
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMedicinal chemistry perspectives of 1,2,3,4-tetrahydroisoquinoline analogs – biological activities and SAR studies
Faheem
 a,
Banoth Karan Kumar
a,
Kondapalli Venkata Gowri Chandra Sekhar
b,
Subhash Chander
c,
Selvaraj Kunjiappan
d and
Sankaranarayanan Murugesan
*a
a,
Banoth Karan Kumar
a,
Kondapalli Venkata Gowri Chandra Sekhar
b,
Subhash Chander
c,
Selvaraj Kunjiappan
d and
Sankaranarayanan Murugesan
*a
aMedicinal Chemistry Research Laboratory, Department of Pharmacy, Birla Institute of Technology and Science-Pilani, Pilani Campus, Pilani-333031, Rajasthan, India. E-mail: murugesan@pilani.bits-pilani.ac.in
bDepartment of Chemistry, Birla Institute of Technology and Science-Pilani, Hyderabad Campus, Jawahar Nagar, Shameerpet Mandal, Medchal Dist., Hyderabad, 500078, Telangana, India
cAmity Institute of Phytomedicine and Phytochemistry, Amity University Uttar Pradesh, Noida-201313, India
dDepartment of Biotechnology, Kalasalingam Academy of Research and Education, Krishnankoil-626126, Tamil Nadu, India
First published on 29th March 2021
Abstract
Isoquinoline alkaloids are a large group of natural products in which 1,2,3,4-tetrahydroisoquinolines (THIQ) form an important class. THIQ based natural and synthetic compounds exert diverse biological activities against various infective pathogens and neurodegenerative disorders. Due to these reasons, the THIQ heterocyclic scaffold has garnered a lot of attention in the scientific community which has resulted in the development of novel THIQ analogs with potent biological activity. The present review provides a much-needed update on the biological potential of THIQ analogs, their structural–activity relationship (SAR), and their mechanism of action. In addition, a note on commonly used synthetic strategies for constructing the core scaffold has also been discussed.
 Faheem | Mr Faheem has a Medicinal Chemistry Graduate degree from the Department of Pharmacy, Birla Institute of Technology, and Science Pilani, Pilani campus, Rajasthan. He completed his Bachelor of Pharmacy at Sri Ramachandra Institute of Higher Education and Research, Tamil Nadu. To his credit, he has published 7 research and review articles in various peer-reviewed journals. In addition to that, he has also presented 5 papers/posters at various international and national conferences. |
