
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe effect on ion channel of different protonation states of E90 in channelrhodopsin-2: a molecular dynamics simulation†
Jie Chenga,
Wenying Zhang*a,
Shuangyan Zhoua,
Xu Rana,
Yiwen Shanga,
Glenn V. Lob,
Yusheng Doub and
Shuai Yuan *a
*a
aChongqing Key Laboratory of Big Data for Bio Intelligence, Chongqing University of Posts and Telecommunications, Chongqing 40065, China. E-mail: zhangwenying@cqupt.edu.cn; yuanshuai@cqupt.edu.cn
bDepartment of Chemistry and Physical Sciences, Nicholls State University, P.O. Box 2022, Thibodaux, LA 70310, USA
First published on 19th April 2021
Abstract
Channelrhodopsin-2 (ChR2) is a cationic channel protein that has been extensively studied in optogenetics. The ion channel is opened via a series of proton transfers and H-bond changes during the photocycle but the detailed mechanism is still unknown. Molecular dynamics (MD) simulations with enhanced sampling were performed on the dark-adapted state (i.e., D470) and two photocycle intermediates (P1500 and P2390) to study the proton transfer path of the Schiff base and the subsequent conformational changes. The results suggest there are two possible proton transfer pathways from the Schiff base to proton acceptors (i.e., E123 or D253), depending on the protonation of E90. If E90 is protonated in the P1500 state, the proton on the Schiff base will transfer to E123. The polyene chain of 13-cis retinal tilts and opens the channel that detours the blocking central gate (CG) and forms a narrow channel through the transmembrane helices (TM) 2, 3, 6 and 7. In contrast, if E90 deprotonates after retinal isomerization, the primary proton acceptor is D253, and an almost-open channel through TM1, 2, 3 and 7 is generated. The channel diameter is very close to the experimental value. The potential mean force (PMF) suggests that the free energy is extremely low for ions passing through this channel.
Introduction
Channelrhodopsin-2 (ChR2), a protein with a photoreceptor and ion channel has been the extensively studied the field of in optogenetics.1,2 ChR2 consists of seven transmembrane helices (TM1–TM7). The retinal chromophore is covalently bound to K257 on TM7 to yield a protonated Schiff base (RSBH+). In the dark-adapted state, the ion channel can be blocked at three positions named the extracellular gate (ECG), central gate (CG) and intracellular gate (ICG).3 The H-bond interactions at these three gates regulate the opening and closing of the channel. Three residues located in the CG play crucial roles in the opening mechanism of the channel,4–9 including the proton transfer of RSBH+ and the change of protonation state of E90.The photocycle of ChR2 involves five intermediate states: D470, P1500, P2390, P3520, and P4480,10–12 as seen in Fig. 1. D470 is a dark-adapted state and the channel is closed.13 After light absorption, the all-trans retinal chromophore isomerizes to the 13-cis conformation and forms the P1500 intermediate,13 which allows water molecules to enter the cavity.9,10 Subsequently the Schiff base is deprotonated to form the P2390 intermediate. It had been reported that even ions could pass through the channel in the late P2390 state.12–14 Due to the inflow of water molecules, the channel expands further and ultimately converts to the fully open P3520 state.12,14,15 The channel persists for approximately 2 ms,16 then either decays to the desensitized P4480 state or directly returns to the D470 state. Proton transfer to the Schiff base is one of the important processes in the P2390 state, but it is still controversial whether E123 or D253 is the acceptor.4,6,10
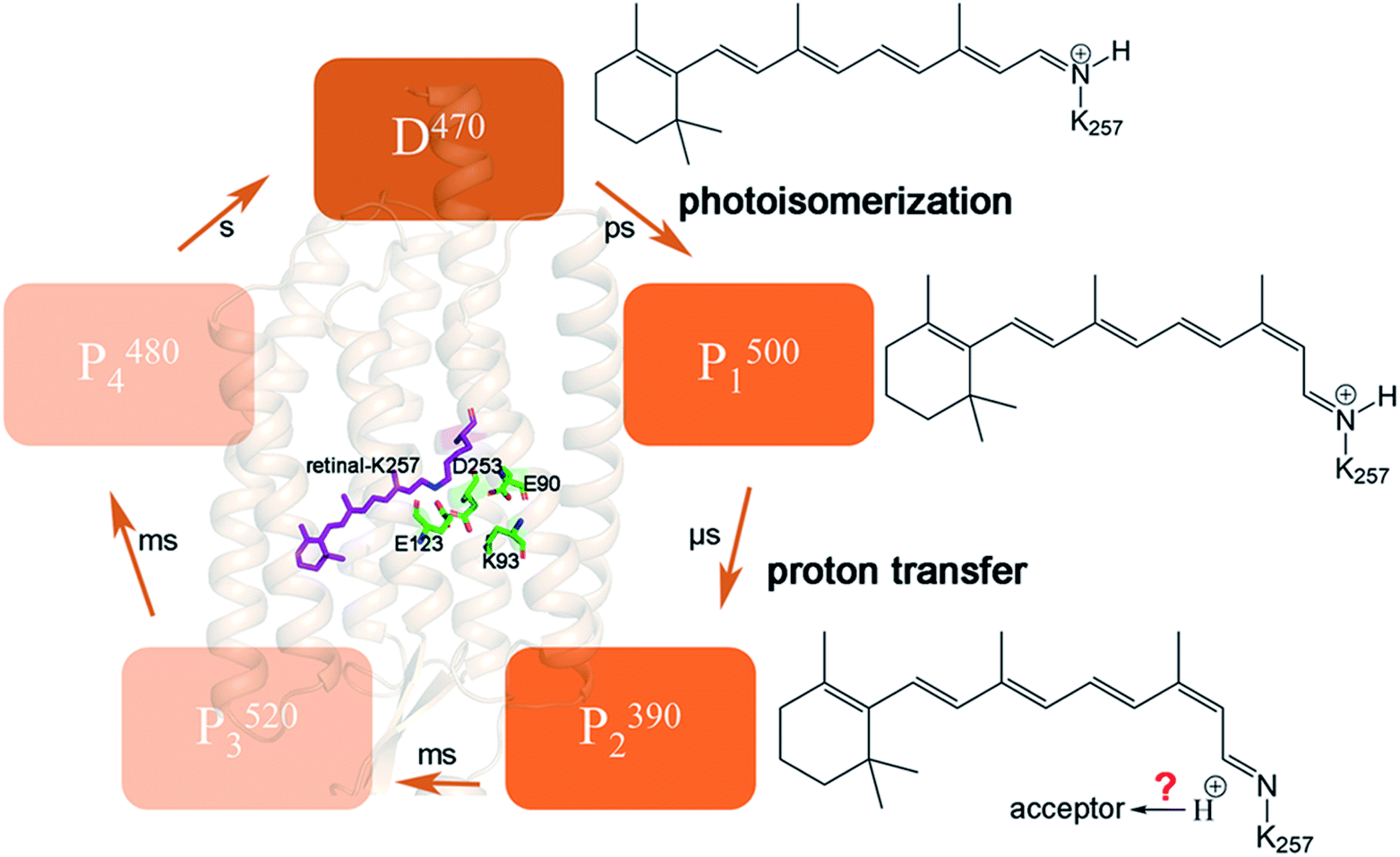 | ||
| Fig. 1 Schematic diagram of the crystal structure of ChR2, and corresponding retinal structure changes in the intermediate states during photocycle. In the D470 state, retinal is in an all-trans conformation. Retinal isomerizes to 13-cis conformation by photoactivation, P1500 state is formed. The proton on retinal is transferred to E123 or D253 to form the P2390 state. | ||
Early studies proposed E123 as the primary acceptor of the RSBH+.15,17 Recently, E123 also was mentioned explicitly as the proton acceptor in more accurate multi-reference QM/MM simulations.6 However, the electrophysiological experiment of Sineshchekov et al.18 and the time-resolved Fourier transform infrared (FTIR) spectroscopy of Lórenz-Fonfría et al.19 suggested that the proton acceptor in ChR2 is D253 (corresponding to D212 for BR), identifying this residue as the RSBH+ counterion. This view was also supported by QM/MM calculation of Adam (corresponding to D292 for C1C2) and Ardevol.4,10 Kuhne et al.9 displayed that both residues E123 and D253 might serve as proton acceptor, either simultaneously or alternately, during formation of the P2390 photocycle intermediate in individual ChR2. Their subsequent study showed that D253 was more favorable as a proton receptor.20 Interestingly, Guo et al.21 also suggest that both E123 and D253 were potential proton acceptors by performing semiempirical quantum mechanics/molecular mechanics (QM/MM) calculation on the excited state potential surface.
