
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of 4-thiol-furanosidic uronate via hydrothiolation reaction†
Shih-Ting Ma,
Chia-Wei Lee and
Wei-Min Liu *
*
Department of Chemistry, Fu Jen Catholic University, No. 510, Zhongzheng Rd., Xinzhuang Dist., New Taipei City 24205, Taiwan, Republic of China. E-mail: 133797@mail.fju.edu.tw
First published on 21st May 2021
Abstract
Uronic acids are not only important building blocks of polysaccharides and oligosaccharides but also are widely used in the food and pharmaceutical industries. Inspired by the structure of natural products, here, we disclosed base-mediated and radical-mediated hydrothiolation reactions for the preparation of thiol-contained uronates. In comparison with base-mediated reaction, radical-mediated hydrothiolation is inefficient due to the electron-withdrawing group on the ethylene group; nevertheless, the adduct had excellent stereoselectivity at both C-4 and C-5 positions. For the alkaline approach, thiols as nucleophiles can regioselectively and stereoselectively attach to the C-4 position of Δ-4,5-unsaturated uronate with moderate to good yields. However, poor stereoselectivity at the C-5 position was observed due to retro thiol-Michael addition. After removing the protecting group of the thiol, the thiol adduct was isomerized to the furanosidic form and the 4-thiol-furanosidic uronate derivative was synthesized for the first time.
1. Introduction
Uronic acids are a group of monosaccharides including L-arabinuronic acid, D-xyluronic acid, D-galacturonic acid, D-glucuronic acid and D-mannuronic acid, and are important building blocks for polysaccharides and oligosaccharides, such as glycosaminoglycans, pectic acid, and alginate etc., which represent an important class of biologically relevant molecules (Fig. 1A).1 For example, these polysaccharides participate in the immune response,2 bacterial pathogenesis,3 and cancer progression and metastasis4 and can be found on cell surfaces as well as in the extracellular matrix of eukaryotes. Among the uronic acids, D-galacturonic acid and D-glucuronic acid, in particular, are widely used in the food and detergent industries as well in medicine.5,6 For instance, D-glucuronic acid can be used as a detergent for removing calcareous and rust deposits from metals or other surfaces owing to its ability for chelating metals. D-Galacturonic-metal complexes can also be used as potential anti-cancer drugs and as cosmetics for protecting skin from the damage of UV radiation.7,8 | ||
| Fig. 1 The structure of uronic acid contained biologically relevant molecules (A) and the general mechanism of the enzymatic β-elimination reaction (B). | ||
Enzymatic digestion and acid hydrolysis were applied for the preparation of valuable uronic acids.9 For the enzymatic approach, two enzymatic reactions, namely hydrolysis, and lytic β-elimination, are involved in the depolymerization of polysaccharides.10 The hydrolytic pathway is utilized by eukaryotic enzymes and lytic β-elimination is employed by the enzymes of bacterial or fungal. The lyase breaks the glycosidic backbone through a β-eliminative cleavage generating an Δ4,5-unsaturated uronic acid (ΔUA) at the non-reducing terminus of digested polysaccharides (Fig. 1B).
This unique feature of ΔUA provides an opportunity for the functionalization of uronic acid via base- or radical-mediated hydrotholation reaction, which are highly efficient click reactions and have been widely used in applied sciences.11 In this way, we can prepare diverse uronic acid analogs, S-linked glycosides in particular, for exploring the application and for understanding the mechanism of enzymatic reaction.12
Generally, S-, N- or C-linked glycosides as carbohydrate mimics are focused synthetic targets in biological studies and leads to drug discovery because that they display lower susceptibility to enzymatic and acid hydrolysis.13,14 However, S-linked glycosides compared with N- and C-linked glycosides are more attractive due to ease preparation.15 Although the bond length of the C–S single bond is longer than the C–O single bond, the C–S–C bond angle is significantly smaller than the C–O–C angle, which often results in relatively small differences between the position of the carbon atoms of the glycosidic linkage.16 Besides, S-linked glycosides represent a similar conformational preference to natural glycosides when in solution and when complexed with a protein.17–20 As a result, we aimed to reveal the reactivity of ΔUA as a starting compound towards the synthesis of complex S-linked glycosyl products.
Herein, both alkaline and radical conditions were applied for the construction of S-linked uronate derivatives. The utility of basic condition with thiophenol (PhSH) or mercaptobenzyl (BnSH) as nucleophiles resulted in moderate to high yields with C-4 stereoselectivity. The poor stereoselectivity of adducts at the C-5 position is due to the retro thiol-Michael addition. In a comparison with the alkaline condition, radical-mediated hydrothiolation reaction gave a product with specific stereoselectivity at both C-4 and C-5 position, however, the overall yield was low. After deprotection of the benzyl group, a 4-thiol-furanosidic uronate derivative was obtained and the preparation of 4-thiol-furanosidic uronate derivative was firstly reported here.
