea Evaluation Institute of Industrial Technology (KEIT) and the Ministry of Trade, Industry & Energy (MOTIE, Republic of Korea). This work has also been supported by Higher Education Commission (HEC), Islamabad, Islamic Republic of Pakistan, under National Research Programme for Universities (NRPU) Grant No. 6840-KP. X-ray crystallography with PLS-II 2D-SMC beamline was supported by MSIP and POSTECH.References
- R. E. Drumright, P. R. Gruber and D. E. Henton, Adv. Mater., 2000, 12, 1841–1846 CrossRef CAS.
- A. Amgoune, C. M. Thomas, T. Roisnel and J.-F. Carpentier, Chem.–Eur. J., 2006, 12, 169–179 CrossRef CAS PubMed.
- R. P. Brannigan and A. P. Dove, Biomater. Sci., 2017, 5, 9–21 RSC.
- M. A. Ghalia and Y. Dahman, J. Polym. Res., 2017, 74, 24–45 Search PubMed.
- E. Chiellini and R. Solaro, Adv. Mater., 1996, 8, 305–313 CrossRef CAS.
- A.-C. Albertsson and I. K. Varma, Biomacromolecules, 2003, 4, 1466–1486 CrossRef CAS PubMed.
- H. Sun, J. S. Ritch and P. G. Hayes, Inorg. Chem., 2011, 50, 8063–8072 CrossRef CAS PubMed.
- R. Petrus and P. Sobota, Organometallics, 2012, 31, 4755–4762 CrossRef CAS.
- B. Gao, X. Li, R. Duan, Q. Duan, Y. Li, X. Pang, H. Zhuang and X. Chen, RSC Adv., 2015, 5, 29412–29419 RSC.
- S. Ghosh, R. R. Gowda, R. Jagan and D. Chakraborty, Dalton Trans., 2015, 44, 10410–10422 RSC.
- G. Li, M. Lamberti, D. Pappalardo and C. Pellecchia, Macromolecules, 2012, 45, 8614–8620 CrossRef CAS.
- C. Bakewell, R. H. Platel, S. K. Cary, S. M. Hubbard, J. M. Roaf, A. C. Levine, A. J. P. White, N. J. Long, M. Haaf and C. K. Williams, Organometallics, 2012, 31, 4729–4736 CrossRef CAS.
- W. Yang, K.-Q. Zhao, T. J. Prior, D. L. Hughes, A. Arbaoui, M. R. J. Elsegood and C. Redshaw, Dalton Trans., 2016, 45, 11990–12005 RSC.
- W. Zhang, Y. Wang, L. Wang, C. Redshaw and W.-H. Sun, J. Organomet. Chem., 2014, 750, 65–73 CrossRef CAS.
- S. Tabthong, T. Nanok, P. Sumrit, P. Kongsaeree, S. Prabpai, P. Chuawong and P. Hormnirun, Macromolecules, 2015, 48, 6846–6861 CrossRef CAS.
- A. Thevenon, C. Romain, M. S. Bennington, A. J. P White, H. J. Davidson, S. Brooker and C. K. Williams, Angew. Chem., Int. Ed., 2016, 55, 8680–8685 CrossRef CAS PubMed.
- T. Rosen, Y. Popowski, I. Goldberg and M. Kol, Chem.–Eur. J., 2016, 22, 11533–11536 CrossRef CAS PubMed.
- D. Jedrzkiewicz, G. Adamus, M. Kwiecień, Ł. John and J. Ejfler, Inorg. Chem., 2017, 56, 1349–1365 CrossRef CAS PubMed.
- A. B. Kremer, R. J. Andrews, M. J. Milner, X. R. Zhang, T. Ebrahimi, B. O. Patrick, P. L. Diaconescu and P. Mehrkhodavandi, Inorg. Chem., 2017, 56, 1375–1385 CrossRef CAS PubMed.
- T. Rosen, I. Goldberg, V. Venditto and M. Kol, J. Am. Chem. Soc., 2016, 138, 12041–12044 CrossRef CAS PubMed.