 Banoth Karan Kumar | Mr Banoth Karan Kumar is currently pursuing his Ph.D. under Dr S. Murugesan at the Department of Pharmacy, Birla Institute of Technology, and Science Pilani, Pilani campus, Rajasthan. Karan has a Pharmaceutical Chemistry Graduate degree from the University College of Pharmaceutical Sciences, Kakatiya University. He has published 14 research and review articles in various peer-reviewed journals of international and national repute. In addition to that, he has also presented 12 papers/posters at various international and national conferences. His current research area includes synthesis, molecular docking, and dynamics studies for infectious diseases like leishmaniasis. |
 Kondapalli Venkata Gowri Chandra Sekhar | Prof. K. V. G. Chandra Sekhar completed his B. Pharm (Hons.), M. Pharm and Ph.D. in synthetic medicinal chemistry at BITS Pilani Rajasthan, India. Since 2015 he has been working as an associate professor at BITS Pilani Hyderabad campus. His areas of interest include medicinal chemistry and drug design. He successfully completed six research projects funded by UGC, DST and DBT, Government of India. Currently Indo-Spain bilateral research is in progress in his lab. He has published over 50 research articles in well renowned international journals. He is an expert reviewer for various journals and has served as an examiner for Ph.D thesis at various universities. |
 Subhash Chander | Dr Subhash Chander is currently working as Assistant Professor II at Amity University Uttar Pradesh, Noida, India. He completed graduation in Pharmacy from GJU S & Tech, Hisar, in the year 2009, followed by a master's degree in Pharmaceutical Chemistry from NIPER, in the year 2011. He worked as Research Associate in a research organization for one and half years. He joined the research group Dr S. Murugesan in 2012 at BITS, Pilani, and completed his PhD in the year 2017. Over the last decade, his team has been working on target specific drug discovery against HIV-1 and other allied opportunistic infections. He and his team have published more than 40 research and review articles, and two book chapters and filed two patents. |
 Selvaraj Kunjiappan | Dr Selvaraj Kunjiappan is working as Assistant Professor of Biotechnology in the School of Bio and Chemical Engineering, Kalasalingam Academy of Research and Education, India. He received his PhD (Chemical Engineering) from Jadavpur University, Kolkata, India. His research interests are health care/pharmaceutically important lead compound design, synthesis and biomaterials isolated from environmentally dumped waste human hair, and waste biological materials like fish shells, and crab shells. The isolated biomaterial is applied to convert into nanoparticle nanocarriers for degenerative disease therapeutic formulations for overcoming multi drug resistance, targeted drug delivery and improved bioavailability. |