E90 plays a curial role in the ion selectivity of the channel and affects the gated mechanism.8–11,22–26 It is also important for proton transfer and the H-bond network.3,20,27 However, there is still no consensus on whether the protonation state of E90 changes during the channel opening process, as well as on the timescale of deprotonation. It has been shown that E90 maintains protonation until channel opening.10,19,28–30 A strong evidence is that the H-bond of E129 in C1C2 (corresponding to E90 in ChR2) was unchanged in Inaguma’ work.28 FTIR experiments suggested E90 deprotonation occurs exclusively in the P4480 state.19 Kuhne et al.20 proposed a “two parallel photocycles” model including anti and syn branches. The anti-cycle and syn-cycle started at the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N-anti and CN-syn retinal conformations, respectively. The former involved successive cation conductance and protonated E90 while the later generated small conductance and deprotonated E90. It seems that only the case where E90 remains protonated can produce high photocurrent in Kuhne's research.20 In addition, the authors stated the E90Q mutation also reduced photocurrents, which is consistent with previous electrophysiological experiments.23,31 However, whether this mutant will affect the kinetics of the channel is still unknown. The protonated E90 is considered to serve as a “proton shuttle” at anti-cycle.20 This means it is possible that it is only transiently deprotonated when the channel is opening. On the other hand, according to the crystal structure of ChR2,3 E90 locates in the CG in the middle of the ion channel, making it difficult rule out its role in the channel opening. Early research of Eisenhauer23 proposed that E90 was already deprotonated in P3520 intermediate, but whether deprotonation takes place in an earlier transition or simultaneously with the appearance of the P3520 is unresolved. The FTIR experiments of Kuhne et al.9 revealed that H-bond changes and deprotonation of E90 within submicroseconds (this time constant is assigned to P1500 intermediate) and cannot be distinguished owing to low signal-to-noise ratio. Many theoretical investigations proposed that early deprotonation of E90 induced water molecules to enter the CG and formed a pre-open channel by MD simulations.7,9 The results showed that photoisomerization of retinal causes the displacement of E90 and permits water molecules to enter the CG, which leads TM2 to tilt and finally results in the channel opening. Conformational changes of some polar residues located on TM2 due to the interactions between K93 and deprotonated E90, occur during channel formation. Thus, the deprotonation of E90 seems to be crucial in the pore preformation in anti-retinal conformation.
N-anti and CN-syn retinal conformations, respectively. The former involved successive cation conductance and protonated E90 while the later generated small conductance and deprotonated E90. It seems that only the case where E90 remains protonated can produce high photocurrent in Kuhne's research.20 In addition, the authors stated the E90Q mutation also reduced photocurrents, which is consistent with previous electrophysiological experiments.23,31 However, whether this mutant will affect the kinetics of the channel is still unknown. The protonated E90 is considered to serve as a “proton shuttle” at anti-cycle.20 This means it is possible that it is only transiently deprotonated when the channel is opening. On the other hand, according to the crystal structure of ChR2,3 E90 locates in the CG in the middle of the ion channel, making it difficult rule out its role in the channel opening. Early research of Eisenhauer23 proposed that E90 was already deprotonated in P3520 intermediate, but whether deprotonation takes place in an earlier transition or simultaneously with the appearance of the P3520 is unresolved. The FTIR experiments of Kuhne et al.9 revealed that H-bond changes and deprotonation of E90 within submicroseconds (this time constant is assigned to P1500 intermediate) and cannot be distinguished owing to low signal-to-noise ratio. Many theoretical investigations proposed that early deprotonation of E90 induced water molecules to enter the CG and formed a pre-open channel by MD simulations.7,9 The results showed that photoisomerization of retinal causes the displacement of E90 and permits water molecules to enter the CG, which leads TM2 to tilt and finally results in the channel opening. Conformational changes of some polar residues located on TM2 due to the interactions between K93 and deprotonated E90, occur during channel formation. Thus, the deprotonation of E90 seems to be crucial in the pore preformation in anti-retinal conformation.
It is worth noting that whether the protonation state of E90 is related to the proton transfer of RSBH+ has not been reported. It is hard to imagine that both E90 and proton acceptors (E123 or D253) are located in CG but are not related to each other. A detailed understanding of the photoactivation mechanism of ChR2 requires the crystal structure of the intermediates in the photocycle, which are currently unavailable. However, it is feasible to further understand the process of the RSBH+ proton transfer and the formation of the H-bond network of related residues. Investigating the changes of the protonation state of E90 and studying the residues responsible for the RSBH+ proton acceptor in photocycle play a key role in understanding the ion channel formation. Detailed exploration of proton transfer and H-bonds changes among the aforementioned key residues in the CG (including E90, E123, D253 and K93 etc.) might shed light on the mechanism.
The timescale for process of channel opening can range from tens to hundreds of milliseconds, and MD simulations would be too costly for such a long timescale. It is also difficult to perform MD for chemical reactions such as proton transfer. Therefore, in this study, we assumed two extreme initial states, viz., that E90 is protonated and deprotonated at P1500 to investigate the subsequent conformation evolution with time by MD simulations. We hypothesized that these equilibrium conformations might represent the structures of intermediates P1500 and P2390. Then Na+ ions passing through the channel in P2390 conformation was simulated by steered molecular dynamics (SMD). Finally, we qualitatively judged the possibility of channels opening by PMF calculation. The details of conformational changes and PMF should provide evidence for proton transfer involving RSBH+ and (de)protonation of E90.
Methods
ChR2 modeling
The crystal structure of ChR2 (PDBID: 6EID)3 was embedded in a pre-balanced 16:0/18:1c9-palmitoyloleyl phosphatidylcholine (POPC) lipid bilayer to build the transmembrane ChR2 structure. ChR2 protein and lipid bilayer were solvated with TIP3P water molecules32 and 0.15 M NaCl solution to mimic a physiological environment. Membrane Builder33 of CHARMM-GUI34,35 was used for generation of lipid bilayer and solvation of protein. The D253 and E123, which serve as the potential counterions of RSBH+, were taken to be negatively charged, as deduced from previous QM/MM simulations and spectroscopic experiments.21,36 The E90, K93 and D156 were protonated while other titratable residues were assumed as neutral for consistency with previous work.9,10,19,21 The equilibrium conformation of dark-adapted ChR2 obtained from a 400 ns MD simulation (i.e. the structure of D470 state) under approximate physiological environment was selected as the starting point of the photocycle instead of crystal structure.Retinal isomerizes from all-trans to 13-cis configuration after absorbing light to form the P1500 state and then the RSBH+ deprotonates to form the P2390 state.10–12,15,20 The 13-cis retinal was picked from the K-intermediate of bacteriorhodopsin (PDB ID: 1IXF).37 The main chain atoms and the hexatomic ring of the 13-cis retinal molecule were superimposed on those of all-trans retinal isomer, then the all-trans retinal in the equilibrated D470 state was replaced with the 13-cis conformation in the P1500 state. This modeling protocol was previously used by Guo,6,21 Takemoto7 and Yang.38 Subsequently, the hybrid QM/MM method39 was used for energy minimization to obtain the stable protein structure in P1500 state with protonated E90 (E90p-P1500). Considering that photoisomerization of all-trans retinal occurs at a subpicosecond timescale, where the protein backbone does not move significantly, we used the aforementioned approach to build the 13-cis model.
