
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEstrogenic activity of lignin-derivable alternatives to bisphenol A assessed via molecular docking simulations†
Alice Amitrano‡
 a,
Jignesh S. Mahajan‡b,
LaShanda T. J. Korleyabc and
Thomas H. Epps III*abc
a,
Jignesh S. Mahajan‡b,
LaShanda T. J. Korleyabc and
Thomas H. Epps III*abc
aDepartment of Chemical and Biomolecular Engineering, University of Delaware, Newark, Delaware 19716, USA. E-mail: thepps@udel.edu
bDepartment of Materials Science and Engineering, University of Delaware, Newark, Delaware 19716, USA
cCenter for Research in Soft matter and Polymers (CRiSP), University of Delaware, Newark, Delaware 19716, USA
First published on 23rd June 2021
Abstract
Lignin-derivable bisphenols are potential alternatives to bisphenol A (BPA), a suspected endocrine disruptor; however, a greater understanding of structure–activity relationships (SARs) associated with such lignin-derivable building blocks is necessary to move replacement efforts forward. This study focuses on the prediction of bisphenol estrogenic activity (EA) to inform the design of potentially safer BPA alternatives. To achieve this goal, the binding affinities to estrogen receptor alpha (ERα) of lignin-derivable bisphenols were calculated via molecular docking simulations and correlated to median effective concentration (EC50) values using an empirical correlation curve created from known EC50 values and binding affinities of commercial (bis)phenols. Based on the correlation curve, lignin-derivable bisphenols with binding affinities weaker than ∼−6.0 kcal mol−1 were expected to exhibit no EA, and further analysis suggested that having two methoxy groups on an aromatic ring of the bio-derivable bisphenol was largely responsible for the reduction in binding to ERα. Such dimethoxy aromatics are readily sourced from the depolymerization of hardwood biomass. Additionally, bulkier substituents on the bridging carbon of lignin-bisphenols, like diethyl or dimethoxy, were shown to weaken binding to ERα. And, as the bio-derivable aromatics maintain major structural similarities to BPA, the resultant polymeric materials should possess comparable/equivalent thermal (e.g., glass transition temperatures, thermal decomposition temperatures) and mechanical (e.g., tensile strength, modulus) properties to those of polymers derived from BPA. Hence, the SARs established in this work can facilitate the development of sustainable polymers that maintain the performance of existing BPA-based materials while simultaneously reducing estrogenic potential.
1. Introduction
Bisphenol A (BPA) is one of the most important precursors to an array of polymeric systems, such as epoxy resins, polysulfones, and polycarbonates, among others.1–4 These polymeric materials are used extensively in numerous applications, particularly for biomedical and food contact products (e.g., dental cement composites, food containers, infant bottles).1,4–6 However, BPA is known to leach from polymeric products and can potentially enter the human body.5,7–9 BPA also is a suspected endocrine disruptor compound because it can mimic the natural estrogen hormone 17β-estradiol (E2) by binding to estrogen receptor alpha (ERα).5,6,9–13 The disruption of the endocrine system can lead to severe human health impacts, including diabetes,14 obesity,15 cardiovascular diseases,16 reproductive disorders,17 and cancer.2,18 Therefore, several countries have restricted or eliminated BPA use in food contact applications.19–21 In fact, the European Chemicals Agency has listed BPA in its “Candidate List of Substances of Very High Concern”.22 In an attempt to address this issue, a next generation of materials was developed and marketed as “BPA-free,” yet these systems often contain BPA alternatives like bisphenol F (BPF), tetramethyl bisphenol A (TMBPA), tetramethyl bisphenol F (TMBPF), or bisphenol S (BPS) that may still have endocrine disruption capability.23 Thus, there remains a need for alternatives to BPA that possess minimal or no estrogenic activity (EA).Lignin is the most abundant feedstock for potential biobased aromatic chemicals and serves as a platform for the development of a wide range of renewable polymers.1,2,4,24–36 Bulk lignin mainly consists of three subunits: p-hydroxyphenyl (H) (no methoxy groups on the aromatic ring), guaiacyl (G) (one methoxy group on the aromatic ring [monomethoxy]), and syringyl (S) (two methoxy groups on the aromatic ring [dimethoxy]) groups.1,2,4,33,37 The compositions of these units can vary as a function of biomass source: softwoods contain a majority of G units, hardwood include a mixture of G and S units, and grasses contain a mixture of H, G, and S units.2,4,37 Additionally, the structural heterogeneity38 and limited reactivity39 of bulk lignin can lead to materials with inconsistent properties, and the odor and dark color of bulk lignin limit its use to lower-value applications.2,38 Several methods, such as pyrolytic, catalytic, and enzymatic depolymerization, can break down bulk lignin into smaller, well-defined aromatics with useful moieties, particularly phenolic hydroxyl and methoxy units.37,40–46 These inherent functionalities of lignin aromatics can be leveraged to generate performance-advantaged polymeric materials (i.e., polymers with the improved properties relative to those of incumbent systems). Moreover, the methoxy groups on lignin-aromatics are likely helpful to mitigate the toxicity concerns of conventional aromatics.5,6
Recently, lignin-derivable bisguaiacols (renewable bisphenols) have been reported as potentially safer alternatives to commercial bisphenols,2,10,11,47–49 with the suggestion being that their methoxy substituents ortho to the hydroxyl group sterically reduce binding to estrogen receptors mediated by those phenolic hydroxyls.5,6,10 Additionally, these bisguaiacol-based polymeric systems retained desirable thermomechanical properties.47 For example, 4,4-methylenedianiline (MDA)-cured diglycidyl ethers of the bisguaiacols exhibited glass transition temperatures (Tgs) between ∼111 °C and ∼151 °C, 5 wt% degradation temperatures (Td5%s) between ∼344 °C and ∼368 °C, and glassy storage moduli (E′) between ∼2.0 GPa and ∼3.2 GPa at 30 °C.47 These thermomechanical properties were comparable to those of the MDA-cured diglycidyl ether of BPF (Tg ∼ 138 °C; Td5% ∼ 375 °C; E′ ∼ 2.4 GPa at 30 °C) likely because of the structural similarities of the bisguaiacols to BPF.47 Notably, these properties (i.e., Tg, Td5%, and E′) were slightly lower than those of the MDA-cured diglycidyl ether of BPA (Tg ∼ 167 °C; Td5% ∼ 381 °C; E′ ∼ 2.5 GPa at 30 °C).47 The differences in thermal and mechanical properties between bisguaiacol-based polymeric systems and BPA-based analogues are likely a result of the absence of substituents on the bridging carbon in common bisguaiacols, allowing free rotation of the resultant polymer backbone.2 By incorporating substituents such as dimethyl, diethyl, or dimethoxy on the bridging carbon, the Tgs of these renewable polymeric systems can be increased to the point of being comparable to those of the BPA variety.2 However, it is also important to investigate the structure–activity relationships (SARs) of the lignin-derivable bisphenols with respect to ERα as a function of both methoxy-group content on the aromatic rings and substituents on the bridging carbon to streamline materials design.
To this end, computational approaches often can be a rapid,50 cost-effective,51 and less materials-intensive method for compound investigation.52 For instance, in silico techniques such as molecular docking are applied widely in the pharmaceutical and food industries to predict ligand–target interactions and SARs.53–55 Molecular docking simulations can follow a structure-based design procedure that uses statistical free-energy perturbation (FEP) calculations.53,54 FEP computations can enable evaluation of the effects of structural differences on the affinity of a bisphenol to ERα.53 The typical outputs are a docking pose for the selected bisphenol and a measurement of the binding strength (or binding affinity) of a bisphenol to binding sites of the receptor.55 A stronger binding to ERα indicates the formation of a more stable complex between the ERα and phenolic hydroxyl groups of the bisphenol due to interactions, such as hydrogen bonding.55–58 Hence, by investigating the binding affinities of bisphenols with ERα, SARs of lignin-derivable bisphenols to ERα can be established.