 Sankaranarayanan Murugesan | Dr S. Murugesan is currently working as an Associate Professor in the Department of Pharmacy, BITS Pilani, Pilani Campus, Pilani, Rajasthan. In the year 2006, he enrolled in a Ph. D. program under the guidance of Dr Swastika Ganguly and acquired his doctorate in the year 2009 from Birla Institute of Technology, Mesra, Ranchi, India. Dr Murugesan is actively involved in research in computer-aided drug design and drug synthesis against HIV and allied opportunistic infections. He has more than 15 years of teaching and research experience. He has guided more than 30 B. Pharm, and 20 M. Pharm students for their dissertation work. Two scholars have already completed a full-time doctorate, while four are being pursued under his guidance. He has completed 3 research projects, including two as major projects from DST-SERB and DBT, India. Dr Murugesan has published more than 100 research papers in various peer-reviewed journals of international and national repute and presented more than 100 papers/posters in various international and national conferences. |
1. Introduction
1,2,3,4-Tetrahydroisoquinoline (THIQ) is a secondary amine with the chemical formula C9H11N. The THIQ nucleus containing alkaloids is widely distributed in nature and forms an essential part of the isoquinoline alkaloids family. The THIQ-based antitumor antibiotics isolated from different natural sources have garnered a lot of attention for over 4 decades starting with the isolation of Naphthyridinomycin 1. Thereafter, several THIQ antitumor antibiotics belonging to different families such as quinocarcin (quinocarcinol 2, tetrazomine 3), saframycins (saframycin A–B 4–5), and naphthyridinomycin (cyanocycline C 6, bioxalomycin α1 7) have been isolated (Fig. 1.). An in-depth review of these antitumor antibiotics has been presented elsewhere.1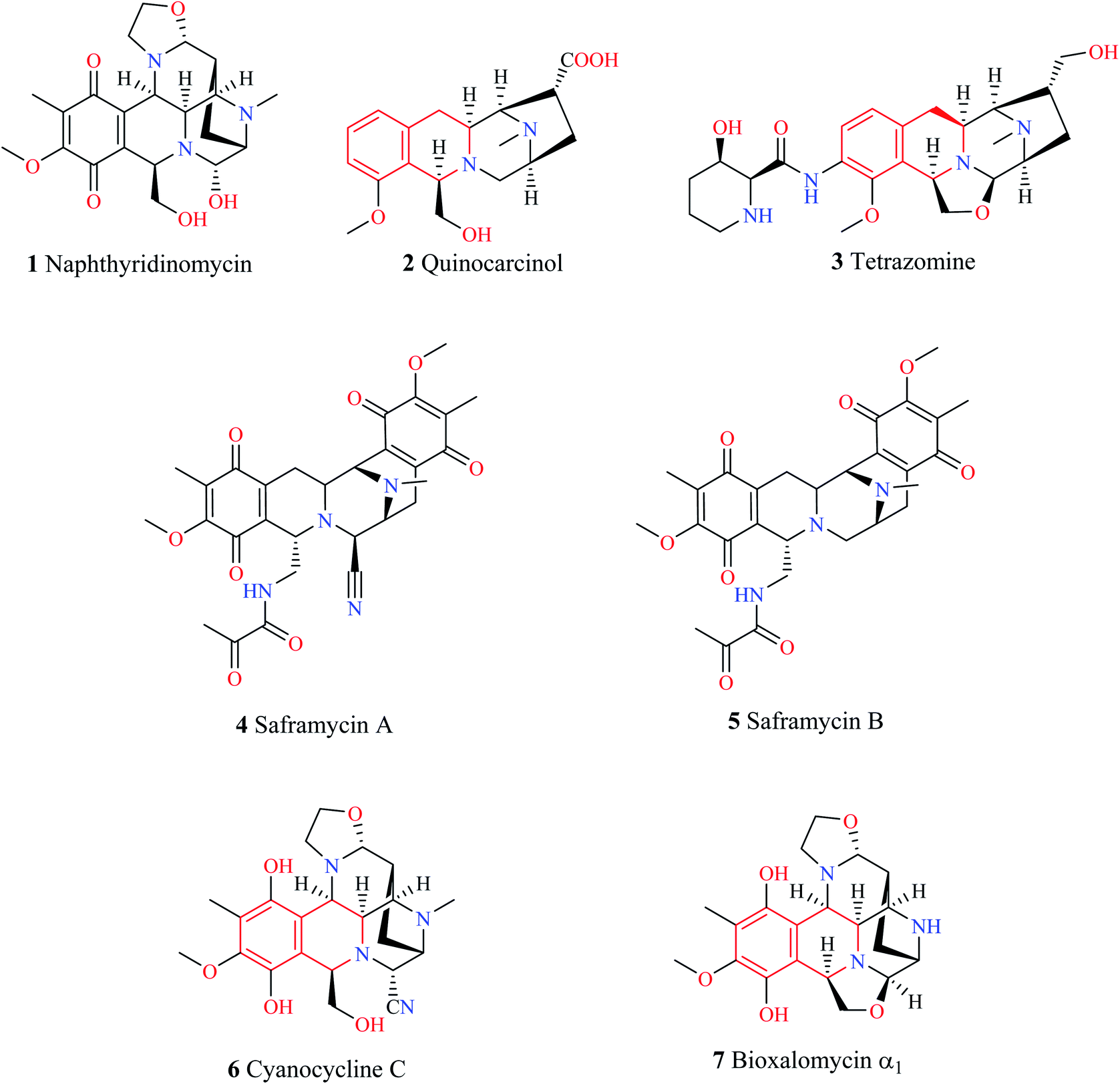 | ||
| Fig. 1 THIQ containing antitumor antibiotics. | ||
THIQ containing synthetic and natural analogs have been reported to possess a wide range of pharmacological activities like anti-inflammatory, anti-bacterial, anti-viral, anti-fungal, anti-leishmanial, anti-cancer, and anti-malarial, among others.2–6 Some of the THIQ analogs that are used clinically are highlighted in Table 1.
| Name | Structure | Application |
|---|---|---|
| Praziquantel | 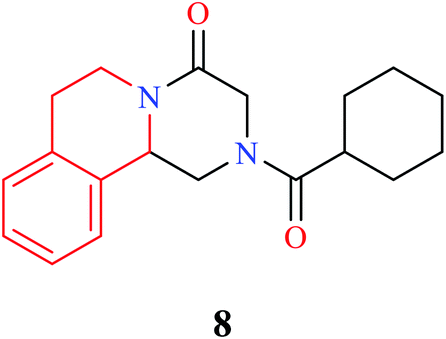 |
Anthelmintic |
| Quinapril | 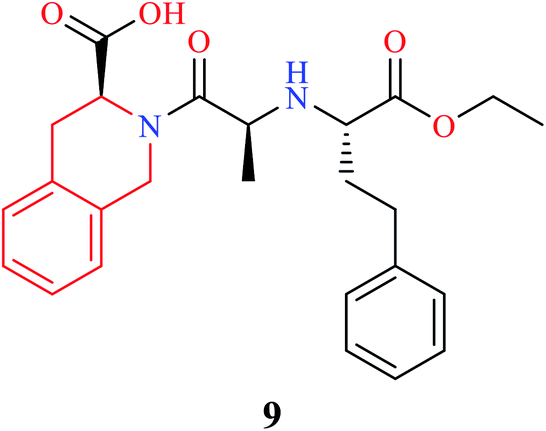 |
Anti-hypertensive |
| Noscapine | 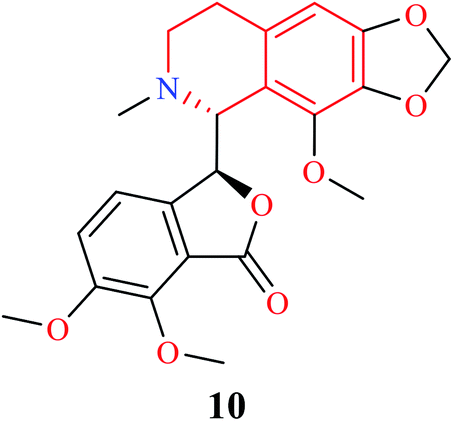 |
Anti-tussive |
| Apomorphine |  |
Anti-parkinsonian |
| Doxacurium |  |
Skeletal muscle relaxant |
| Mivacurium |  |
Skeletal muscle relaxant |
| Tubocurarine |  |
Skeletal muscle relaxant |
| Solifenacin |  |
Used to treat overactive bladder |
| Trabectedin |  |
Anti-cancer |
| Lurbinectedin |  |