After a 400 ns simulation, the proton of the RSBH+ was pointing towards E123 in E90p-P1500 state. We artificially moved the proton on RSBH+ to E123 based on the final structure of the E90p-P1500 state to get the P2390 state (E90p-P2390). To discuss the effect of E90 protonated state on the proton acceptor RSBH+, we also assumed that E90 is deprotonated in the P1500 state (E90d-P1500). For the E90 deprotonated state, the E90d-P1500 and E90d-P2390 were obtained using a similar scheme. The equilibrated structure of E90d-P1500 exhibited that the proton of the Schiff base was oriented towards D253. Then the proton of RSBH+ was transferred to D253 to generate the E90d-P2390 state.
The QM region was composed of E123, D253, and retinal chromophore covalently bonded with K257. The remaining part of the protein, lipid, and solution were set as the MM region. The QM area was calculated using the self-consistent charge density-functional-based tight-binding (SCC-DFTB) method40 in the Amber 16 package.41 The SHAKE algorithm was used to freeze the bonds containing hydrogen atoms during the minimization. PME was described by the long-distance static electricity with a cut-off radius of 8 Å. Then the SHAKE restriction on hydrogen bond was removed and a constant-temperature simulation was run by QM/MM approach. The time step was 0.02 fs per step, and the trajectories were saved every 100 fs, and 1 ns of the production run was performed.
The non-standard residue, composed of a retinal molecule and linked K257, was described by the deprotonated retinal force field parameters published by Zhu et al.,42 while the other protein, lipid membrane and ions of D470, P1500 and P2390 state were described by the CHARMM36 force field.43 Similar simulation protocols have been reported in recent ref. 44 and 45.
MD simulation
MD and SMD simulations were run in NAMD 2.12 software.46 A grid-based fast Fourier transforms method, Particle-Mesh–Ewald (PME),47 was used to evaluate the long-range electrostatic interactions. The grid spacing is less than 1 Å, and the cut-off radius of short-range non-bonding interaction is set to 12 Å in PME. The bonds containing hydrogen atoms were frozen using the SHAKE constraint algorithm.48 The steepest descent method was used to minimize the energy of the system, and the system was heated to a temperature of 310 K. The Langevin Piston algorithm to keep the pressure constant at 1 bar (NVT) was used in this process. Finally, a 400 ns run was carried out under the constant temperature and pressure (NPT) of 310 K and 1 bar without restraint at 2 fs per step and the trajectories were saved every 5 ps. The last equilibrated 50 ns trajectory was selected for analysis. We carried out 5 parallel simulation trajectories for each intermediate.SMD and umbrella sampling
There are still some limitations on the timescale for dynamics simulation. It is impossible to complete the simulation of the ion passing through the ion channel with timescale of milliseconds to seconds.5,49 In addition, in MD simulation, there is a high probability that sampling will fall into the local minimum in the conformation space, which makes it difficult to simulate processes with a high energy barrier such as ion transmembrane conduction. Therefore, SMD is used in this investigation to obtain detailed information about atomic coordinates during the ion conduction process by artificially adding an imaginary external force along the Na+ path in the simulation. The system sometimes cannot relax to equilibrium when constructing the potential mean force (PMF) of the stretching process because of the imaginary external force. In order to reduce the impact of the imaginary external force, umbrella sampling50,51 is applied to construct PMF in this work. Umbrella sampling consists of some individual “windows” that run the reaction coordinate simultaneously. It can find the equilibrium state in each window along the reaction coordinates, and calculate the free energy change of each window. Then, the Weighted Histogram Analysis Method (WHAM)52 or some other method is used to combine the windows according to the reaction coordinates for constructing PMF.All the SMD simulations were performed with constant-velocity.53 In the SMD simulations, Na+ was steered with a velocity of 0.2 Å·ns−1 to simulate the quasi-static process. To ensure constant velocity steering, the spring coefficient k was set to 4 kcal mol−1·Å−2.38 To avoid protein structure changes during the SMD simulation, a 5 kcal mol−1 Å−2 of Hamiltonian limit was added to the amino acid residues around the helix.
Umbrella sampling used the z-direction along the channel axis and the reaction coordinate was divided into the windows in units of 1 Å. To ensure the interval of each window in the z-direction, a harmonic potential with a force constant of 2.5 kcal mol−1·Å−1 was set and the width of translocation colvar was set as 0.1 Å. The WHAM was used to construct the PMF based on the umbrella sampling simulation.
Results and discussions
The dark-adapted state of ChR2