The aim of this study is to investigate the SARs of lignin-derivable bisphenols with respect to ERα to understand how the EA is impacted by the methoxy-group content on the aromatic rings and by the nature of the bridging group substituents of these bisphenolic compounds. To achieve this goal, first, an empirical correlation curve was developed to predict the median effective concentration (EC50) of lignin-derivable compounds using the known EC50 values of several petroleum-derivable (bis)phenols. The appropriate data were obtained from an in vitro study,59 and the associated binding affinities were calculated via molecular docking simulations. Second, the influence of methoxy-group content on bio-derivable bisphenol interactions with ERα was evaluated to elucidate the significance of phenolic aromatics (hardwood-derivable versus softwood-derivable) on EA. Furthermore, the role of the substituents on the bridging carbon (e.g., dimethyl, diethyl, or dimethoxy) of these bisphenols on EA was studied to assess their effect on toxicity. The EAs of the lignin-derivable bisphenols were examined along with those of BPA and commercial alternatives, such as TMBPA, TMBPF, and BPS, to benchmark the toxicity of lignin-bisphenols. Finally, the implications with respect to structure–property relationships in next-generation, renewable polymers are briefly discussed to guide the design of new materials.
2. Experimental
2.1. Compounds analyzed
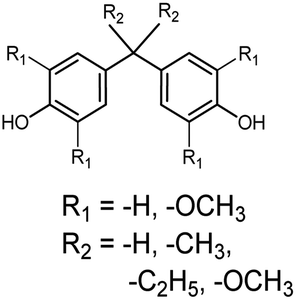
Bisphenols as a function of methoxy-group content (from 0 to 4) and with an unsubstituted, dimethyl-substituted, diethyl-substituted, or dimethoxy-substituted bridging carbon were analyzed, and are summarized in Table 1. The nomenclature of the compounds in this study describes the number of methoxy substituents on the aromatic rings and the type of bridging functional group. For example, BP(0,1)(Un) denotes a bisphenol with zero methoxy groups on the right-most aromatic ring, one methoxy group on the left-most aromatic ring, and an unsubstituted bridging carbon. Similarly, BP(1,2)(Me) indicates a bisphenol with one methoxy group on the right-most aromatic ring, two methoxy groups on the left-most ring, and a dimethyl-substituted bridging carbon. Additional molecules are described in a similar fashion using Et to represent diethyl substituents and MeO to represent dimethoxy substituents on the bridge.| General structure
|
Unsubstituted (Un) bridging carbon | Dimethyl-substituted (Me) bridging carbon | Diethyl-substituted (Et) bridging carbon | Dimethoxy-substituted (MeO) bridging carbon |
|---|---|---|---|---|
| Base bisphenols | 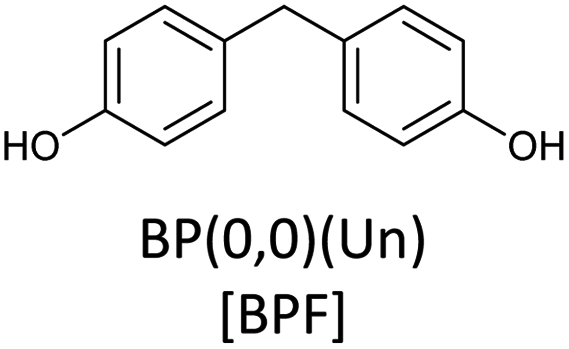 |
 |
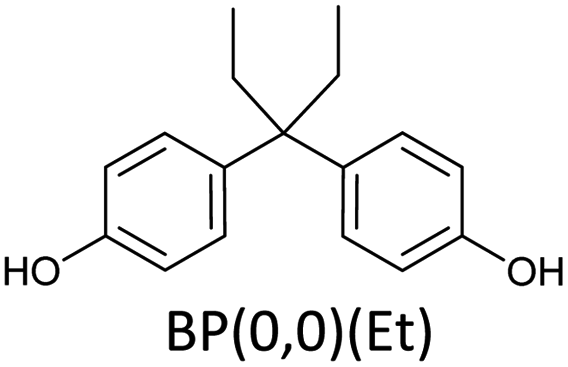 |
 |
| Lignin-derivable bisphenols | 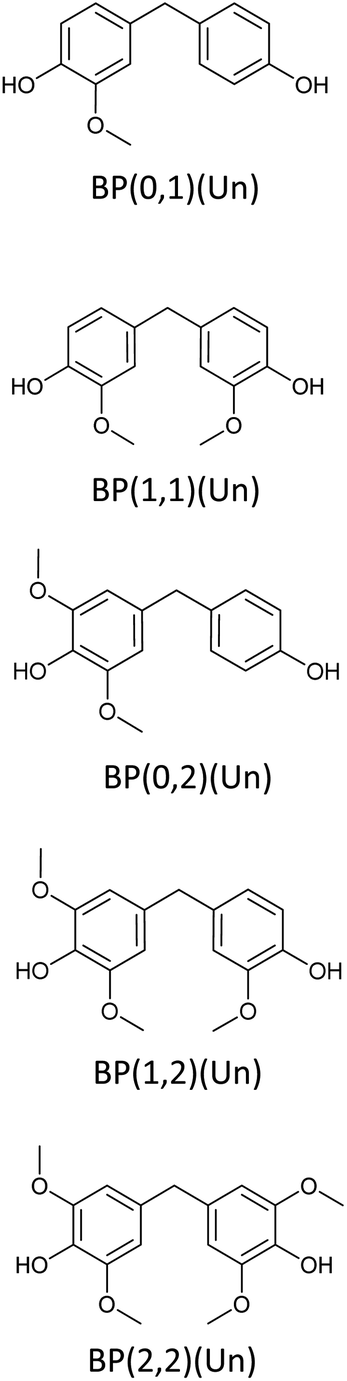 |
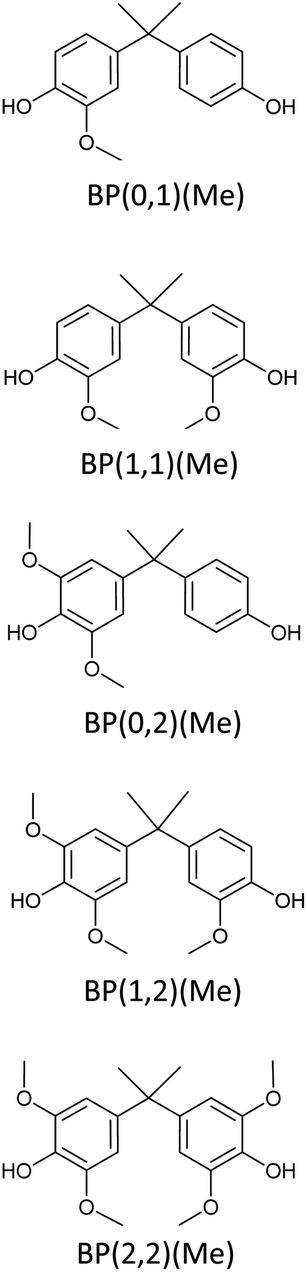 |
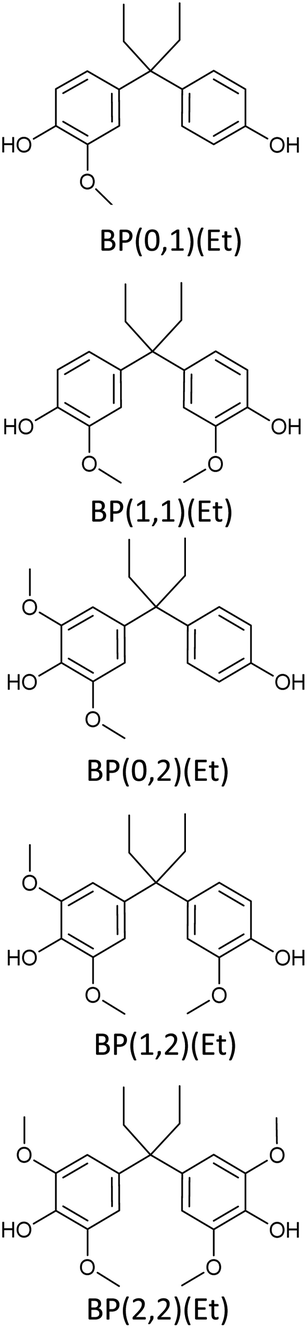 |
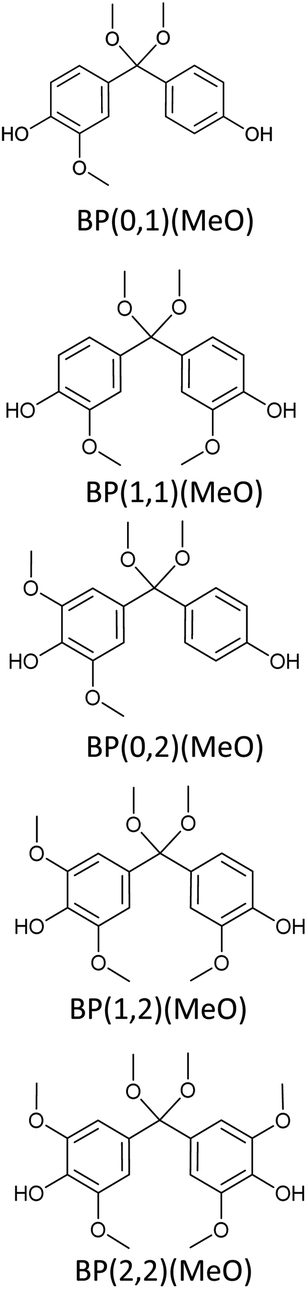 |
2.2. Receptor and ligand preparation
The structure of ERα was obtained from the Protein Data Bank (1A52)60–62 as a PDB file, and its bonding ligand, E2, was deleted from the receptor using the software, Chimera 1.15.63 The ERα was prepared for docking using AutoDockTools 1.5.6,64 and water molecules were deleted from the estrogen receptor to avoid distortion of the pose search.65 Subsequently, polar hydrogens were added to the ERα, as the AutoDock Vina 4.0 software66 uses the United Atom Model to represent molecules, which considers polar hydrogens for calculations.65–67 Additionally, all Gasteiger charges were computed because the software scoring function was calibrated using Gasteiger charges on the receptor.65 The PDB file was converted to a PDBQT format that could be used later to calculate the binding affinities.Common ligands were downloaded from the Protein Data Bank62 and saved as PDBQT files using AutoDockTools.64 For cases in which the necessary molecules were not available in the Protein Data Bank, the molecules were drawn using the ChemDraw 19.0 software package68 or ACD ChemSketch Freeware Version,69 converted to three-dimensional structures, and saved as PDBQT files using Open Babel 2.3.1.70 The respective molecular structures then were opened in AutoDockTools, and the polar hydrogen atoms were added to the respective structures.67 Subsequently, the Gasteiger charges were computed so that the software's scoring function could be calibrated.67 Additionally, the torsion tree was defined to set the rigid and rotatable portions of the molecule because AutoDock Vina requires that information for the docking algorithm.71 Finally, the structures of the respective bisphenols were saved as PDBQT files.
2.3. Molecular docking calculations