Anti-cancer |
| Atracurium |  |
Skeletal muscle relaxant |
To the best of our knowledge, there is a paucity of review articles for corroborating the biological significance of the THIQ analogs. Ysern et al. reviewed this class of compounds for the first time in the year 1981.7 Scott et al. provided an in-depth analysis of the antitumor antibiotics,1 whereas Singh et al. provided a patent review of THIQs in the field of therapeutics.8 The present review, therefore, is aimed to provide a critical update on the biological activities of THIQ analogs along with their structure–activity relationship (SAR) in order to shed light on the impact of various functional groups that are responsible for dictating the desired activity. A note on the commonly used synthetic strategies for the construction of the THIQ core has also been discussed.
2. Strategies for synthesizing THIQ core
2.1 Pictet–Spengler condensation
Pictet–Spengler condensation is a commonly used reaction for the synthesis of heterocyclic scaffolds such as THIQ and tetrahydro β-carboline. The reaction was first described by Pictet and Spengler in the year 1911, wherein phenylethylamine 19 and dimethoxymethane in the presence of aq. HCl at 100 °C to afford THIQ 20 in 40% yield (Scheme 1).9 Later, researchers have replaced dimethoxymethane with aldehydes to give one substituted THIQs.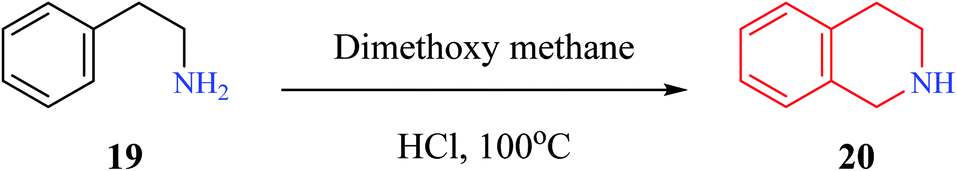 | ||
| Scheme 1 THIQ synthesis by Pictet and Spengler. | ||
A generalized mechanism (Scheme 2) consists of an initial condensation between a phenylethylamine derivative 21 and an aldehyde 22 to give an intermediate iminium 23. The resulting imine is then activated using a Brønsted or Lewis acid, which undergoes cyclization to give the one substituted THIQ product 24.10
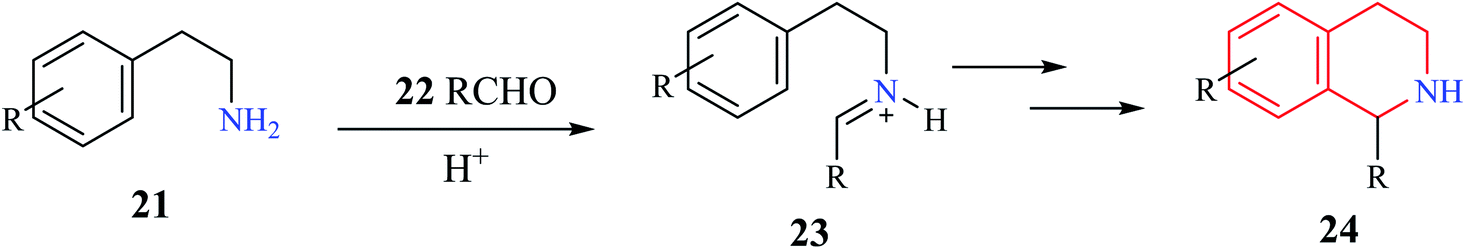 | ||
| Scheme 2 Mechanism of Pictet–Spengler condensation. | ||
Different variants of Pictet–Spengler condensation are currently available which are currently used for the construction of racemic as well as asymmetric THIQ derivatives. One such variant of the Pictet–Spengler reaction for the synthesis of 1-substituted THIQ derivatives is shown in Scheme 3.11 This reaction involved the synthesis of N-acetyl intermediate 26 from the starting material 2-(3,4-dimethoxyphenyl)-ethylamine 25. Then intermediate compound 26 was converted into N-acylcarbamates 27. The reduction of N-acylcarbamates 27 by diisobutyl aluminum hydride (DIBAL-H) followed by simultaneous cyclization mediated by BF3·OEt2 leads to the titled compound 28. The synthetic potential of this method was illustrated by the synthesis of tetrahydroisoquinoline alkaloids laudanosine 29, xylopinine 30, and isoindoloisoquinolone 31.