2. Materials and methods
2.1 General experimental procedures
Solvents and chemicals were purchased from commercial suppliers and used as received. Compounds 1β were prepared according to literature procedures and the detailed information was shown in the ESI.†21 1H and 13C NMR spectra were recorded with a Bruker Avance III 300 MHz and a Bruker Avance III HD-600 MHz instrument. NMR spectra were recorded in CDCl3, and chloroform signals (δ = 7.26 ppm in 1H NMR; δ = 77.16 ppm in 13C NMR) were used as an internal standard. Splitting patterns are reported as following: s: singlet, d: doublet, t: triplet, q: quartet, m: multiplet. Coupling constants (J) are reported in Hz. IR spectra were recorded with a PerkinElmer Spectrum 100 FTIR spectrometer and data are reported in cm−1. High-resolution mass spectrometry (HRMS) was carried out with an AB SCIE X QSTAR® XL spectrometer (ESI-MS). Liquid chromatography-mass spectrometry (LC-Mass) was measured on a Thermo TSQ-20002 Triple-Q spectrometer. Optical rotations were measured with a Horiba SEPA-300 digital polarimeter. TLC was carried out on pre-coated sheets (Merck Art. 60 F254, 0.25 mm). Reaction products were isolated by flash chromatography on FUJI silysia chemistry MB-70-40/75 silica gel. Reverse-phase high-performance liquid chromatography (Shimadzu LC-20AD) was employed to determine the ratio of products by using Platisil 5 μm PH column (150 mm × 4.6 mm). Yields of products refer to chromatographically purified products unless otherwise stated. Dichloromethane was dried with CaH2 (5% wt.) then distilled under N2. N,N-Dimethylformamide, acetone, toluene, and methanol were dried with 4 Å or 3 Å molecular sieves. All reactions were carried out under N2 or Ar.2.2 General protocol for base-mediated thiol-Michael addition
To a solution of Δ4,5-unsaturated uronate derivative, 1β (1 equiv.) in CH3CN at a final concentration of 0.2 M was added bases (1 equiv.) and thiols (10 equiv.). The reaction was stirred at 60 or 80 °C for 18 h. Then, the reaction mixture was concentrated under reduced pressure and purified by flash column chromatography giving desired products.It is worth notice that the starting material 1β was not fully consumed during the reaction. The reaction condition leading to the best conversion yield of 1β was around 80% and that could be recovered after flash column chromatography. The ratio of desired products was determined by high-performance liquid chromatography (HPLC) analysis (0.1% formic acid and a 50 to 100% acetonitrile gradient on Platisil 5 μm PH column).
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :n-Hex = 1:2 v/v).; [α]25589 = +17.0° (c = 0.5, CHCl3).; 1H NMR (300 MHz, CDCl3, 297 K): δ = 7.47–7.44 (m, 2H, Ar), 7.31–7.23 (m, 3H, Ar), 5.43 (dd, J1 = 10.1 Hz, J2 = 7.9 Hz, 1H, H2), 5.10 (dd, J1 = 10.1 Hz, J2 = 4.4 Hz, 1H, H3), 4.50 (d, J = 2.1 Hz, 1H, H5), 4.40 (d, J = 7.9 Hz, 1H, H1), 4.23 (dd, J1 = 4.3 Hz, J2 = 2.1 Hz, 1H, H4), 3.79 (s, 3H, OMe), 3.58 (s, 3H, OMe), 2.08 (s, 3H, OAc), 1.62 (s, 3H, OAc) ppm.; 13C NMR (75 MHz, CDCl3, 295 K): δ = 170.60, 169.46, 167.60, 134.80, 132.44, 129.15, 127.67, 102.51, 73.88, 73.24, 69.15, 57.12, 52.74, 52.51, 20.94, 20.24 ppm.; IR (KBr): 3003, 2853, 1759, 1744, 1481, 1377, 1234, 1111743 cm−1.; HRMS (ESI) m/z: calcd for C18H22O8SNa+ [M + Na]+: 