
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe fabrication of TiO2-supported clinoptilolite via F− contained hydrothermal etching and a resultant highly energetic {001} facet for the enhancement of its photocatalytic activity†
Anadil Gul‡
,
Raza Ullah‡,
Jihong Sun *,
Tallat Munir and
Shiyang Bai*
*,
Tallat Munir and
Shiyang Bai*
Beijing Key Laboratory for Green Catalysis and Separation, Department of Environmental and Chemical Engineering, Beijing University of Technology, Beijing 100124, P. R. China. E-mail: jhsun@bjut.edu.cn; sybai@bjut.edu.cn
First published on 18th May 2021
Abstract
TiO2-supported clinoptilolite (TiO2/CP) was synthesized in the presence of F− ions. Various characterizations demonstrated that the particle size of loaded TiO2 increased linearly with an increase in the temperature and concentration of F− ions. In particular, the additive F− ions were favored to produce the mutually independent co-exposed {001} and {101} facets of loaded TiO2, while TiO2/CPs synthesized in the absence of F− ions were dominated by the thermodynamically stable {101} facet. As photocatalysts for the removal of crystal violet or methyl orange dyes under UV-irradiation in aqueous solutions, TiO2/CPs (ACP6) synthesized in the presence of F− ions significantly improved the degradation efficiency, as compared to ACP3 obtained in the absence of F− ions. These results elucidated that the highly energetic {001} exposed facet, large particle size and fine dispersion of loaded TiO2 in TiO2/CP accounts for its best photocatalytic performance. The effected mechanism of operational parameters on the degradation performances is proposed.
1. Introduction
With the increasing demands of energy- and environment-related problems, photocatalysis, as a light-driven photochemical process over the surface of a photocatalyst, has attracted increasing attention over the last few decades due to its potential applications in the fields of wastewater treatment and the generation of renewable energy.1,2 Although TiO2-based photocatalysis has been over-exploited in the past few decades, there are still some challenges related to the reaction efficiency, complicated treatment of catalyst recovery step, electron–hole recombination rate and electron–hole transport mechanism of TiO2, which restrain the large-scale practical application of the process.3–5 To achieve higher efficiency in the practical application of TiO2, the tailoring of its physical properties, such as phase composition, exposed crystal facets, average particle size, surface area, crystallinity and porosity, are of key importance.6–9 The immobilization of TiO2 onto the surface of some suitable support is an encouraging alternative to the inherent limitations of unsupported TiO2.The preparation of the TiO2/zeolite composite photocatalyst has attracted increasing attention over recent years, as the composite configuration tailors the photocatalytic properties of loaded TiO2 by controlling the particle size, bandgap, surface area, loading content and porosity of loaded TiO2. Moreover, the physical and electronic properties of the composite photocatalysts can be regulated by interfacial interactions between TiO2 and zeolite. The use of zeolite as a supporting matrix for TiO2 nanoparticles has also been found to enhance the adsorption capacity10 and dispersion of titania,11 which in turn improve the photoactivity of the system. Generally, the immobilization of TiO2 onto the surface of zeolite can be achieved by numerous methods including sol–gel,10 hydrothermal,12 impregnation,11 and solid-state dispersion methods.13 Among the aforementioned methods, the hydrothermal method is an effective route for immobilizing TiO2 onto the surface of zeolite with high surface area, good crystallinity and variant particle size,14 which offer several advantages such as the formation of the anatase phase at relatively low temperature, low agglomeration between particles, defect-free nano-crystals with high surface area, and narrow particle size distribution.15,16
Clinoptilolite (CP), being one of the most abundantly occurring zeolites in nature, is regarded as the best candidate as a support for TiO2.17–19 Though significant research is available on the synthesis and application of TiO2/CP composites, the literature still lacks data relating to the loading of anatase TiO2 onto CP with controlled particle size and highly reactive exposed crystal facets. Recently, intensive research interest has been focused on controlling the crystal particle size and exposed facets of TiO2 to enhance its photocatalytic performance.20,21 There exists an optimum particle size for excellent photocatalytic performance of TiO2 due to the competing effects of the effective particle size on light absorption and scattering efficiency. Almquist and Biswas22 synthesized anatase TiO2 particles in the range of 5 to 165 nm and found that an optimum particle size of 25 to 40 nm exhibited excellent photoactivity. The tailoring of anatase TiO2 crystals with the termination of specific facets has received great interest for many years. As reported by Liu et al.,9 the anatase phase {001} facet was found to be the most reactive with an average surface energy of 0.90 J mol−2; therefore, the photocatalytic performance of anatase TiO2 depends not only on its particle size, but also on the type of exposed crystal facets. However, unfortunately, most available anatase TiO2 is dominated by the thermodynamically stable {101} facet during the crystallization process due to its relatively low surface energy, i.e. 0.44 J mol−2.23 The breakthrough in controlling the synthesis of crystals with a high percentage of reactive high energy {001} facets was not made until 2008.24 Roy et al.25 demonstrated that among the various facets, {101} was the least active towards methyl orange (MO) degradation, whilst an optimum {001}/{101} ratio resulted in excellent performance because of the reduced electron–hole recombination rate. In order to enhance the photocatalytic performance of TiO2, a large group of researchers are making efforts to synthesize anatase TiO2 with highly reactive exposed facets, i.e. {001}. Hence, TiO2/CPs with highly reactive exposed facets i.e. {001} are still highly desired and pursued in the field of heterogeneous photocatalysis. Numerous methods have been developed to fabricate anatase TiO2 nanocrystals with {001} exposed facets, in which surface fluorination is the most effective in stabilizing {001} facets based on first-principles calculations.23,24
Herein, TiO2/CP photocatalysts with controlled anatase TiO2 particle size and exposed highly reactive crystal facets were synthesized under different hydrothermal treatment temperatures and concentrations of fluoride ions (F− ions) in an aqueous solution of TiCl4. The effect of hydrothermal treatment temperature and additive amount of F− ions on the photocatalytic activity of TiO2/CP was investigated in detail via the degradation of crystal violet (CV) and methyl orange (MO) dyes under UV-irradiation in aqueous media. F− ions were used as tailoring agents to expose the highly energetic {001} facet of the loaded TiO2 in order to enhance its photocatalytic activity. The as-prepared TiO2/CP catalysts were characterized via X-ray diffraction (XRD), scanning electron microscopy (SEM), transmission electron microscopy (TEM), inductively coupled plasma-optical emission spectrometry (ICP-OES), X-ray Photoelectron Spectroscopy (XPS), Fourier Transform Infrared (FT-IR), UV-visible spectroscopy and BET-isotherm. Finally, the degradation kinetics of CV and MO dye in the aqueous solutions were elucidated.
2. Experimental
2.1. Materials
Natural CP with a Si/Al ratio of 10.63 was supplied by GuoTouShengShi Science and Technology Co. Ltd. TiCl4 (99% purity), NH4F (96.0%), CV dye (high purity biological stain), and MO dye were obtained from J & K Co. Ltd. All solutions were made in deionized water with a resistivity of 18.25 MΩ cm.2.2. Synthesis of TiO2/CP catalysts
The finely divided powdered natural CP was first purified by heating its suspension in deionized water (10 g L−1) at 70 °C under continuous vigorous stirring for 8 hours. The resulting suspension was then allowed to settle down prior to filtration and then washed thoroughly with deionized water. TiO2/CP photocatalysts were synthesized using a procedure similar to that reported by Ullah et al.14 In a typical procedure, 1.00 g of CP was dispersed in 25 mL of 0.25 M TiCl4 solution made in ice-cold water and the suspension was then autoclaved on oil baths under continuous vigorous stirring for 5 hours at 100, 150, and 200 °C. The precipitate was then separated by filtration, washed thoroughly with deionized water and dried in an oven at 80 °C overnight. The obtained products were ground to a fine powder and named ACP1, ACP2, and ACP3, corresponding to the hydrothermal temperature at 100, 150, and 200 °C, respectively.Keeping the other conditions constant as used for the first set of three catalysts, the second set of four catalysts were synthesized at 200 °C by adding 0.08, 0.15, 0.25, and 0.35 M NH4F to 0.25 M TiCl4 solution, and named ACP4, ACP5, ACP6, and ACP7, respectively. Pure TiO2 was also made by the same procedure using 0.25 M F− ions without adding CP.