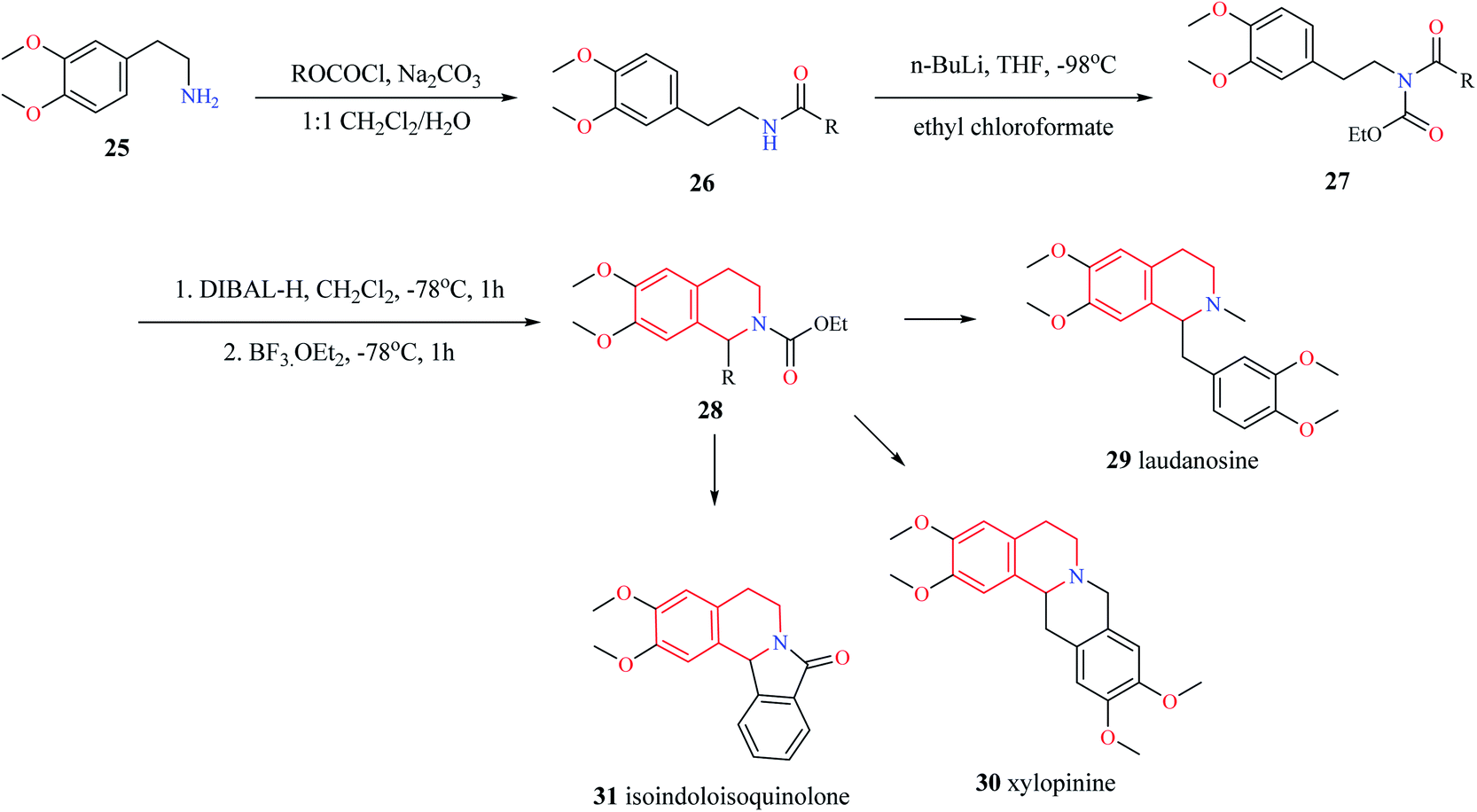 | ||
| Scheme 3 Synthesis of compound 28. | ||
Gremmen and co-workers reported the synthesis of enantiopure THIQs using a chiral auxiliary (1R,2S,5R)-(−) menthyl-(S)-p-toluene sulfinate (Andersen reagent).12 This method (Scheme 4) involves the reaction of 2-(3,4-dimethoxyphenyl)-ethylamine 25 with n-butyllithium, followed by reaction with Andersen reagent 32 to give N-p-tolyl sulfinyl phenylethylamine 33. The intermediate compound 33 then undergoes cyclization with different aldehydes via Pictet–Spengler condensation in the presence of BF3·OEt2 to give the derivatives of THIQ 34. The chiral auxiliary is then removed upon treating 34 with HCl in ethanol at 0 °C to give the final THIQ 35. The