The most common binding sites of ERα to E2 were selected from the literature.72,73 The binding sites were confirmed with the Schrodinger 2020-3 software package,74 which identifies the ligand sites of a specific protein once a molecule is inserted in the estrogen receptor. These binding sites corresponded to: methionine (MET) 343, leucine (LEU) 346, threonine (THR) 347, LEU 349, alanine (ALA) 350, glutamic acid (GLU) 353, tryptophan (TRP) 383, LEU 384, LEU 387, MET 388, LEU 391, arginine (ARG) 394, phenylalanine (PHE) 404, MET 421, LEU 428, and LEU 525. A grid box large enough to cover all active binding sites, 54 Å × 62 Å × 56 Å, was built in AutoDockTools, and a text file containing all of the grid box coordinates was created. The binding affinities in kcal mol−1 were calculated with AutoDock Vina. The software displayed nine binding affinity values, which ranged from the lowest to the highest root mean square distance (RMSD) between a hydroxyl group on the bisphenol and the binding site on the ERα. The binding affinity with the lowest RMSD value was chosen as it represents the most favorable binding interaction.75,76 Finally, the binding positions and orientations were visualized using PyMOL 4.6,77 by opening the output PDBQT file created from AutoDock Vina.66,772.4. Correlation curve to relate binding affinities to toxicity
Toxicity data on the EA of E2, petroleum-based phenols, and bisphenols were collected from the literature.59 The EC50 values of E2, BPA, BPF, 4,4-cyclohexidelene bisphenol, 4,4-ethylidenebisphenol, 4,4-dihidroxybenzophenone, 4-(1-adamantyl)phenol, 4-tert-octylphenol, 4-benzyloxyphenol, 4-hydroxyoctanophenone, 4-octylphenol, 4-butoxyphenol, 4-hydroxypropiophenone, 4-propoxyphenol, and 4-propylphenol are listed in Table S1.†59 Additionally, the binding affinities of these compounds, were calculated using molecular docking simulations in AutoDock Vina. The specific binding affinity values of E2 and these (bis)phenols also are provided in Table S1.† The logarithms of the inverse of the EC50 [log(EC50−1)] data were plotted against the binding affinities for all the compounds, and a linear relationship was obtained, as shown in Fig. 1a. This empirical curve was used to predict the log(EC50−1) of the lignin-derivable bisphenols from the binding affinities calculated via molecular docking, as shown in Fig. 1b.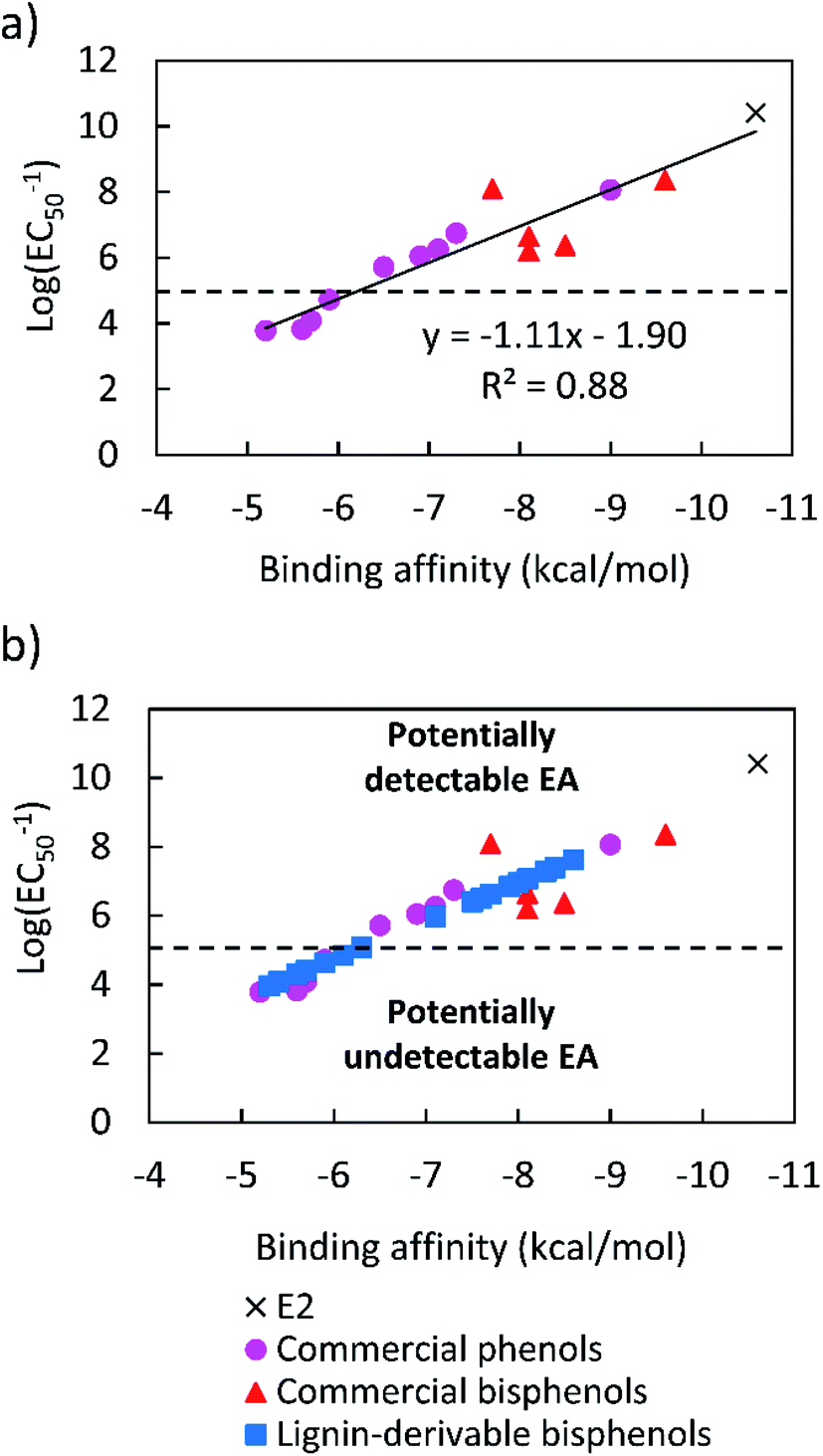 | ||
| Fig. 1 (a) Correlation curve for log(EC50−1) values of E2, commercial phenols, and bisphenols generated from in vitro data,59 and (b) predicted log(EC50−1) values of lignin-derivable bisphenols estimated from their binding affinities, which were calculated from the linear relationship shown in panel a. The dashed line at log(EC50−1) = 5, represents a cutoff value of 10 μm. The region above the dashed line shows compounds with estrogenic potential, and the region below the dashed line indicates potentially ‘safe’ compounds.59,78 | ||
2.5. SARs of lignin-derivable bisphenols
The binding affinities of several bisphenol compounds were computed using molecular docking simulations. The targets were chosen by varying the number of aromatic methoxy-substituents from 0 to 4, as well as the nature of the bridging carbon substituents (Fig. 2a). First, the binding affinities of compounds lacking methoxy groups on the aromatic rings [i.e. (0,0)] were calculated to set a benchmark for commercial bisphenols, such as BPF and BPA. Second, lignin-derivable bisphenols with a methoxy group on one ring or both rings [i.e. (0,1), (1,1)] adjacent to the phenolic hydroxyl groups were examined to determine the impact of monomethoxy aromatics on the reduction of EA. Third, the influence on binding affinities of lignin-derivable bisphenols with two methoxy moieties on one aromatic ring [i.e. (0,2)] was studied to elucidate the significance of dimethoxy aromatics on estrogenic potential. Fourth, by keeping one hydroxyl group hindered and further increasing the methoxy-group content around the other phenolic hydroxyl [i.e. (1,2)], the effect of the increased methoxy groups on the EA was evaluated. Fifth, the binding affinity of bisphenols with dimethoxy substituents around both phenolic hydroxyl groups [i.e. (2,2)] was assessed to provide insights about the steric bulk created near binding sites. Finally, the role of the substituents on the bridging carbon (e.g., unsubstituted, dimethyl, diethyl, or dimethoxy) of these bisphenols on EA was investigated within each of the above-mentioned parameter sets to understand the function of these bridging substituents on estrogen binding. | ||
| Fig. 2 (a) The binding affinities of bisphenols as a function of methoxy-group content from 0 to 4: (b) unsubstituted bridging carbon, (c) dimethyl-substituted bridging carbon, (d) diethyl-substituted bridging carbon, and (e) dimethoxy-substituted bridging carbon. The dashed line at −8.0 kcal mol−1 represents compounds with a potential for significant EA, and the dashed line at −6.0 kcal mol−1 indicates potentially ‘safe’ compounds. | ||