421.0933, found: 421.0936.:n-Hex = 1:2 v/v).; [α]25589 = −103.5° (c = 0.5, CHCl3).; 1H NMR (300 MHz, CDCl3, 294 K): δ = 7.44–7.40 (m, 2H, Ar), 7.34–7.27 (m, 3H, Ar), 5.17 (dd, J1 = J2 = 3.5 Hz, 1H, H3), 4.92 (dd, J1 = 3.7 Hz, J2 = 1.7 Hz, 1H, H2), 4.70 (d, d, J = 1.7 Hz, 1H, H1), 4.65 (d, J = 10.1 Hz, 1H, H5), 3.76 (dd, J1 = 10.1 Hz, J2 = 3.3 Hz, 1H, H4), 3.61 (s, 3H, OMe), 3.41 (s, 3H, OMe), 2.11 (s, 3H, OAc), 2.08 (s, 3H, OAc) ppm.; 13C NMR (75 MHz, CDCl3, 296 K): δ = 170.00, 169.52, 169.38, 133.27, 132.61, 129.29, 128.10, 98.59, 69.46, 69.37, 67.78, 56.26, 52.54, 46.96, 21.10, 20.81 ppm.; IR (ATR source): 2952, 1745, 1477, 1371, 1216, 1120, 731 cm−1.; HRMS (ESI) m/z: calcd for C18H22O8SNa+ [M + Na]+: 421.0933, found: 421.0934.:n-Hex = 1:3 v/v).; [α]25589 = −60.7° (c = 0.5, CHCl3).; 1H NMR (300 MHz, CDCl3, 297 K): δ = 7.31–7.20 (m, 5H, Ar), 5.22 (dd, J1 = 10.2 Hz, J2 = 7.8 Hz, 1H, H2), 5.08 (dd, J1 = 10.2 Hz, J2 = 4.4 Hz, 1H, H3), 4.33–4.31 (m, 2H, H1, H5), 3.68–3.61 (m, 5H, OMe, CH2Ph), 3.57 (dd, J1 = 4.4 Hz, J2 = 2.0 Hz, 1H, H4), 3.52 (s, 3H, OMe), 2.05 (s, 3H, OAc), 1.96 (s, 3H, OAc) ppm.; 13C NMR (75 MHz, CDCl3, 298 K): δ = 170.38, 169.44, 167.40, 137.91, 129.29, 128.73, 127.42, 102.23, 74.18, 72.94, 69.70, 56.99, 52.61, 46.78, 37.66, 20.89 ppm.; IR (ATR source): 2953, 1739, 1495, 1368, 1218, 1097, 731 cm−1.; HRMS (ESI) m/z: calcd for C19H24O8SNa+ [M + Na]+: 435.1192, found: 435.1190.:n-Hex = 1:3 v/v).; [α]25589 = −56.6° (c = 0.5, CHCl3).; 1H NMR (300 MHz, CDCl3, 297 K): δ = 7.32–7.25 (m, 5H, Ar), 4.78–4.76 (m, 2H, H2, H3), 4.62 (d, J = 0.5 Hz, 1H, H1), 4.55 (d, J = 10.9 Hz, 1H, H5), 3.84 (s, 3H, OMe), 3.72 (d, J = 1.4 Hz, 2H, CH2Ph), 3.37 (s, 3H, OMe), 3.19 (dd, J1 = 10.9 Hz, J2 = 2.6 Hz, 1H, H4), 2.09 (s, 3H, OAc), 2.02 (s, 3H, OAc) ppm.; 13C NMR (75 MHz, CDCl3, 296 K): δ = 169.99, 169.93, 169.33, 137.30, 129.38, 128.69, 127.51, 98.47, 69.47, 68.85, 67.33, 56.13, 52.73, 40.98, 36.57, 20.97, 20.89 ppm.; IR (ATR source): 2952, 1745, 1602, 1495, 1218, 1093, 1025, 739 cm−1.; HRMS (ESI) m/z: calcd for C19H24O8SNa+ [M + Na]+: 435.1192, found: 435.1192.
:n-Hex = 1:2 v/v).; [α]25589 = +17.0° (c = 0.5, CHCl3).; 1H NMR (300 MHz, CDCl3, 297 K): δ = 7.47–7.44 (m, 2H, Ar), 7.31–7.23 (m, 3H, Ar), 5.43 (dd, J1 = 10.1 Hz, J2 = 7.9 Hz, 1H, H2), 5.10 (dd, J1 = 10.1 Hz, J2 = 4.4 Hz, 1H, H3), 4.50 (d, J = 2.1 Hz, 1H, H5), 4.40 (d, J = 7.9 Hz, 1H, H1), 4.23 (dd, J1 = 4.3 Hz, J2 = 2.1 Hz, 1H, H4), 3.79 (s, 3H, OMe), 3.58 (s, 3H, OMe), 2.08 (s, 3H, OAc), 1.62 (s, 3H, OAc) ppm.; 13C NMR (75 MHz, CDCl3, 295 K): δ = 170.60, 169.46, 167.60, 134.80, 132.44, 129.15, 127.67, 102.51, 73.88, 73.24, 69.15, 57.12, 52.74, 52.51, 20.94, 20.24 ppm.; IR (KBr): 3003, 2853, 1759, 1744, 1481, 1377, 1234, 1111743 cm−1.; HRMS (ESI) m/z: calcd for C18H22O8SNa+ [M + Na]+: 421.0933, found: 421.0936.:n-Hex = 1:2 v/v).; [α]25589 = −103.5° (c = 0.5, CHCl3).; 1H NMR (300 MHz, CDCl3, 294 K): δ = 7.44–7.40 (m, 2H, Ar), 7.34–7.27 (m, 3H, Ar), 5.17 (dd, J1 = J2 = 3.5 Hz, 1H, H3), 4.92 (dd, J1 = 3.7 Hz, J2 = 1.7 Hz, 1H, H2), 4.70 (d, d, J = 1.7 Hz, 1H, H1), 4.65 (d, J = 10.1 Hz, 1H, H5), 3.76 (dd, J1 = 10.1 Hz, J2 = 3.3 Hz, 1H, H4), 3.61 (s, 3H, OMe), 3.41 (s, 3H, OMe), 2.11 (s, 3H, OAc), 2.08 (s, 3H, OAc) ppm.; 13C NMR (75 MHz, CDCl3, 296 K): δ = 170.00, 169.52, 169.38, 133.27, 132.61, 129.29, 128.10, 98.59, 69.46, 