- C. -Y. Li, S. -J. Hsu, C. -I. Lin, C. -Y. Tsai, J. -H. Wang, B. -T. Ko, C. -H. Lin and H. -Y. Huang, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 3840–3849 CrossRef CAS.
- S. Fortun, P. Daneshmand and F. Schaper, Angew. Chem., Int. Ed., 2015, 54, 13669–13672 CrossRef CAS PubMed.
- J. Zhang, B. Wang, L. Wang, J. Sun, Y. Zhang, Z. Cao and Z. Wu, Appl. Organomet. Chem., 2017, 4077–4085 Search PubMed.
- D. M. Lyubov, A. O. Tolpygin and A. A. Trifonov, Coord. Chem. Rev., 2019, 392, 83–145 CrossRef CAS.
- M. Hu, F. Han, W. Zhang, W. Ma, Q. Deng, W. Song, H. Yan and G. Dong, Catal. Sci. Technol., 2017, 7, 1394–1403 RSC.
- M. J. Stanford and A. P. Dove, Chem. Soc. Rev., 2010, 39, 486–494 RSC.
- R. H. Platel, L. M. Hodgson and C. K. Williams, Polym. Rev., 2008, 48, 11–63 CrossRef CAS.
- W. A. Munzeiwa, V. O. Nyamori and B. Omondi, Appl. Organomet. Chem., 2018, 32, 4247–4256 CrossRef.
- T. J. J. Whitehorne and F. Schaper, Inorg. Chem., 2013, 52, 13612–13622 CrossRef CAS PubMed.
- J. Cho, M. K. Chun, S. Nayab and J. H. Jeong, Transition Met. Chem., 2019, 44, 175–185 CrossRef CAS.
- K. S. Kwon, S. Nayab and J. H. Jeong, Polyhedron, 2017, 130, 23–29 CrossRef CAS.
- K. D. Pressing, J. H. Lehr, M. E. Pratt, L. N. Dawe, A. A. Sarjeant and C. M. Kozak, Dalton Trans., 2015, 44, 12365–12375 RSC.
- L. Chen, W. Li, D. Yuan, Y. Zhang, Q. Shen and Y. Yao, Inorg. Chem., 2015, 54, 4699–4708 CrossRef CAS PubMed.
- C. Kan, J. Ge and H. Ma, Dalton Trans., 2016, 45, 6682–6695 RSC.
- G. Xiao, B. Yan, R. Ma, W. J. Jin, X. Q. Lu, L. Q. Ding, C. Zeng, L. L. Chen and F. Bao, Polym. Chem., 2011, 2, 659–664 RSC.
- M. P. F. Pepels, M. Bouyahyi, A. Heisea and R. Duchateau, Macromolecules, 2013, 46, 4324–4334 CrossRef CAS.
- K. S. Kwon, S. Nayab and J. H. Jeong, Polyhedron, 2015, 85, 615–620 CrossRef CAS.
- N. W. Attandoh, S. O. Ojwach and O. Q. Munro, Eur. J. Inorg. Chem., 2014, 3053–3064 CrossRef CAS.
- S. O. Ojwach, T. T. Okemwa, N. W. Attandoh and B. Omondi, Dalton Trans., 2013, 42, 10735–10745 RSC.
- M. Zikode, S. O. Ojwach and M. P. Akerman, Appl. Organomet. Chem., 2017, 31, e3556 CrossRef.
- M. Zikode, S. O. Ojwach and M. P. Akerman, J. Mol. Catal. A: Chem., 2016, 413, 24–31 CrossRef CAS.
- X. F. Yu, C. Zhang and Z. X. Wang, Organometallics, 2013, 32, 3262–3268 CrossRef CAS.
- A. John, M. M. Shaikh, R. J. Butcher and P. Ghosh, Dalton Trans., 2010, 39, 7353–7363 RSC.
- F. Xue, J. Zhao and T. S. A. Hor, Dalton Trans., 2013, 42, 5150–5158 RSC.
- R. Mukherjee, Coord. Chem. Rev., 2000, 203, 151–218 CrossRef CAS.