2.6. Water/octanol partition coefficient (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) P) estimations
P) estimations
The chemical structure of each individual bisphenol was an input to the online platform http://chemicalize.org by ChemAxon 2020,79 which displays atoms in MarvianSpace.79 Subsequently, the log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P value for the respective bisphenol was generated. Similarly, the logP value for each isomer was predicted after inputting the respective chemical structure. The logP results in http://chemicalize.org were predicted from a pool of predefined group/fragments, as per existing data in Viswanadhan et al.80
P value for the respective bisphenol was generated. Similarly, the logP value for each isomer was predicted after inputting the respective chemical structure. The logP results in http://chemicalize.org were predicted from a pool of predefined group/fragments, as per existing data in Viswanadhan et al.80
3. Results and discussion
3.1. Correlation curve to relate in silico binding affinities with EC50 values
A correlation curve was generated from known EC50 values,59 and the binding affinities of commercial (bis)phenols were calculated using AutoDock Vina software (Fig. 1a). As the binding affinities are empirically related to the EC50 values, the correlation curve enabled the establishment of an EC50 cutoff value of 10 μM (∼25 times higher than that of BPA [EC50 = 0.42 μM]), above which, the EA could be considered undetectable.59,78,81 As such, binding affinities weaker than ∼−6.0 kcal mol−1 are related to EC50 values higher than 10.0 μM.59,78,81 Molecular docking simulations were used to calculate the binding affinities of twenty bio-derivable bisphenols to ERα. All compounds bonded to the ERα via their hydroxyl group, primarily at the ARG and GLU ligand sites, as presented in Table S2.† The results, i.e., the binding affinities of bisphenols, were placed on the correlation curve, and the EC50 values of lignin-derivable bisphenols were estimated as shown in Fig. 1b. It is worth noting that BPA had a binding affinity of −8.5 kcal mol−1, whereas eight of the lignin-derivable bisphenols, i.e., BP(1,2)(Me), BP(2,2)(Me), BP(0,2)(Et), BP(1,2)(Et), BP(2,2)(Et), BP(0,2)(MeO), BP(1,2)(MeO), and BP(2,2)(MeO), had binding affinities between ∼−6.0 kcal mol−1 and −5.3 kcal mol−1. These affinities corresponded to EC50 values that were greater than 10.0 μM [i.e., log(EC50−1) lower than 5.0], suggesting that these bio-derivable biphenolic monomers are most likely to exhibit undetectable EA in in vitro studies.59,78,81 With this framework, we examine the SARs of lignin-derivable to understand how the EA was impacted by the methoxy-group content and the bridging group substituents of these monomers.3.2. SARs of lignin-derivable bisphenols
Bisphenols with no methoxy groups on the rings, such as BP(0,0)(Un) [BPF], BP(0,0)(Me) [BPA], BP(0,0)(Et), and BP(0,0)(MeO), had binding affinities to ERα of −7.7 kcal mol−1, −8.5 kcal mol−1, −8.3 kcal mol−1, and −7.6 kcal mol−1, respectively. These compounds were used to assess how changing the methoxy-group content of the bisphenols affected binding affinities. The binding affinities to ERα for bisphenols with (0,1) and (1,1) methoxy groups on the aromatic rings were nearly equivalent to their (0,0) analogues. For example, BP(0,0)(Un) had a binding affinity of −7.7 kcal mol−1, BP(0,1)(Un) had a binding affinity of −7.9 kcal mol−1, and BP(1,1)(Un) had a binding affinity of −8.0 kcal mol−1. The different substituents at the bridging carbon of these compounds also did not significantly affect the binding affinity. For instance, BP(0,1)(Un), BP(0,1)(Me), BP(0,1)(Et), and BP(0,1)(MeO) had binding affinities of −7.9 kcal mol−1, −8.4 kcal mol−1, −8.1 kcal mol−1 and −7.7 kcal mol−1, respectively. Therefore, bisphenols with one methoxy group on the rings [i.e. (0,1), (1,1)] likely may not possess enough steric hindrance around the phenolic hydroxyl groups to limit access to the appropriate regions within ERα.Interestingly, the compounds with two methoxy groups on a single aromatic ring [e.g., (0,2)] showed significantly weaker binding to ERα than those with one methoxy group on each aromatic ring [e.g., (1,1)]. For instance, BP(0,2)(Un) had a binding affinity of −7.1 kcal mol−1, and BP(1,1)(Un) had a binding affinity of −8.0 kcal mol−1 (Fig. 2b). As another example, BP(0,2)(Me) had a binding affinity of −7.6 kcal mol−1 and BP(1,1)(Me) had a binding affinity of −8.6 kcal mol−1 (Fig. 2c). Similarly, BP(0,2)(Et) possessed a binding affinity of −5.9 kcal mol−1, whereas BP(1,1)(Et) had a binding affinity of −7.7 kcal mol−1 (Fig. 2d), and BP(0,2)(MeO) exhibited a binding affinity of −5.7 kcal mol−1, while BP(1,1)(MeO) had a binding affinity of −7.5 kcal mol−1 (Fig. 2e).