69.37, 67.78, 56.26, 52.54, 46.96, 21.10, 20.81 ppm.; IR (ATR source): 2952, 1745, 1477, 1371, 1216, 1120, 731 cm−1.; HRMS (ESI) m/z: calcd for C18H22O8SNa+ [M + Na]+: 421.0933, found: 421.0934.:n-Hex = 1:3 v/v).; [α]25589 = −60.7° (c = 0.5, CHCl3).; 1H NMR (300 MHz, CDCl3, 297 K): δ = 7.31–7.20 (m, 5H, Ar), 5.22 (dd, J1 = 10.2 Hz, J2 = 7.8 Hz, 1H, H2), 5.08 (dd, J1 = 10.2 Hz, J2 = 4.4 Hz, 1H, H3), 4.33–4.31 (m, 2H, H1, H5), 3.68–3.61 (m, 5H, OMe, CH2Ph), 3.57 (dd, J1 = 4.4 Hz, J2 = 2.0 Hz, 1H, H4), 3.52 (s, 3H, OMe), 2.05 (s, 3H, OAc), 1.96 (s, 3H, OAc) ppm.; 13C NMR (75 MHz, CDCl3, 298 K): δ = 170.38, 169.44, 167.40, 137.91, 129.29, 128.73, 127.42, 102.23, 74.18, 72.94, 69.70, 56.99, 52.61, 46.78, 37.66, 20.89 ppm.; IR (ATR source): 2953, 1739, 1495, 1368, 1218, 1097, 731 cm−1.; HRMS (ESI) m/z: calcd for C19H24O8SNa+ [M + Na]+: 435.1192, found: 435.1190.:n-Hex = 1:3 v/v).; [α]25589 = −56.6° (c = 0.5, CHCl3).; 1H NMR (300 MHz, CDCl3, 297 K): δ = 7.32–7.25 (m, 5H, Ar), 4.78–4.76 (m, 2H, H2, H3), 4.62 (d, J = 0.5 Hz, 1H, H1), 4.55 (d, J = 10.9 Hz, 1H, H5), 3.84 (s, 3H, OMe), 3.72 (d, J = 1.4 Hz, 2H, CH2Ph), 3.37 (s, 3H, OMe), 3.19 (dd, J1 = 10.9 Hz, J2 = 2.6 Hz, 1H, H4), 2.09 (s, 3H, OAc), 2.02 (s, 3H, OAc) ppm.; 13C NMR (75 MHz, CDCl3, 296 K): δ = 169.99, 169.93, 169.33, 137.30, 129.38, 128.69, 127.51, 98.47, 69.47, 68.85, 67.33, 56.13, 52.73, 40.98, 36.57, 20.97, 20.89 ppm.; IR (ATR source): 2952, 1745, 1602, 1495, 1218, 1093, 1025, 739 cm−1.; HRMS (ESI) m/z: calcd for C19H24O8SNa+ [M + Na]+: 435.1192, found: 435.1192.2.3 General protocol for radical-mediated hydrothiolation reaction
To a solution of Δ4,5-unsaturated uronate derivate, 1β (1 equiv.) in toluene at a final concentration of 0.2 M was added photoinitiators (5 equiv.) and BnSH (3 equiv.). The solution was deoxygenated and irradiated at ambient temperature for 1 hour. The addition of photoinitiators and irradiation was repeated three times more. Then, the reaction mixture was concentrated under reduced pressure and purified by flash column chromatography giving desired product. The isolated yield of desired compound was around 30%.2.4 Compound V: methyl (methyl-2,3-di-O-acetyl-4-thiol-α-D-galactofuranosid) uronate
To a solution of IV (30.4 mg, 0.07 mmol) in 0.3 mL dry toluene was added AlCl3 (34.8 mg, 0.3 mmole). The reaction was stirred at room temperature for 48 h. When the reaction was completed, it was quenched with cold demi water and the pH value was adjusted to 1 with 1 N HCl(aq). The aqueous layer was extracted with EtOAc three times. The organic layer was combined, dried over MgSO4, concentrated under reduced pressure and purified by flash column chromatography yielding desired products as yellow oil. Rf = 0.29 (EtOAc:n-Hex = 1:1.5 v/v).; 1H NMR (300 MHz, CDCl3, 298 K): 5.89 (dd, J1 = 9.1 Hz, J2 = 6.5 Hz, 1H, H3), 5.19 (dd, J1 = 9.1 Hz, J2 = 4.2 Hz, 1H, H2), 4.96 (d, J = 4.2 Hz, 1H, H1), 4.45 (d, J = 4.8 Hz, 1H, H5), 3.81 (s, 3H, OMe), 3.61 (dd, J1 = 6.4 Hz, J2 = 4.9 Hz, 1H, H4), 3.31 (s, 3H, OMe), 2.09 (s, 3H, OAc), 2.08 (s, 3H, OAc) ppm.; 13C NMR (75 MHz, CDCl3, 302 K): 172.25, 170.59, 84.35, 78.71, 72.42, 56.48, 52.91, 48.37, 21.04, 20.91 ppm; IR (KBr): 3469, 2958, 1739, 1634, 1439, 1239, 1118, 912, 739 cm−1.; HRMS (ESI) m/z: calcd for C12H18O8SNa+ [M + Na]+: 345.0615, found: 345.0621.