The MD simulation of the crystal structure was performed for 400 ns and the root-mean-square deviation (RMSD) is shown in Fig. S1.† The isomerization of retinal and E90 deprotonation in CG lead to the breaking of H-bonds, with the retinal twist causing an increase in the steric interaction with the surrounding residues. There is an obvious change in the RMSD curve in E90d-P1500 state. The last 50 ns segments of the equilibrated trajectories were picked for cluster analysis, using the K-means algorithm in the MMTSB toolset54 to get the representative conformations of D470 state. In the ChR2 crystal structure,3 the two counterion carboxylates, E123 and D253 are almost symmetrically located on two sides of the RSBH+ respectively and stabilize the charge of RSBH+. It is very similar to the C1C2 chimera.11 Compared with the crystal structure, the protein backbone of D470 has not significantly changed except for some local conformations. For example, the polyene chain of retinal that was originally close to a planar conformation ended up bent by a few degrees after the equilibration run (as seen in Fig. S2†). However, a small amount of water has entered the ECG after kinetic equilibrium, as shown in Fig. 2. This indicates that the opening of the ECG causes the K93 moving and forming an H-bond with E123, as well as an H-bond network with D253 mediated by a water molecule. The position of D253 is almost unchanged while the carboxyl of E123 tilts toward the Schiff base, which destroys the original electrostatic balance. Therefore, the Schiff base reoriented so that the proton could face toward D253 to electrostatically rebalance in the D470 state. This conformation of D470 is opposite to that found by Kuhne and Ardevol,9,10 in which the Schiff base pointed towards the anionic form of E123. In their work, the structure of ChR2 was constructed by homology modeling based on the crystal structure of the C1C2 chimera. Although the homology modeling is very similar to crystal structure of ChR2, it cannot be exactly the same. Perhaps the slight difference between homology modeling and crystal structure leads to the different orientation of the RSBH+.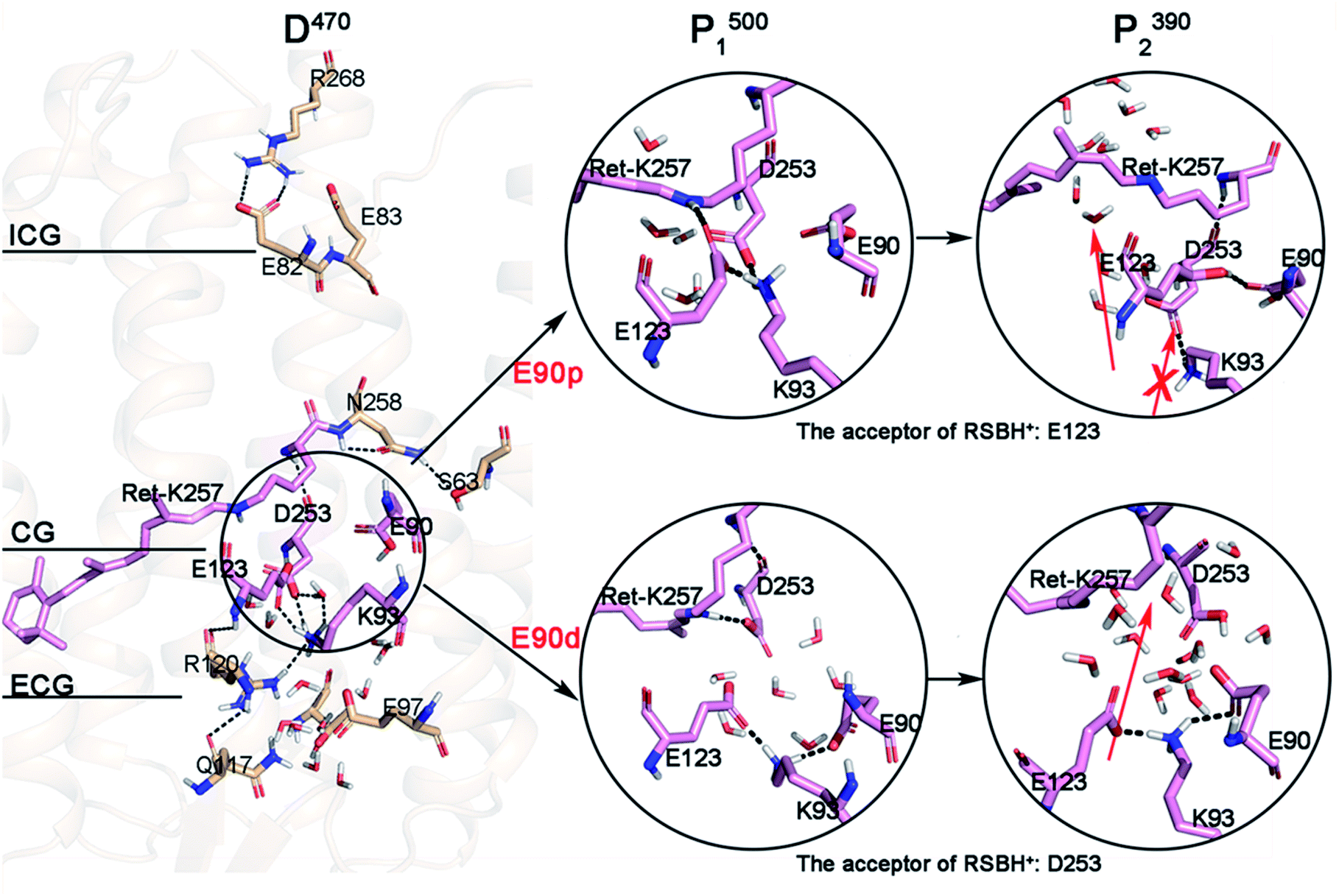 | ||
| Fig. 2 The structure of gates in the dark-adapted state and the structural changes of CG in the photocycle under different E90 protonation states. The red arrows represent potential channels; the arrow with cross represent an inaccessible channel. | ||
Structural changes of intermediates during photocycle
The simulation results suggest that the integral structure of the protein does not change significantly during the photocycle when E90 stays protonated in the E90p-P1500 and E90p-P2390 states over the production runs of 400 ns (Fig. S3a and S3b†). The intracellular segment of TM3 is tilted 3.9 Å to TM4 at the E90p-P500 state. However, the same segment of TM3 is tilted 2.4 Å to TM2 at the E90p-P2390 state.It was found that the ECG is opening in the D470 state and the CG is blocked by the formation of salt bridge between positive K93 and negative E123 (Fig. 2). However, since the side chain amino of K93 is bridge-linked to E123, it is difficult to form an H-bond with E90, in which the side chain carboxyl is oriented towards the peptide bond of K93. This local conformation of D470 leads K93 to be far away from E90, while being attracted by the negative charge of D253. Pushed by K93, the carboxyl of E123 tilts toward the RSBH+ and finally forms an H-bond with it in E90p-P1500. This suggests that the primary proton acceptor of the RSBH+ in the next step of the photocycle is most probably E123, which is consistent with latest theoretical work.6 Obviously, the conformational transition from proton being oriented towards D253 in the D470 state to being directed towards E123 in the P1500 intermediate arises from the isomerization of retinal. Meanwhile, the isomerization of retinal leads the indole group of W223 in TM6 to shift 3.7 Å towards TM5 (Fig. S4†), and results in a cavity near the polyene chain, possibly forming a novel channel that was also observed in C1C2.5,7 The proton of the RSBH+ artificially transfers to E123 and forms the E90p-P2390 state according to conformation of E90p-P1500. It can be seen that the retinal distorts more dramatically, which will result in a larger cavity in the E90p-P2390 state. On other hand, K93 still maintains electrostatic interaction with D253 and blocks CG. Thus, a novel channel located next to the Schiff base was suggested when E90 is protonated in P2390, and labeled by red arrow in Fig. 2. The arrow with cross represents an inaccessible channel.
The MD simulation of 400 ns were also carried out for the E90d-P1500 and E90d-P2390 states respectively. Previous studies have shown that the photoactivation of ChR2 is always associated with the conformational changes of TM2, TM6, and TM7,7,11,55,56 and the outward tilt of TM2 helps water molecules permeate near TM2.9,55 We saw similar results. It can be seen from Fig. S3c† that the extracellular segment of TM2 tilted outward by 4 Å in the E90d-P1500 state. In contrast, the integral TM2 tilted outward with an average of 3.2 Å in the E90d-P2390 state (Fig. S3d†).
The cluster analysis demonstrates that there are significant changes at the three gates of the E90d-P1500 and E90d-P2390 intermediates. Compared to D470, the interactions patterns around K93 at CG in E90d-P1500 state changes significantly because of the isomerization of retinal and the deprotonation of E90. In the E90d-P1500 state, E90 and K93 form an intrahelical salt bridge8–10 which leads TM2 to move outward.9,55,56 Therefore, the connection between K93 and D253 in the D470 state is cut off because K93 approached E90. Simultaneously, the negative E123 on TM3 was attracted by the positive K93 and leaves RSBH+ with K93. Then, D253 flips towards RSBH+ to form an electrostatic interaction. As seen in Fig. 2, D253 is the only possible proton acceptor of RSBH+ in E90d-P2390. E123, K93 and E90 form a tight H-bond network on the side of the cavity. As a result, the cavities become larger and more water molecules are able to enter.