A representative scheme showing the preparation procedures and the structure of TiO2/CPs is illustrated in Fig. S1 of the ESI† section.
2.3. Characterizations
The phase compositions and crystalline structures of the TiO2/CP catalysts were analyzed by X-ray diffractometer (Beijing Purkinje General Instrument Co. Ltd), having a CuKα irradiation source, and 2θ (diffraction angle) ranging from 5–75°. The supported TiO2, crystallite size was calculated with the help of Scherrer's equation (D = kλ/β![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cosθ), where D is the estimated crystallite size (nm), k is a constant (0.9), λ is the X-ray wavelength (0.15432 nm), β is the peak width at half of the maximum height, θ is the diffraction angle. ICP-OES (Perkin Elmer Optima 2000-DV) was used to analyze the elemental composition and weight % of TiO2 in the synthesized samples (TiO2/CP). The chemical structures of the samples were examined via FT-IR spectrophotometer in the range of 400–4000 cm−1. N2 adsorption–desorption isotherms were used to determine the specific surface area of synthesized samples at −196 °C, with the help of a JW-BK300 (Beijing Sci. & Techno. Co. Ltd). The samples were degassed at 120 °C for 6 hours prior to each measurement in order to remove all the adsorbed gas molecules in the samples. The structural morphology and particle size of the synthesized samples (TiO2/CP) were examined using scanning and transmission electron microscopy (JEOLJEM-200 and JEOL-2010, respectively). The operating voltage was 15.0 kV for SEM and 200 kV for TEM. The optical bandgaps of the samples were determined using a Shimadzu UV-2600 spectrophotometer. The pHpzc (point of zero charge) was measured with the help of a Zeta-sizer (Malvern Instrument Ltd, UK) through light scattering at different pH values. Surface chemical analysis of the synthesized samples (TiO2/CP) was performed by XPS spectra (ESCALAB 250Xi, Germany). The total organic carbon (TOC) removal efficiencies of CV and MO dye via photocatalytic degradation were investigated via vario TOC (UK).
cosθ), where D is the estimated crystallite size (nm), k is a constant (0.9), λ is the X-ray wavelength (0.15432 nm), β is the peak width at half of the maximum height, θ is the diffraction angle. ICP-OES (Perkin Elmer Optima 2000-DV) was used to analyze the elemental composition and weight % of TiO2 in the synthesized samples (TiO2/CP). The chemical structures of the samples were examined via FT-IR spectrophotometer in the range of 400–4000 cm−1. N2 adsorption–desorption isotherms were used to determine the specific surface area of synthesized samples at −196 °C, with the help of a JW-BK300 (Beijing Sci. & Techno. Co. Ltd). The samples were degassed at 120 °C for 6 hours prior to each measurement in order to remove all the adsorbed gas molecules in the samples. The structural morphology and particle size of the synthesized samples (TiO2/CP) were examined using scanning and transmission electron microscopy (JEOLJEM-200 and JEOL-2010, respectively). The operating voltage was 15.0 kV for SEM and 200 kV for TEM. The optical bandgaps of the samples were determined using a Shimadzu UV-2600 spectrophotometer. The pHpzc (point of zero charge) was measured with the help of a Zeta-sizer (Malvern Instrument Ltd, UK) through light scattering at different pH values. Surface chemical analysis of the synthesized samples (TiO2/CP) was performed by XPS spectra (ESCALAB 250Xi, Germany). The total organic carbon (TOC) removal efficiencies of CV and MO dye via photocatalytic degradation were investigated via vario TOC (UK).
2.4. Photocatalytic degradation experiments
The photocatalytic evaluation of the obtained TiO2/CPs for the degradation of CV and MO dyes was carried out in a 250 mL glass beaker containing 100 mL aqueous solution at room temperature. A set of three 20 W high-pressure UVC Hg lamps enclosed in a rectangular steel box, was used as the irradiation source. Before being subjected to the UV-light source, the reaction mixture was stirred for 30 min continuously in the dark to reach the adsorption–desorption equilibrium. The amount of both the dyes adsorbed was less than 5% for all the photocatalysts synthesized, which indicates that adsorption has a negligible role in the removal process. Therefore, the degradation process was mainly focused in the removal of these dyes from the reaction mixture. At a set time, the aliquots of used samples were withdrawn, centrifuged prior to the analysis to remove the suspended catalyst particles and then analyzed using a UV-vis spectrophotometer. Each experiment was carried out in duplicate in order to validate the results.The following eqn (1) was used for the percentage degradation (X%) calculation of CV and MO dyes, as follows:
 | (1) |
The TOC removal percentage (%) was estimated using the following expression (2):
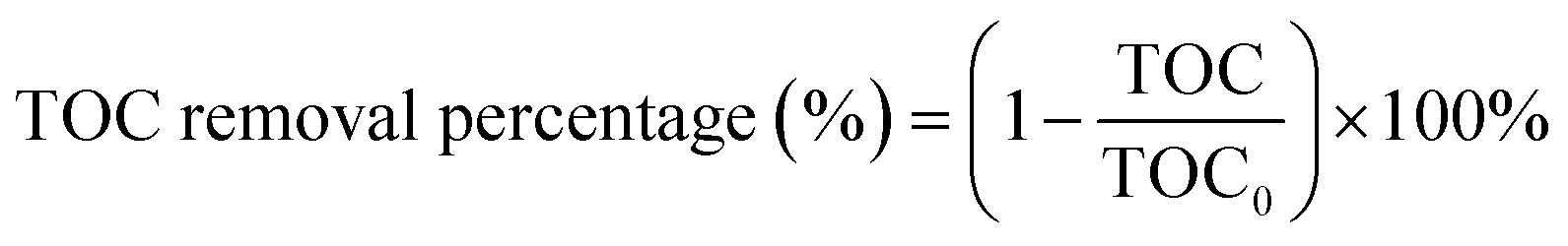 | (2) |
3. Results and discussion
3.1. Characterization of TiO2/CPs