3. Results and discussion
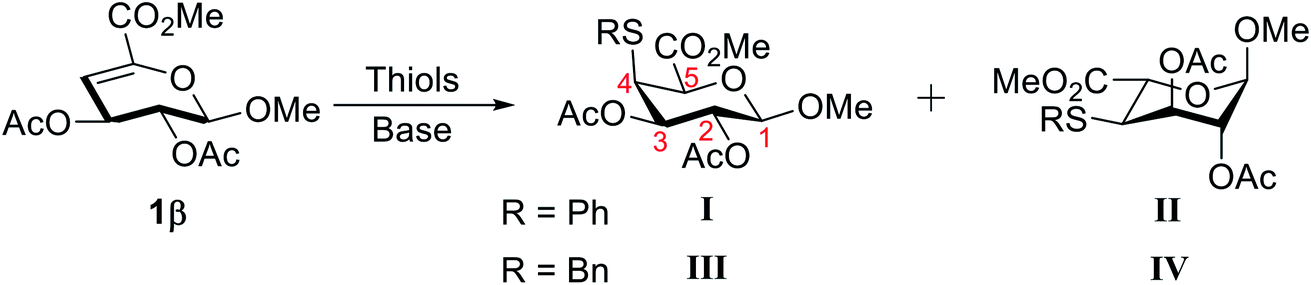
To investigate the feasibility of hydrothiolation reaction, Δ4,5-unsaturated methyl uronate 1β was used as glycosyl donor and PhSH or BnSH were chosen as the model thiol reagents. Initially, 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) was applied as a base to mediate the thiol-Michael addition. The reaction mixture was stirred at 80 °C for 6 hours yielding two PhSH attached products, namely I and II, in a lower yield, and the ratio of these two products is 1.8:1 (Table 1, entry 1). The structures of compounds I and II were characterized by detailed 1D and 2D NMR spectroscopy (see ESI†). The NOESY spectrum (Fig. S1A†) of I shows a strong NOE interaction between the H-5, H-1, and H-3, which indicates that these protons are located on the axial positions of the six-membered ring and confirms that the product I in the 4C1 conformation. In contrast, a NOE effect was observed between the H-5 and H-2 of II, which indicated that the H-5 and H-2 of II were spatially near each other and showed that the product II in the 1C4 conformation (Fig. S1B†). Encouraged by this initial result, various bases were subsequently surveyed to optimize the reaction condition (Table 1, entries 2–5). 4-Dimethylaminopyridine (DMAP) resulted in the highest yield among these bases, although the ratio of these two PhSH attached products was slightly decrease compared to the result from the use of DBU. To improve the stereoselectivity of the C-5 position, the reactions were carried out at different temperatures (Table 1, entries 5–7). The stereoselective at the C-5 position was somewhat improved at a lower temperature. Metallic Lewis acids and alcohols with steric hindrance were also have been tried to ameliorate the stereoselectivity (Table S1†),22,23 however, the ratio of these two thiol attached products remains around 1:1. Owing to air oxidation of thiols to form disulfide, an excess amount of PhSH was required to obtain a decent yield (Table 1, entries 8–10). To determine whether any other solvent could be suitable for this methodology, a survey of reaction media was conducted. The reaction was performed in different solvents, including acetonitrile, toluene, chloroform, and tetrahydrofuran. The results revealed that acetonitrile was the optimal solvent for this reaction (Table 1, entry 8). No reaction occurred when tetrahydrofuran was employed (Table 1, entry 11) and both the reactivity and selectivity were decreased when toluene or chloroform were used (Table 1, entries 12 and 13). Next, BnSH as a nucleophile based on the best condition was also tested in this system. Surprisingly, the ratio of two BnSH adducts is increased to 17:1 with a reasonable yield (Table 1, entry 14). It is worth noticing that 1β was not fully consumed under these reaction conditions even with elongated reaction time and that could be recovered after purification.
|
||||||
|---|---|---|---|---|---|---|
| Entry | Base | Solvent | Thiol (equiv.) | Temp. | Time | Resultb |
| a General procedure: 1β (1 equiv.) and bases (1 equiv.) were dissolved in the indicated solvent and then the thiols were added under various temperatures. After stirring, the reaction mixture was worked up, concentrated under reduced pressure, and purified by flash column chromatography.b Yield percentage and the ratio of products were determined by HPLC analysis.c When the reaction time was elongated to 12 h, the yield was up to 86%.; N.D. = not detected. | ||||||
| 1 | DBU | CH3CN | PhSH (10) | 80 °C | 6 h | I/II (28%, 1.8:1) |
| 2 | TEA | CH3CN | PhSH (10) | 80 °C | 6 h | I/II (52%, 1.5:1) |
| 3 | Quinine | CH3CN | PhSH (10) | 80 °C | 6 h | I/II (40%, 1.3:1) |
| 4 | DBACO | CH3CN | PhSH (10) | 80 °C | 6 h | I/II (45%, 0.6:1) |
| 5c | DMAP | CH3CN | PhSH (10) | 80 °C | 6 h | I/II (65%, 1.3:1) |
| 6 | DMAP | CH3CN | PhSH (10) | 60 °C | 6 h | I/II (36%, 1.5:1) |
| 7 | DMAP | CH3CN | PhSH (10) | 40 °C | 18 h | I/II (42%, 1.8:1) |
| 8 | DMAP | CH3CN | PhSH (10) | 60 °C | 18 h | I/II (72%, 1.6:1) |
| 9 | DMAP | CH3CN | PhSH (5) | 60 °C | 18 h | I/II (58%, 1:1) |
| 10 | DMAP | CH3CN | PhSH (1.5) | 60 °C | 18 h | I/II (N.D, N.D.) |
| 11 | DMAP | THF | PhSH (10) | 60 °C | 18 h | I/II (N.D, N.D.) |
| 12 | DMAP | Toluene | PhSH (10) | 60 °C | 18 h | I/II (39%, 1.2:1) |
| 13 | DMAP | CHCl3 | PhSH (10) | 60 °C | 18 h | I/II (62%, 0.7:1) |
| 13 | DMAP | CH3CN | BnSH (10) | 80 °C | 12 h | III/IV (60%, 17:1) |
In a comparison with BnSH, PhSH has a lower pKa value, as a consequence, a retro thiol-Michael addition may more likely to occur resulting base-catalyzed equilibration between I and II. To investigate this hypothesis, compound I was treated with DMAP under the best-optimized condition. After few hours, an appreciable amount of elimination product, 1β, was observed (Table 2, entries 1–3). Upon addition of PhSH, the rate of elimination was increased and led to equilibration with 1β, I, and II (Table 2, entries 4–6). A similar outcome was obtained, when III was treated with DMAP and BnSH. However, the rate of retro thiol-Michael addition was slower owing to the higher pKa value of BnSH (Table S2†). These results confirmed the occurrence of retro thiol-Michael addition under alkaline conditions.