Thus, the two methoxy moieties on one aromatic ring appear to be important in the reduction of binding affinities to ERα, and these dimethoxy aromatics are a signature of hardwood biomass. As highlighted in the above results, the two methoxy groups on one aromatic ring (0,2) may severely hinder the accessibility of one phenolic hydroxyl group to the docking ligands. On the other hand, in the case of one methoxy group on each aromatic ring (1,1), both the phenolic groups can still be accessible to ERα. However, it is noted that for BP(0,2)(Un), the lack of substitution at the bridging carbon may leave the molecule sterically unencumbered enough to allow the remaining phenolic hydroxyl access to the binding pockets. Therefore, BP(0,2)(Un) [binding affinity of −7.1 kcal mol−1] had a significantly stronger binding with ERα in comparison to bisphenols with bulkier substituents, such as BP(0,2)(Et) [binding affinity of −5.9 kcal mol−1] or BP(0,2)(MeO) [binding affinity of −5.7 kcal mol−1].
To probe the impact of the increased methoxy-group content on the EA, the binding affinities of bisphenols with (0,2) methoxy groups on the aromatic rings were compared with those of their (1,2) analogues. The results suggested that, binding affinities of compounds with (0,2) methoxy groups were stronger than those of BP(1,2)(Me), BP(1,2)(Et), and BP(1,2)(MeO), respectively. In contrast, bisphenols with an unsubstituted bridging carbon showed an opposite trend. For example, BP(0,2)(Un) had a binding affinity of −7.1 kcal mol−1, and BP(1,2)(Un) a binding affinity of −7.5 kcal mol−1.
Following from the above results, an increase in methoxy-group content on the aromatic rings should increase steric bulk around phenolic hydroxyl groups and thus reduce the interaction with binding sites. However, in the case of bisphenols with no bridging substitution, the incorporation of a single methoxy substituent on the left-most ring produces insufficient steric hindrance [i.e., BP(1,2)(Un)] to prevent phenolic access to ERα. This reasoning could explain why BP(1,2)(Un) did not follow a similar trend of reduced binding affinity with increasing methoxy-group content in comparison to the other (0,2) analogues with a substituted bridging carbon.
In support of the above points, lignin-derivable bisphenols with (2,2) methoxy groups on the aromatic rings showed significantly reduced binding affinities versus bisphenols with (1,2) methoxy groups on the rings (Fig. 2a–e). For example, BP(2,2)(Un) had a binding affinity of −6.3 kcal mol−1, whereas BP(1,2)(Un) had a binding affinity of −7.5 kcal mol−1 (Fig. 2b). Moreover, BP(1,2)(Un) exhibited a greater binding affinity than comparable bisphenols with bulkier substituents (like dimethyl, diethyl, or dimethoxy) on the bridging carbon. To expound, BP(1,2)(Me), BP(1,2)(Et), and BP(1,2)(MeO) had binding affinities of −6.1 kcal mol−1, −5.4 kcal mol−1, and −5.6 kcal mol−1, respectively (Fig. 2c–e, respectively). Additionally, bisphenols with (2,2) methoxy groups, i.e., BP(2,2)(Un), BP(2,2)(Me), (BP(2,2)(Et), and BP(2,2)(MeO), showed binding affinities between −5.6 kcal mol−1 and −5.4 kcal mol−1. Together, the presence of two methoxy groups (2,2) adjacent to both the phenolic hydroxyl groups on both of the rings could be crucial to restrict access to the binding pockets via a steric hindrance pathway.
3.3. Effect of isomers on binding affinities with ERα
It is important to consider the impact of isomeric structures (i.e., the position of hydroxyl groups on bisphenols) on EA, as estrogen binding most likely occurs at phenolic hydroxyl groups. Therefore, the binding affinities of several isomers of bisphenol, such as p,p′, m,p′, o,p′, m,m′, o,o′, and o,m′, were calculated. As reported in Table S3,† many of the isomers had a difference of less than 0.6 kcal mol−1 in their binding affinities within the same chemical-formula family. However, in the case of bisphenols with at least one of the methoxy moieties on the rings, there were a few exceptions, e.g., those with more than a 1.5 kcal mol−1 difference in binding affinities, such as between m,p′-BP(1,1)(Et) [binding affinity of −8.6 kcal mol−1] and o,m′-BP(1,1)(Et) [binding affinity of −6.9 kcal mol−1]. As another example, p,p′-BP(1,1)(Un) [binding affinity of −8.0 kcal mol−1] and m,p′-BP(1,1)(Un) [binding affinity of −7.1 kcal mol−1, each] had ∼1.0 kcal mol−1 variation in binding affinities. This behavior also is in agreement with an in vitro study,6 in which an isomeric mixture of BP(1,1)(Un) exhibited different EA as per relative content of p,p′, m,p′, and o,p′-isomers.6 Additionally, as per an in vitro study conducted using an MCF-7 cell proliferation assay and a VM7Luc4E2 transactivation (reporter gene) test,6 BP(1,1)(Un) likely resulted in lower EA in comparison to BPA owing to the inherent methoxy groups on lignin-aromatics. A few additional isomer sets with significant differences in binding affinities can be found in Table S3.† Thus, certain lignin-derivable bisphenols can influence estrogen binding depending upon the position of phenolic hydroxyl groups. As there was no general trend of a particular isomer being less or more toxic, it is difficult to comment on the exact mechanism from the dataset herein.3.4. Impact of logP values on binding affinities with ERα
The logP values reflect the hydrophobicity of the bisphenols, which also may play a role in interactions with ERα.82–85 More positive logP values are associated with greater hydrophobicity, and more negative logP values are suggestive of stronger hydrophilicity.86–89 It is expected that the more hydrophobic a bisphenol, the greater the probability that it will interact with ERα.84,90 Therefore, the logP values of all the bisphenols and their respective isomers were calculated using http://chemicalize.org by ChemAxon79 and are listed in Table S3.† First, the influence of methoxy groups on the rings (from 0 to 4) on the logP values of the bisphenols was analyzed. As the number of methoxy groups on the rings increased from 0 to 4, the logP value of the respective bisphenols decreased (see Fig. S1†). For instance, the logP values for BP(0,0)(Un) and BP(2,2)(Un) were 3.4 and 2.8, respectively. Similarly, BP(0,0)(Et) had a logP value of 4.9 versus a logP value of 4.3 for BP(2,2)(Et). Additional examples can be found in Table S3.† These data suggest that the decreased hydrophobicity of these bisphenols is in agreement with the lessening of the binding affinity to ERα as per molecular docking simulations. Second, the role of the different bridging substituents on the logP values was investigated. Bisphenols with a dimethyl-substituted bridging carbon exhibited higher logP values than those with an unsubstituted bridging carbon. The logP value for BP(0,0)(Un) was 3.4, whereas BP(0,0)(Me) had a logP value of 4.0. Furthermore, bisphenols with a diethyl-substituted bridging carbon showed higher logP values than their analogues with a dimethyl-substituted bridging carbon. For example, BP(0,0)(Me) had a logP value of 4.0, and BP(0,0)(Et) had a logP value of 4.9. Thus, bisphenols with a diethyl-substituted bridging carbon are more hydrophobic than bisphenols with an unsubstituted or a dimethyl-substituted bridging carbon. Although diethyl-substituted bisphenols exhibited the highest logP values (in comparison to other bisphenols), the increased hydrophobicity alone did not significantly impact the binding affinities, as the bisphenols with diethyl-substituted bridging carbon had lower binding affinities than those with a dimethyl-substituted bridging carbon. This behavior suggests that the docking mechanism may not be dictated by hydrophobicity alone,91 without the consideration of the steric effects that appear to be the major factor in the reduction of EA.