The XRD patterns of parent natural CP, TiO2, and TiO2/CPs (ACP1, ACP3, ACP4, and ACP6) are shown in Fig. 1. The heulandite (HEU) structure of CP and the crystal phase of TiO2 were confirmed by comparison with the Joint Committee on Powder Diffraction Standards (JCPDSs), in which, the characteristic diffraction peaks at 2θ values of 9.98, 11.27, 22.29, 26.82, 30.07, and 32.23° belonged to the major components of the CP (as shown in Fig. 1a), and those at 25.5, 38.2, 48.3, 54.2, 55.4, and 62.5° corresponded to anatase phase TiO2 (as shown in Fig. 1f), which are indexed as (101), (001), (200), (105), (211), and (204) crystal planes, respectively. With the improved hydrothermal temperatures from 100 °C for ACP1 (Fig. 1b) to 200 °C for ACP3 (Fig. 1c), or increased concentrations of used F− ions form 0.08 M for ACP4 (Fig. 1d) to 0.25 M for ACP6 (Fig. 1e), the decline in peak intensity of CP characteristic phases, but the enhancement in the intensity of anatase peaks may be because TiO2 particles effectively covered the surfaces of CP during composite formation and hindered peaks of CP from being detected by XRD patterns. Another reason may be related to the etching effect of used F− ions at a high hydrothermal temperature on the excessive dealumination of CP structures.26 Furthermore, the crystallite size of loaded TiO2 calculated using Scherrer's equation, as shown in Table 1, presented the increasing tendency from 7.8 to 17.7 nm along with enhancements of temperature and concentrations of used F− ions. As can be seen in Fig. 1, the intensity of the (004) diffraction peak, being used (I004/I101) to represent the highly energetic {001} facet in TiO2/CP,9 remarkably improved with enhancements of temperature and concentrations of used F− ions, and was also higher than that of pure anatase TiO2 (ca. 0.30, as shown in Table S1 of the ESI† section, corresponding to the reference standard (JCPDS. 21-1272) which presented I004/I101 of (0.2)9). These observations suggest the preferential orientation growth along the {001} facet direction for TiO2/CPs (ACP4-ACP7); in other words, the existence of both F− ions and CPs plays a significant role in exposing the highly energetic {001} facet of loaded TiO2. However, in the absence of CP, the hydrothermal treatment of the TiO2 precursor in HF solution easily leads to the formation of pure anatase particles with a relatively low intensity of {004} diffraction peaks (as seen from the I004/I101 values in Table S1 of the ESI† section). This unique co-existence of highly energetic {001} and thermodynamically stable {101} facets in TiO2/CP (ACP6) is responsible for the synergistic removal of CV and MO dyes from aqueous solution under UV-irradiation.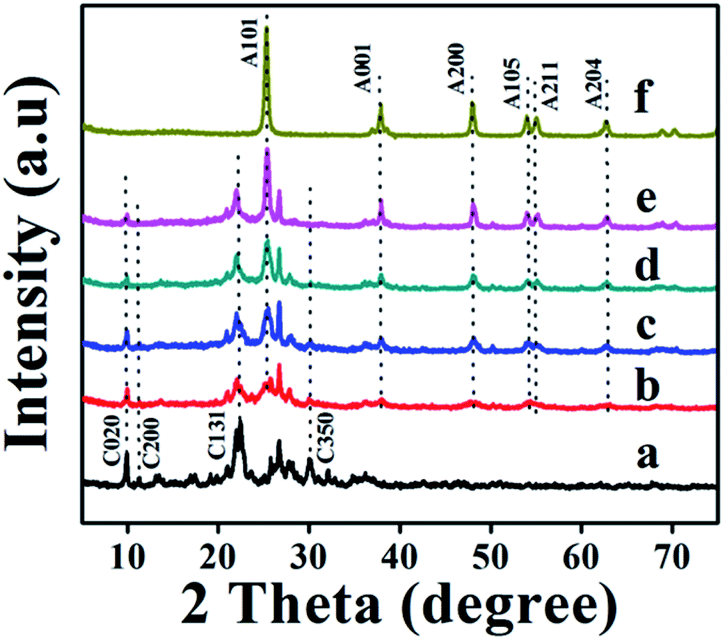 | ||
| Fig. 1 XRD patterns of (a) natural CP, (b) ACP1, (c) ACP3, (d) ACP4, (e) ACP6, and (f) pure TiO2. | ||
| Sample | Formula | TiO2 amounta (wt%) | TiO2 phase | TiO2 particle size (nm) | Si/Ald (molar ratio) | SBETe (m2 g−1) | Bandgap (eV) | ||
|---|---|---|---|---|---|---|---|---|---|
| XRDb | SEMc | n = 2 | n = 1/2 | ||||||
| a Determined from ICP data.b Determined from XRD patterns according to Schererr's equation.c Determined from SEM images.d Determined from ICP data.e BET surface area determined from N2 adsorption–desorption isotherms. | |||||||||
| CP | Na0.75K1.89Al8.06Si85.70O184.81 | — | — | — | — | 10.63 | 15.8 | — | — |
| ACP1 | Ti21.57Na1.36K1.23Al6.12Si88.24O230.10 | 27.31 | Anatase | 7.80 | 4.7 | 14.41 | 193.7 | 2.73 | 3.33 |
| ACP2 | Ti23.14Na1.45K1.12Al5.07Si101.47O258.11 | 29.29 | Anatase | 8.77 | 6.2 | 20.02 | 116.3 | 2.72 | 3.33 |
| ACP3 | Ti23.85Na1.44K0.27Al3.31Si138.35O130.22 | 30.20 | Anatase | 9.25 | 10.4 | 41.79 | 50.1 | 2.81 | 3.32 |
| ACP4 | Ti24.49Na1.59K0.61Al4.80Si132.93O330.22 | 31.01 | Anatase | 10.01 | 14.5 | 27.68 | 51.3 | 2.91 | 3.43 |
| ACP5 | Ti24.29Na1.47K0.35Al2.86Si108.83O271.44 | 30.76 | Anatase | 14.92 | 19.0 | 38.11 | 44.8 | 2.85 | 3.22 |
| ACP6 | Ti33.93Na1.64K0.21Al2.79Si129.96O332.96 | 42.95 | Anatase | 17.36 | 16.0 | 46.48 | 43.6 | 2.80 | 3.28 |
| ACP7 | Ti36.87Na1.57Al1.52Si137.46O351.80 | 46.68 | Anatase | 17.72 | 20.0 | 90.62 | 35.6 | 2.50 | 3.57 |
All the TiO2/CP samples formed in the presence of F− ions exhibited much sharper and higher characteristic peaks of loaded TiO2, which may be ascribed to their highly crystalline characteristics. Table S1† also shows that the crystallinity degrees of CP support decreased, while that of loaded TiO2 increased along with the enhancement of the temperature and concentration of F− ions during hydrothermal treatments. The XRD patterns of ACP2, ACP5 and ACP7 displayed similar observations as shown in Fig. S2A of the ESI† section.
The SEM images of natural CP, TiO2 and TiO2/CPs (ACP1, ACP3, ACP4, and ACP6) are illustrated in Fig. 2. As can be seen, the pure natural CP (Fig. 2a) revealed smooth sheet-like structures, while that of TiO2/CPs (Fig. 2 from b to e) presented rough surfaces with nanoscale particles of TiO2 with a size of around 4.7–20.0 nm determined from ImageJ software27 (as shown in Table 1), which were dispersed on the surface of the CP support. These nano-scale TiO2 particles loaded onto CP support are beneficial to provide more active sites for synergistic effects in the photocatalytic degradation of CV and MO dyes. As can be seen in Fig. 2b and c, both ACP1 and ACP3 prepared in the absence of F− ions maintained the sheet-like structure of the CP support. However, Fig. 2d and e indicated that the TiO2 loadings in ACP4 and ACP6 were relatively uniform but there were no apparent sites of unloaded CP. Thermal shocks at high hydrothermal temperature (200 °C) and the existence of F− ions would cause the excessive dealumination and etching phenomena of the CP supports in ACP3-ACP7, leading to the partial dissolution of the CP framework and good dispersions of the loaded TiO2, similar to the XRD results. The SEM image of bare TiO2 (Fig. 2f) exhibited obvious aggregation and therefore indicated that CP supports could act as dispersants to avoid the typical tendency of TiO2 to form agglomerates.
 | ||
| Fig. 2 SEM images of (a) natural CP, (b) ACP1, (c) ACP3, (d) ACP4, (e) ACP6, and (f) pure TiO2. | ||
The SEM images of ACP2, ACP5 and ACP7 as shown in Fig. S3 of the ESI† section exhibited almost similar information to that of ACP1, ACP4 and pure TiO2, respectively. The use of relatively high concentrations of F− ions (up to 0.35 M) easily led to the severe aggregation of loaded TiO2 as seen in ACP7 (Fig. S3c†), which looks like pure TiO2 (Fig. 2f).
