
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Green synthesis of new lawsone enaminones and their Z/E(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e001.gif) C)-isomerization induced by organic solvent†
C)-isomerization induced by organic solvent†
Abolfazl Olyaei *,
Amir Mohamadi and
Nilufar Rahmani
*,
Amir Mohamadi and
Nilufar Rahmani
Department of Chemistry, Payame Noor University (PNU), PO BOX 19395-4697, Tehran, Iran. E-mail: Olyaei_a@pnu.ac.ir; Fax: +98-28-33374081; Tel: +98-28-33376366
First published on 6th April 2021
Abstract
The synthesis of a new class of lawsone enaminone derivatives by using lawsone, triethyl orthoformate and aromatic amines in the presence of guanidinium chloride under solvent-free conditions has been developed. Investigation of 1H-NMR spectra indicated that lawsone enaminones exist in the ketoenamine tautomeric form and undergo Z/E-isomerization with respect to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond in DMSO-d6 at room temperature. Furthermore, intramolecular hydrogen bonds have been observed in the synthesized compounds. This method has some advantages including short reaction times, high to excellent yields, simple work-up procedure and very easy purification of products by non-chromatographic methods.
C bond in DMSO-d6 at room temperature. Furthermore, intramolecular hydrogen bonds have been observed in the synthesized compounds. This method has some advantages including short reaction times, high to excellent yields, simple work-up procedure and very easy purification of products by non-chromatographic methods.
Introduction
Enaminone derivatives are versatile readily obtainable reagents and their chemistry has received considerable attention in recent years. They have demonstrated a potential as multipurpose synthetic intermediates in organic synthesis such as various heterocyclic compounds,1–5 synthons for the synthesis of various biologically active compounds such as antitumor, anti-epileptic, anti-inflammatory, antibacterial, anticonvulsant, and other therapeutic agents6–13and also a substructure that frequently occurs in natural products.14 They can also be used as starting materials for the stereoselective preparation of γ-aminoalcohols (by reduction).15 A literature survey reveals the availability of numerous catalysts and methods for the synthesis of these compounds. The most important and straightforward method involves the direct condensation of β-dicarbonyl compounds with amines using various catalysts.16–20 Enaminones have a pronounced tendency to undergo various isomeric transformations in solvents with different polarities. This property is particularly important for applications of enaminones and plays a major role in determining chemical, biological, and therapeutic activities of these compounds. According to the 1H- and 13C-NMR spectroscopy, they have feature intramolecular hydrogen bonds of O–H⋯N and N–H⋯O type, tautomerism between the enol imine and ketoenamine forms, and Z/E-isomerization of the ketoenamine form in respect to the CC bond.21
2-Hydroxy-1,4-naphthoquinone, or lawsone, or hennotannic acid, is one of the simplest naturally occurring naphthoquinones and is useful for many applications in various scientific and technological fields. It has been used as the starting material for the synthesis of a variety of biologically active compounds and materials with interesting properties.22–25 In recent decades, it has been used in many reactions for the synthesis of various organic compounds.26 Moreover, synthesis of aminonaphthoquinone Mannich base derivatives, as an interesting class of naphthoquinones, by multicomponent reactions of lawsone with a non-enolizable aldehyde and a primary or secondary amine via the Mannich reaction and their metal complexes using different conditions, and applications of these compounds has been reviewed.27
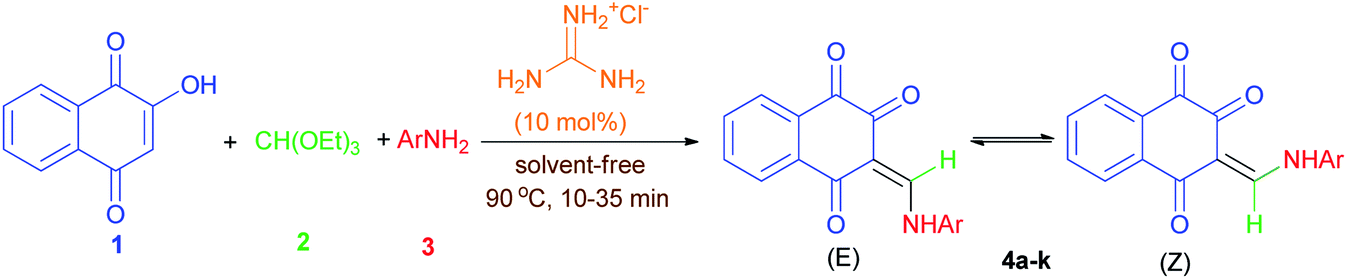
Considering the above points, and also in continuation of our interest on the synthesis of different kinds of heterocyclic compounds based on MCRs,28 herein we wish to report one-pot three-component synthesis of new lawsone enaminones from lawsone, triethyl orthoformate and aromatic amines in the presence of guanidinium chloride under solvent-free conditions and investigation of their Z/E(CC)-isomerization in DMSO-d6 at room temperature by 1H-NMR spectroscopy. To the best of our knowledge, there are no reports available for the synthesis of lawsone enaminone derivatives in the literature.