3.5. Toxicity comparison of lignin-bisphenols and commercial BPA alternatives
Finally, the EAs of the lignin-derivable bisphenols were benchmarked against those of BPA and commercial alternatives, such as TMBPA, TMBPF, and BPS, which had binding affinities between −8.3 kcal mol−1 and −7.9 kcal mol−1 (Fig. 3). Several renewable bisphenols, such as BP(1,2)(Me), BP(2,2)(Me), BP(0,2)(Et), BP(1,2)(Et), BP(2,2)(Et), BP(0,2)(MeO), BP(1,2)(MeO), and BP(2,2)(MeO), exhibited much lower binding affinities, between ∼−6.0 kcal mol−1 and −5.3 kcal mol−1, than their commercial counterparts. The reduced binding strength, and resulting decreased EA potential, in the lignin-derivable compounds is likely because of the restricted interactions of ERα with phenolic hydroxyl groups as a result of the methoxy moieties adjacent to those phenolic hydroxyls. Similarly, bulkier substituents on the bridging carbon of lignin-bisphenols, like diethyl or dimethoxy, could provide additional steric bulk to lower the binding affinities to ERα. Therefore, increased steric hindrance in lignin-bisphenols may be a necessary factor in the design of potentially “safe” BPA alternatives. Additionally, as most of these lignin-derivable aromatics are structural analogues to BPA, resulting next-generation polymers may offer comparable/equivalent thermal (Tg, Td5%) and mechanical properties (E′) to polymers derived from commercial bisphenols.2,47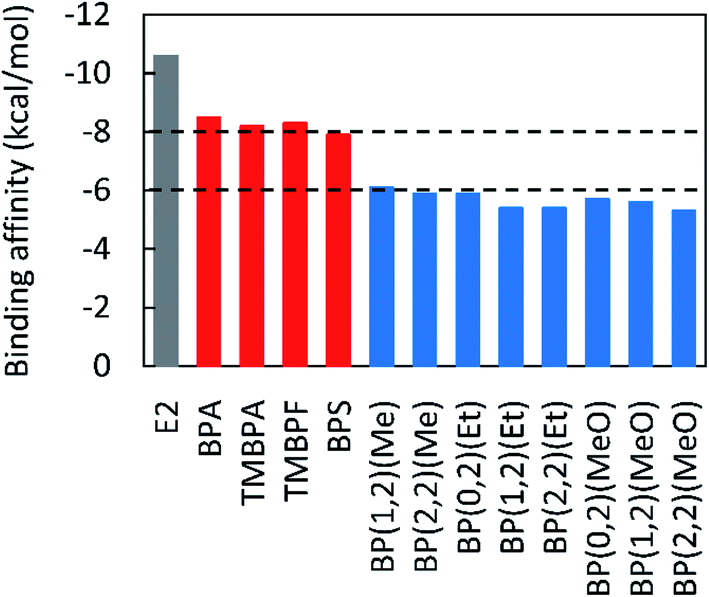 | ||
| Fig. 3 Binding affinities of E2 (grey), BPA and commercially available BPA alternatives (red), and lignin-derivable bisphenols (blue). The dashed line at −8.0 kcal mol−1 represents compounds with a potential of EA, and the dashed line at −6.0 kcal mol−1 indicates potentially ‘safe’ compounds. | ||
4. Conclusion
This work discusses the SARs of lignin-derivable bisphenols as a function of inherent methoxy-group content on the aromatic rings and with various substitutions at the bridging carbon linkages. An empirical correlation curve enabled estimation of the EC50 values of lignin-derivable monomers using their binding affinities calculated via molecular docking simulations. From the correlation curve, eight of the bio-derivable bisphenols had binding affinities weaker than ∼−6.0 kcal mol−1 suggesting that they would have non-existent or minimal EA (relative to BPA), as dictated by their empirically generated EC50 values. Analysis of the SARs suggests that hardwood-derivable aromatics can be crucial in designing BPA alternatives as two methoxy groups on a single aromatic ring significantly lowered bisphenolic binding affinities to ERα. This effect likely is a result of the increased steric hindrance around binding pockets (introduced by the methoxy groups) that restricted the phenolic hydroxyl moieties from binding with the estrogen receptor. Additionally, the incorporation of dimethoxy or diethyl substituents on the bridging carbon of the lignin-bisphenols similarly reduced the binding affinities, also likely as a result of the steric effects. As these bio-derivable bisphenols maintain major structural similarities with BPA, the resultant polymeric materials should possess comparable/equivalent thermochemical properties relative to BPA-based systems. Hence, these potentially safer, lignin-based bisphenols are promising candidates as replacements for commercial bisphenols.Abbreviations
| ALA | Alanine |
| ARG | Arginine |
| BP | Bisphenol |
| BPA | Bisphenol A |
| BPF | Bisphenol F |
| BPS | Bisphenol S |
| E′ | Storage modulus |
| EA | Estrogenic activity |
| EC50 | Median effective concentration |
| Et | Diethyl-substituted |
| ERα | Estrogen receptor alpha |
| E2 | 17β-estradiol |
| FEP | Free-energy perturbation |
| G | Guaiacyl |
| GLU | Glutamic acid |
| H | p-hydroxyphenyl |
| LEU | Leucine |
| log(EC50−1) | Logarithm of the inverse of EC50 |
| logP | Octanol/water partition coefficient |
| MDA | 4,4-Methylenedianiline |
| Me | Dimethyl-substituted |
| MeO | Dimethoxy-substituted |
| MET | Methionine |
| PHE | Phenylalanine |
| RMSD | Root mean square distance |
| S | Syringyl |
| Td5% | Temperature of 5% weight loss (degradation) |
| Tg | Glass transition temperature |
| THR | Threonine |
| TMBPA | Tetramethyl bisphenol A |
| TMBPF | Tetramethyl bisphenol F |
| TRP | Tryptophan |
| Un | Unsubstituted |
Author contributions
A. Amitrano and J. S. Mahajan contributed equally.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors are grateful to the National Science Foundation (NSF GCR CMMI 1934887) awarded to T. H. E. and L. T. J. K. for financial support.References
- A. Llevot, E. Grau, S. Carlotti, S. Grelier and H. Cramail, Macromol. Rapid Commun., 2016, 37, 9–28 CrossRef CAS PubMed
.