The micro/nanostructures of the ACP3 and ACP6 were further characterized by TEM images. As manifested in Fig. 3a and b, the grain micro-photos of ACP3 and ACP6 were composed of a large amount of TiO2 nanocrystals with an average size of about 3 and 2.7 μm, respectively. The high magnification TEM image of ACP6 exhibited relatively large and highly crystalline TiO2 particles with more uniform and independent distributions on the surface of the CP support (Fig. 3d), which is very useful for improving its photocatalytic properties, as compared to that of ACP3 (Fig. 3c), in which the severe aggregation of loaded TiO2 appeared. The mean sizes of the loaded TiO2 nano-particles in ACP3 and ACP6 determined from high magnification TEM image using ImageJ software11,28 were found to be around 10.4 nm (Fig. 3c (inset)) and 19 nm (Fig. 3d (inset)), respectively, consistent with that calculated from SEM images (as shown in Fig. 2c and e) and XRD patterns (as shown in Fig. 1c and e). The high-resolution TEM image of ACP3, as shown in Fig. 3e, revealed that the lattice spaces of 0.35 nm supporting the exposed facets are {101} of the anatase TiO2. The high-resolution TEM image of ACP6 (Fig. 3f) presented a clear lattice with well-defined lattice fringes of 0.24 and 0.35 nm, corresponding to {001} and {101} exposed facets, respectively, which confirmed the anatase phase of TiO2.9 These observations further proved that the existence of F− ions could be beneficial for the etching effect and formation of the co-exposed {001} and {101} facets of TiO2 in TiO2/CPs.9
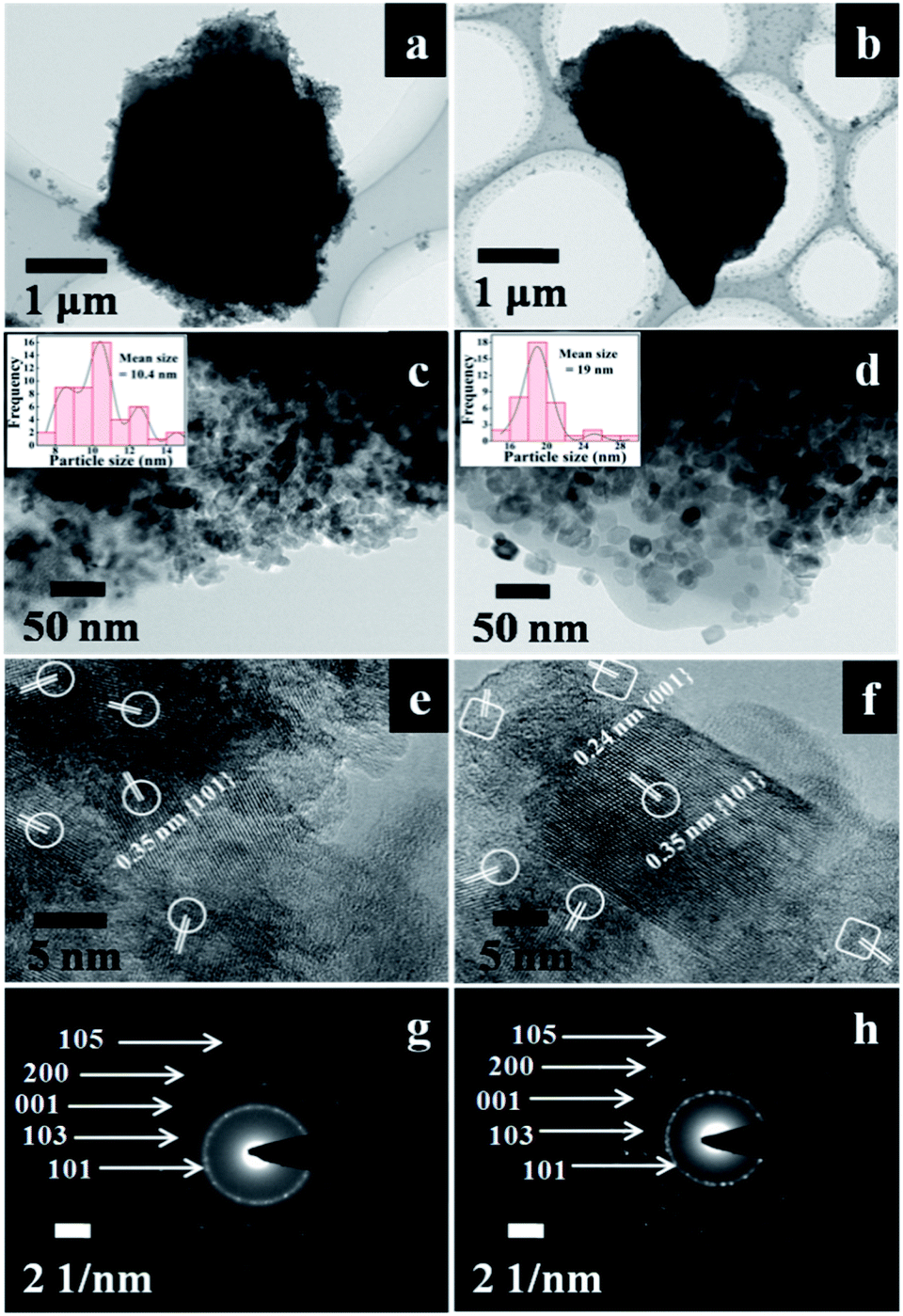 | ||
| Fig. 3 (a) and (b) TEM images of ACP3 and APC6, (c) and (d) high-magnification TEM images of ACP3 and APC6, (e) and (f) high-resolution TEM images of ACP3 and APC6, (the selected areas in elliptical shapes represent {101} facets, while those in box shapes represent {001} facets) and (g) and (h) the SAED pattern of ACP3 and APC6. The histograms of TiO2 particle size distribution determined from the high-magnification TEM images of ACP3 and ACP6 are given in the inset of (c) and (d). | ||
The different crystallographic exposed facets of TiO2 nanocrystals are controlled by altering their relative stability during the crystal growth, which is intrinsically determined by their surface energies. The capping agents (F− ions) adsorbed on the surface of TiO2 interact differently with different crystalline facets leading to various dominant reactive facets.29 The capping agents (F− ions) play a crucial role of selectively adsorbing on the surface of TiO2 and reducing the surface free energy with more active {001} facets of loaded TiO2, which results in preserving the highly reactive facets. The selected areas in the elliptical shape represent {101} facets, while that in the box shape represents the {001} facets. According to Wolff's construction principle,9 more than 90% of the exposed facets consist of the thermodynamically stable {101} facet during the crystal growth process of TiO2. The co-existence of the reactive high energy {001} facet (0.90 J m−2) and the thermodynamically stable {101} facet (0.44 J m−2) may be beneficial to produce a number of thermal intermediate interfaces with a reduction of their Gibbs free energy,30 and thereby enhance the synergistic removal of CV and MO dyes from aqueous solution.
The corresponding SAED patterns of ACP3 and ACP6, as shown in Fig. 3g and h, respectively, further demonstrated the existence of the anatase phase of loaded TiO2. The SAED pattern of ACP6 (Fig. 3h) exhibited more bright circular fields, indicating its high crystalline characteristics, as compared to that of ACP3 (Fig. 3g).