synthetic potential of this method was corroborated by the synthesis of THIQ natural product, (+) salsolidine 36.
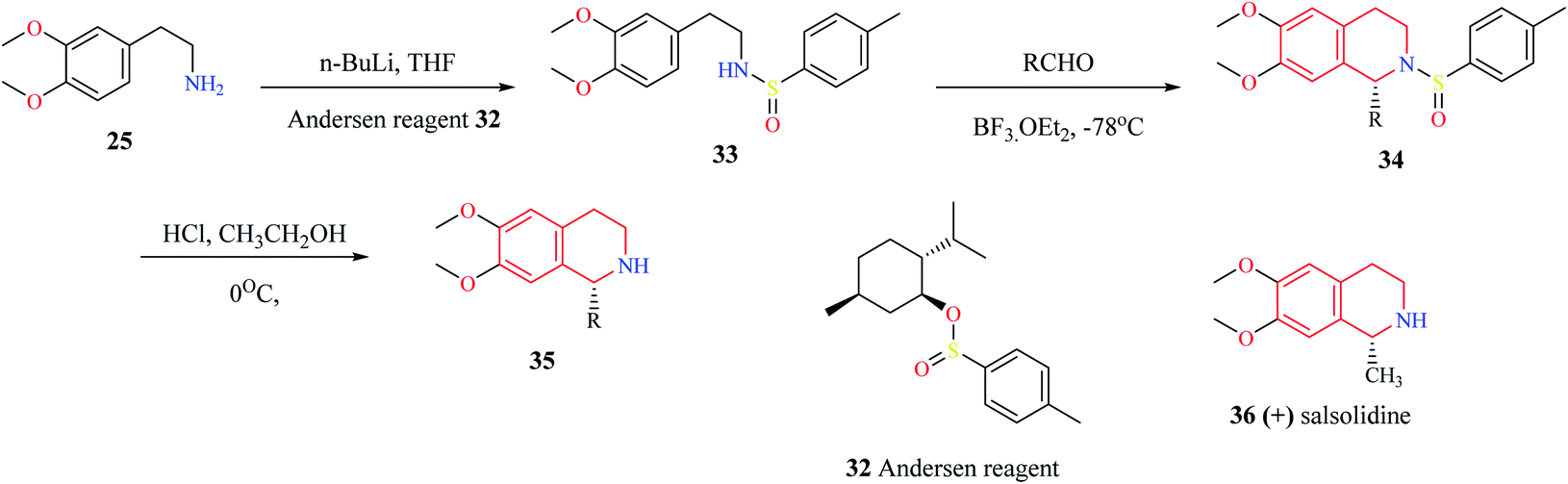 | ||
| Scheme 4 Enantioselective synthesis of compound 35. | ||
Mons et al. employed chiral catalysts (R)-TRIP 37 and (S)-BINOL 38 for the enantioselective synthesis of 1-substituted THIQs (Scheme 5).13 The 2-(3-hydroxy-4-methoxy phenyl)-ethylamine 39 is first converted to intermediate compound 41 by reacting with o-nitrophenyl sulfenylchloride (NpsCl) 40. Then, intermediate compound 41 undergoes asymmetric Pictet–Spengler reaction with different aldehydes to give the corresponding THIQs 42. This methodology was employed for the synthesis of (R)-(+)-crispine A 46 (Scheme 6).