Results and discussion
To find an appropriate reaction condition for the synthesis of lawsone enaminone derivatives, a one-pot three-component reaction between lawsone (1) (1.0 mmol), triethyl orthoformate (2) (1.0 mmol) and 2-aminopyrimidine (3a) (1.0 mmol) was selected as a model reaction and was studied by employing guanidinium chloride as organocatalyst. Initially, we commenced the above three-component reaction using protic and aprotic solvents such as ethanol, water, acetonitrile and tetrahydrofuran under reflux conditions in the presence of guanidinium chloride (10 mol%) as the catalyst. As indicated in Table 1, the reaction proceed in CH3CN and resulted the desired product 4a in 45% yield after 90 min (Table 1, entry 2). Moreover, the reaction couldn't proceed smoothly in EtOH, H2O and THF after 90 min (Table 1, entries 1, 3 and 4). Increasing the reaction time did not improve the results. Then, we performed the same reaction under solvent-free conditions at 70, 80, 90 °C, and observed that the reaction took place smoothly at 90 °C, affording the desired product 4a in 85% yield at 15 min (Table 1, entry 7). Thus, 90 °C was selected as the promising condition for further screening of the appropriate loading amount of catalyst (0, 5, 15 and 20 mol%). When the reaction was performed under solvent and catalyst-free condition, the corresponding product 4a obtained in 30% yield after 60 min (Table 1, entry 8). The reaction with 5 mol% of the catalyst required a longer reaction time and produced only a 75% yield of the desired product 4a (Table 1, entry 9). Also, increasing the catalyst loading to 15 and 20 mol% did not affect the progress of the reaction