Thereafter, a radical initiated hydrothiolation reaction was surveyed and BnSH was used as an example. Although a thiol–ene radical reaction could be generated without any initiator, azobis(isobutyronitrile) (AIBN), dibenzoyl peroxide (DPO), and 2,2-dimethoxy-2-phenylacetophenone (DPAP) were used here to promote the efficiency of this reaction. At the beginning, thermally activated initiators, AIBN and DPO, were utilized (Table 3, entries 1 and 2). However, there was no adduct was observed under these reaction conditions, and the starting material was recovered even excessive amount of thermal initiators and BnSH were applied. Then, the reaction was carried out with DPAP as photoinitiator in toluene with UVA lamp at 365 nm. Equivalent amount of BnSH and a catalytic amount of DPAP were initially utilized, however, only a trace amount of product was obtained (Table 3, entry 3). By contrast, disulfide-linked BnSH and incomplete conversion of the starting 1β were indicated by TLC. To improve the yield, the reaction time was prolonged and an excess amount of DPAP and BnSH were used (Table 3, entry 4). Under this condition, the desired adduct was obtained in 21% yield. Nevertheless, the further increase amount of BnSH and changing solvent system did not be able to improve the yield of this reaction (Table 3, entries 5 and 6). The low conversion yield might be attributed by the electron-withdrawing capability of COOMe that affects the electron density of the double bonds decreasing the reactivity of this reaction system.24 Besides, internal alkenes are significantly less reactive towards radical hydrothiolation in a comparison with terminal alkenes25 and a reversible equilibrium between thiyl radical and 4,5-disubstituted ene causing the low yield.26 The position of thiol attached carbon (regioselectivity) and the conformation of adduct were confirmed by fully assigned 1-D and 2-D NMR spectra.
|
||||||
|---|---|---|---|---|---|---|
| Entry | Initiator | Solvent | BnSH (equiv.) | Temp. | Time | Resultc |
| a Method A: the reaction was carried out with thermal initiators (10 equiv.) in a sealed tube and was heated in an oil bath.b Method B: the reactions were carried out by irradiation at λmax 365 nm UVA lamp (8 W) for 1 × 4 h in the presence of DPAP (4 × 0.1 equiv.).c The yield is isolated yield. N.R. = no reaction and starting material was recovered. | ||||||
| 1a | AIBN, Δ | Toluene | 3 | 82 °C | 8 h | N.R. |
| 2a | DPO, Δ | Toluene | 3 | 70 °C | 8 h | N.R. |
| 3b | DPAP, hν | Toluene | 1 | r.t. | 4 h | Trace |
| 4b | DPAP, hν | Toluene | 3 | r.t. | 4 h | III (21%) |
| 5b | DPAP, hν | Toluene | 5 | r.t. | 4 h | III (18%) |
| 6b | DPAP, hν | Toluene:MeOH (5:1) |
3 | r.t. | 4 h | III (16%) |

According to the results of alkaline/radical-mediated hydrothiolation reactions, plausible mechanisms were proposed and described in Fig. 2. Unsaturated uronic acid and its derivatives are mostly represented by half-chair form.10,27,28 However, 1H2 conformer is less stable than 2H1 due to the steric hindrance between the anomeric methoxyl group and the acetate at the C-3 position. The exo-face attachment at the C-4 position yields a chair-like intermediate, but the endo-face attack gives a higher energy twist-boat-like intermediate. As a consequence, the thiol groups were attached to unsaturated uronate with axial-selectivity at the C-4 position. The following protonation of adducts at the C-5 position provides two different final products. These two products can be converted to each other via retro thiol-Michael addition (Fig. 2A). For radical-mediated reaction, photo-irradiative generated thiyl-radical was adding from the exo-face of 1β and the subsequent hydrogen abstraction by the C-5 radical of the substrate took place from the axial-position resulting S-linkage product III (Fig. 2B).