Fig. 4A illustrates the FT-IR spectra of parent CP and TiO2/CPs (ACP3 and ACP6) in the wavenumber range of 400–4000 cm−1. As can be seen, all samples exhibited almost the same peak profiles, suggesting that the synthesis method did not cause significant destruction of the HEU microstructure. However, the intensity of all peaks in TiO2/CPs related to CP support became weak, consistent with that of XRD analysis. The most intense peak at around 1030 cm−1 was associated with the asymmetric stretching vibration of O–Si(Al)–O, showing high sensitivity to the degree of dealumination.31 This band became weak and shifted to a relatively higher wavenumber (1100 cm−1) in TiO2/CPs (Fig. 4A-b and c), as compared to that of parent CP (as shown in Fig. 4A-a). This decrease in intensity and the slight shift in the position of the absorption band around 1030 cm−1 may be due to the elution of a portion of the Al3+/Si4+ intra-framework on hydrothermal treatment.32 The peaks that appeared at around 465, 605, and 795 cm−1 were attributed to the stretching vibrations of SiO4 and AlO4 tetrahedral atoms present in the zeolite structure.33 The absorption band at around 1640 cm−1 in CP (Fig. 4A-a) was assigned to the –OH bending vibration of physically adsorbed water. This band became very weak in TiO2/CPs, which was due to the desorption of physically adsorbed water during hydrothermal treatment. The intensity of all peaks below 1000 cm−1 belonging to CP features decreased after loading TiO2 in the HEU structures, which may be due to the overlapping of the Si–O–Si band of CP and Ti–O band of TiO2.34 However, the existence of peaks between 400 and 1200 cm−1 in all the TiO2/CPs reflected that the skeleton structure of CP was not destroyed after hydrothermal treatment, thus supporting XRD analysis.
 | ||
| Fig. 4 (A) FT-IR spectra of (a) CP, (b) ACP3, (c) ACP6. (B) Zeta potential of (a) CP, (b) ACP6, and (c) TiO2. | ||
The FT-IR spectra of ACP1, ACP2, ACP4, ACP5 and ACP7 are shown in Fig. S2B of the ESI† section, exhibiting almost the same information as that of ACP3 and ACP6. The absorption bands centered at 430 and 740 cm−1 in TiO2 (Fig. S2B-f†) may be attributed to the bending vibration of the Ti–O–Ti bonds.34
The zeta potentials of the parent CP, bare TiO2 and TiO2/CP (ACP6) as a function of equilibrium pH of the solution in the range of 2–10 are shown in Fig. 4B. The observed variation in the zeta potentials of all samples with different pH values is due to the acid and base used to adjust the pH values of the media. The zeta potential of bare CP was found to be highly negative in the pH range studied, which is consistent with that reported by Ullah et al.14 The surface charges of TiO2 and ACP6 were found to be positive at low pH value due to the effect of highly acidic TiCl4 aqueous solution during their synthesis. The pH values of point of zero charge (pHpzc) for CP (Fig. 4B-a), bare TiO2 (Fig. 4B-c), and ACP6 (Fig. 4B-b) were found to be 2.3, 5.9, and 6.9, respectively.
The surfaces of these samples at a pH value above their pHpzc were negatively charged, whereas they were positively charged at pH below pHpzc. Therefore, it was concluded that the surface of bare CP is highly negatively charged, while the hydrothermal treatment of CP under highly acidic TiCl4 aqueous solution leads to a relatively higher value of pHpzc, i.e. 6.9 of the TiO2/CP.
The XPS spectra of ACP3 and ACP6 are shown in Fig. 5. In Fig. 5A, the XPS spectra of the survey scan exhibited that the peaks of Ti, O, C and Si were common in both ACP3 and ACP6 samples, whereas a weak extra peak of F appeared in the spectrum of ACP6. The two strong peaks at 458.22 and 463.96 eV in the high-resolution XPS spectra of Ti2p (Fig. 5B) correspond to Ti4+2p3/2 and Ti4+2p1/2, respectively, consistent with the values of Ti4+ in the TiO2 lattice. The high-resolution XPS spectra of O1s (Fig. 5C) presented two clear peaks at 529.55 and 532.96 eV, which were assigned to the metal–oxygen bond (lattice oxygen) and oxygen defects sites, respectively.35 The peak at 532.96 eV representing oxygen vacancy was stronger in ACP6 (Fig. 5C-b) as compared to ACP3 (Fig. 5C-a), indicating the significant role of F− ions in creating oxygen vacancies. Additionally, the C1s peak (Fig. 5A) arises from the adventitious hydrocarbon present in the high vacuum of the XPS instrument. The ACP6 sample (Fig. 5D-b) clearly revealed two distinct peaks at 683.96 and 687.36 eV, which correspond to the F1s level, whereas ACP3 (Fig. 5D-a) contained no such peaks. The F1s binding energy of 684 eV in Fig. 5D-b corresponds to F− adsorbed on TiO2 in ACP6, whereas the binding energy of 687 eV showed signs of F ions in the lattice of TiO2 in ACP6.36
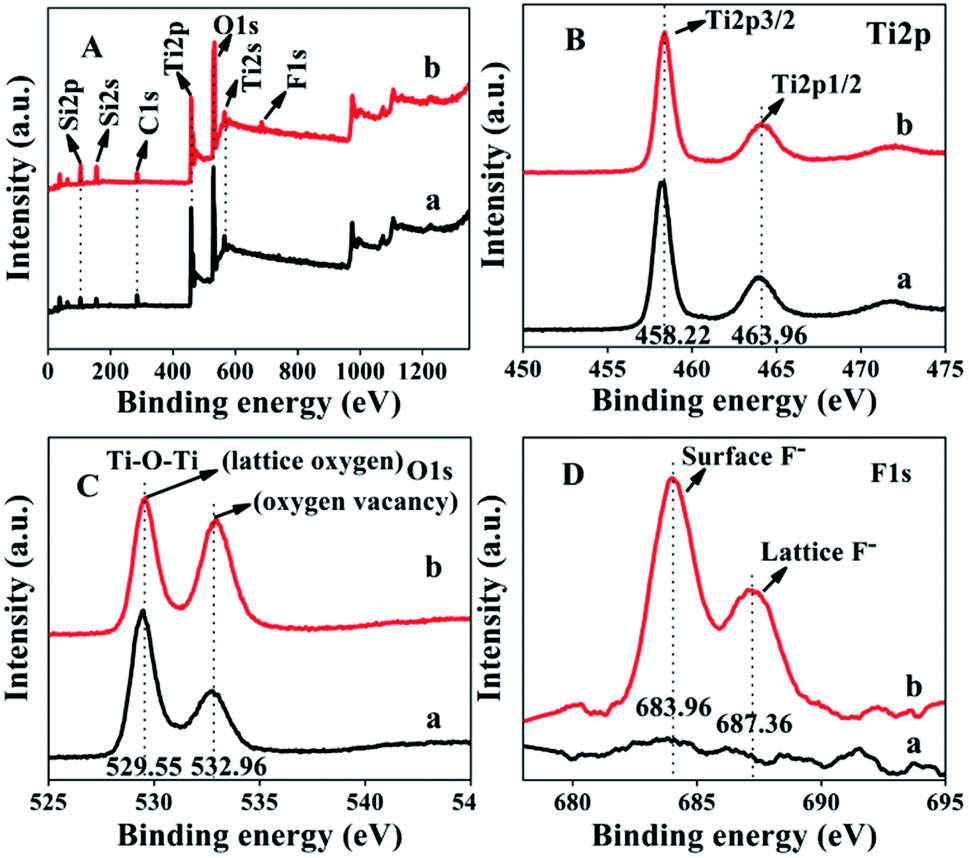 | ||
| Fig. 5 XPS spectra of the survey scan (A), Ti2p (B), O1s (C) and F1s (D). (a) ACP3 and (b) ACP6. | ||
UV-vis absorption spectra of the TiO2/CPs were obtained to estimate the optical bandgaps, which were determined using the Tauc relation given in eqn (3).
| (Ahν)n = C(hν − Eg) | (3) |
The chemical formulas of TiO2/CPs and the TiO2-loaded amount determined from ICP are summarized in Table 1. The increased Si/Al ratios along with the enhancement of the hydrothermal treatment temperature and concentrations of used F− ions are due to the excessive dealumination of the CP supports. As can be seen, although the prepared TiO2/CPs had a higher surface area than the bare CP, their BET surface areas decreased gradually as the particle size of TiO2 loaded on the surface of CPs increased from 7.8 to 17.7 nm.