markedly. As a result, it was found that 10 mol% of catalyst was enough to promote the reaction efficiently. Finally, the optimal conditions were determined as lawsone (1.0 mmol), triethyl orthoformate (1.0 mmol) and 2-aminopyrimidine (1.0 mmol) under solvent-free condition at 90 °C for 15 min.
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Catalyst (mol%) | Temperature (°C) | Time (min) | Yield (Z/E)b (%) |
| a Reaction conditions: 1.0 mmol of 1, 1.0 mmol of 2, 1.0 mmol of 3a, solvent (5 ml), catalyst (guanidinium chloride).b Isolated yield. | |||||
| 1 | EtOH | 10 | Reflux | 90 | Trace |
| 2 | CH3CN | 10 | Reflux | 90 | 45 |
| 3 | H2O | 10 | Reflux | 90 | Trace |
| 4 | THF | 10 | Reflux | 90 | Trace |
| 5 | Neat | 10 | 70 | 40 | 78 |
| 6 | Neat | 10 | 80 | 25 | 80 |
| 7 | Neat | 10 | 90 | 15 | 85 |
| 8 | Neat | — | 90 | 60 | 30 |
| 9 | Neat | 5 | 90 | 25 | 75 |
| 10 | Neat | 15 | 90 | 15 | 86 |
| 11 | Neat | 20 | 90 | 15 | 86 |
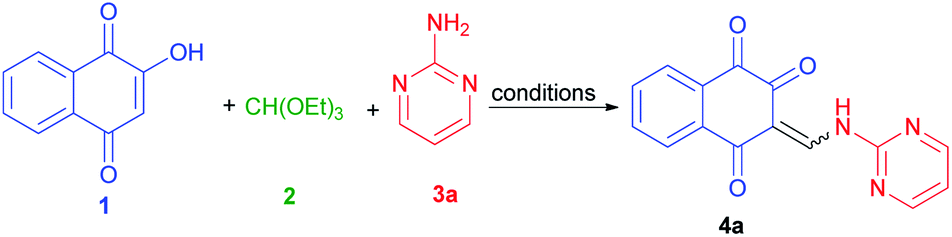
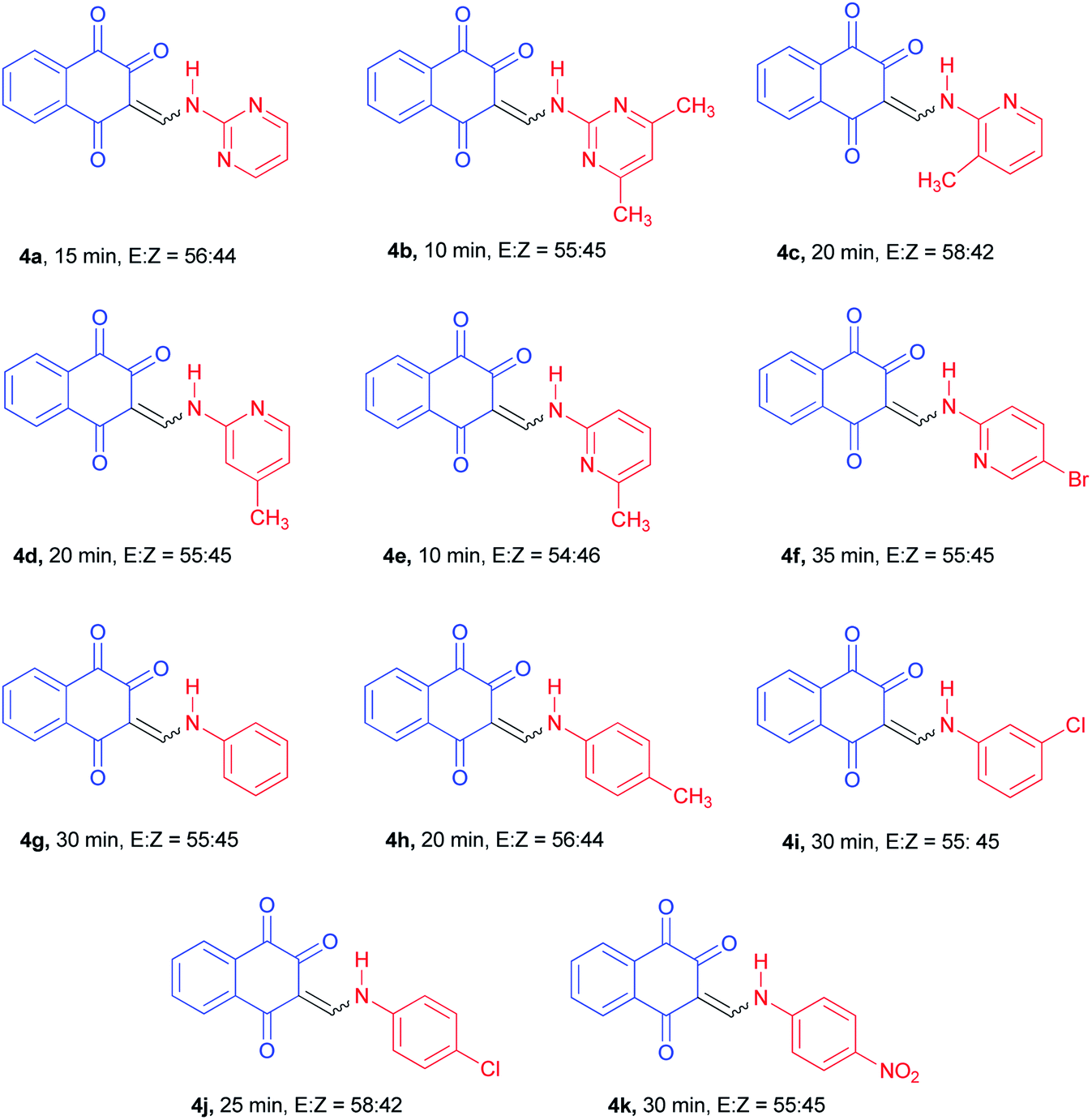
With the optimized conditions in hand, the scope and generality of this protocol were next examined by employing various aromatic amines. As shown in Table 2, the reaction with different aryl/heteroaryl amines bearing electron-donating or electron-withdrawing substituents proceeded smoothly in 10–35 min to afford the desired products 4a–k in high to excellent yields (75–87%). Meanwhile, we also found that the aromatic amines bearing electron-donating substituents such as methyl reacted rapidly, while those with bearing electron-withdrawing substituents such as chloro and nitro required longer reaction times, but products were obtained in comparable yields.