 | ||
| Fig. 2 Proposed mechanism of base-mediated (A) and radical-mediated (B) hydrothiolation reactions. | ||
Owing to the low yield of photo-irradiated hydrothiolation reaction, we following focused on the base-mediated approach for investigating the substrate scope between unsaturated methyl uronate 1β with various thiol partners. Unfortunately, only a trace amount of adducts were observed either use a thiosugar or a cysteine derivative. The adducts were difficult to be isolated due to the low conversion yield even after a prolonged reaction time (data not shown). The low conversion yield could be caused by the steric hindrance at the C-3 position. In this context, we change our strategy to prepared S-linked glycosides. The plan was to use 4-thiol uronate derivatives to attach glycosyl bromide to make S-linked disaccharide. As a consequence, the thiolbenzyl functionalized IV was de-protected using aluminum chloride.29 Unexpectedly, α-furanosidic form of 4-thiol-uronate was obtained. During the reaction, aluminum(III) catalyzed the six-member ring opening. Subsequently, the sulfur was attached to the anomeric carbonium ion generating a sulfonium intermediate with a five-member ring molecular structure. After workup, thiol-furanoside V was obtained (Scheme 1). To the best of our knowledge, the thiol-furanoside V was synthesized for the first time.
 | ||
| Scheme 1 The use of AlCl3 for the deprotection of S-linked uronate IV. | ||
4. Conclusions
In summary, both base- and radical-mediated hydrothiolation reactions were applied for the synthesis of S-linked uronate derivatives, while the radical-mediated reaction was inefficient due to the electron-withdrawing group on ethylene group. For the alkaline approach, PhSH or BnSH as nucleophiles could regioselectively and stereoselectively attach to the C-4 position of unsaturated uronate. However, a poor stereoselectivity at the C-5 position was observed owing to retro thiol-Michael addition. When the use of AlCl3 to remove the benzyl protecting group, 4-thiol-furanosidic uronate was synthesized for the first time.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is financially supported by the Ministry of Science and Technology, Taiwan (MOST109-2113-M-030-001) and the Department of Chemistry, Fu Jen Catholic University. We also thank the Instrumentation Center of National Taiwan Normal University for NMR measurements and for acquisition of HRESIMS spectra.References
- S. R. Herron, J. A. E. Benen, R. D. Scavetta, J. Visser and F. Jurnak, Structure and function of pectic enzymes: Virulence factors of plant pathogens, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 8762–8769 CrossRef CAS PubMed.
- S. S. Ferreira, C. P. Passos, P. Madureira, M. Vilanova and M. A. Coimbra, Structure-function relationships of immunostimulatory polysaccharides: A review, Carbohydr. Polym., 2015, 132, 378–396 CrossRef CAS PubMed.
- M. Otto, Bacterial evasion of antimicrobial peptides by biofilm formation, Curr. Top. Microbiol. Immunol., 2006, 306, 251–258 CAS.
- S. M. Smorenburg and C. J. Van Noorden, The complex effects of heparins on cancer progression and metastasis in experimental studies, Pharmacol. Rev., 2001, 53, 93–105 CAS.
- V. H. Pomin, W. P. Vignovich, A. V. Gonzales, A. A. Vasconcelos and B. Mulloy, Galactosaminoglycans: Medical Applications and Drawbacks, Molecules, 2019, 24, 8203 CrossRef PubMed.
- C. Martinez-Villaluenga, J. Frias and C. Vidal-Valverde, Alpha-galactosides: antinutritional factors or functional ingredients?, Crit. Rev. Food Sci. Nutr., 2008, 48, 301–316 CrossRef CAS PubMed.
- A. Mirescu and U. Prüße, A new environmental friendly method for the preparation of sugar acids via catalytic oxidation on gold catalysts, Appl. Catal., B, 2007, 70, 644–652 CrossRef CAS.
- A. Attanzio, M. Ippolito, M. A. Girasolo, F. Saiano, A. Rotondo, S. Rubino, L. Mondello, M. L. Capobianco, P. Sabatino, L. Tesoriere and G. Casella, Anti-cancer activity of di- and tri-organotin(IV) compounds with D-(+)-Galacturonic acid on human tumor cells, J. Inorg. Biochem., 2018, 188, 102–112 CrossRef CAS PubMed.
- C. I. Gama and L. C. Hsieh-Wilson, Chemical approaches to deciphering the glycosaminoglycan code, Curr. Opin. Chem. Biol., 2005, 9, 609–619 CrossRef CAS PubMed.
- L. Jin, M. Hricovíni, J. A. Deakin, M. Lyon and D. Uhrín, Residual dipolar coupling investigation of a heparin tetrasaccharide confirms the limited effect of flexibility of the iduronic acid on the molecular shape of heparin, Glycobiology, 2009, 19, 1185–1196 CrossRef CAS PubMed.
- D. P. Nair, M. Podgórski, S. Chatani, T. Gong, W. Xi, C. R. Fenoli and C. N. Bowman, The Thiol-Michael Addition Click Reaction: A Powerful and Widely Used Tool in Materials Chemistry, Chem. Mater., 2014, 26, 724–744 CrossRef CAS.
- K. Yamamoto, N. Watanabe, H. Matsuda, K. Oohara, T. Araya, M. Hashimoto, K. Miyairi, I. Okazaki, M. Saito, T. Shimizu, H. Kato and T. Okuno, Design, synthesis, and enzymatic property of a sulfur-substituted analogue of trigalacturonic acid, Bioorg. Med. Chem. Lett., 2005, 15, 4932–4935 CrossRef CAS PubMed.
- R. A. A. Al-Shuaeeb, D. Montoir, M. Alami and S. Messaoudi, Synthesis of (1 → 2)-S-Linked Saccharides and S-Linked Glycoconjugates via a Palladium-G3-XantPhos Precatalyst Catalysis, J. Org. Chem., 2017, 82, 6720–6728 CrossRef CAS PubMed.