- J. S. Mahajan, R. M. O'Dea, J. B. Norris, L. T. J. Korley and T. H. Epps, III, ACS Sustainable Chem. Eng., 2020, 8, 15072–15096 CrossRef CAS
- F. Liguori, C. Moreno-Marrodan and P. Barbaro, Chem. Soc. Rev., 2020, 49, 6329–6363 RSC
- R. M. O'Dea, J. A. Willie and T. H. Epps, III, ACS Macro Lett., 2020, 9, 476–493 CrossRef
- Y. Peng, K. H. Nicastro, T. H. Epps, III and C. Wu, Food Chem., 2021, 338, 1–9 CrossRef PubMed
- Y. Peng, K. H. Nicastro, T. H. Epps, III and C. Wu, J. Agric. Food Chem., 2018, 66, 11775–11783 CrossRef CAS PubMed
- A. V. Krishnan, P. Stathis, S. F. Permuth, L. Tokes and D. Feldman, Endocrinology, 1993, 132, 2279–2286 CrossRef CAS PubMed
- T. Geens, L. Goeyens and A. Covaci, Int. J. Hyg. Environ. Health, 2011, 214, 339–347 CrossRef CAS PubMed
- Z. Ji, J. Liu, S. Sakkiah, W. Guo and H. Hong, ACS Sustainable Chem. Eng., 2021, 9, 2433–2446 CrossRef CAS
- S.-F. Koelewijn, D. Ruijten, L. Trullemans, T. Renders, P. Van Puyvelde, H. Witters and B. F. Sels, Green Chem., 2019, 21, 6622–6633 RSC
- S. F. Koelewijn, S. Van den Bosch, T. Renders, W. Schutyser, B. Lagrain, M. Smet, J. Thomas, W. Dehaen, P. Van Puyvelde, H. Witters and B. F. Sels, Green Chem., 2017, 19, 2561–2570 RSC
- E. Zago, E. Dubreucq, J. Lecomte, P. Villeneuve, F. Fine, H. Fulcrand and C. Aouf, New J. Chem., 2016, 40, 7701–7710 RSC
- C. A. Sutton, A. Polykarpov, K. Jan van den Berg, A. Yahkind, L. J. Lea, D. C. Webster and M. P. Sibi, ACS Sustainable Chem. Eng., 2020, 8, 18824–18829 CrossRef CAS
- A. Shankar and S. Teppala, J. Clin. Endocrinol. Metab., 2011, 96, 3822–3826 CrossRef CAS PubMed
- F. S. Vom Saal, S. C. Nagel, B. L. Coe, B. M. Angle and J. A. Taylor, Mol. Cell. Endocrinol., 2012, 354, 74–84 CrossRef CAS PubMed
- D. Melzer, P. Gates, N. J. Osborn, W. E. Henley, R. Cipelli, A. Young, C. Money, P. McCormack, P. Schofield, D. Mosedale, D. Grainger and T. S. Galloway, PLoS One, 2012, 7, 1–7 CrossRef
- E. Jašarević, P. T. Sieli, E. E. Twellman, T. H. Welsh, T. R. Schachtman, R. M. Roberts, D. C. Geary and C. S. Rosenfeld, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 11715–11720 CrossRef PubMed
- S. Jenkins, J. Wang, I. Eltoum, R. Desmond and C. A. Lamartiniere, Environ. Health Perspect., 2011, 119, 1604–1609 CrossRef CAS PubMed
- NCSL Policy Update: State Restrictions on Bisphenol A (BPA) in Consumer Products, National Conference of State Legislatures, https://www.ncsl.org/research/environment-and-natural-resources/ncsl-policy-update-state-statutes-on-chemical-safety.aspx, accessed June 3, 2021 Search PubMed.
- J. C. Worch and A. P. Dove, ACS Macro Lett., 2020, 9, 1494–1506 CrossRef CAS
- C. Jehanno, J. Demarteau, D. Mantione, M. C. Arno, F. Ruipérez, J. L. Hedrick, A. P. Dove and H. Sardon, ACS Macro Lett., 2020, 9, 443–447 CrossRef CAS
- European Chemicals Agency, https://echa.europa.eu/candidate-list-table, accessed June 3, 2021.
- G. D. Bittner, M. S. Denison, C. Z. Yang, M. A. Stoner and G. He, Environ. Health, 2014, 13, 1–18 CrossRef
- S. Zhao, X. Huang, A. J. Whelton and M. M. Abu-Omar, ACS Sustainable Chem. Eng., 2018, 6, 7600–7608 CrossRef CAS
- S. Zhao and M. M. Abu-Omar, ACS Sustainable Chem. Eng., 2016, 4, 6082–6089 CrossRef CAS
- A. Llevot, P.-K. Dannecker, M. von Czapiewski, L. C. Over, Z. Söyler and M. A. R. Meier, Chem.–Eur. J., 2016, 22, 11510–11521 CrossRef CAS PubMed
- C.-H. Chen, S.-H. Tung, R.-J. Jeng, M. M. Abu-Omar and C.-H. Lin, Green Chem., 2019, 21, 4475–4488 RSC
- S. A. Miller, ACS Macro Lett., 2013, 2, 550–554 CrossRef CAS
- S. A. Miller, Polym. Chem., 2014, 5, 3117–3118 RSC
- J. Sternberg, O. Sequerth and S. Pilla, Prog. Polym. Sci., 2020, 2021(113), 1–15 Search PubMed
- M. Shen and M. L. Robertson, ACS Sustainable Chem. Eng., 2021, 9, 438–447 CrossRef CAS
- C. Gioia, G. Lo Re, M. Lawoko and L. Berglund, J. Am. Chem. Soc., 2018, 140, 4054–4061 CrossRef CAS PubMed
- C. Gioia, M. Colonna, A. Tagami, L. Medina, O. Sevastyanova, L. A. Berglund and M. Lawoko, Biomacromolecules, 2020, 21, 1920–1928 CrossRef CAS PubMed
- A. Duval and M. Lawoko, React. Funct. Polym., 2014, 85, 78–96 CrossRef CAS
- M. Fache, B. Boutevin and S. Caillol, Green Chem., 2016, 18, 712–725 RSC
- S. Fadlallah, P. Sinha Roy, G. Garnier, K. Saito and F. Allais, Green Chem., 2021, 23, 1495–1535 RSC
- W. Schutyser, T. Renders, S. Van den Bosch, S. F. Koelewijn, G. T. Beckham and B. F. Sels, Chem. Soc. Rev., 2018, 47, 852–908 RSC
- A. Vishtal and A. Kraslawski, Bioresources, 2011, 6, 3547–3568 Search PubMed
- J. Podschun, B. Saake and R. Lehnen, Eur. Polym. J., 2015, 67, 1–11 CrossRef CAS
- A. J. Ragauskas, G. T. Beckham, M. J. Biddy, R. Chandra, F. Chen, M. F. Davis, B. H. Davison, R. A. Dixon, P. Gilna, M. Keller, P. Langan, A. K. Naskar, J. N. Saddler, T. J. Tschaplinski, G. A. Tuskan and C. E. Wyman, Sci, 2014, 344, 1–12 CrossRef PubMed
- S. P. S. Chundawat, G. T. Beckham, M. E. Himmel and B. E. Dale, Annu. Rev. Chem. Biomol. Eng., 2011, 2, 121–145 CrossRef CAS PubMed
- Z. Sun, B. Fridrich, A. de Santi, S. Elangovan and K. Barta, Chem. Rev., 2018, 118, 614–678 CrossRef CAS PubMed
- H. Nguyen, R. F. DeJaco, N. Mittal, J. I. Siepmann, M. Tsapatsis, M. A. Snyder, W. Fan, B. Saha and D. G. Vlachos, Annu. Rev. Chem. Biomol. Eng., 2017, 8, 115–137 CrossRef PubMed
- M. S. Mettler, D. G. Vlachos and P. J. Dauenhauer, Energy Environ. Sci., 2012, 5, 7797–7809 RSC
- M. J. Mehta, A. Kulshrestha, S. Sharma and A. Kumar, Green Chem., 2021, 23, 1240–1247 RSC
- X. Liu, F. P. Bouxin, J. Fan, V. L. Budarin, C. Hu and J. H. Clark, ChemSusChem, 2020, 13, 4296–4317 CrossRef CAS PubMed