|
|---|
 |
All isolated products were new and fully characterized based on their analytical data and detailed spectral studies including Fourier transform infrared (FT-IR), 1H-NMR and mass spectroscopy. Despite of heating the samples in DMSO-d6 prior to 13C-NMR measurement, the low solubility of such compounds makes it difficult to receive a clean 13C-NMR spectrum.
The 1H-NMR spectra indicated that all of the synthesized compounds 4a–k existed in DMSO-d6 solution as a mixture of E- and Z-keto enamine isomers. For example, a characteristic feature of the 1H-NMR spectrum of compound 4a is the presence of two downfield signals corresponding to the protons of the NH group at 12.66 and 12.56 ppm. As indicated in Fig. 1, the more downfield signal corresponds to the Z-isomer of compound 4a which is stabilized by a strong chelate type intramolecular hydrogen bond with the oxygen of the C(2)O group. The signal for the proton of the NH group in the more upfield region corresponds to the E-isomer. The formation of another intramolecular hydrogen bond with oxygen atom of the C(4)O fragment is possible in this isomer. This hydrogen bond is significantly weaker than the hydrogen bond between the 12-NH and C(2)O groups. Moreover, in the isomer E, due to the resonance of the electron–nitrogen pair with the C(2)O, a S-trans structure is formed, which is more stable than the S-cis structure formed in the isomer Z (Scheme 1). With such an assignment the integral intensities of the 12-NH signals allow quantitative determination of the corresponding isomer content in the equilibrium mixtures: the Z-isomer from the signal at 12.66 ppm (44%) and the E-isomer from the signal at 12.56 ppm (56%). The difference between the 11-CH proton chemical shift values for the two geometric isomers is substantially smaller, and the 11-CH signal from the Z-isomer is located upfield. It is interesting to note the values of the vicinal spin–spin coupling constants J11,12 of the 11-CH and 12-NH protons for the two isomeric forms are 13.2 Hz, indicating a transoid arrangement of the N(12)–H and C(11)–H bonds. It should be noted that there is no signal of hydroxyimine tautomer in the 1H-NMR spectra of the synthesized compounds. In the IR spectrum of compound 4a, the sharp peaks at 3397, 1690, 1665 and 1608 cm−1 are due to the stretching frequencies of N–H and CO, respectively. The E![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Z ratio of isomers 4a–k are listed in Table 2.
:Z ratio of isomers 4a–k are listed in Table 2.