Water distributions and position of channel
It has been widely reported that water molecules play an important role in the channel formation in ChR2.4,9,10 Water molecules can enter from the extracellular side to form a preopen pore in the P1500 state.10 The grid water density was used to calculate the water distribution of each intermediate in the photocycle, as shown in Fig. 3. It was found that the continuous water distribution only exists at the ECG in the D470 state (Fig. 3a). Previous studies have shown that the protonated E90 at the CG acts as a hydrophobic barrier in the dark state to prevent water from entering through the CG and ICG.3,23,24 In the E90p-P1500 state (Fig. 3b), the water distribution is found in the vicinity of the retinal instead of the blocked CG. From the E90p-P1500 state to E90p-P2390 state (Fig. 3c), the retinal tilts remarkably and permits water molecules to enter the cavity near the retinal, just like the result of new distributions of water molecules appear near the retinal and H173 in C1C2 protein found by Cheng et al.5 For ChR2, however, only one hydrophobic residue (W260) is located between the Schiff base and the ICG, which hinders further entry of water molecules, as shown in Fig. 3c.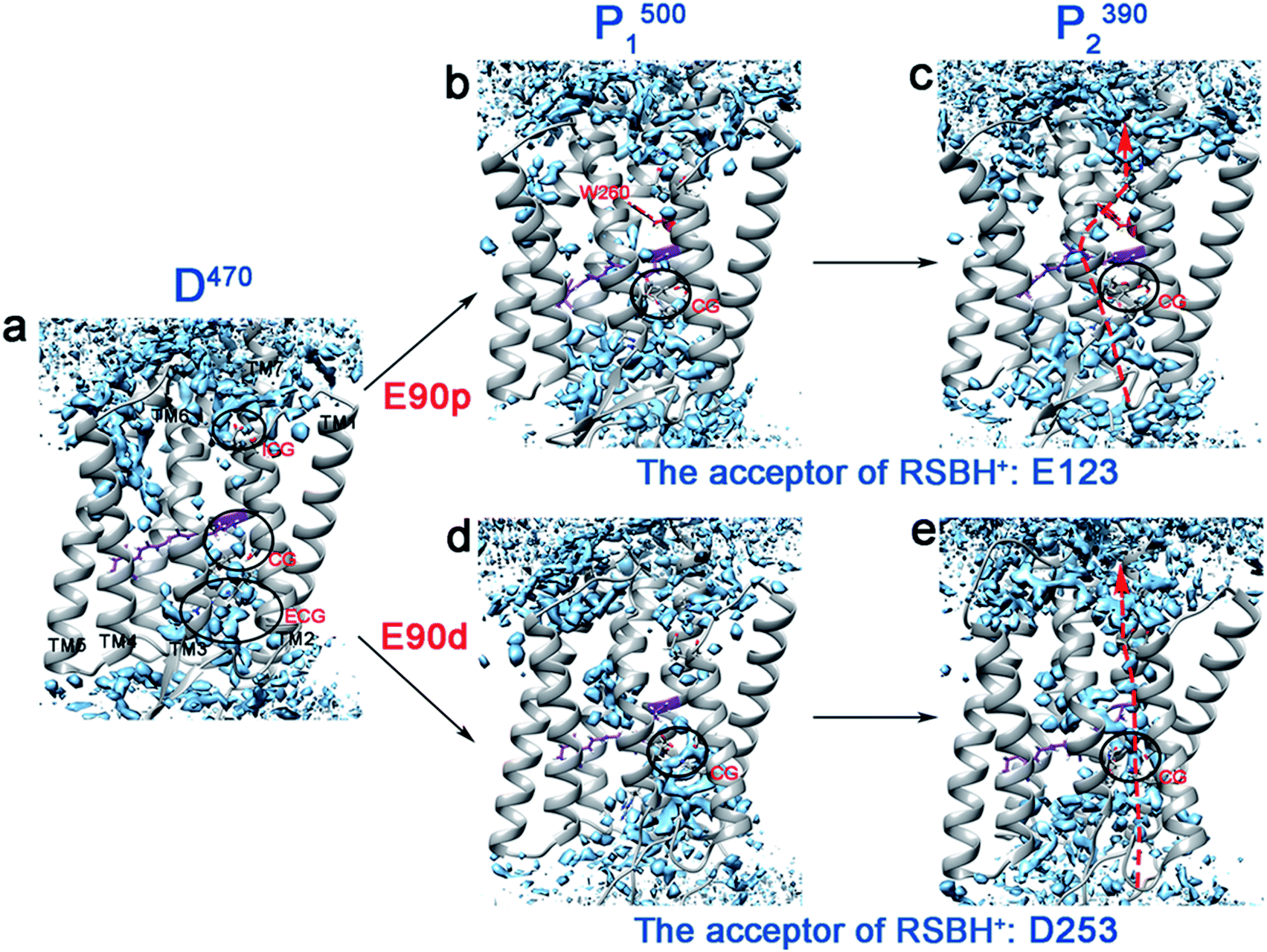 | ||
| Fig. 3 (a–e) The water distributions of the photocycle intermediates. The water molecules are shown in blue. The arrows indicate potential locations of ion channel. | ||
The deprotonated E90 can open the CG and lead to rearrangement of the H-bond network. It aids the ICG opening and the ion channel formation.7–9,23 As seen in Fig. 3d, both CG and ICG are filled with water molecules in the E90d-P1500 state. However, the water distribution is not continuous, which suggests that blockages exist between CG and ICG in the E90d-P1500 state. The proton transferred from the RSBH+ to the D253 forms the E90d-P2390 state further changing the hydrogen bonding patterns in the channel. Consequently, the water molecules enter the channel in the E90d-P2390 state (Fig. 3e) and leads to a pre-opened state. The degree of hydration of the TM1, 2, 3, 6, and 7 in the P2390 state calculated is consistent with the water distribution of the channel (Fig. S5†). It should be pointed out that the continuous water distribution only means that water molecules can enter, and does not mean that the ion can penetrate freely.
The static structure of the potential ion channel of the E90p-P2390 state was calculated by The CAVER 3.0 plug-in.57,58 A visualization of the pore along the TM3 is shown in Fig. 4a, and the higher helix hydration of pore is displayed in Fig. S5.† This channel passes through the slit between R120 and H249 at the ECG, enters the cavity formed by the retinal, W223, S22, and S256 (Fig. 4c), and then crosses the gap between W260, F219, and I131 (Fig. 4b). It finally enters the ICG. The average diameter of the channel is 2.5 Å to 2.7 Å from the ECG to the ICG, which suggests that the E90p-P2390 state is not a completely open state. The displacement of other relevant residues in the bottleneck may increase the size of channel pore in the subsequent photocycle.
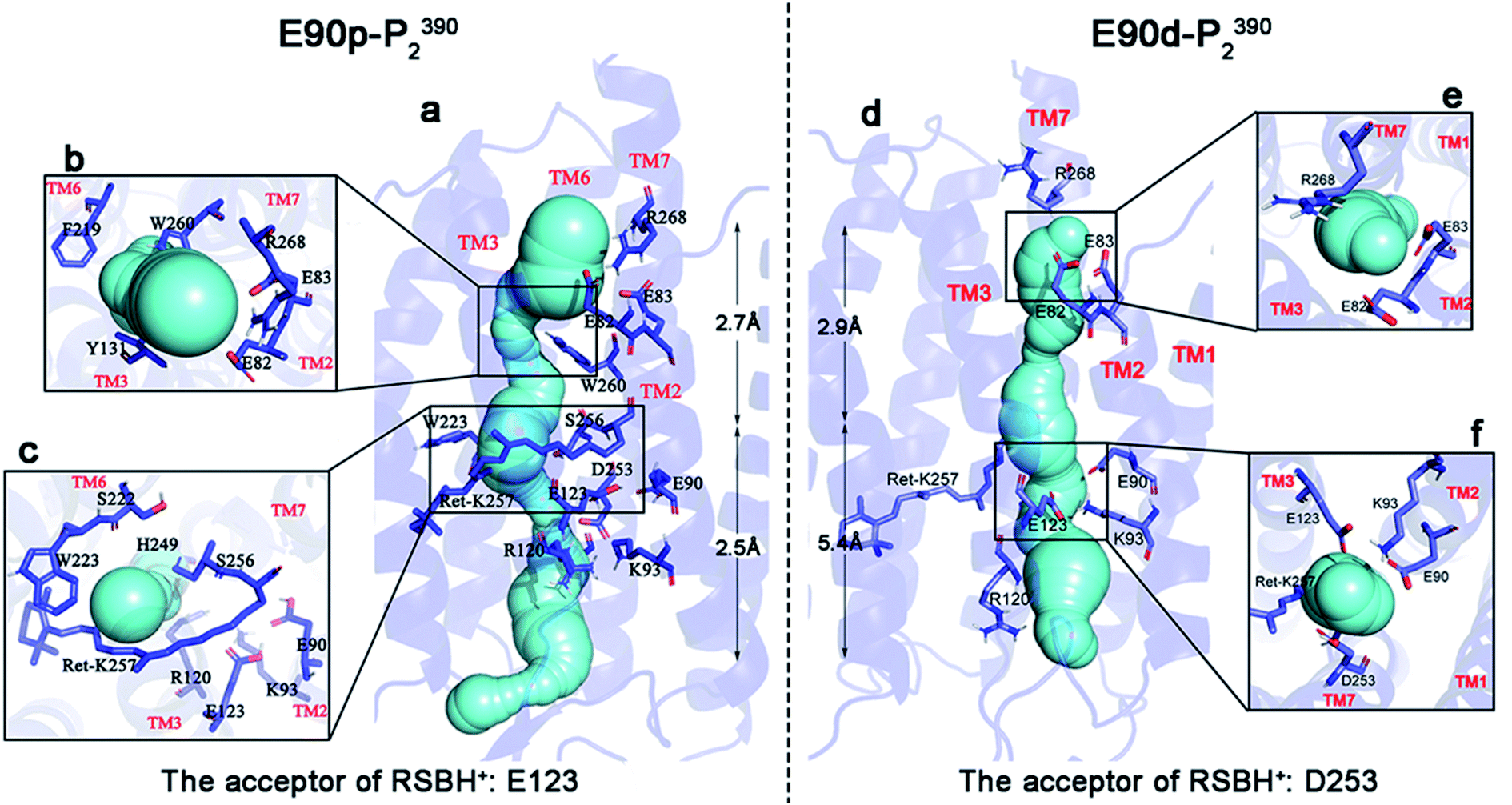 | ||
| Fig. 4 The detailed views and key sites of the channels in the E90p-P2390 and E90d-P2390 states. The channels are shown in cyan (a and d). In the figure, (b) is the top view at ECG in the E90p-P2390 state, (c) the top view near the retinal in the E90p-P2390 state, (e) the top view at ECG in the E90d-P2390 state, and (f) the top view at CG in E90d-P2390 state. | ||
The static structure of the pre-opened ion channel of the E90d-P2390 intermediate is shown in Fig. 4d. The channel starts from the ECG, passes through the CG, then enters the ICG. This channel forms along TM2, consistent with the potential channel position in previous study.38 TM2 has a higher helix hydration than that in the E90p-P2390 state, as shown in Fig. S5.† Fig. 4f shows a top view of the distribution of the key residues at CG. It is noticeable that K93 forms hydrogen bonds with E90 and E123 respectively on side of the channel. At ICG (Fig. 4e), R268 flips over and ICG turns on. The average diameter of the channel from ECG to CG is 5.4 Å, close to the experimental result of 6.2 Å.1,59 The average diameter from CG to ICG is only 2.9 Å. This distance may increase further in the subsequent photocycle.