Pure CP has a relatively low BET surface area of around 15.8 m2 g−1. The BET surface area was first increased in TiO2/CPs formed at a relatively low hydrothermal temperature, i.e. ACP1 and ACP2 with 193.7 and 116.3 m2 g−1, due to the relatively low particle size of loaded TiO2 (around 4.7 and 6.2 nm) in these samples. As the particle size of loaded TiO2 increased, the BET surface area decreased abruptly due to the fact that surface area and particle size were inversely related.
The pore size distribution curves of pure CP and TiO2/CPs are shown in Fig. S4 of the ESI† section. Pure CP bears pores of both micro- (pore size < 2 nm) and meso-dimensions (pore size > 2 nm), while all of TiO2/CPs were mesoporous in nature. This is due to the fact that the micropores of CP are filled by the small loaded-TiO2 nanoparticles. The broadly distributed mesopores in TiO2/CPs are due to aggregations of intra-particles during the hydrothermal procedures.
3.2. Photodegradation performances
To demonstrate the application of fabricated samples, their photocatalytic activities for the synergistic removal of CV or MO dyes from aqueous solutions were investigated under UV-irradiation. Prior to photocatalytic degradation, the dye solutions containing photocatalysts were stirred continuously for 30 minutes in the dark to reach the adsorption–desorption equilibrium. The amount of each dye adsorbed was found to be less than 5% for all the synthesized TiO2/CPs. However, it was observed that bare CP exhibited good adsorption properties for CV dye, with 51% removal, while there was poor adsorption with 2% removal for the MO dye. The effect of UV-light on the photodegradation behavior of CV and MO dyes was investigated by carrying out a blank experiment under UV-irradiation in the absence of photocatalyst. Only about 5% degradation of each dye was obtained under UV-irradiation within 180 minutes. The efficiency of the photocatalytic reaction was affected by the characteristic properties of the synthesized TiO2/CPs, such as crystal structure, particle size, surface charge, surface area, and reaction conditions like the pH of the solution, dose of catalyst, and concentration of pollutant, etc. Therefore, to elucidate the effect of operational parameters on the photodegradation performance, the reaction conditions were kept constant. The influences of various parameters on the photocatalytic activity of CV and MO dyes are thoroughly investigated and given in the following results and discussion section.The photocatalytic activities of ACP1, ACP3, ACP4, ACP6 and pure TiO2 are shown in Fig. 6, while those of ACP2, ACP5 and ACP7 are shown in Fig. S5 of the ESI† section. As can be seen, the degradation efficiencies were higher using TiO2/CPs as photocatalysts, as compared with the photolysis alone and bare CP (as shown in Fig. S6†). The photocatalytic activity increased as the hydrothermal temperature increased, in the following order: ACP1 < ACP2 < ACP3, or ACP4 < ACP5 < ACP7 < ACP6 when the concentration of F− ions (as shown in Fig. 6 and S5†) was enhanced, which was also evidenced by the values of the pseudo-first-order rate constants calculated for these catalysts (as shown in Table S2 of the ESI† section). The possible explanation for the marked differences in their photocatalytic behaviors is related to the particle size of the loaded-TiO2 and their exposed crystal facets. In detail, the ACP1, ACP2, and ACP3 exhibited relatively low photocatalytic activities, which may be due to the smaller particle size of loaded TiO2 (as shown in Table 1, calculated from XRD patterns and SEM images). Furthermore, the high-resolution TEM image of ACP3 (Fig. 3e) depicted that the exposed surfaces were dominated by the thermodynamically stable low energy {101} facet, which also accounts for their low photocatalytic activity. ACP4–ACP6 showed a linear increase in photocatalytic activity with the increase in the particle size of loaded TiO2 (as shown in Table 1). Although ACP7 had a high particle size of loaded-TiO2, as compared to ACP6, it exhibited relatively low photocatalytic activity, mainly due to the severe aggregation of loaded TiO2, as seen in SEM images (Fig. S3c†). ACP6 showed the best photocatalytic performance among all the synthesized photocatalysts with 98.7 and 96.8% degradation of CV and MO dyes, respectively. The probable reasons for the best photocatalytic performance of ACP6 are due to the more uniform distributions, suitable size of the loaded-TiO2 particles, and particularly the dominant reactive {001} exposed facet of the nanocrystals, which effectively enhanced the charge separation of the photocatalyst. Moreover, the relatively high number of oxygen vacancies in ACP6 produced by the use of F− ions, as demonstrated in XPS results (Fig. 5C-b), is another important factor for its enhanced photocatalytic performance. Therefore, ACP6 was selected as the optimum photocatalyst for further exploration.
 | ||
| Fig. 6 The effects of various catalysts on the adsorption equilibrium and photocatalytic degradation of CV (A) and MO (B) dyes: (a) ACP1, (b) ACP3, (c) ACP4, (d) ACP6, (e) pure TiO2, and (f) pure CP. Conditions: initial concentration of dye = 0.0245 mM, catalyst dose = 0.5 g L−1, pH = 6.0, room temperature. | ||
 | ||
| Fig. 7 The effects of the dose of ACP6 catalyst (A) and the initial pH of the solution (B) on the photocatalytic degradation of CV (a) and MO dyes (b). Conditions: initial concentration of dye = 0.0245 mM, time = 60 min, pH in dose study = 6.0, or catalyst dose in pH study = 0.75 g L−1, and room temperature. | ||
The pH of the reaction medium is also an important factor due to its strong influences on the charge of the catalyst.39 While keeping other conditions constant, the effect of the pH value on the photocatalytic degradation of CV and MO dyes was investigated. As shown in Fig. 7B, the degradation efficiency of both dyes first increased by raising the solution pH from 2 to 8 and thereafter decreased. In detail, the photocatalytic degradation was around 57 and 48% for CV and MO, respectively, at a low pH value of around 2.0, but gradually increased to 85 and 79% for CV and MO with increasing the initial pH (up to 8) of the solution. As shown in Fig. 7B-b, the pHpzc value of 6.9 determined for ACP6 had a direct influence on the photocatalytic degradation of dyes. The ACP6 surface at pH < pHpzc is positively charged, which hinders the adsorption of cationic CV and MO dyes, leading to the low degradation rate.40 Furthermore, at pH < pHpzc, there is competition between the cationic dyes and H+ ions for the adsorption sites of ACP6; therefore, small amounts of CV and MO dyes with relatively low positive charges were adsorbed on the surface of the ACP6, consequently reducing the degradation efficiency.