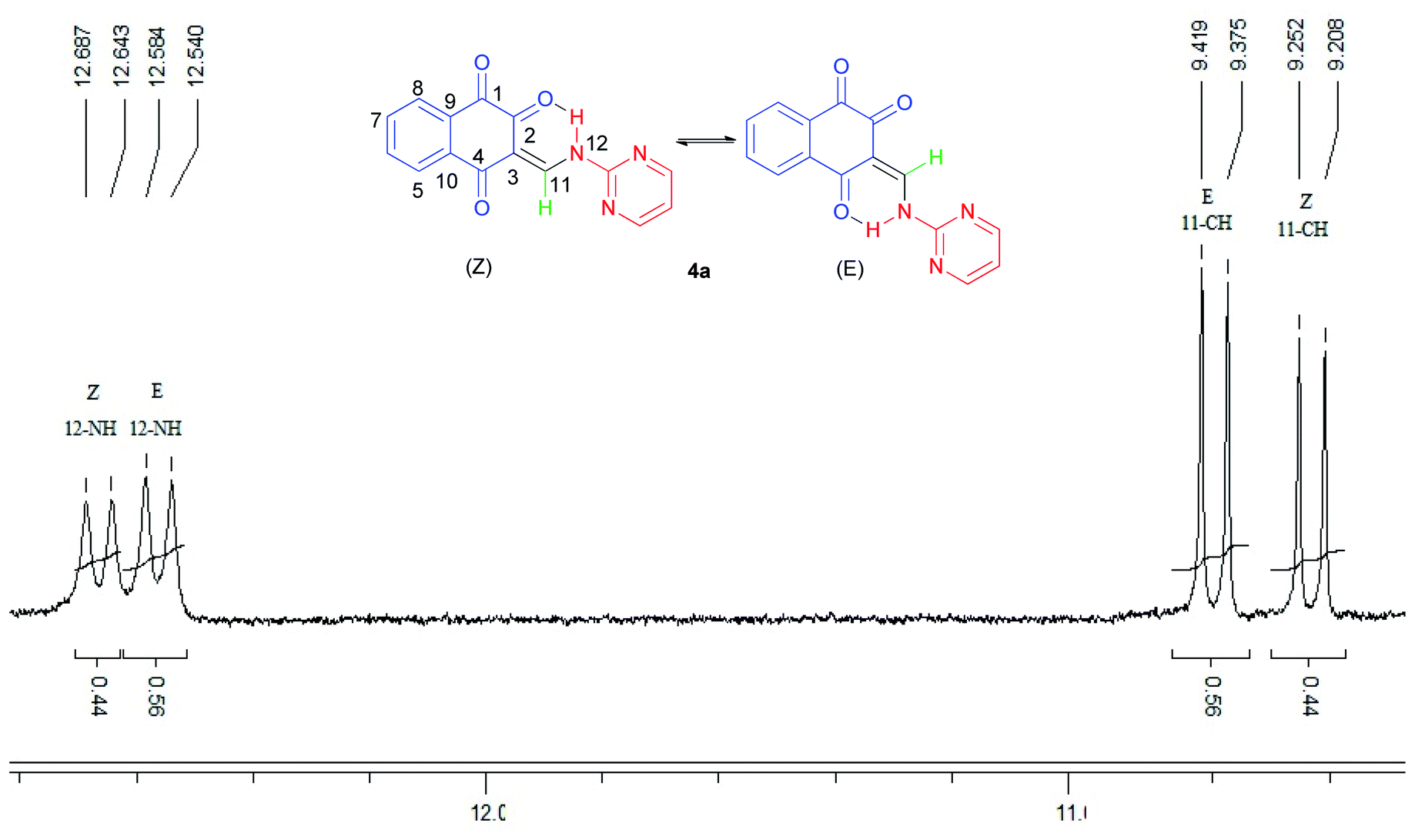 | ||
| Fig. 1 1H-NMR spectrum of compound 4a. | ||
 | ||
| Scheme 1 Structures of compound 4a. | ||
The plausible mechanism that could be accounted for this three-component reaction is depicted in Scheme 2. Firstly, the reaction proceeds through the in situ formation of intermediate 5 by the nucleophilic addition of amine 3 to triethyl orthoformate (2) which lose two molecules of EtOH in the presence of guanidine hydrochloride as catalyst. Subsequently lawsone (1) reacts with imine 5 to form another intermediate 6 which undergoes elimination of another EtOH molecule to give the desired product 4.
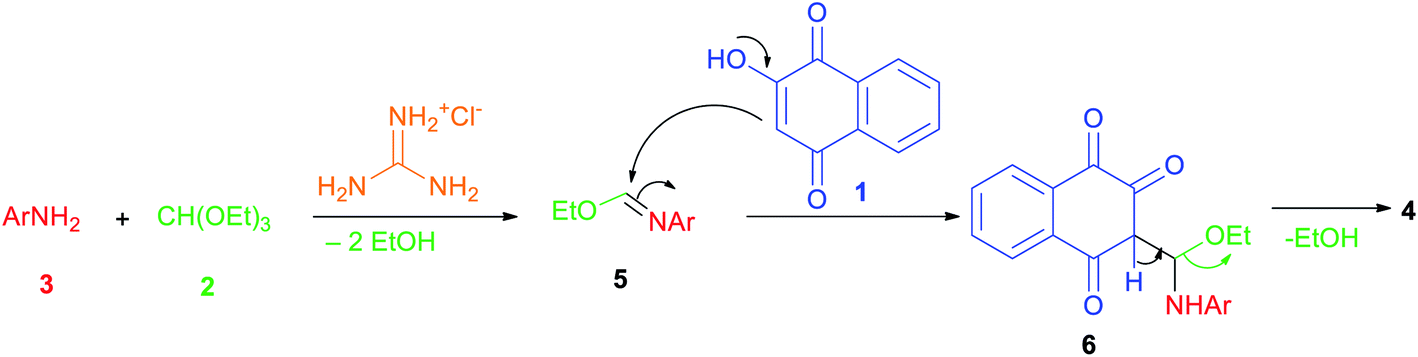 | ||
| Scheme 2 Possible reaction mechanism. | ||
Conclusions
In conclusion, we presented an environmentally benign synthetic and one-pot protocol for the synthesis of lawsone enaminone derivatives in high to excellent yields via the three-component reactions of lawsone, triethyl orthoformate and aromatic amines in the presence of guanidinium chloride under solvent-free condition at 90 °C. 1H-NMR spectra of synthetic compounds indicated that lawsone enaminones exist in the ketoenamine tautomeric form and undergo Z/E-isomerization in respect to the CC bond in DMSO-d6 at room temperature. Moreover, the attractive features of this protocol are simple reaction procedure, short reaction time, easy product separation, simple purification, high to excellent yields of products and this is the first report on the synthesis of lawsone enaminones.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Research Council of Payame Noor University for financial support.References
- Y. Huang and R. W. Hartmann, Synth. Commun., 1998, 28, 1197–1200 CrossRef CAS.
- C. Balakrishna, R. Gudipati, V. Kandula, S. Yennam, P. U. Devi and M. Behera, New J. Chem., 2019, 43, 2458–2463 RSC.
- C. P. Cartaya-Marin, D. G. Henderson and R. W. Soeder, Synth. Commun., 1997, 27, 4275–4283 CrossRef CAS.
- Z. Yang, L. Hu, T. Cao, L. An, L. Li, T. Yang and C. Zhou, New J. Chem., 2019, 43, 16441–16444 RSC.
- N. D. Eddington, D. S. Cox, R. R. Roberts, J. P. Stables, C. B. Powell and K. R. Scott, Curr. Med. Chem., 2000, 7, 417–436 CrossRef CAS PubMed.
- I. O. Edafiogho, K. V. V. Ananthalakshmi and S. B. Kombian, Bioorg. Med. Chem., 2006, 14, 5266–5272 CrossRef CAS PubMed.
- M. Khurana, N. N. Salama, K. R. Scott, N. N. Nemieboka, K. S. Bauer and N. D. Eddington, Biopharm. Drug Dispos., 2003, 24, 397–407 CrossRef CAS PubMed.
- Y. F. Wang, T. Izawa, S. Kobayashi and M. Ohno, J. Am. Chem. Soc., 1982, 104, 6465–6466 CrossRef CAS.
- J. P. Michael, C. B. De Koning, G. D. Hosken and T. V. Stanbury, Tetrahedron, 2001, 57, 9635–9648 CrossRef CAS.
- I. O. Edafiogho, M. S. Alexander, J. A. Moore, V. A. Farrar and K. R. Scott, Curr. Med. Chem., 1994, 1, 159–175 CAS.
- D. L. Boger, T. Ishizaki, R. J. Wysocki, S. A. Munk, P. A. Kitos and O. Suntornwat, J. Am. Chem. Soc., 1989, 111, 6461–6463 CrossRef CAS.
- J. D. White and D. C. Ihle, Org. Lett., 2006, 8, 1081–1084 CrossRef CAS PubMed.
- I. O. Edafiogho, S. B. Kombian, K. V. V. Ananthalakshmi, N. N. Salama, N. D. Eddington, T. L. Wilson, M. S. Alexander, P. L. Jackson, C. D. Hanson and K. R. Scott, J. Pharm. Sci., 2007, 96, 2509–2531 CrossRef CAS PubMed.
- H. Zang and K. S. Gates, Biochemistry, 2000, 39, 14968–14975 CrossRef CAS PubMed.
- C. Cimarelli, G. Palmieri and E. Volpini, Tetrahedron Lett., 2004, 45, 6629–6631 CrossRef CAS.
- J. V. Greenhill, Chem. Soc. Rev., 1977, 6, 277–294 RSC.
- B. Govindh, B. S. Diwakar and Y. L. N. Murthy, Org. Commun., 2012, 5, 105–119 Search PubMed.
- A. E. Filkale and C. Pathak, New J. Chem., 2020, 44, 15109–15121 RSC.
- H. M. Gaber, M. C. Bagley, Z. A. Muhammad and S. M. Gomha, RSC Adv., 2017, 7, 14562–14610 RSC.
- A. Z. A. Elassar and A. A. El-Khair, Tetrahedron, 2003, 59, 8463–8480 CrossRef CAS.
- V. F. Traven, I. V. Ivanov, V. S. Lebedev, B. G. Milevskii, T. A. Chibisova, N. P. Soloveva, V. I. Polshakov, O. N. Kazheva, G. G. Alexandrov and O. A. Dyachenko, Mendeleev Commun., 2009, 19, 214–216 CrossRef CAS.
- A. K. Jordão, M. D. Vargas, A. C. Pinto, F. d. C. da Silva and V. F. Ferreira, RSC Adv., 2015, 5, 67909–67943 RSC.
- S. R. Patil, A. S. Choudhary and N. Sekar, New J. Chem., 2016, 40, 6803–6811 RSC.
- D. Choudhari, S. Salunke-Gawali, D. Chakravarty, S. R. Shaikh, D. N. Lande, S. P. Gejji, P. K. Rao, S. Satpute, V. G. Puranik and R. Gonnade, New J. Chem., 2020, 44, 6889–6901 RSC.
- A. da R. Louvis, N. A. A. Silva, F. S. Semaan, F. d. C. da Silva, G. Saramago, L. C. S. V. de Souza, B. L. A. Ferreira, H. C. Castro, J. P. Salles, A. L. A. Souza, R. X. Faria, V. F. Ferreira and D. d. L. Martins, New J. Chem., 2016, 40, 7643–7656 RSC.
- A. K. Jordao, M. D. Vargas, A. C. Pinto, F. d. C. da Silva and V. F. Ferreira, RSC Adv., 2015, 5, 67909–67943 RSC.
- A. Olyaei, M. Sadeghpour and M. Khalaj, RSC Adv., 2020, 10, 30265–30281 RSC.
- (a) A. Olyaei, M. S. Shahsavari and M. Sadeghpour, Res. Chem. Intermed., 2018, 44, 943–956 CrossRef CAS; (b) A. Olyaei, R. Mohammad Ebrahimi, A. Adl and M. Sadeghpour, Chem. Heterocycl. Compd., 2019, 55, 1104–1110 CrossRef; (c) F. Noruzian, A. Olyaei and R. Hajinasiri, Res. Chem. Intermed., 2019, 45, 4383–4394 CrossRef CAS; (d) R. Khoeiniha, A. Olyaei and M. Saraei, J. Heterocycl. Chem., 2017, 54, 1746–1750 CrossRef CAS; (e) R. Khoeiniha, A. Olyaei and M. Saraei, Synth. Commun., 2018, 48, 155–160 CrossRef CAS; (f) A. Olyaei, H. Ramezanipour Moghadam and M. Sadeghpour, J. Heterocycl. Chem., 2020, 57, 3029–3036 CrossRef CAS; (g) A. Olyaei, S. Javarsineh and M. Sadeghpour, Chem. Heterocycl. Compd., 2018, 54, 934–939 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization data of products, IR, 1H-NMR and mass. See DOI: 10.1039/d1ra01858b |
| This journal is © The Royal Society of Chemistry 2021 |