Ion steering and PMF in the channel
To understand the basic principle of the penetration of Na+ through the ion channel, the kinetics of the ionic transmembrane process in the P2390 state was simulated by SMD, and the PMF was calculated by umbrella sampling. In our simulations, the different protonation states of E90 form two different P2390 intermediates, resulting in two different channels during the photocycle. The PMFs of two channels are shown in Fig. 5.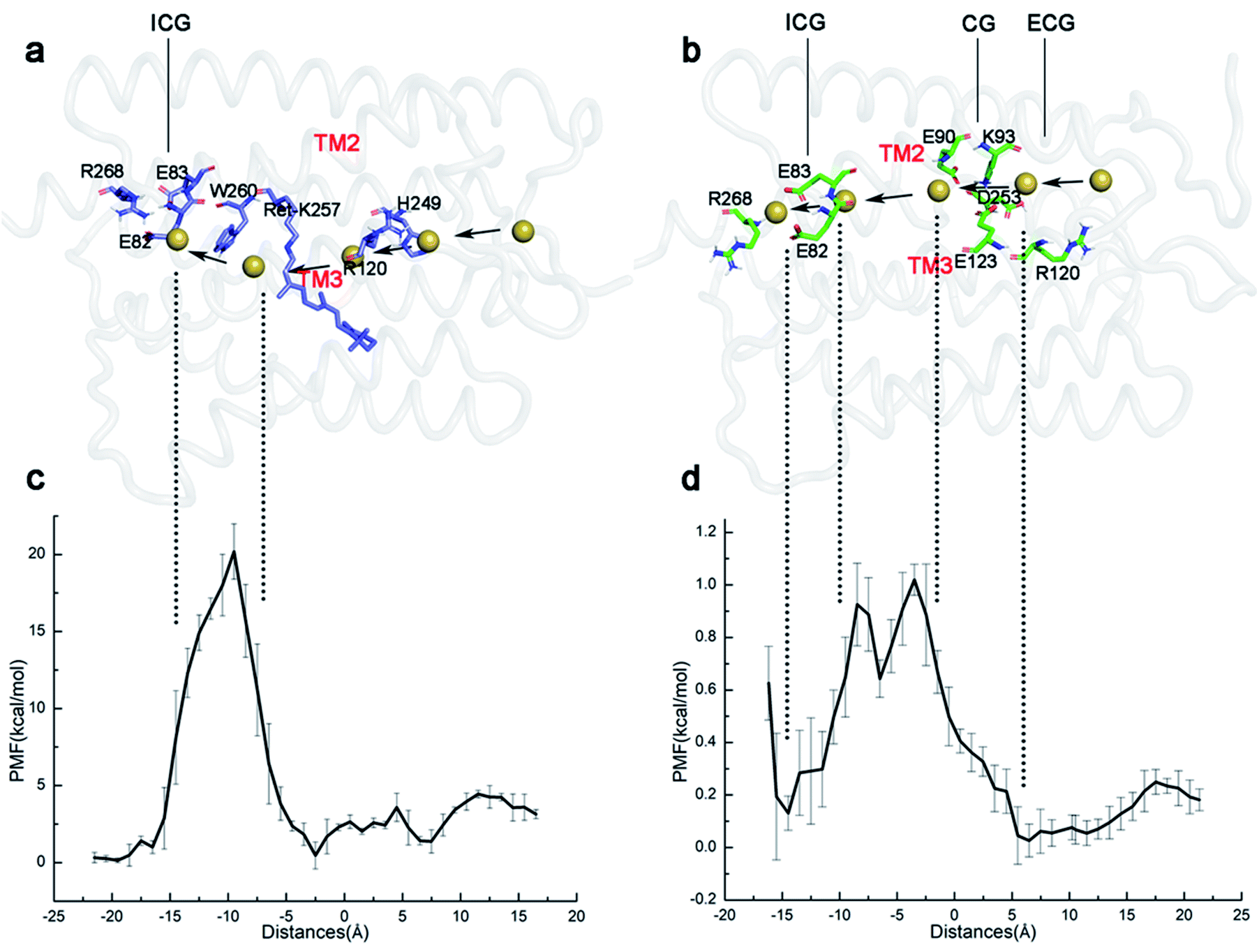 | ||
| Fig. 5 (a and b) The schematic diagrams of the simulated trajectory of ions passing through the channel in the E90p-P2390 and E90d-P2390 states, respectively. Na+ is represented by a small yellow ball. (c and d) Umbrella sampling is used to reconstruct the PMF of ion permeability. | ||
The average diameter of the ion channel in E90p-P2390 state is only 2.6 Å. This does not allow multiple ions to pass through the channel simultaneously. As seen in Fig. 5c, an energy barrier of 4.1 kcal mol−1 needs to be overcome as Na+ enters the ECG. When ions pass through the gap formed by R120 and H249 (10 Å–5 Å), the PMF will drop to about 1.4 kcal mol−1 at first. Then, the PMF stays below 3 kcal mol−1, and then drops to around 1 kcal mol−1 near the retinal chromophore. The PMF rises sharply because of the W260, approaching 20 kcal mol−1. Since the E90p-P2390 state is not an open state, the free energy is relatively high for ions to pass through the potential channel. However, the positions of the related residues might change in the subsequent photocycle to allow ions passing freely.
As shown in Fig. 5d, in the E90d-P2390 state, the PMF of Na+ conduction in the channel is very low. The Na+ enters the CG through the ECG and the PMF reaches a minimum energy value (approximately 0 kcal mol−1) near the CG. The electronegative residues E123 and E90 in CG electrostatically attract the Na+ cations consequently, the vicinity of CG is a binding site for Na+. An energy barrier of 1.0 kcal mol−1 in the PMF suggests that there is a small bottleneck between CG and ICG (Fig. 4d). The PMF drops rapidly to 0.1 kcal mol−1 at ECG because E82 and E83 in the ICG attract ions Na+ electrostatically and consequently. The ICG becomes another potential binding site for Na+. The small barrier also implies that the P3520 may completely open because of the modification of the conformations of some residues at this site.