Both dyes presented the highest degradation efficiency at pH of 8 and then a small decrease in degradation efficiency occurred at pH 10. These results are similar to those reported by Liu et al.41 The decreased degradation efficiency at pH > 8 should be due to the appearance of repulsive forces between the highly negatively charged surface of ACP6 and free electron pairs of the CV and MO dyes. Therefore, relatively fewer molecules of dyes can reach the ACP6 surface where the highly reactive ˙OH radicals are generated. Furthermore, two types of reactions might occur, such as (i) the reaction of ˙OH with ˙OH due to the presence of greater amount of ˙OH radicals;41 (ii) the reaction of ˙OH with −OH, at high pH values producing relatively less reactive species,42 and consequently reducing the percentage degradation.
The extent of mineralization of dyes can be also determined by its TOC removal efficiency analysis because complete decolorization does not mean the complete mineralization of the dye. However, on using ACP6 as a catalyst, the relatively low TOC removal (81.1%) of the CV dye after 180 minutes as compared to the decolorization percentage (98.7%) indicated that the complete mineralization of CV dye needed a longer time. Similarly, the TOC removal percentage (78.4%) of MO dye after 180 minutes was also lower than that its decolorization percentage (96.8%). The rapid decrease in the colour removal of the dye might be due to the cleavage of the chromophores, while the low percentage of TOC removal might be due to the formation of intermediates due to the presence of N atoms in both CV and MO dyes.43
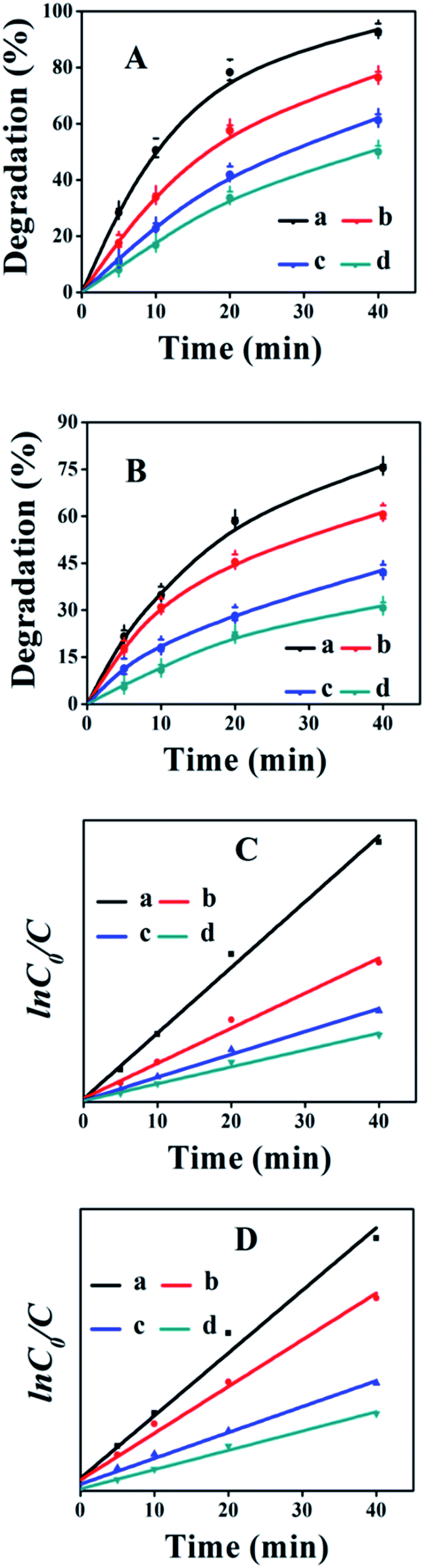 | ||
| Fig. 8 The effects of initial concentration on the photocatalytic degradation of the CV dye (A) and its corresponding kinetic behavior (C), as well as MO dye (B) and its corresponding kinetic behavior (D). (a) 0.0122, (b) 0.0245, (c) 0.0368, and (d) 0.0490 mM. Conditions: catalyst dose = 1.0 g L−1, pH = 10, room temperature. | ||
The kinetic behavior of heterogeneous photocatalytic reactions is generally described by the Langmuir Hinshelwood equation, which states that the initial degradation rate (r) of organic compounds is proportional to the surface coverage (θ),44 as follows:
| r = −dc/dt = kθ = k(KC/1 + KC) | (4) |
|
lnC0/C = kapp·t
| (5) |
The photocatalytic degradation process is initiated by the generation of ˙HO and O2˙− radicals. When the reaction mixture is illuminated by UV-visible irradiation, the electrons in the conduction band become excited and shift to the valance band. Thus, created holes in the valance band react with water molecules and form ˙HO radicals, while the electrons from the conduction band react with absorbed oxygen and generate O2˙− radicals.45 These generated reactive species (˙HO and O2˙−) are responsible for the degradation of CV dye. According to the literature,46 the degradation of CV dye mainly follows two processes, as shown in Fig. S9a,† namely (i) the demethylation pathway, and (ii) chromophore cleavage. The CV molecule undergoes demethylation and chromophores cleavage and forms DLPM (bis(4-(dimethylamino)phenyl)methanone), the main intermediate product of CV dye degradation, due to the destruction of the p–π conjugate structure of CV.46 The dihydroxylation of DLPM generates aromatic compounds, such as benzoic acid, 3,6 dimethyl benzoic acid, and hydroxybenzoic acid, which undergo a ring-opening reaction and generate aliphatic compounds such as ethylene glycol, carbonic acid, and oxalic acid. The final degradation products (CO2 and H2O) were formed via the mineralization of aliphatic compounds.
According to the literature,47,48 the proposed degradation pathway of MO dye is shown in Fig. S9b.† MO dye degradation is initiated with the attack of reactive species on the chromophore (–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–), and the cleavage of aromatic rings occurs. The aromatic rings undergo ring-opening reaction and form aliphatic small compounds that further mineralize into CO2, H2O, SO3− and NO3−.
N–), and the cleavage of aromatic rings occurs. The aromatic rings undergo ring-opening reaction and form aliphatic small compounds that further mineralize into CO2, H2O, SO3− and NO3−.
| Dye | % Degradation | ||||
|---|---|---|---|---|---|
| 1st cycle | 2nd cycle | 3rd cycle | 4th cycle | 5th cycle | |
| CV | 96 | 92 | 85 | 81 | 78 |
| MO | 93 | 90 | 84 | 79 | 76 |
4. Conclusions
The effects of the hydrothermal treatment temperature and concentration of F− ions on the structure, morphology, particle size, exposed crystal facets and photocatalytic activity of TiO2-supported clinoptilolite (TiO2/CP) were assessed. The various characterizations of obtained TiO2/CPs demonstrated that increasing temperature and concentration of F− ions caused a net decrease in their surface areas and an increase in the particle size of the loaded TiO2 along with their enhanced crystallinity degree. The presence of F− ions plays an important role in hydrothermal etching and exposing highly energetic {001} facets of the supported TiO2. The photocatalytic activity of TiO2/CPs was evaluated for the synergistic removal of CV and MO dyes under UV-irradiation in aqueous media. The high degree of crystallinity, exposed highly energetic {001} facets, uniform and fine dispersion of TiO2 in TiO2/CP (ACP6) caused by the use of F− ions reflected the higher surface density of the active sites and the separation efficiency of electron–hole pairs, which account for its best photocatalytic performance. Mutually independent exposed {001} and {101} facets of TiO2 loaded onto the CP supports provide new insight into the design and fabrication of advanced photocatalytic materials.Author contributions
Anadil Gul and Raza Ullah: equal contribution, investigation, writing original draft preparation; Tallat Munir: data curation; Jihong Sun: supervision, conceptualization, methodology; Shiyang Bai: formal analysis, validation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21878006).References
- L. Soares and A. Alves, Mater. Lett., 2018, 211, 339–342 CrossRef CAS.