- K. H. Nicastro, C. J. Kloxin and T. H. Epps, III, ACS Sustainable Chem. Eng., 2018, 6, 14812–14819 CrossRef CAS
- E. D. Hernandez, A. W. Bassett, J. M. Sadler, J. J. La Scala and J. F. Stanzione, ACS Sustainable Chem. Eng., 2016, 4, 4328–4339 CrossRef CAS
- K. H. S. Reno, J. F. Stanzione III, R. P. Wool, J. M. Sadler, J. J. LaScala and E. D. Hernandez, US Pat. 15313656, 2014
- P. D. Lyne, Drug Discovery, 2002, 7, 1047–1055 CrossRef CAS
- E. Yildirim, D. Dakshinamoorthy, M. J. Peretic, M. A. Pasquinelli and R. T. Mathers, Macromolecules, 2016, 49, 7868–7876 CrossRef CAS
- K. A. Ford, ILAR J., 2017, 57, 226–233 CrossRef
- T. T. Schug, R. Abagyan, B. Blumberg, T. J. Collins, D. Crews, P. L. DeFur, S. M. Dickerson, T. M. Edwards, A. C. Gore, L. J. Guillette, T. Hayes, J. J. Heindel, A. Moores, H. B. Patisaul, T. L. Tal, K. A. Thayer, L. N. Vandenberg, J. C. Warner, C. S. Watson, F. S. vom Saal, R. T. Zoeller, K. P. O'Brien and J. P. Myers, Green Chem., 2013, 15, 181–198 RSC
- T. Clymer, V. Vargas, E. Corcoran, R. Kleinberg and J. Kostal, Green Chem., 2019, 21, 1935–1946 RSC
- J. Dakka, M. Turilli, D. W. Wright, S. J. Zasada, V. Balasubramanian, S. Wan, P. V. Coveney and S. Jha, BMC Bioinf., 2018, 19, 482 CrossRef CAS PubMed
- Ajay and M. A. Murcko, J. Med. Chem., 1995, 38, 4953–4967 CrossRef CAS PubMed
- C. Berto-Júnior, A. P. Santos-Silva, A. C. F. Ferreira, J. B. Graceli, D. P. de Carvalho, P. Soares, N. C. Romeiro and L. Miranda-Alves, Environ. Sci. Pollut. Res., 2018, 25, 26916–26926 CrossRef
- G. Ren, H. Sun, G. Li, J. Fan, Y. Wu and G. Cui, J. Mol. Struct., 2019, 1195, 369–377 CrossRef CAS
- T. W. Schultz, G. D. Sinks and M. T. D. Cronin, Environ. Toxicol., 2002, 17, 14–23 CrossRef CAS PubMed
- D. M. Tanenbaum, Y. Wang, S. P. Williams and P. B. Sigler, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 5998–6003 CrossRef CAS PubMed
- Protein Data Bank, Estrogen receptor alpha ligand-binding domain complexed to estradiol, https://www.rcsb.org/structure/1A52, accessed June 3, 2021 Search PubMed.
- RCSB Protein Data Bank, https://www.rcsb.org/, accessed June 3, 2021.
- UCSF Chimera, An extensible molecular docking system, https://www.cgl.ucsf.edu/chimera/, accessed June 3, 2021 Search PubMed
- AutoDockTools, http://autodock.scripps.edu/, accessed June 3, 2021
- Should I always use polar hydrogens?, http://autodock.scripps.edu/faqs-help/faq/should-i-always-use-polar-hydrogens, accessed June 3, 2021 Search PubMed.
- AutoDock Vina 4.0, http://vina.scripps.edu/, accessed June 3, 2021
- How should I prepare a ligand for docking with AutoDock?, http://autodock.scripps.edu/faqs-help/faq/how-should-i-prepare-a-ligand-for-docking-with-autodock, accessed June 3, 2021 Search PubMed.
- ChemDraw, https://www.perkinelmer.com/category/chemdraw, accessed June 3, 2021
- ACD/ChemSketch, https://www.acdlabs.com/resources/freeware/chemsketch/download.php, accessed June 3, 2021
- Open Babel 2.3.1, https://openbabel.org/docs/dev/Installation/install.html, accessed June 2, 2021
- What is the format of a PDBQT file?, http://autodock.scripps.edu/faqs-help/faq/faqsection_view?section=Technical%20Questions, accessed June 2, 2021 Search PubMed.
- L. Celik, J. D. D. Lund and B. Schiøtt, Chem. Res. Toxicol., 2008, 21, 2195–2206 Search PubMed
- S. Sengupta, I. Obiorah, P. Maximov, R. Curpan and V. Jordan, Br. J. Pharmacol., 2013, 169, 167–178 CrossRef CAS PubMed
- Schrödinger, https://www.schrodinger.com/, accessed June 3, 2021
- AutoDock, Lowest energy or Largest Cluster? How to evaluate docking results, http://autodock.scripps.edu/faqs-help/faq/lowest-energy-or-largest-cluster-how-to-evaluate-docking-results, accessed June 2, 2021 Search PubMed
- D. Yusuf, A. M. Davis, G. J. Kleywegt and S. Schmitt, J. Chem. Inf. Model., 2008, 48, 1411–1422 CrossRef CAS PubMed
- PyMOL, https://pymol.org/2/, accessed June 3, 2021
- V. Breinholt and J. C. Larsen, Chem. Res. Toxicol., 1998, 11, 622–629 Search PubMed
- ChemAxon, Software solutions and services for chemistry and biology, https://chemaxon.com/, accessed June 3, 2021 Search PubMed
- V. N. Viswanadhan, A. K. Ghose, G. R. Revankar and R. K. Robins, J. Chem. Inf. Comput. Sci., 1989, 29, 163–172 CrossRef CAS
- X. Ji, N. Li, S. Yuan, X. Zhou, F. Ding, K. Rao, M. Ma and Z. Wang, Ecotoxicol. Environ. Saf., 2019, 175, 208–214 CrossRef CAS PubMed
- Y. Endo, T. Iijima, Y. Yamakoshi, M. Yamaguchi, H. Fukasawa and K. Shudo, J. Med. Chem., 1999, 42, 1501–1504 CrossRef CAS PubMed
- S. Fujii, Y. Miyajima, H. Masuno and H. Kagechika, J. Med. Chem., 2013, 56, 160–166 CrossRef CAS PubMed
- W. Tong, R. Perkins, L. Xing, W. J. Welsh and D. M. Sheehan, Endocrinology, 1997, 138, 4022–4025 CrossRef CAS PubMed
- S. Zhuang, C. Zhang and W. Liu, Chem. Res. Toxicol., 2014, 27, 1769–1779 Search PubMed
- G. Tse and S. I. Sandler, J. Chem. Eng. Data, 1994, 39, 354–357 CrossRef CAS
- S.-T. Lin and S. I. Sandler, Ind. Eng. Chem. Res., 1999, 38, 4081–4091 CrossRef CAS
- A. J. D. Magenau, J. A. Richards, M. A. Pasquinelli, D. A. Savin and R. T. Mathers, Macromolecules, 2015, 48, 7230–7236 CrossRef CAS
- J. C. Foster, I. Akar, M. C. Grocott, A. K. Pearce, R. T. Mathers and R. K. O'Reilly, ACS Macro Lett., 2020, 9, 1700–1707 CrossRef CAS PubMed
- J. A. Katzenellenbogen, Environ. Health Perspect., 1995, 7, 99–101 Search PubMed
- M. P. Gleeson, J. Med. Chem., 2007, 50, 101–112 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra02170b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |