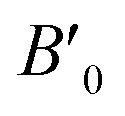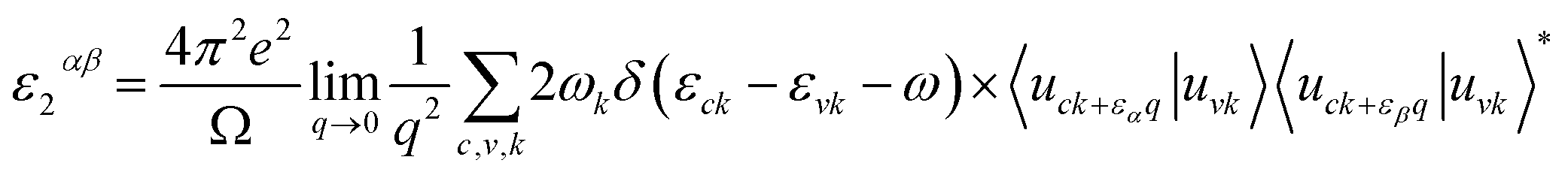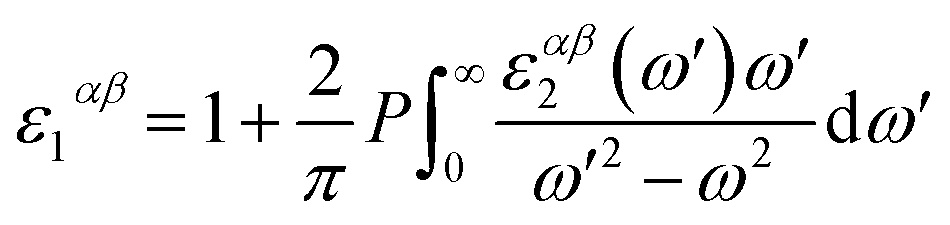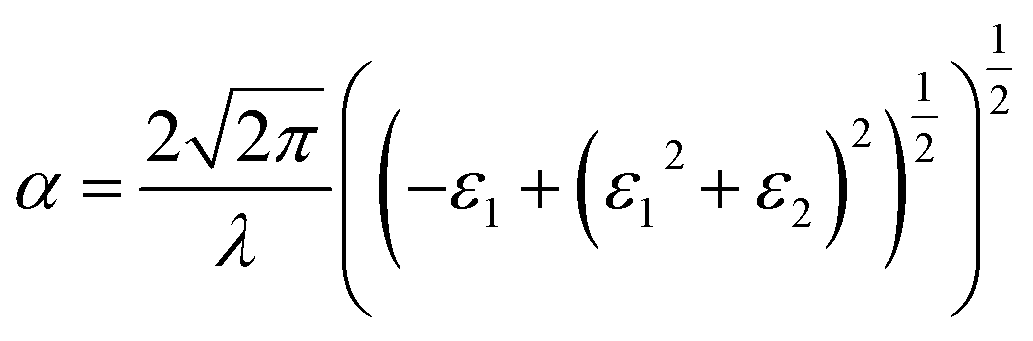DOI:
10.1039/D1RA01586A
(Paper)
RSC Adv., 2021,
11, 22264-22272
Electronic and optical properties of orthorhombic (CH3NH3)BX3 (B = Sn, Pb; X = F, Cl, Br, I) perovskites: a first-principles investigation†
Received
27th February 2021
, Accepted 12th June 2021
First published on 23rd June 2021
Abstract
Lead halide perovskites have generated considerable interest in solar cell, sensor, and electronics applications. While great focus has been placed on (CH3NH3)PbI3, an organic–inorganic hybrid perovskite, comparatively little work has been done to understand some of its existing crystal phases and analogous materials after substituting with Sn and/or other halogens in the framework. Here, first-principles density functional theory calculations are performed to comprehensively evaluate the electronic and optical properties of (CH3NH3)BX3 (B = Sn, Pb; X = F, Cl, Br, I) in a low-temperature orthorhombic phase. Bulk modulus, electronic structures, and several optical properties of these perovskite systems are further calculated. The obtained results are first confirmed by comparing with existing perovskite systems in literature. The shifting trends on those physical properties when extending to other barely studied systems of (CH3NH3)BX3 is further revealed. The band gap of these perovskites is found to decrease when varying halogen anion in “X” sites from F to I, and/or substituting Pb cations with Sn in “B” sites. Notably, the less toxic Sn-containing perovskites, (CH3NH3)SnI3 in particular, display higher absorption coefficients in the visible light range than their Pb-containing counterparts. An orthorhombic (CH3NH3)PbF3 is predicted to exist at low temperature, and adsorb strongly UV energy. Our systematical examination efforts on the two groups of perovskites provide valuable physical insights in these materials, and the accompanied new findings warrant further investigation on such subjects.
1. Introduction
There is an ever-increasing environmental and social need to reduce carbon emissions which originate largely from fossil fuel burning. Utilizing clean and abundant solar energy by photovoltaic technologies is one major effort scientists are working on to accomplish these missions. Although silicon-based photovoltaic dominates current market share with an efficiency as high as ∼21%, perovskites-based photovoltaics (PVs) demonstrates the fastest improvement on power conversion efficiency (PCE) in single-junction solar cells, reaching 25.2% in only ten years. Besides their promising PCE, perovskite thin films can be easily synthesized and integrated into devices and the prepared PVs also can tolerate much higher defect concentration than their Si-based counterparts. Besides their promising long-term solution to the climate crisis, perovskite materials can also play important roles in short-term solutions that make the current fossil fuel dominated energy supply greener.1–5 For example, the maintenance of appropriate fuel-to-air ratio can not only maximize combustion efficiency of fossil fuels, but also minimize the emission of toxic gases such as NOx to the environment. This requires gas sensors suitable for extreme environmental conditions (temperature, pressure, and potential exposure to free radicals).6 The semiconductive and tunable optical properties of perovskite materials grant them interests as optical and photoelectrochemical sensors in combustion processes to detect ppm-range NOx signal in emission gas at high-temperature.7–9
In a common metal halide perovskites structure, ABX3, A is an organic (e.g., methyl ammonium and formamidinium) or inorganic cation (e.g., cesium), B is a metal cation such as lead, and X is a halogen anion. One big drawback of such perovskite materials lies on the involvement of lead, which causes safety concerns during synthesis and applications. With similar valence electrons for bonding, tin is considered as a potential replacement for lead in the metal halide perovskite structure to mitigate toxicity.10 Another challenge in perovskite applications comes from their short storage and operation lifetime. Perovskites face great stability and degradation issues when exposed to environmental (e.g., moisture, oxygen, and ultraviolet light), accompanied with consequent hygroscopicity, thermal instability, and ion migration issues.4 Different degradation mechanisms are proposed, and great efforts have been made for their mitigations or solutions. One effective strategy is to replace elements in the ABX3 structure of perovskite by partially substituting some majority halogen and/or metal ions or using their mixture, which could not only alleviate those stability issues, but also finely tune the electronic and optical properties of the perovskite crystals.11 For example, as the A site in perovskites is typically occupied by a larger cation than the B site, Pb and Sn containing perovskites normally have the A site occupied by these relatively large metal atoms. However, by selection of a comparatively large organic cation, the metal can be made to occupy the B position.1 An abnormally high absorption coefficient in the visible light range allows the material to function as a thin absorbing layer in photovoltaic cells to mitigate efficiency loss occurring mainly due to electron–hole recombination. These results have fueled hopes in perovskite applications and interests in new physical insights on the roles different metal and halide ions play in perovskite structure.1–5
Another effective strategy comes from the retention of stable phases of perovskite crystals. Depending on the temperature, (MA)BX3 (MA = CH3NH3; B = Sn, Pb; X = Cl, Br, I) perovskites exist in several different phases.12–14 At high temperatures, they are in the cubic phase (Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m). For example, the transition temperatures of (MA)PbX3 (X = Cl, Br, I) are 177 K, 236 K, and 330 K respectively. Below these temperatures, (MA)PbX3 perovskites stay in the tetragonal phase (I4/mcm). Under further low temperatures (i.e., below 172 K, 149–154 K, and 161 K for (MA)PbX3 (X = Cl, Br, I respectively)), these perovskites are in the form of an orthorhombic phase (Pnma).12 In the literature, the electronic and optical properties of the high-temperature cubic phase (MA)BX3 (B = Sn, Pb; X = Cl, Br, I) were widely studied both by computational calculations and experimental measurements.5,15–31 However, few studies look into these perovskites in their low-temperature phases, particularly the orthorhombic phase. One possible reason lies in the fact that many of these structures may only be stable at very low temperatures. Employing density functional theory (DFT), Ali et al.17 explored the structural, mechanical, electronic, and thermoelectric properties of the low-temperature orthorhombic perovskite phase of (MA)PbI3. They obtained a 1.57 eV band gap for (MA)PbI3 and found its lattice thermal conductivity is anisotropic, and the lattice thermal conductivity at 150 K was found to be 0.189, 0.138, and 0.530 W m−1 K−1 along the a, b, and c directions; however, there was no reporting on its optical properties. Using the non-local optB86b + vdWDF functional, Wang et al.32 predicted the crystal structure and band gap of (MA)PbI3 in excellent agreement with experimental data. Using HSE06 hybrid functional approach, Feng and Xiao33 explored the effective masses and electronic and optical properties of orthorhombic (MA)SnX3 (X = Cl, Br, and I) as solar energy absorbers. Their results indicated that both the effective masses and band gaps of (MA)SnX3 perovskites decrease when the “X” sites change from chlorine to iodine, but their optical dielectric constants do not go down monotonically. No report was yet found in the literature on the structures and optical properties of (MA)BF3 (B = Sn, Pb). Hence, a question arises if such structures could exist, even at low temperatures. Before we could evaluate the performance of perovskites (MA)BX3 (MA = CH3NH3; B = Sn, Pb; X = F, Cl, Br, I) as either solar cell or sensor candidate materials, much work is still needed to be done to understand the electronic and optical properties of these perovskites in their stable phases at different temperatures. In this study, we performed extensive investigations on the electronic and optical properties of perovskite crystals of (MA)BX3 in the low-temperature orthorhombic phase and make comparisons with the corresponding phases stable in high temperatures. This study serves as the first step to evaluate this organic–inorganic hybrid halide perovskite group in low-temperature phase to provide physical insights on their potentials.
m). For example, the transition temperatures of (MA)PbX3 (X = Cl, Br, I) are 177 K, 236 K, and 330 K respectively. Below these temperatures, (MA)PbX3 perovskites stay in the tetragonal phase (I4/mcm). Under further low temperatures (i.e., below 172 K, 149–154 K, and 161 K for (MA)PbX3 (X = Cl, Br, I respectively)), these perovskites are in the form of an orthorhombic phase (Pnma).12 In the literature, the electronic and optical properties of the high-temperature cubic phase (MA)BX3 (B = Sn, Pb; X = Cl, Br, I) were widely studied both by computational calculations and experimental measurements.5,15–31 However, few studies look into these perovskites in their low-temperature phases, particularly the orthorhombic phase. One possible reason lies in the fact that many of these structures may only be stable at very low temperatures. Employing density functional theory (DFT), Ali et al.17 explored the structural, mechanical, electronic, and thermoelectric properties of the low-temperature orthorhombic perovskite phase of (MA)PbI3. They obtained a 1.57 eV band gap for (MA)PbI3 and found its lattice thermal conductivity is anisotropic, and the lattice thermal conductivity at 150 K was found to be 0.189, 0.138, and 0.530 W m−1 K−1 along the a, b, and c directions; however, there was no reporting on its optical properties. Using the non-local optB86b + vdWDF functional, Wang et al.32 predicted the crystal structure and band gap of (MA)PbI3 in excellent agreement with experimental data. Using HSE06 hybrid functional approach, Feng and Xiao33 explored the effective masses and electronic and optical properties of orthorhombic (MA)SnX3 (X = Cl, Br, and I) as solar energy absorbers. Their results indicated that both the effective masses and band gaps of (MA)SnX3 perovskites decrease when the “X” sites change from chlorine to iodine, but their optical dielectric constants do not go down monotonically. No report was yet found in the literature on the structures and optical properties of (MA)BF3 (B = Sn, Pb). Hence, a question arises if such structures could exist, even at low temperatures. Before we could evaluate the performance of perovskites (MA)BX3 (MA = CH3NH3; B = Sn, Pb; X = F, Cl, Br, I) as either solar cell or sensor candidate materials, much work is still needed to be done to understand the electronic and optical properties of these perovskites in their stable phases at different temperatures. In this study, we performed extensive investigations on the electronic and optical properties of perovskite crystals of (MA)BX3 in the low-temperature orthorhombic phase and make comparisons with the corresponding phases stable in high temperatures. This study serves as the first step to evaluate this organic–inorganic hybrid halide perovskite group in low-temperature phase to provide physical insights on their potentials.
2. Theoretical methods
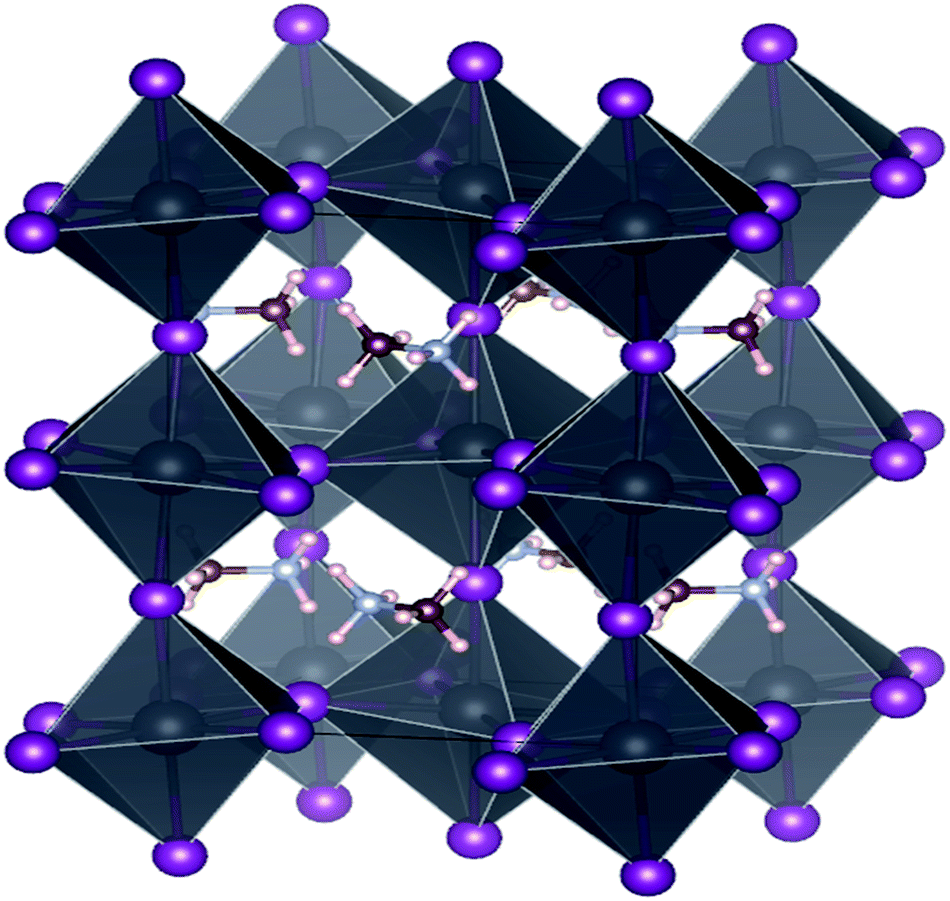
Calculations here were performed via first-principles density functional theory (DFT), accomplished via the Vienna ab initio simulation package (VASP),34–36 using the plane-wave basis set and projector-augmented wave (PAW) method.37 Exchange and correlation effects were taken into account in a generalized gradient approximation (GGA) by Perdew–Burke–Ernzerhof (PBE) functional.38 The cutoff energy for plan waves was 520 eV and the electronic energy convergence was set to 10−4 eV. As shown in Fig. 1, the unit cells of the orthorhombic phase (Z = 4) with space group Pnma (#62) were used to model all eight structures. K-point sampling grid of 9 × 6 × 9 was used to sample the (MA)PbX3 and (MA)SnX3 perovskites. The ionic positions were relaxed until the Hellmann–Feynman force on each atom was less than 0.01 eV Å−1.
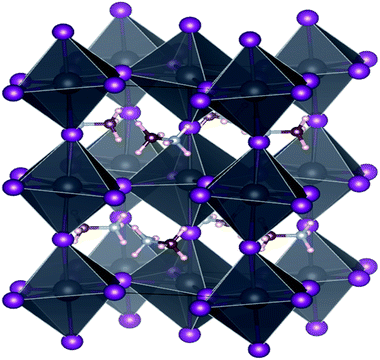 |
| | Fig. 1 The unit cell of (CH3NH3)BX3 (B = Sn, Pb, X = F, Cl, Br, I) with Pnma (#62) nspace symmetry. Pink spheres are hydrogen atoms, brown ones are for carbon, light gray for nitrogen, purple for halides, and dark gray for metal Sn or Pb. | |
3. Results and discussion
3.1 Geometric and mechanical properties
The structures of (MA)BX3 were first relaxed geometrically by minimizing their energies. We performed the optimizations for Pnma (#62) symmetries of (MA)BX3 with the received crystal constants listed in Table 1. While studies of some individual perovskites of those or others that contain a mixture of two halogens can be found in scattered literature, a comprehensive comparison between the perovskites in this class is yet available to reveal trends in their physical properties with various halogen and/or metal substitutions. In contrast to the rich available data for the cubic phase of this halide perovskite family which has been published in the literatures,5,12,15,16,18–31 only a few computational studies focus on the orthorhombic phase of (MA)PbI3 and (MA)SnX3 (X = Cl, Br, and I).1,17,32,33 As an example, Table 2 lists our optimized atomic fractional coordinates of MAPbI3 in comparison with the reported structures in Materials Project,39 while the optimized atomic coordinates of the rest of the seven structures of (MA)BX3 are provided in Tables S1–S7 of the ESI.† Overall, as one can see from Tables 1, 2 and S1–S7,† our optimized structures of (MA)BX3 are in good agreement with available experimental data and other theoretical modeling results. For (MA)SnX3 (X = Cl, Br, I), our optimized crystal constants and volumes are slightly bigger than those calculated by HSE06 functional.33 For (MA)PbI3, our geometry simulation results (Tables 1 and 2) agree well with that of using PBE.17,39 However, the PBEsol appears to better approximate the b and c lattice constants as compared to the experimental results, though it is outperformed by PBE for the a lattice constant.17 For the available Sn-containing perovskite sources, the Grimme van der Waals correction was used to account for the dispersion interactions, resulting in lower lattice constants than ours.32,33 This lack of dispersion interaction correction and the precision of exchange-correlation functional (PBE over HSE06) likely explain the systematic over-estimation of lattice constants in our results. But when focusing on trends of crystal structure changes with different halogen anions, larger halogens lead to more volume expansion of the crystal framework in the orthorhombic phase perovskites as expected. As the consequence, a decreased band gap is found with their values becoming more favorable to the adsorption of visible light (a rationale for this behavior being laid out in Section 3.2). Similar trends are also found when large metal element (e.g., Pb over Sn) occupy the B sites of (MA)BX3. However, such changes are more pronounced when varying halogen anions in the X sites of the perovskite structure.
Table 1 The optimized crystal structures and volumes of orthorhombic (MA)BX3 (B = Sn, Pb; X = F, Cl, Br, I) perovskites
| Perovskite |
a (Å) |
b (Å) |
c (Å) |
V (Å3) |
E (eV per formula unit) |
| From ref. 40. From ref. 1. From ref. 17. From ref. 39. |
| (MA)SnF3 |
6.3623 |
9.6369 |
7.4482 |
456.67 |
−59.13799 |
| (MA)SnCl3 |
7.5518 (7.550)a |
11.3363 (11.067)a |
8.4178 (7.905)a |
720.64 (660.5)a |
−54.21088 |
| (MA)SnBr3 |
7.9478 (7.813)a |
11.8729 (11.546)a |
8.7304 (8.144)a |
823.83 (734.7)a |
−52.58994 |
| (MA)SnI3 |
8.5025 (8.326)a |
12.6634 (12.428)a |
9.1863 (8.556)a |
989.10 (885.4)a |
−50.82122 |
| (MA)PbF3 |
6.3889 |
9.8163 |
7.5376 |
472.72 |
−59.39415 |
| (MA)PbCl3 |
7.6084 |
11.5334 |
8.5153 |
747.23 |
−54.41641 |
| (MA)PbBr3 |
8.0029 |
12.0780 |
8.8423 |
854.70 |
−52.75472 |
| (MA)PbI3 |
8.5536 (8.5551)b (8.581)c (8.557)d |
12.9027 (12.5804)b (12.908)c (12.964)d |
9.2975 (8.8362)b (9.346)c (9.250)d |
1026.12 |
−50.94207 |
Table 2 The calculated fractional coordinates of atoms in orthorhombic (MA)PbI3. The corresponding data from Materials Project39 are given in brackets for comparison
| Element |
Fractional coordinates |
| x |
y |
z |
| H |
0.4977 (0.4943) |
0.3151 (0.3146) |
0.858 (0.8570) |
| H |
0.4905 (0.4948) |
0.3197 (0.3192) |
0.119 (0.1201) |
| H |
0.3395 (0.3381) |
0.2500 (0.2500) |
0.9198 (0.9214) |
| H |
0.6576 (0.6598) |
0.2500 (0.2500) |
0.0526 (0.0503) |
| C |
0.5300 (0.5324) |
0.2500 (0.2500) |
0.0624 (0.0622) |
| N |
0.4610 (0.4596) |
0.2500 (0.2500) |
0.9149 (0.9151) |
| I |
0.8096 (0.8161) |
0.9755 (0.9779) |
0.3012 (0.3072) |
| I |
0.5404 (0.5377) |
0.2500 (0.2500) |
0.4846 (0.4874) |
| Pb |
0.5000 (0.5000) |
0.0000 (0.0000) |
0.5000 (0.5000) |
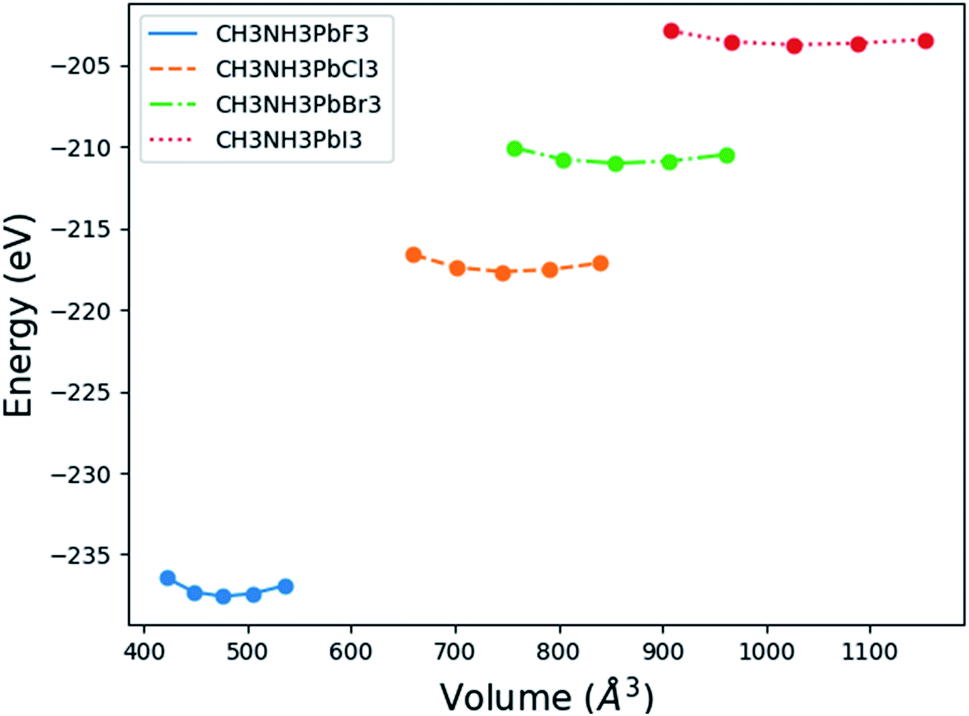
Fig. 2 displays the total energy versus volume change for (MA)PbX3 (X = F, Cl, Br, I) perovskites. The corresponding data for (MA)SnX3 (X = F, Cl, Br, I) are provided in Fig. S1 of the ESI.† For either Pb or Sn in the B sites, the perovskites involving smaller halogen anions have a significantly higher change rate of the total energy with respect to volume near the minimum energy, indicating a higher bulk modulus. This is confirmed by fitting the simulation data of the total energy (E) versus volume (V) in the form of Birch–Murnaghan equation of states (EOS) using eqn (1),
| |
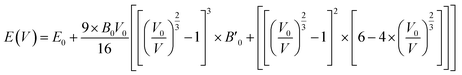 | (1) |
where a subscript of 0 indicates the equilibrium value of that parameter.
B0 refers to the base-level bulk modulus, with
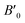
referring to the first order variation of the bulk modulus,
B, with pressure,
P, all according to
eqn (2).
P is taken to be 1 atm.
| |
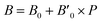 | (2) |
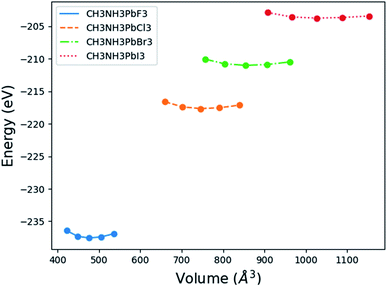 |
| | Fig. 2 Energy versus volume for (MA)PbX3 perovskites. | |
The received parameters are listed in Table 3, together with the coefficients of determination of the regression fitting of the EOS models in those systems. Besides minimum fitting error, our bulk modulus result for (MA)PbI3 (15 GPa) agrees well with the only available literature data (15.45 GPa in ref. 17). As halogen anion varies from F to Br in the X sites, the bulk modulus of perovskites decreases, which is consistent with observations from others. This can be attributed to the reduction of electronegativity when large halogen anions are present in the crystal. Smaller halogen anions in the X sites of perovskites provide larger resistance to the volume change, particularly to compression, as compared to larger halogens, due to the stronger interactions between protons and electrons. Such energy upshift from the equilibrium state is reflected in the fitting values of the bulk modulus change listed in Table 3. As for Sn and Pb in the B sites of the crystal structure, Sn-containing perovskites carry higher bulk moduli. The presence of f-orbitals in Pb's electronic configuration may enable more occupiable states for electrons. The changes of B0 values in our results reflect such lower energy deviations with an increase in pressure, while an outlier exists for I-containing perovskites, which have significantly different 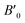 values than the other six perovskites examined.
values than the other six perovskites examined.
Table 3 The fitted parameters (B0, 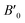 ) and bulk modulus (B) of (MA)BX3 by fitting to the Birch–Murnaghan equation of state (E–V)
) and bulk modulus (B) of (MA)BX3 by fitting to the Birch–Murnaghan equation of state (E–V)
| Perovskite |
B0 (eV Å−3) |
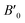
(dimensionless) |
B (GPa) |
R2 (dimensionless) |
| From ref. 17. |
| (MA)SNF3 |
0.29533 |
6.02412 |
32.0 |
1.0000 |
| (MA)SNCL3 |
0.21958 |
6.75097 |
24.0 |
0.9998 |
| (MA)SNBR3 |
0.18370 |
7.06354 |
20.2 |
0.9998 |
| (MA)SNI3 |
0.26958 |
4.85373 |
29.1 |
1.0000 |
| (MA)PBF3 |
0.18086 |
6.74740 |
19.9 |
1.0000 |
| (MA)PBCL3 |
0.13778 |
6.57428 |
15.3 |
0.9999 |
| (MA)PBBR3 |
0.11688 |
6.30812 |
13.1 |
0.9999 |
| (MA)PBI3 |
0.13023 |
11.24429 |
15.0 (15.45)a |
0.9983 |
3.2 Electronic structure properties
Due to the DFT approximations, which underestimate the excited state energies, the calculated band gap is usually smaller than the experimental measurement.41 The beyond-DFT methods are usually used to amend such band gap underestimations. As shown in Table 4, with the HSE06 functional approach, Feng and Xiao33,40 obtained larger band gaps of (MA)SnX3 (X = Cl, Br, I). With inclusion of the GW many-body correction, Chiarella et al.42 calculated the band gaps of the cubic phase of two Sn-containing perovskites ((MA)SnCl3: 3.44 eV; (MA)SnBr3: 1.90 eV), which are relatively close to the experimental measured values of 3.69 eV and 2.15 eV respectively.43 Besides, Lang et al.5 reported that the spin-orbital coupling (SOC) could induce a band gap decrease of about 0.35 eV and 1.13 eV for B = Sn and Pb, respectively.
Table 4 The calculated band gaps (Egap: without including SOC, SOC_Egap: with including SOC) and the formation energies (Eform) of orthorhombic (MA)BX3 (B = Sn, Pb; X = F, Cl, Br, I) perovskites
| Perovskites |
Egap (eV) |
SOC Egap (eV) |
Eform (eV per formula unit) |
| From ref. 42. From ref. 33. From ref. 16. From ref. 1. From ref. 17. From ref. 39. |
| (MA)SnF3 |
2.325 |
2.122 |
0.11890 |
| (MA)SnCl3 |
1.494 (1.94)a (2.8)b |
1.193 |
−0.16782 |
| (MA)SnBr3 |
1.186 (1.04)a (2.0)b |
0.804 |
−0.18245 |
| (MA)SnI3 |
0.969 (1.7)b |
0.520 |
−0.12346 |
| (MA)PbF3 |
3.466 |
2.651 |
−0.17052 |
| (MA)PbCl3 |
2.685 |
1.614 |
−0.20019 |
| (MA)PbBr3 |
2.265 (2.3)c |
1.171 |
−0.18966 |
| (MA)PbI3 |
2.030 (1.61)d (1.80)e (1.76)f |
0.808 |
−0.14134 |
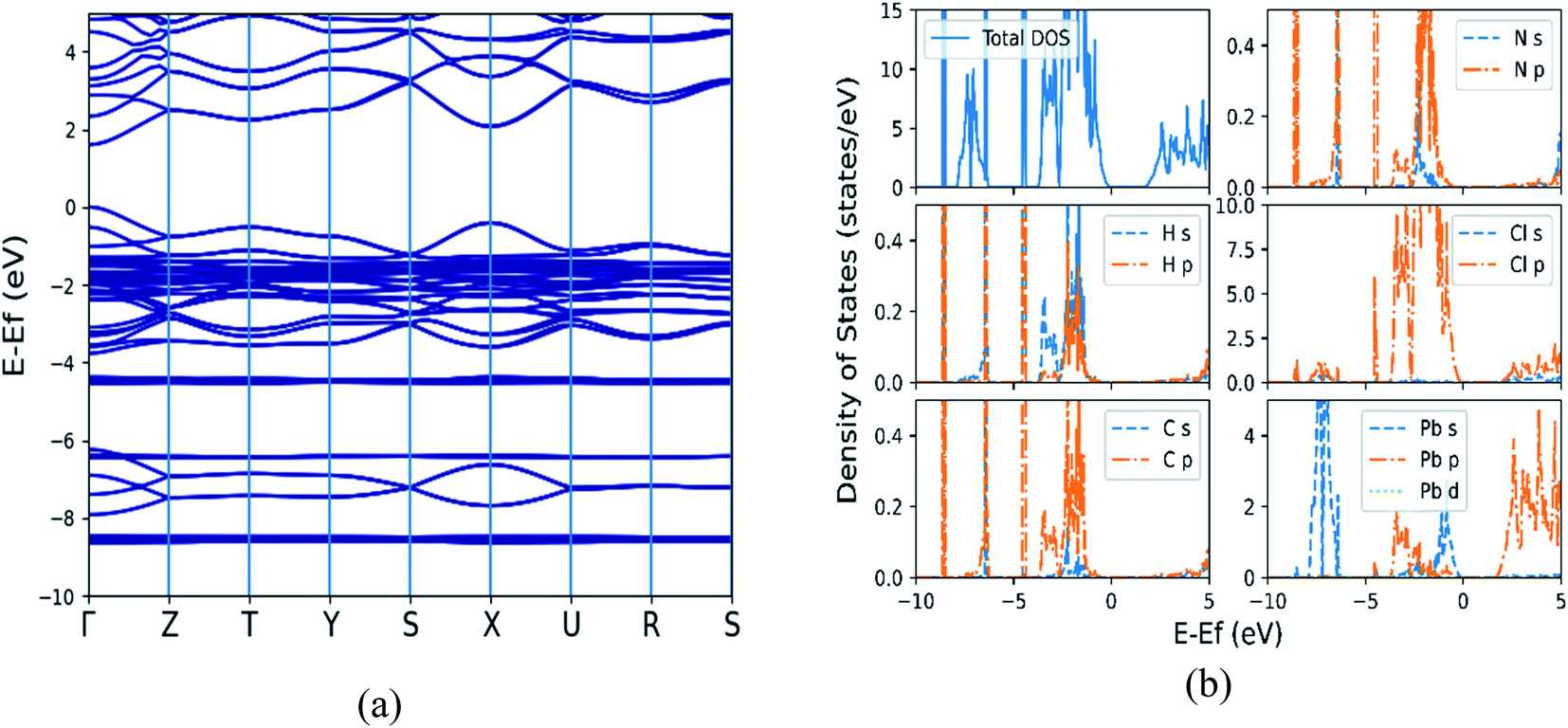
As SOC is known to be important for modeling electronic properties of heavier elements,44 the electronic structure properties of all orthorhombic (MA)BX3 (B = Sn, Pb; X = F, Cl, Br, I) perovskites are further calculated by GGA + SOC. As an example, Fig. 3 shows the calculated band structure as well as the total and partial density of states (DOS) of (MA)PbCl3, while that of the rest of the seven systems are provided in Fig. S2–S8 of the ESI.†
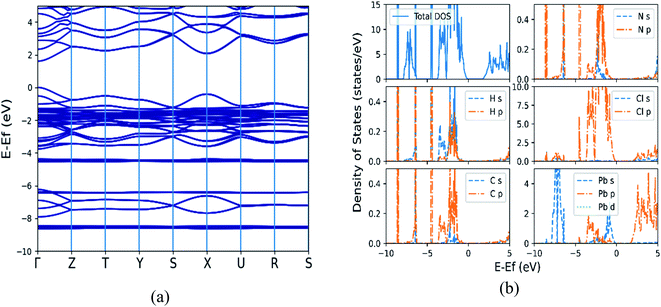 |
| | Fig. 3 The calculated electronic structural properties for (MA)PbCl3 by including spin orbit coupling (SOC). (a) Band structure with a direct band gap of 1.614 eV (Γ point); (b) total and partial density of states. | |
From Fig. 3(a) and S2(a)–S8(a),† the direct band gap nature of semiconductors is confirmed in these perovskites with their valence band maxima and conduction band minima located at the Γ point of the Brillouin zone. Table 4 summarizes our calculated band gaps of orthorhombic (MA)BX3 (B = Sn, Pb; X = F, Cl, Br, I) perovskites, which are in good agreement with available theoretical results of (MA)PbI3 and (MA)SnX3 (X = Cl, Br, and I).1,17,32,33 Comparing with the GGA + PBE calculations without including SOC (2.685 eV for (MA)PbCl3) shown in Fig. S9 of the ESI,† a much lower band gap (1.614 eV) was obtained (but still stay as direct band-gap semiconductors) which is consistent with the band-gap decrease predicted by Lang et al.5 In addition, the contribution of the p-orbitals is much lower when SOC is included. The calculated band gap values in Table 4 do show some deviations from the experimental data, with the results from the calculations including SOC being farther from experiment in most cases. Although the absolute values of the calculated band gaps vary with the adopted methods, their shifting trends when altering metal cations or halogen anions in these perovskites stays the same. The band gaps become smaller as large halogen and/or metal ions are included in the (MA)BX3 structure as shown in Table 4, with the two most extreme cases of (MA)SnI3 and (MA)PbF3. When substituting Sn with Pb in B sites, larger band gaps are received for (MA)BX3 with the same type of halogen.
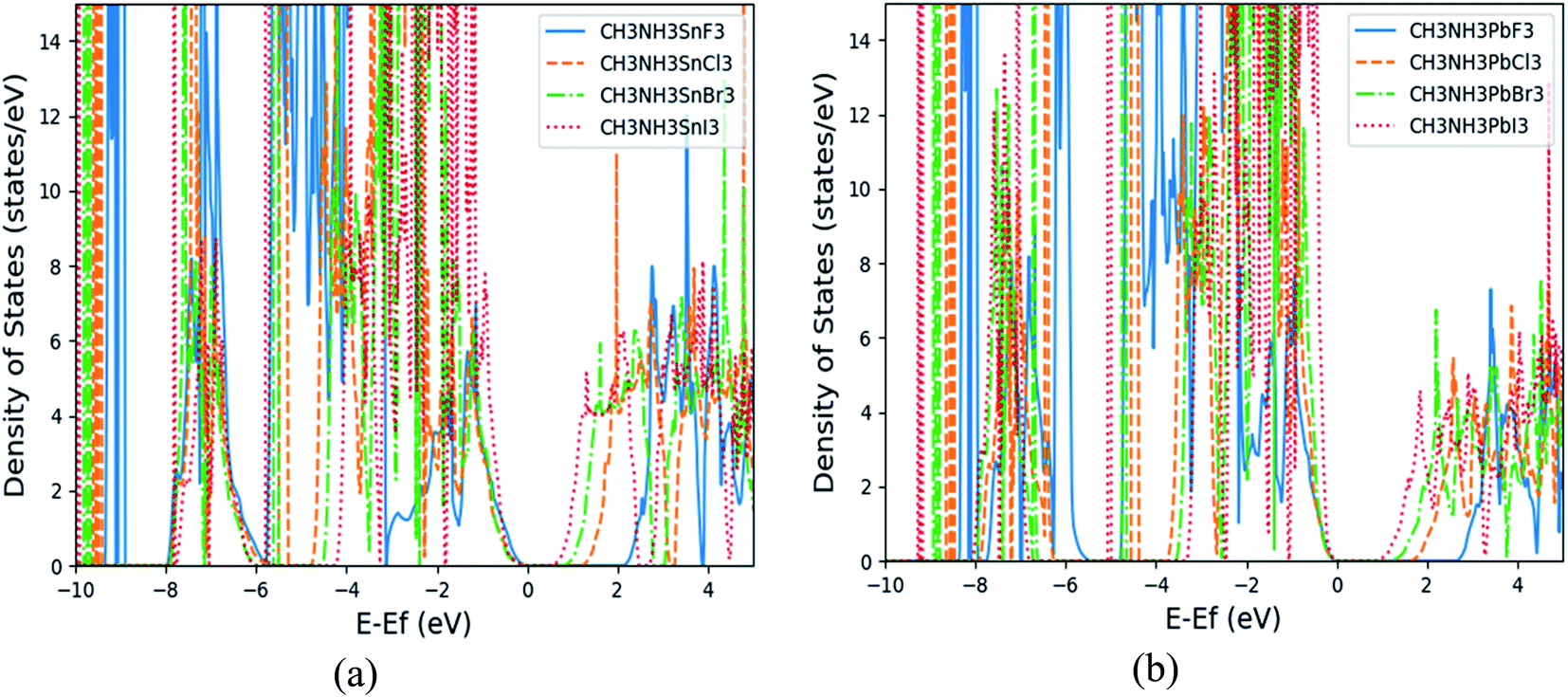
From the total DOS (TDOS) and the partial DOS (PDOS) for each element in the (MA)BX3 shown in Fig. 3(b) and S2(b)–S8(b),† it suggests that methyl ammonium ion contributes little to the region around the Fermi level. Taking (MA)PbCl3 as an example, the halogen Cl p-orbital contributes primarily to the valence band (VB) and the metal p-orbital to the conduction band (CB), as shown in the subpanels of Fig. 3(b). Cl has its p-orbital PDOS shifted to further above the Fermi energy, moving the relevant perovskites' CBs in the same manner. Conversely, larger halogens appear to shift the CB closer to the Fermi energy (note: possibly by raising the Fermi energy). Drifting away from the Fermi energy enlarges the band gap, which explains why these two types of perovskites (i.e., (MA)SnI3 and (MA)PbF3) have the smallest and largest band gaps, respectively, as shown in the TDOS comparison in Fig. 4. Compared to available literature trends, e.g. (MA)SnI3 versus (MA)PbI3, our results do match well with the literature.33
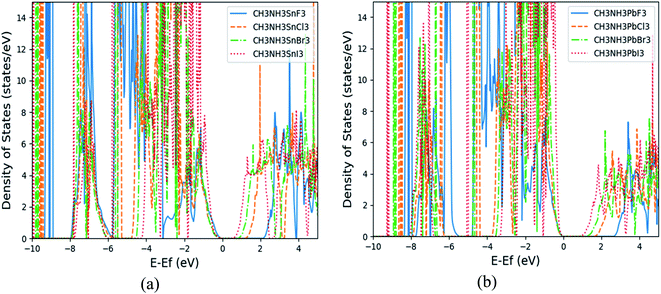 |
| | Fig. 4 The calculated total density of states (TDOS) of (MA)SnX3 (a) and (MA)PbX3 (b). | |
Based on the electronic structural properties, the formation energies (Eform) of orthorhombic (MA)BX3 (B = Sn, Pb; X = F, Cl, Br, I) perovskites are evaluated by
| | |
Eform,ABX = E0,ABX − E0,AX − E0,BX
| (3) |
where
E0,ABX refers to the energy of a particular perovskite per formula unit as listed in
Table 1,
E0,AX refers to the energy per formulation of the (MA)X salt formed from the relevant halogen of the perovskite, and
E0,BX refers to the energy per formulation of the BX
2 salt formed from the relevant halogen and metal of the perovskite. The calculated
E0,AX and
E0,BX are listed in Table S8 of the ESI.
† AX and BX
2 are important to reference against when quantifying thermodynamic stability as they are the phases typically in thermodynamic equilibrium with ABX
3 perovskites. The calculated
Eform of (MA)BX
3 perovskites are given in
Table 4. The formation energy of all perovskites of (MA)BX
3 is negative in their orthorhombic phase, with an exception of (MA)SnF
3. This suggests that all seven perovskites could exist in
Pnma phase, including the least studied (MA)PbF
3. Unlike (MA)SnF
3, whose formation energy is positive,
Pnma phase (MA)PbF
3 perovskite could be favorable under low temperature conditions, although this has not yet been discovered experimentally to our best knowledge. To further verify the stability, additional computational work on the phonon properties of these fluorine-containing perovskites is needed to examine the stability and dynamics of their crystal structures.
3.3 Optical properties
To study the optical properties, the frequency-dependent dielectric matrix in the long wavelength limit (q → 0) can be calculated using the sum over states approach.45 A brief description of our calculation methods is summarized here while detailed can be found in our previous publications.46–49 The formula for the imaginary part of the dielectric constant is a 3 × 3 Cartesian tensor:| |
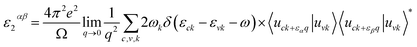 | (4) |
where the summation is over indices c (CB states), v (VB states), and k (k-points). e is elementary charge, Ω is volume of the Brillouin zone, ωk is weight of the k-point vector, εck and εvk are energy levels, uck is the periodic portion of the orbital at k-point k. Vector eα is made of unit vectors for the Cartesian directions and α/β refer to axis x, y, and z. From the imaginary part, the real part can be obtained by a Kramers–Kronig transformation.| |
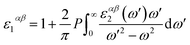 | (5) |
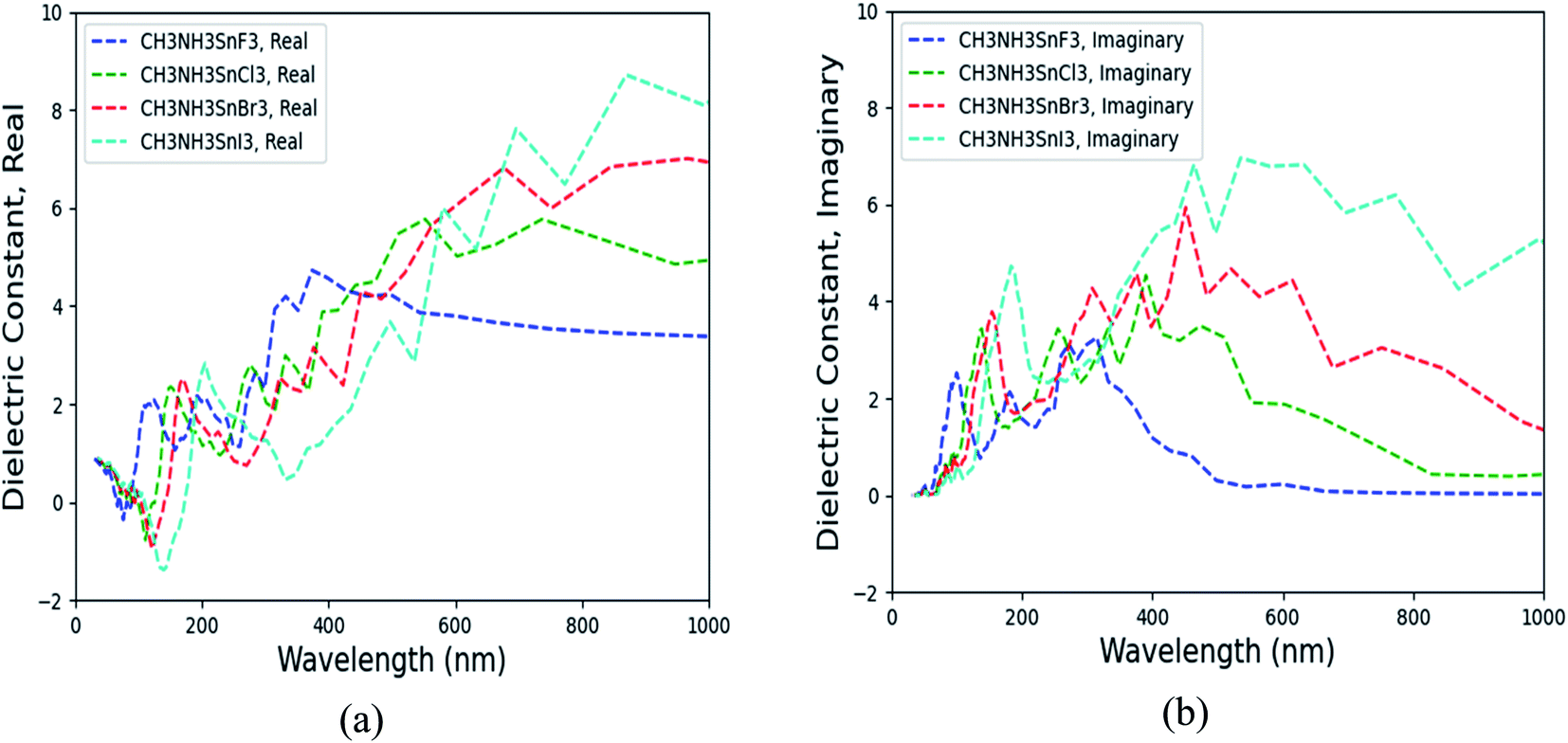
By including SOC, Fig. 5 shows the calculated ε1αβ and ε2αβ versus wavelength for (MA)SnX3 (X = F, Cl, Br, I). The results of (MA)PbX3 (X = F, Cl, Br, I) are shown in Fig. S10 of the ESI.† Due to the symmetrical and isotropic features of these crystals, the off-diagonal elements of dielectric tensor are zero and the diagonal elements (εxx, εyy, εzz) are close to each other, but not identical. As shown in Fig. 5, we take the average of these three diagonal elements as the overall dielectric constants in further calculation of their optical properties. Comparing Fig. 5 with the results of the GGA + PBE calculations without including SOC shown in Fig. S11,† the main difference is seen in the imaginary dielectric constant, for which the heavier halogen perovskites do not fall off as quickly against wavelength.
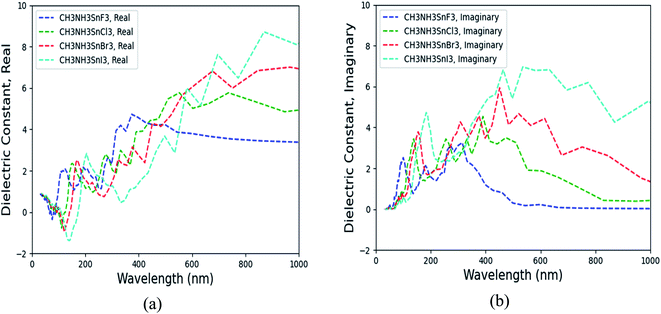 |
| | Fig. 5 The calculated dielectric matrix of (MA)SnX3 perovskites by taking the average of diagonal elements (εxx, εyy, εzz). (a) Real part ε1, (b) imaginary part ε2. | |
As promising candidate materials in optical sensor applications, the most interesting wavelength range is from ultraviolet (UV) (10–400 nm) through the visible spectrum (400–800 nm) to near infrared and infrared (IR) (>800 nm), particularly, around the visible and near infrared range (∼400 to 1000 nm).46 As shown in Fig. 5 and S9,† with halogen anions varying from F to I, both real and imaginary parts of the dielectric matrix in the frequency range of 400–1000 nm are increased. Other optical properties (e.g., the index of refraction, extinction coefficient, reflectivity, optical conductivity, absorption coefficient) derived from the dielectric matrix change correspondingly. According to eqn (4) and (5), the dielectric matrix depends on the differences of CB and VB states. When the halogen anion changes from F to I, the band gap of (MA)BX3 is decreased, as shown in Table 4. The small band gap slows down the decay of the imaginary portion of the dielectric constant. Therefore, (MA)SnI3 possesses higher ε2αβ over (MA)SnF3 with longer wavelength, as shown in Fig. 5. Similarly, when substituting Sn for Pb, the corresponding (MA)PbX3 possesses a higher band gap as shown in Table 4 and hence its dielectric matrix is smaller in comparison with that of (MA)SnX3. Comparing Fig. 5 with Fig. S10,† at visible light wavelength range (>400 nm), (MA)SnI3 has the highest ε1αβ and ε2αβ values while (MA)PbF3 possesses the lowest ε2αβ and ε2αβ, which are consistent with trends of their electronic properties as described in Section 3.2.
Based on the calculated ε1αβ and ε2αβ values from eqn (4) and (5), the absorption coefficient, α, is further calculated at a particular wavelength, λ, as shown in eqn (6).
| |
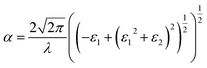 | (6) |
where
c refers to the speed of light. Other optical properties (
e.g., index of refraction, extinction coefficient, reflectivity, optical conductivity,
etc.) also can be derived from dielectric matrix and are provided in the ESI.
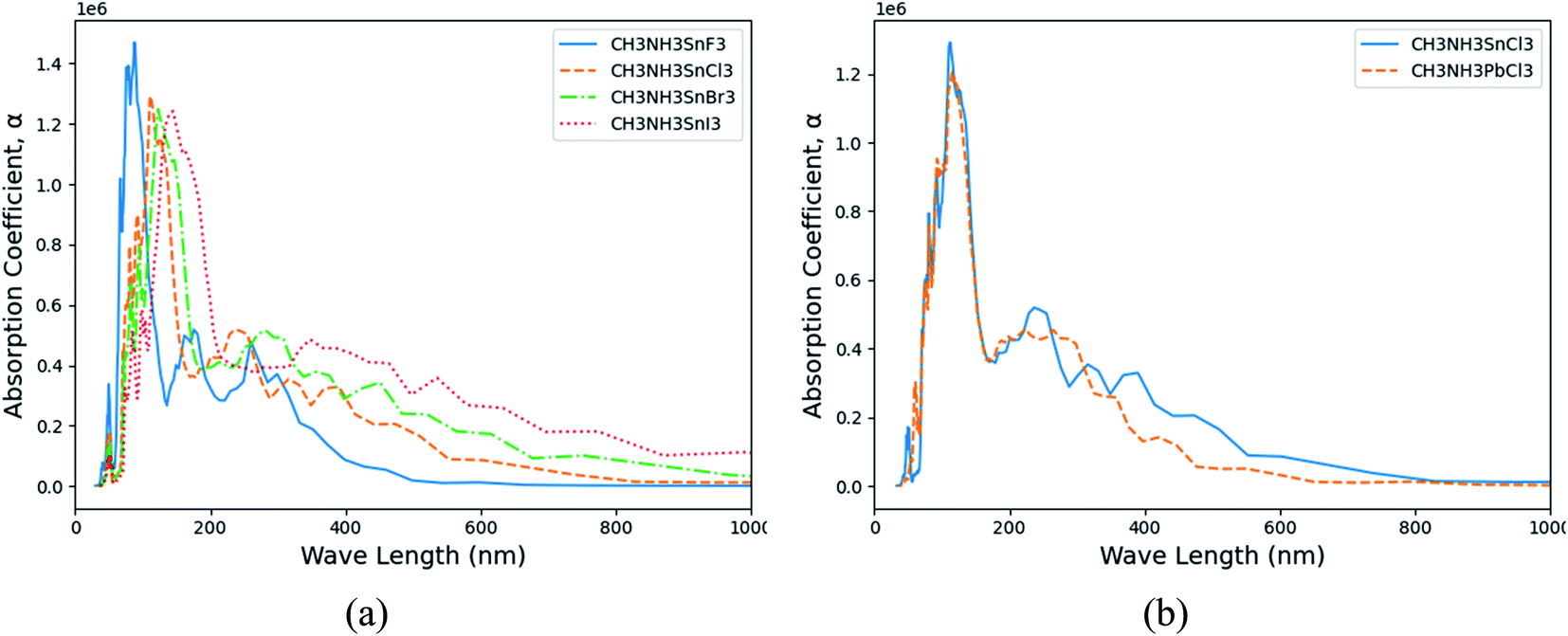
† The calculated absorption coefficients of (MA)SnX
3 and (MA)PbX
3 are shown in
Fig. 6(a) and S12 of the ESI,
† respectively. Notably from these figures, perovskites containing heavier halogen anions exhibit higher absorption coefficients, particularly in the visible light range (400–800 nm) – a key parameter for both optical sensor and photovoltaic cell applications. Such shifts are a directly reflection of the change trends on their electronic structure properties. Despite Pb-containing perovskites' prevalence over Sn counterparts, (MA)BCl
3 (B = Sn, Pb) as an example, the Sn-containing perovskites display a higher absorption coefficient in the visible light range, as shown in
Fig. 6(b). This suggests another important benefit the Sn-based perovskites could contribute to the performance improvement in the aforementioned applications besides their environmental benign advantages over the Pb-based counterparts.
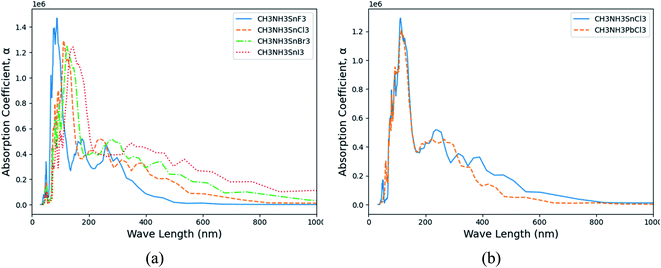 |
| | Fig. 6 The calculated absorption coefficient (α) versus frequency in wavelength. (a) (MA)SnX3 (X = F, Cl, Br, I); (b) (MA)BCl3 (B = Sn, Pb). | |
Other optical properties such as extinction coefficient, index of refraction, and reflectivity are also calculated with detailed results placed in Fig. S13–S19 of the ESI.† From these results, similar change trends are also observed when varying the X site anions of perovskites from F to I and/or substituting Sn with Pb cations in the B sites. Hence, to achieve better optical performance (e.g., higher absorption coefficient) among orthorhombic perovskites (MA)BX3 (B = Sn, Pb; X = F, Cl, Br, I), our results suggest that (MA)SnX3 are better candidates than (MA)PbX3. Overall, with the same metal (Sn or Pb) cations, iodine anion contributes the strongest optical absorption among all halogen types while fluorine makes the least. As for high optical absorption performance in the visible light range, we predict that (MA)SnI3 is the best candidate while (MA)PbF3 is least favorable. However, (MA)PbF3, if stable, could serve in UV detection applications for its strong adsorption in the 10–400 nm range.
4. Conclusions
In this work, the electronic and optical properties of orthorhombic (MA)BX3 (B = Sn, Pb; X = F, Cl, Br, I) perovskites were calculated by first-principles density functional theory. The structures were relaxed to obtain the energy-minimized geometry and formation energies. The bulk modulus was evaluated by fitting the Birch–Murnaghan equation-of-state and in good agreement with available data. Deviations from literature structure simulations exist, but a definite trend is still observed. Notably, the (MA)PbF3 perovskite is found to have a negative formation energy, indicating its possible presence at low temperature conditions. The electronic structures of all eight perovskite systems were analyzed to evaluate the band gaps and contributors to the band gaps, with electronegativity suspected to be a major driving factor. Variations in the p-orbitals of the halogen anions is believed to decide the different levels in the valence band of these perovskite systems, and that of the metal cations contributing to the conduction band level. Hence, the band gaps of (MA)BX3 are decreased with halogen element X varying from F to I, and larger band gaps are found in (MA)PbX3 than corresponding (MA)SnX3. Our calculations on the optical properties of these orthorhombic perovskites show the adsorption enhancement in the visible light range when the halogen anions vary from F to I, so does the substitution of Pb with Sn cation in the B sites, reflecting the similar trends as in the partial DOS shifting. Tin-based halide perovskites (particularly (MA)SnI3) are shown to – among those examined – have comparably better absorbance of visible light energy, suggesting additional benefits in their replacement of the lead-based counterparts in broad perovskites applications.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
S. N. thanks the Department of Energy, Mickey Leland Energy Fellowship (MLEF) for the Summer Internship at National Energy Technology Laboratory (NETL) as well as the Louisiana Board of Regents (LBR) for supporting his research through the LBR Graduate Fellowship. T. J. thanks the NETL Research and Innovation Center (R&IC) for providing Computational resources administered by the Oak Ridge Institute for Science and Education (ORISE). S. W. thanks Louisiana Harrelson Family Professorship for supporting this research. Revision computations were performed on computational resources provided by the Louisiana Optical Network Infrastructure (LONI). This report was prepared as an account of work sponsored by an agency of the United States Government. Neither the United States Government nor any agency thereof, nor any of their employees, makes any warranty, express or implied, or assumes any legal liability or responsibility for the accuracy, completeness, or usefulness of any information, apparatus, product, or process disclosed, or represents that its use would not infringe privately owned rights. Reference herein to any specific commercial product, process, or service by trade name, trademark, manufacturer, or otherwise does not necessarily constitute or imply its endorsement, recommendation, or favoring by the United States Government or any agency thereof. The views and opinions of authors expressed herein do not necessarily state or reflect those of the United States Government or any agency hereof.
References
- T. Baikie, Y. Fang, J. M. Kadro, M. Schreyer, F. Wei, S. Mhaisalkar, M. Graetzel and T. J. White, J. Mater. Chem. A, 2013, 5628–5641 RSC
 .
. - G. M. Dalpian, Q. Liu, C. C. Stoumpos, A. P. Douvalis, M. Balasubramanian, M. G. Kanatzidis and A. Zunger, Phys. Rev. Mater., 2017, 1, 1–10 Search PubMed .
- J. H. Noh, S. H. Im, J. H. Heo, T. N. Mandal and S. I. Seok, Nano Lett., 2013, 13, 1764–1769 CrossRef CAS .
- C. Eames, J. M. Frost, P. R. F. Barnes, B. C. O'Regan, A. Walsh and M. S. Islam, Nat. Commun., 2015, 6, 2–9 Search PubMed .
- L. Lang, J. H. Yang, H. R. Liu, H. J. Xiang and X. G. Gong, Phys. Lett. A, 2014, 378, 290–293 CrossRef CAS .
- R. Prasanna, T. Leijtens, S. P. Dunfield, J. A. Raiford, E. J. Wolf, S. A. Swifter, J. Werner, G. E. Eperon, C. de Paula, A. F. Palmstrom, C. C. Boyd, M. F. A. M. van Hest, S. F. Bent, G. Teeter, J. J. Berry and M. D. McGehee, Nat. Energy, 2019, 4, 939–947 CrossRef CAS .
- S. Ruan, J. Lu, N. Pai, H. Ebendorff-Heidepriem, Y.-B. Cheng, Y. Ruan and C. R. McNeill, J. Mater. Chem. C, 2018, 6, 6988–6995 RSC .
- X. Yang, Y. Gao, Z. Ji, L. B. Zhu, C. Yang, Y. Zhao, Y. Shu, D. Jin, Q. Xu and W. W. Zhao, Anal. Chem., 2019, 91, 9356–9360 CrossRef CAS PubMed .
- G. Kakavelakis, E. Gagaoudakis, K. Petridis, V. Petromichelaki, V. Binas, G. Kiriakidis and E. Kymakis, ACS Sens., 2018, 3, 135–142 CrossRef CAS PubMed .
- F. Hao, C. C. Stoumpos, D. H. Cao, R. P. H. Chang and M. G. Kanatzidis, Nat. Photonics, 2014, 8, 489–494 CrossRef CAS .
- A. Jaffe, Y. Lin, C. M. Beavers, J. Voss, W. L. Mao and H. I. Karunadasa, ACS Cent. Sci., 2016, 2, 201–209 CrossRef CAS PubMed .
- T. Oku, in Solar Cells – New Approaches and Reviews, ed. L. A. Kosyachenko, InyechOpen, 2015, pp. 77–101 Search PubMed .
- N. Onodayamamuro, T. Matsuo and H. Suga, J. Phys. Chem. Solids, 1990, 51, 1383–1395 CrossRef CAS .
- D. Weber, Z. Naturforsch., B: J. Chem. Sci., 1978, 33, 1443–1445 CrossRef .
- M. Afsari, A. Boochani and F. Shirdel, Optik, 2019, 199, 163360 CrossRef CAS .
- A. O. Al Ghaithi, S. A. Aravindh, M. N. Hedhili, T. K. Ng, B. S. Ooi and A. Najar, ACS Omega, 2020, 5, 12313–12319 CrossRef CAS PubMed .
- I. O. A. Ali, D. P. Joubert and M. S. H. Suleiman, Eur. Phys. J. B, 2018, 91, 263 CrossRef CAS .
- C. Bernal and K. S. Yang, J. Phys. Chem. C, 2014, 118, 24383–24388 CrossRef CAS .
- K. T. Butler, J. M. Frost and A. Walsh, Mater. Horiz., 2015, 2, 228–231 RSC .
- D. O. Demchenko, N. Izyumskaya, M. Feneberg, V. Avrutin, U. Ozgur, R. Goldhahn and H. Morkoc, Phys. Rev. B, 2016, 94, 075206 CrossRef .
- Y. Q. Huang, J. Su, Q. F. Li, D. Wang, L. H. Xu and Y. Bai, Phys. B, 2019, 563, 107–112 CrossRef CAS .
- R. Lindblad, N. K. Jena, B. Philippe, J. Oscarsson, D. Q. Bi, A. Lindblad, S. Mandal, B. Pal, D. D. Sarma, O. Karis, H. Siegbahn, E. M. J. Johansson, M. Odelius and H. Rensmo, J. Phys. Chem. C, 2015, 119, 1818–1825 CrossRef CAS .
- A. Maalej, Y. Abid, A. Kallel, A. Daoud, A. Lautie and F. Romain, Solid State Commun., 1997, 103, 279–284 CrossRef CAS .
- J. S. Park, S. Choi, Y. Yan, Y. Yang, J. M. Luther, S. H. Wei, P. Parilla and K. Zhu, J. Phys. Chem. Lett., 2015, 6, 4304–4308 CrossRef CAS PubMed .
- S. Park, Y. S. Seo, C. W. Ahn, W. S. Woo, J. Kyhm, S. A. Lee, I. W. Kim and J. Hwang, J. Phys. D: Appl. Phys., 2019, 52, 335302 CrossRef CAS .
- L. P. Peng, Y. Hu and L. Xu, Sci. Adv. Mater., 2016, 8, 2223–2230 CrossRef CAS .
- L. P. Peng and L. Xu, Sci. Adv. Mater., 2018, 10, 1519–1527 CrossRef CAS .
- P. Umari, E. Mosconi and F. De Angelis, Sci. Rep., 2014, 4, 4467 CrossRef PubMed .
- L. J. Wu, Y. Q. Zhao, C. W. Chen, L. Z. Wang, B. Liu and M. Q. Cai, Chin. Phys. B, 2016, 25, 107202 CrossRef .
- M. Zhong, W. Zeng, H. Tang, L. X. Wang, F. S. Liu, B. Tang and Q. J. Liu, Sol. Energy, 2019, 190, 617–621 CrossRef CAS .
- C. Li, J. Wei, M. Sato, H. Koike, Z. Z. Xie, Y. Q. Li, K. Kanai, S. Kera, N. Ueno and J. X. Tang, ACS Appl. Mater. Interfaces, 2016, 8, 11526–11531 CrossRef CAS PubMed .
- Y. Wang, T. Gould, J. F. Dobson, H. M. Zhang, H. G. Yang, X. D. Yao and H. J. Zhao, Phys. Chem. Chem. Phys., 2014, 16, 1424–1429 RSC .
- J. Feng and B. Xiao, J. Phys. Chem. C, 2014, 118, 19655–19660 CrossRef CAS .
- G. Kresse and J. Furthmuller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS .
- G. Kresse and J. Furthmuller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS .
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed .
- P. E. Blochl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed .
- J. P. Perdew, M. Ernzerhof and K. Burke, J. Chem. Phys., 1996, 105, 9982–9985 CrossRef CAS .
- A. Jain, S. P. Ong, G. Hautier, W. Chen, W. D. Richards, S. Dacek, S. Cholia, D. Gunter, D. Skinner, G. Ceder and K. A. Persson, APL Mater., 2013, 1, 011002 CrossRef .
- J. Feng and B. Xiao, J. Phys. Chem. Lett., 2014, 5, 1278–1282 CrossRef CAS PubMed .
- Y. Duan, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 045332 CrossRef .
- F. Chiarella, A. Zappettini, F. Licci, I. Borriello, G. Cantele, D. Ninno, A. Cassinese and R. Vaglio, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 045129 CrossRef .
- I. Borriello, G. Cantele and D. Ninno, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 235215 CrossRef .
- P. Mavropoulos, I. Galanakis, V. Popescu and P. H. Dederichs, J. Phys.: Condens. Matter, 2004, 16, S5759–S5762 CrossRef CAS .
- M. Gajdos, K. Hummer, G. Kresse, J. Furthmuller and F. Bechstedt, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 045112 CrossRef .
- Y. Duan, P. Ohodnicki, B. Chorpening and G. Hackett, J. Solid State Chem., 2017, 256, 239–251 CrossRef CAS .
- T. Jia, P. Ohodnicki, B. Chorpening, J. Lekse, G. Hackett and Y. Duan, Phys. Chem. Chem. Phys., 2019, 21, 26117–26122 RSC .
- T. Jia, Z. Zeng, H. Q. Lin, Y. Duan and P. Ohodnicki, RSC Adv., 2017, 7, 38798–38804 RSC .
- Y. N. Wu, W. A. Saidi, J. K. Wuenschell, T. Tadano, P. Ohodnicki, B. Chorpening and Y. Duan, J. Phys. Chem. Lett., 2020, 11, 2518–2523 CrossRef CAS PubMed .
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra01586a |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a
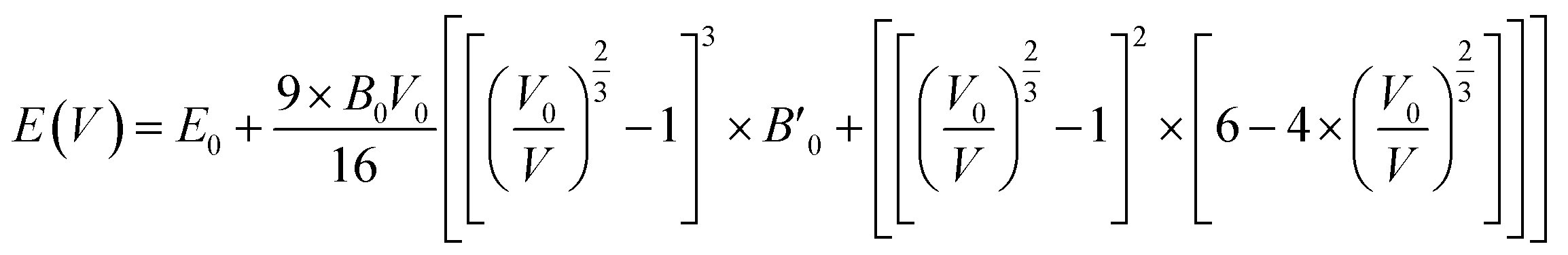
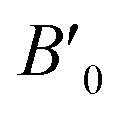 referring to the first order variation of the bulk modulus, B, with pressure, P, all according to eqn (2). P is taken to be 1 atm.
referring to the first order variation of the bulk modulus, B, with pressure, P, all according to eqn (2). P is taken to be 1 atm.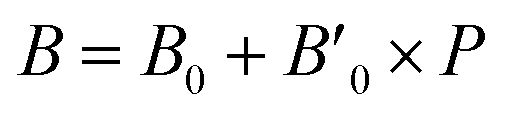
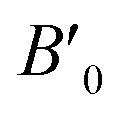 values than the other six perovskites examined.
values than the other six perovskites examined.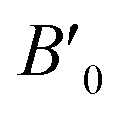 ) and bulk modulus (B) of (MA)BX3 by fitting to the Birch–Murnaghan equation of state (E–V)
) and bulk modulus (B) of (MA)BX3 by fitting to the Birch–Murnaghan equation of state (E–V)