- Q. Wu, H. Yang, H. Zhu and Z. Gao, Optik, 2019, 179, 195–206 CrossRef CAS.
- B. Paul, W. N. Martens and R. L. Frost, Appl. Clay Sci., 2012, 57, 49–54 CrossRef CAS.
- J. Hu, Y. Cao, K. Wang and D. Jia, RSC Adv., 2017, 7, 11827–11833 RSC.
- X. Liu, Y. Liu, S. Lu, W. Guo and B. Xi, Chem. Eng. J., 2018, 350, 131–147 CrossRef CAS.
- O. O. Prieto-Mahaney, N. Murakami, R. Abe and B. Ohtani, Chem. Lett., 2009, 38, 238–239 CrossRef CAS.
- H. Cheng, J. Wang, Y. Zhao and X. Han, RSC Adv., 2014, 4, 47031–47038 RSC.
- K. Tanaka, M. F. Capule and T. Hisanaga, Chem. Phys. Lett., 1991, 187, 73–76 CrossRef CAS.
- H. Liu, S. Liu, Z. Zhang, X. Dong and T. Liu, Sci. Rep., 2016, 6, 33839 CrossRef CAS PubMed.
- D. Kanakaraju, J. Kockler, C. A. Motti, B. D. Glass and M. Oelgemoller, Appl. Catal., B, 2015, 166, 45–55 CrossRef.
- Z. Mehrabadi and H. Faghihian, Spectrochim. Acta, Part A, 2018, 204, 248–259 CrossRef CAS PubMed.
- M. Khatamian, S. Hashemian and S. Sabaee, Mater. Sci. Semicond. Process., 2010, 13, 156–161 CrossRef CAS.
- R. Mohamed, A. Ismail, I. Othman and I. Ibrahim, J. Mol. Catal. A: Chem., 2005, 238, 151–157 CrossRef CAS.
- R. Ullah, C. Liu, H. Panezai, A. Gul, J. Sun and X. Wu, Arabian J. Chem., 2020, 13, 4092–4101 CrossRef CAS.
- A. H. Mamaghani, F. Haghighat and C. S. Lee, J. Photochem. Photobiol., A, 2019, 378, 156–170 CrossRef CAS.
- B. G. T. Keerthana, T. Solaiyammal, S. Muniyappan and P. Murugakoothan, Mater. Lett., 2018, 220, 20–23 CrossRef.
- R. Ullah, J. Sun, A. Gul and S. Bai, J. Environ. Chem. Eng., 2020, 8, 103852 CrossRef CAS.
- H. Dzinun, M. H. D. Othman and A. Ismail, Chemosphere, 2019, 228, 241–248 CrossRef CAS PubMed.
- A. Ahmadpour, A. H. Asl and N. Fallah, Part. Sci. Technol., 2018, 36, 791–798 CrossRef CAS.
- C. Z. Wen, J. Z. Zhou, H. B. Jiang, Q. H. Hu, S. Z. Qiao and H. G. Yang, Chem. Commun., 2011, 47, 4400–4402 RSC.
- M. A. Behnajady, N. Modirshahla, M. Shokri, H. Elham and A. Zeininezhad, J. Environ. Sci. Health, Part A, 2008, 43, 460–467 CrossRef CAS PubMed.
- C. B. Almquist and P. Biswas, J. Catal., 2002, 212, 145–156 CrossRef CAS.
- X. Han, X. Wang, S. Xie, Q. Kuang, J. Ouyang, Z. Xie and L. Zheng, RSC Adv., 2012, 2, 3251–3253 RSC.
- H. G. Yang, C. H. Sun, S. Z. Qiao, J. Zou, G. Liu, S. C. Smith, H. M. Cheng and G. Q. Lu, Nature, 2008, 453, 638–641 CrossRef CAS PubMed.
- N. Roy, Y. Sohn and D. Pradhan, ACS Nano, 2013, 7, 2532–2540 CrossRef CAS PubMed.
- R. A. Sene, S. Sharifnia and G. Moradi, Int. J. Hydrogen Energy, 2018, 43, 695–707 CrossRef CAS.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671 CrossRef CAS PubMed.
- H. Zabihi-Mobarakeh and A. Nezamzadeh-Ejhieh, J. Ind. Eng. Chem., 2015, 26, 315–321 CrossRef CAS.
- W. J. Ong, L. L. Tan, S. P. Chai, S. T. Yong and A. R. Mohamed, Nanoscale, 2014, 6, 1946–2008 RSC.
- T. R. Gordon, M. Cargnello, T. Paik, F. Mangolini, R. T. Weber, P. Fornasiero and C. B. Murray, J. Am. Chem. Soc., 2012, 134, 6751–6761 CrossRef CAS PubMed.
- M. Trujillo, D. Hirales, M. Rincon, J. Hinojosa, G. Leyva and F. Castillon, J. Mater. Sci., 2013, 48, 6778–6785 CrossRef CAS.
- F. Rahmani, M. Haghighi and M. Amini, J. Ind. Eng. Chem., 2015, 31, 142–155 CrossRef CAS.
- N. Davari, M. Farhadian, A. R. S. Nazar and M. Homayoonfal, J. Environ. Chem. Eng., 2017, 5, 5707–5720 CrossRef CAS.
- S. Bagheri, Z. A. Mohd Hir, A. Termeh Yousefi and S. B. Abd Hamid, Desalin. Water Treat., 2016, 57, 10859–10865 CrossRef CAS.
- L. Wu, L. Shi, S. Zhou, J. Zhao, X. Miao and J. Guo, Energy Technol., 2018, 6, 2350–2357 CrossRef CAS.
- H. Park and W. Choi, J. Phys. Chem. B, 2004, 108, 4086–4093 CrossRef CAS.
- A. Nezamzadeh-Ejhieh and H. Zabihi-Mobarakeh, J. Ind. Eng. Chem., 2014, 20, 1421–1431 CrossRef CAS.
- A. Nezamzadeh-Ejhieh and Z. Banan, Desalination, 2012, 284, 157–166 CrossRef CAS.
- A. Nezamzadeh-Ejhieh and M. Amiri, Powder Technol., 2013, 235, 279–288 CrossRef CAS.
- A. Nezamzadeh-Ejhieh and A. Shirzadi, Chemosphere, 2014, 107, 136–144 CrossRef CAS PubMed.
- A. L. Liu, K. Wang, W. Chen, F. Gao, Y. S. Cai, X. H. Lin, Y. Z. Chen and X. H. Xia, Electrochim. Acta, 2012, 63, 161–168 CrossRef CAS.
- A. Nezamzadeh-Ejhieh and S. Moeinirad, Desalination, 2011, 273, 248–257 CrossRef CAS.
- M. R. Abhilash, G. Akshatha and S. Srikantaswamy, RSC Adv., 2019, 9, 8557–8568 RSC.
- Z. Shams-Ghahfarokhi and A. Nezamzadeh-Ejhieh, Mater. Sci. Semicond. Process., 2015, 39, 265–275 CrossRef CAS.
- Y. Ju, J. Fang, X. Liu, Z. Xu, X. Ren, C. Sun, S. Yang, Q. Ren, Y. Ding and K. Yu, J. Hazard. Mater., 2011, 185, 1489–1498 CrossRef CAS.
- N. Couselo, F. S. García Einschlag, R. J. Candal and M. Jobbágy, J. Phys. Chem. C, 2008, 112, 1094–1100 CrossRef CAS.
- N. Bahrudin and M. Nawi, J. Water Process Eng., 2019, 31, 100843 CrossRef.
- S. Xie, P. Huang, J. J. Kruzic, X. Zeng and H. Qian, Sci. Rep., 2016, 6, 1–10 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra02269e |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2021 |