- J. R. Rich, A. Szpacenko, M. M. Palcic and D. R. Bundle, Glycosyltransferase-Catalyzed Synthesis of Thiooligosaccharides, Angew. Chem., Int. Ed., 2004, 43, 613–615 CrossRef CAS PubMed.
- D. R. Bundle, J. R. Rich, S. Jacques, H. N. Yu, M. Nitz and C.-C. Ling, Thiooligosaccharide Conjugate Vaccines Evoke Antibodies Specific for Native Antigens, Angew. Chem., Int. Ed., 2005, 44, 7725–7729 CrossRef CAS PubMed.
- H. Yuasa and H. Hashimoto, Recent Advances in the Development of Unnatural Oligosaccharides Conformation and Bioactivity, Trends Glycosci. Glycotechnol., 2001, 13, 31–55 CrossRef.
- E. Montero, A. García-Herrero, J. L. Asensio, K. Hirai, S. Ogawa, F. Santoyo-González, F. J. Cañada and J. Jiménez-Barbero, The Conformational Behaviour of Non-Hydrolizable Lactose Analogues: The Thioglycoside, Carbaglycoside, and Carba-Iminoglycoside Cases, Eur. J. Org. Chem., 2000, 2000, 1945–1952 CrossRef.
- T. Weimar, U. C. Kreis, J. S. Andrews and B. M. Pinto, Conformational analysis of maltoside heteroanalogues using high-quality NOE data and molecular mechanics calculations. Flexibility as a function of the interglycosidic chalcogen atom, Carbohydr. Res., 1999, 315, 222–233 CrossRef CAS.
- B. Aguilera, J. Jiménez-Barbero and A. Fernández-Mayoralas, Conformational differences between Fuc(α1–3)GlcNAc and its thioglycoside analogue, Carbohydr. Res., 1998, 308, 19–27 CrossRef CAS PubMed.
- E. Montero, M. Vallmitjana, J. A. Pérez-Pons, E. Querol, J. Jiménez-Barbero and F. J. Cañada, NMR studies of the conformation of thiocellobiose bound to a β-glucosidase from Streptomyces sp, FEBS Lett., 1998, 421, 243–248 CrossRef CAS PubMed.
- S. A. K. Jongkees and S. G. Withers, Glycoside Cleavage by a New Mechanism in Unsaturated Glucuronyl Hydrolases, J. Am. Chem. Soc., 2011, 133, 19334–19337 CrossRef CAS PubMed.
- W.-J. Tsai, Y.-T. Lin and B.-J. Uang, Asymmetric Michael addition of thiols to (1R,2R,4R)-(−)-2,10-N-enoylcamphorsultam, Tetrahedron: Asymmetry, 1994, 5, 1195–1198 CrossRef CAS.
- E. Emori, T. Arai, H. Sasai and M. Shibasaki, A Catalytic Michael Addition of Thiols to α,β-Unsaturated Carbonyl Compounds: Asymmetric Michael Additions and Asymmetric Protonations, J. Am. Chem. Soc., 1998, 120, 4043–4044 CrossRef CAS.
- L. Lázár, L. Juhász, G. Batta, A. Borbás and L. Somsák, Unprecedented β-manno type thiodisaccharides with a C-glycosylic function by photoinitiated hydrothiolation of 1-C-substituted glycals, New J. Chem., 2017, 41, 1284–1292 RSC.
- A. B. Lowe, Thiol-ene “click” reactions and recent applications in polymer and materials synthesis, Polym. Chem., 2010, 1, 17–36 RSC.
- L. Lázár, M. Csávás, Á. Hadházi, M. Herczeg, M. Tóth, S. László, T. Barna, P. Herczegh and A. Borbás, Systematic study on free radical hydrothiolation of unsaturated monosaccharide derivatives with exo- and endocyclic double bonds, Org. Biomol. Chem., 2013, 11, 5339–5350 RSC.
- D. Mikhailov, J. R. Linhardt and H. K. Mayo, NMR solution conformation of heparin-derived hexasaccharide, Biochem. J., 1997, 328, 51–61 CrossRef CAS PubMed.
- A. Silipo, Z. Zhang, F. J. Cañada, A. Molinaro, R. J. Linhardt and J. Jiménez-Barbero, Conformational Analysis of a Dermatan Sulfate-Derived Tetrasaccharide by NMR, Molecular Modeling, and Residual Dipolar Couplings, ChemBioChem, 2008, 9, 240–252 CrossRef CAS PubMed.
- R. Romagnoli, P. G. Baraldi, M. D. Carrion, C. L. Cara, D. Preti, F. Fruttarolo, M. G. Pavani, M. A. Tabrizi, M. Tolomeo, S. Grimaudo, A. Di Cristina, J. Balzarini, J. A. Hadfield, A. Brancale and E. Hamel, Synthesis and Biological Evaluation of 2- and 3-Aminobenzo[b]thiophene Derivatives as Antimitotic Agents and Inhibitors of Tubulin Polymerization, J. Med. Chem., 2007, 50, 2273–2277 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed 1D and 2D NMR spectroscopic data, NOESY experiments and HPLC analysis. See DOI: 10.1039/d1ra02110a |
| This journal is © The Royal Society of Chemistry 2021 |