The variations of the PMF suggest that a very high energy barrier exists for ions Na+ passing and therefore the W260 blocks the channel in the E90p-P2390 state. In the photocycle of the E90 deprotonation, the aperture of the channel becomes bigger and its PMF is close to the open state. Therefore, the deprotonation of E90 is more conducive to the formation of the channel.
In the case where E90 remains protonated, the CG is still blocked. Lateral movement of retinal polyene chain gets out of the way of a new narrow channel, which is very similar with what Cheng5 found in C1C2. If E90 is deprotonated, the changes in the hydrogen bond network result in the CG blockage site opening, and forms the widely accepted “broad channel”, which is conducive to the complete opening of the channel. Furthermore, the pKa of E90 in different states was calculated. As shown in Table S1,† the pKa values imply that E90 in ChR2 is protonated in D470 state but deprotonated in subsequent states at neutral environment. This result supports the notion that E90 tends to deprotonate after retinal isomerization. In other words, E90 deprotonation is more beneficial to channel opening.
Conclusions
In this study, the photocycle intermediates of ChR2 during the formation of the channel were studied by molecular dynamics simulation. To understand the role of E90 in the formation of channel, both protonated and deprotonated E90 were assumed in the P1500 intermediate.When E90 remains protonated, K93 is located in the center of CG and hardly forms H-bond with E90. The unconstrained K93 moves to E123 and pushes E123 closer to RSBH+, resulting in a “RSBH+–E123−” pattern. In this case, E123 acts as the proton acceptor of the RSBH+. Meanwhile, with the tilting of polyene chain, a novel narrow channel detoured CG forms among TM2, TM3, TM6, and TM7 after proton transfers to E123. The variation of the PMF suggests that a higher energy barrier needs to be overcome for Na+ to pass through this channel. It indicates that the channel is still closed. On the other hand, when E90 is deprotonated in P1500 state, E90 and K93 concertedly pull TM2 outward, pushes E123 far away from RSBH+, and ultimately leads to formation of “RSBH+–D253−” pattern. In this case, the proton acceptor of RSBH+ is D253. An average diameter of 5.4 Å from ECG to CG forms among TM1, TM2, TM3 and TM7 and the PMF calculations suggest an approximately opened channel.
Our simulations show that protonation state of E90 can influence the structure of CG, which is related to the primary proton acceptor of RSBH+. Different protonation states of E90 affect the position of K93, and then affect the orientations of E123 and D253 that generate the corresponding proton acceptor of RSBH+. The subsequent conformational changes might lead to different potential channels. Based on the simulation results, the deprotonation of E90 seems to be more favorable for the opening of the channel in the late P2390 state.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by Natural Science Foundation of Chongqing, China (cstc2020jcyj-msxmX0653) and Chongqing Education Commission Foundation (KJQN201900618).References
- G. Nagel, T. Szellas, W. Huhn, S. Kateriya, N. Adeishvili, P. Berthold, D. Ollig, P. Hegemann and E. Bamberg, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 13940–13945 CrossRef CAS PubMed.
- E. S. Boyden, F. Zhang, E. Bamberg, G. Nagel and K. Deisseroth, Nat. Neurosci., 2005, 8, 1263–1268 CrossRef CAS PubMed.
- O. Volkov, K. Kovalev, V. Polovinkin, V. Borshchevskiy, C. Bamann, R. Astashkin, E. Marin, A. Popov, T. Balandin, D. Willbold, G. Büldt, E. Bamberg and V. Gordeliy, Science, 2017, 358, eaan8862 CrossRef PubMed.
- S. Adam and A. N. Bondar, PLoS One, 2018, 13, e0201298 CrossRef PubMed.
- C. Cheng, M. Kamiya, M. Takemoto, R. Ishitani, O. Nureki, N. Yoshida and S. Hayashi, Biophys. J., 2018, 115, 1281–1291 CrossRef CAS PubMed.
- Y. Guo, F. E. Wolff, I. Schapiro, M. Elstner and M. Marazzi, Phys. Chem. Chem. Phys., 2018, 20, 27501–27509 RSC.
- M. Takemoto, H. E. Kato, M. Koyama, J. Ito, M. Kamiya, S. Hayashi, A. D. Maturana, K. Deisseroth, R. Ishitani and O. Nureki, PLoS One, 2015, 10, e0131094 CrossRef PubMed.
- J. C. D. Kaufmann, B. S. Krause, C. Grimm, E. Ritter, P. Hegemann and F. J Bartl, J. Biol. Chem., 2017, 292, 14205–14216 CrossRef CAS PubMed.
- J. Kuhne, K. Eisenhauer, E. Ritter, P. Hegemann, K. Gerwert and F. Bartl, Angew. Chem., Int. Ed. Engl., 2015, 54, 4953–4957 CrossRef CAS PubMed.
- A. Ardevol and G. Hummer, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 3557–3562 CrossRef CAS PubMed.
- F. Schneider, C. Grimm and P. Hegemann, Annu. Rev. Biophys., 2015, 44, 167–186 CrossRef CAS PubMed.
- V. A. Lórenz-Fonfría and J. Heberle, Biochim. Biophys. Acta, 2014, 1837, 626–642 CrossRef PubMed.
- M. K. Verhoefen, C. Bamann, R. Blöcher, U. Förster, E. Bamberg and J. Wachtveitl, ChemPhysChem, 2010, 11, 3113–3122 CrossRef CAS PubMed.
- T. Kottke, V. A. Lórenz-Fonfría and J. Heberle, J. Phys. Chem. B, 2017, 121, 335–350 CrossRef CAS PubMed.
- C. Bamann, T. Kirsch, G. Nagel and E. Bamberg, J. Mol. Biol., 2008, 375, 686–694 CrossRef CAS PubMed.
- M. Nack, I. Radu, B. J. Schultz, T. Resler, R. Schlesinger, A. N. Bondar, C. Del Val, S. Abbruzzetti, C. Viappiani and C. Bamann, FEBS Lett., 2012, 586, 1344–1348 CrossRef CAS PubMed.
- A. Berndt, O. Yizhar, L. A. Gunaydin, P. Hegemann and K. Deisseroth, Nat. Neurosci., 2009, 12, 229–234 CrossRef CAS PubMed.
- O. A. Sineshchekov, E. G. Govorunova, J. Wang, H. Li and J. L. Spudich, Biophys. J., 2013, 104, 807–817 CrossRef CAS PubMed.
- V. A. Lórenz-Fonfría, T. Resler, N. Krause, M. Nack, M. Gossing, G. Fischer von Mollard, C. Bamann, E. Bamberg, R. Schlesinger and J. Heberle, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E1273–E1281 CrossRef PubMed.
- J. Kuhne, J. Vierock, S. A. Tennigkeit, M.-A. Dreier, J. Wietek, D. Petersen, K. Gavriljuk, S. F. El-Mashtoly, P. Hegemann and K. Gerwert, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 9380 CrossRef CAS PubMed.
- Y. Guo, F. E. Beyle, B. M. Bold, H. C. Watanabe, A. Koslowski, W. Thiel, P. Hegemann, M. Marazzi and M. Elstner, Chem. Sci., 2016, 7, 3879–3891 RSC.
- J. Y. Lin, M. Z. Lin, P. Steinbach and R. Y. Tsien, Biophys. J., 2009, 96, 1803–1814 CrossRef CAS PubMed.
- K. Eisenhauer, J. Kuhne, E. Ritter, A. Berndt, S. Wolf, E. Freier, F. Bartl, P. Hegemann and K. Gerwert, J. Biol. Chem., 2012, 287, 6904–6911 CrossRef CAS PubMed.
- D. Gradmann, A. Berndt, F. Schneider and P. Hegemann, Biophys. J., 2011, 101, 1057–1068 CrossRef CAS PubMed.