- E. Ocansey, J. Darkwa and B. C. E. Makhubela, RSC Adv., 2018, 8, 13826–13834 RSC.
- A. Budhai, B. Omondi, S. O. Ojwach, C. Obuah, E. Y. Osei-Twum and J. Darkwa, Catal. Sci. Technol., 2013, 3, 3130–3135 RSC.
- Y. K. Kang, J. H. Jeong, N. Y. Lee, Y. T. Lee and H. Lee, Polyhedron, 2010, 29, 2404–2408 CrossRef CAS.
- S. Shin, H. Cho, H. Lee, S. Nayab and Y. Kim, J. Coord. Chem., 2018, 71, 556–584 CrossRef CAS.
- S. Shin, S. Nayab and H. Lee, Polyhedron, 2018, 141, 309–321 CrossRef CAS.
- H. Cho, S. Choe, D. Kim, H. Lee and S. Nayab, Polyhedron, 2020, 187, 114641–114650 CrossRef CAS.
- I. Dvoretzky and G. H. Richter, J. Org. Chem., 1950, 15, 1285–1288 CrossRef CAS.
- F. Xue, J. Zhao and T. S. A. Hor, Dalton Trans., 2011, 40, 8935–8940 RSC.
- S. Choi, Cobalt(II) and cadmium(II) complexes containing N′-substituted N,N′,N-bis((1H-pyrazol-1-yl)methyl)amine: Synthesis, structure and methyl methacrylate polymerization, Thesis for the degree of Master of Science, Kyungpook National University, 2014 Search PubMed.
- T. J. J. Whitehorne and F. Schaper, Inorg. Chem., 2013, 52, 13612–13622 CrossRef CAS PubMed.
- M. Cheng, A. B. Attygalle, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 1999, 121, 11583–11584 CrossRef CAS.
- B. M. Chamberlain, M. Cheng, D. R. Moore, T. M. Ovitt, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2001, 123, 3229–3238 CrossRef CAS PubMed.
- M. T. Zell, B. E. Padden, A. J. Paterick, K. A. M. Thakur, R. T. Kean, M. A. Hillmyer and E. J. Munson, Macromolecules, 2002, 35, 7700–7707 CrossRef CAS.
- J. W. Shin, K. Eom and D. Moon, J. Synchrotron Radiat., 2016, 23, 369–373 CrossRef PubMed.
- Z. Otwinowski and W. Minor, Methods Enzymol., 1997, 276, 307–326 CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 2015, 71, 3–8 CrossRef PubMed.
- SMART and SAINT-Plus v 6.22, Bruker AXS Inc., Madison, Wisconsin, USA, 2000 Search PubMed.
- G. M. Sheldrick, SADABS v 2.03, University of Göttingen, Germany, 2002 Search PubMed.
- SHELXTL v 6.10, Bruker AXS Inc, Madison, Wisconsin, USA, 2000 Search PubMed.
- M. H. Sadhu and S. B. Kumar, J. Mol. Struct., 2018, 15, 239–247 CrossRef.
- J. Seo, J. Lee, E. Kim, H. Lee, J. Jeon and S. Nayab, Inorg. Chim. Acta, 2019, 496, 119071–119078 CrossRef CAS.
- M. E. Castro, M. J. Percino, V. M. Chapela, M. Ceron, G. Soriano-Moro, J. Lopez-Cruz and F. J. Melendez, Int. J. Mol. Sci., 2013, 14, 4005–4029 CrossRef CAS PubMed.
- E. Kim, H. Y. Woo, S. Kim, H. Lee, D. Kim and H. Lee, Polyhedron, 2012, 42, 135–141 CrossRef CAS.
- S. Shin, S. H. Ahn, M. J. Jung, S. Nayab and H. Lee, J. Coord. Chem., 2016, 69, 2391–2402 CrossRef CAS.
- M. E. Kodadi, F. Malek, R. Touzani, A. Ramdani, S. E. Kadiri and D. Eddike, Molecules, 2003, 8, 780–787 CrossRef.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- S. Toyota and M. Oki, Bull. Chem. Soc. Jpn., 1992, 65, 1832–1840 CrossRef CAS.