- W. Zhang, T. Yang, S. Zhou, J. Cheng, S. Yuan, G. V. Lo and Y. Dou, Biomolecules, 2019, 9, 852 CrossRef CAS PubMed.
- M. R. VanGordon, G. Gyawali, S. W. Rick and S. B. Rempe, Biophys. J., 2017, 112, 943–952 CrossRef CAS PubMed.
- H. E. Kato, F. Zhang, O. Yizhar, C. Ramakrishnan, T. Nishizawa, K. Hirata, J. Ito, Y. Aita, T. Tsukazaki and S. Hayashi, Nature, 2012, 482, 369–374 CrossRef CAS PubMed.
- A. Inaguma, H. Tsukamoto, H. E. Kato, T. Kimura, T. Ishizuka, S. Oishi, H. Yawo, O. Nureki and Y. Furutani, J. Biol. Chem., 2015, 290, 11623–11634 CrossRef CAS PubMed.
- P. Nogly, T. Weinert, D. James, S. Carbajo, D. Ozerov, A. Furrer, D. Gashi, V. Borin, P. Skopintsev, K. Jaeger, K. Nass, P. Båth, R. Bosman, J. Koglin, M. Seaberg, T. Lane, D. Kekilli, S. Brünle, T. Tanaka, W. Wu, C. Milne, T. White, A. Barty, U. Weierstall, V. Panneels, E. Nango, S. Iwata, M. Hunter, I. Schapiro, G. Schertler, R. Neutze and J. Standfuss, Science, 2018, 361, eaat0094 CrossRef PubMed.
- V. A. Lórenz-Fonfría, C. Bamann, T. Resler, R. Schlesinger, E. Bamberg and J. Heberle, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E5796–E5804 CrossRef PubMed.
- K. Ruffert, B. Himmel, D. Lall, C. Bamann, E. Bamberg, H. Betz and V. Eulenburg, Biochem. Biophys. Res. Commun., 2011, 410, 737–743 CrossRef CAS PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- E. L. Wu, X. Cheng, S. Jo, H. Rui, K. C. Song, E. M. Dávila-Contreras, Y. Qi, J. Lee, V. Monje-Galvan, R. M. Venable, J. B. Klauda and W. Im, J. Comput. Chem., 2014, 35, 1997–2004 CrossRef CAS PubMed.
- S. Jo, T. Kim, V. G. Iyer and W. Im, J. Comput. Chem., 2008, 29, 1859–1865 CrossRef CAS PubMed.
- J. Lee, X. Cheng, J. M. Swails, M. S. Yeom, P. K. Eastman, J. A. Lemkul, S. Wei, J. Buckner, J. C. Jeong, Y. Qi, S. Jo, V. S. Pande, D. A. Case, C. L. Brooks 3rd, A. D. MacKerell Jr, J. B. Klauda and W. Im, J. Chem. Theory Comput., 2016, 12, 405–413 CrossRef CAS PubMed.
- H. C. Watanabe, K. Welke, D. J. Sindhikara, P. Hegemann and M. Elstner, J. Mol. Biol., 2013, 425, 1795–1814 CrossRef CAS PubMed.
- Y. Matsui, K. Sakai, M. Murakami, Y. Shiro, S. Adachi, H. Okumura and T. Kouyama, J. Mol. Biol., 2002, 324, 469–481 CrossRef CAS PubMed.
- T. Yang, W. Zhang, J. Cheng, Y. Nie, Q. Xin, S. Yuan and Y. Dou, Int. J. Mol. Sci., 2019, 20, 3780 CrossRef CAS PubMed.
- R. C. Walker, M. F. Crowley and D. A. Case, J. Comput. Chem., 2008, 29, 1019–1031 CrossRef CAS PubMed.
- G. D. M. Seabra, R. C. Walker, M. Elstner, D. A. Case and A. E. Roitberg, J. Phys. Chem. A, 2007, 111, 5655–5664 CrossRef PubMed.
- D. A. Case, R. M. Betz, D. S. Cerutti, T. E. Cheatham III, T. A. Darden, R. E. Duke, T. J. Giese, H. Cohlke, A. W. Goetz and N. Homeyer, et al., AMBER 2016, University of California, San Francisco, 2016 Search PubMed.
- S. Zhu, M. F. Brown and S. E. Feller, J. Am. Chem. Soc., 2013, 135, 9391–9398 CrossRef CAS PubMed.
- J. Huang and A. D. MacKerell Jr, J. Comput. Chem., 2013, 34, 2135–2145 CrossRef CAS PubMed.
- K. Kovalev, R. Astashkin, I. Gushchin, P. Orekhov, D. Volkov, E. Zinovev, E. Marin, M. Rulev, A. Alekseev, A. Royant, P. Carpentier, S. Vaganova, D. Zabelskii, C. Baeken, I. Sergeev, T. Balandin, G. Bourenkov, X. Carpena, R. Boer, N. Maliar, V. Borshchevskiy, G. Büldt, E. Bamberg and V. Gordeliy, Nat. Commun., 2020, 11, 2137 CrossRef CAS PubMed.
- H. E. Kato, Y. S. Kim, J. M. Paggi, K. E. Evans, W. E. Allen, C. Richardson, K. Inoue, S. Ito, C. Ramakrishnan, L. E. Fenno, K. Yamashita, D. Hilger, S. Y. Lee, A. Berndt, K. Shen, H. Kandori, R. O. Dror, B. K. Kobilka and K. Deisseroth, Nature, 2018, 561, 349–354 CrossRef CAS PubMed.
- J. C. Phillips, R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid, E. Villa, C. Chipot, R. D. Skeel, L. Kalé and K. Schulten, J. Comput. Chem., 2005, 26, 1781–1802 CrossRef CAS PubMed.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- K. D. Hammonds and J. P. Ryckaert, Comput. Phys. Commun., 1991, 62, 336–351 CrossRef CAS.
- M. Saita, F. P. Sellnau, T. Resler, R. Schlesinger, J. Heberle and V. A. Lórenz-Fonfría, J. Am. Chem. Soc., 2018, 140, 9899–9903 CrossRef CAS PubMed.
- J. Kästner, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 932–942 Search PubMed.
- P. Akhshi and G. Wu, Phys. Chem. Chem. Phys., 2017, 19, 11017–11025 RSC.
- S. Kumar, J. M. Rosenberg, D. Bouzida, R. H. Swendsen and P. A. Kollman, J. Comput. Chem., 1992, 13, 1011–1021 CrossRef CAS.
- B. Isralewitz, M. Gao and K. Schulten, Curr. Opin. Struct. Biol., 2001, 11, 224–230 CrossRef CAS PubMed.
- M. Feig, J. Karanicolas and C. L. Brooks, J. Mol. Graphics Modell., 2004, 22, 377–395 CrossRef CAS PubMed.
- M. Müller, C. Bamann, E. Bamberg and W. Kühlbrandt, J. Biol. Chem., 2015, 427, 341–349 Search PubMed.
- T. Sattig, C. Rickert, E. Bamberg, H. J. Steinhoff and C. Bamann, Angew. Chem., Int. Ed., 2013, 52, 9705–9708 CrossRef CAS PubMed.
- A. Jurcik, D. Bednar, J. Byska, S. M. Marques, K. Furmanova, L. Daniel, P. Kokkonen, J. Brezovsky, O. Strnad, J. Stourac, A. Pavelka, M. Manak, J. Damborsky and B. Kozlikova, Bioinformatics, 2018, 34, 3586–3588 CrossRef CAS PubMed.
- M. Petřek, M. Otyepka, P. Banáš, P. Košinová, J. Koča and J. Damborský, BMC Bioinf., 2006, 7, 316 CrossRef PubMed.
- R. Richards and R. E. Dempski, PLoS One, 2012, 7, e50018 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra01879e |
| This journal is © The Royal Society of Chemistry 2021 |