- H. Hopfl, J. Organomet. Chem., 1999, 581, 129–149 CrossRef CAS.
- J. Vela, J. Cirera, J. M. Smith, R. J. Lachicotte, C. J. Flaschenriem, S. Alvarez and P. L. Holland, Inorg. Chem., 2007, 46, 60–71 CrossRef CAS PubMed.
- D. Kim, S. Kim, H. Y. Woo, H.-J. Lee and H. Lee, Appl. Organomet. Chem., 2014, 28, 445–453 CrossRef CAS.
- L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 9, 955–964 RSC.
- M. A. Masuelli, J. Polym. Biopolym. Phys. Chem., 2014, 2, 37–43 CAS.
- I. Peckermann, A. Kapelski, T. P. Spaniol and J. Okuda, Inorg. Chem., 2009, 48, 5526–5534 CrossRef CAS PubMed.
- A. Stopper, T. Rosen, V. Venditto, I. Goldberg and M. Kol, Chem.–Eur. J., 2017, 23, 11540–11548 CrossRef CAS.
- S. Bian, S. Abbina, Z. Lu, E. Kolodka and G. Du, Organometallics, 2014, 33, 2489–2495 CrossRef CAS PubMed.
- B.-H. Huang, C.-Y. Tsai, C.-T. Chen and B.-T. Ko, Dalton Trans., 2016, 45, 17557–17580 RSC.
- L. Falivene, R. Credendino, A. Poater, A. Petta, L. Serra, R. Oliva, V. Scarano and L. Cavallo, Organometallics, 2016, 35, 2286–2293 CrossRef CAS . For online program, see https://www.molnac.unisa.it/OMtools/sambvca2.0/index.html.
- Y. Cui, C. Chen, Y. Sun, J. Wu and X. Pan, Inorg. Chem. Front., 2017, 4, 261–269 RSC.
- N. Ajellal, D. M. Lyubov, M. A. Sinenkov, G. K. Fukin, A. V. Cherkasov, C. M. Thomas, J.-F. Carpentier and A. A. Trifonov, Chem.–Eur. J., 2008, 14, 5440–5448 CrossRef CAS PubMed.
- M.-S. Kanga, J. Cho, S. Nayab and J. H. Jeong, Polyhedron, 2019, 158, 135–143 CrossRef.
- J. Lee, M. Yoon, H. Lee and S. Nayab, RSC Adv., 2020, 10, 16209–16220 RSC.
- S. Nayab and J. H. Jeong, Polyhedron, 2013, 59, 138–143 CrossRef CAS.
- U. Herber, K. Hegner, D. Wolters, R. Siris, K. Wrobel, A. Hoffmann, C. Lochenie, B. Weber and S. H. Pawlis, Eur. J. Inorg. Chem., 2017, 1341–1354 CrossRef CAS.
- H. C. Pradhan, S. Mantri, A. Routaray, T. Maharaba and A. K. Sutar, J. Chem. Sci., 2020, 132, 25–31 CrossRef CAS.
- P. Marin, M. J.-L. Tschan, P. Haquette, T. Roisnel, I. del Rosal, L. Maron and C. M. Thomas, Eur. Polym. J., 2019, 120, 109208 CrossRef CAS.
- B. Rajashekhar and D. Chakraborty, Polym. Bull., 2014, 71, 2185–2203 CrossRef CAS.
- M. Mandal, U. Monkowius and D. Chakraborty, J. Polym. Res., 2016, 23, 1–9 CrossRef CAS.
- J. Lee, H. Lee, S. Nayab and K. B. Yoon, Polyhedron, 2019, 158, 432–440 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Scheme, 1H NMR, 13C NMR, FTIR, EA, homodecoupled 1H NMR, GPC, ball and stick model, crystal data and structure refinement, general experimental procedures. CCDC 2068377–2068382 contains the supplementary crystallographic data for [LACoCl2], [LAZnCl2], [LACdBr2], [LBCoCl2], [LBZnCl2], and [LBCdBr2], respectively. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra02365a |
| This journal is © The Royal Society of Chemistry 2021 |