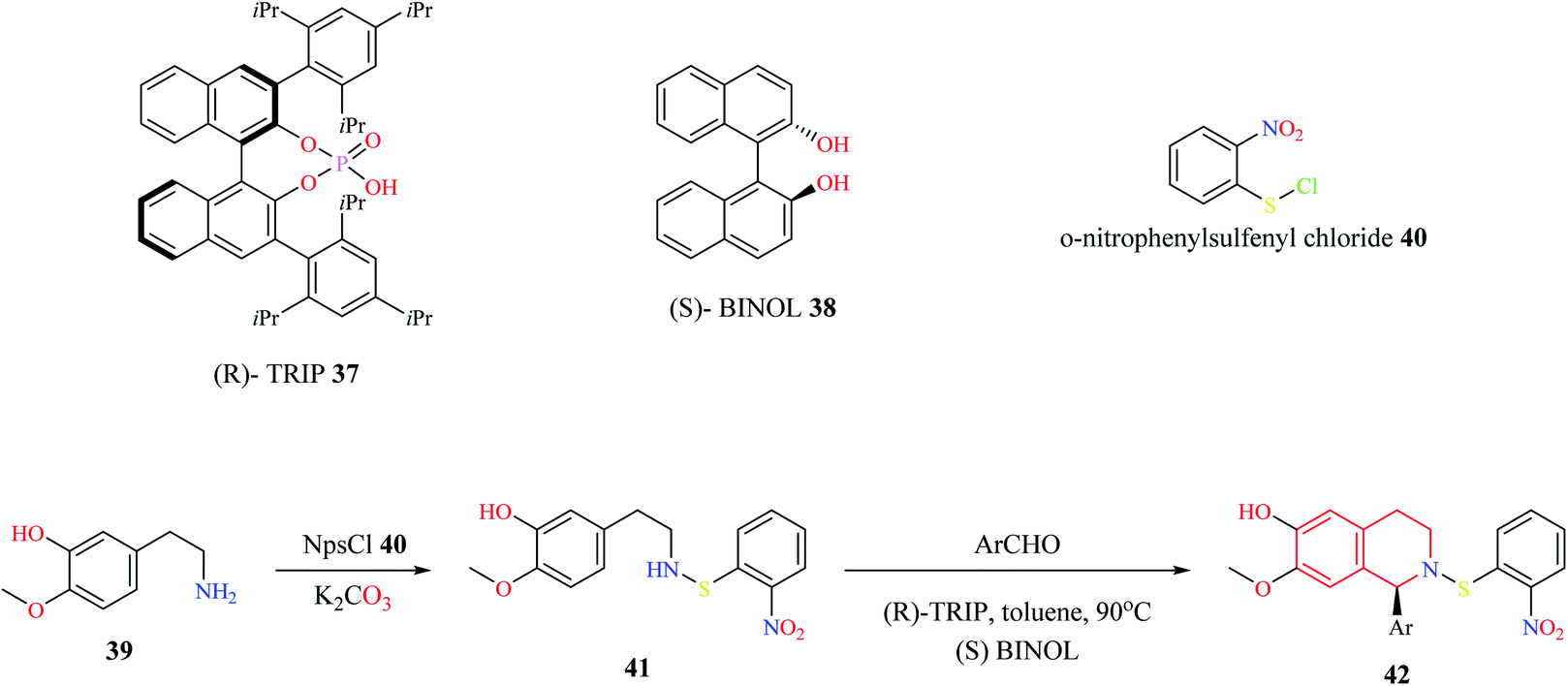 | ||
| Scheme 5 Enantioselective synthesis of compound 42. | ||
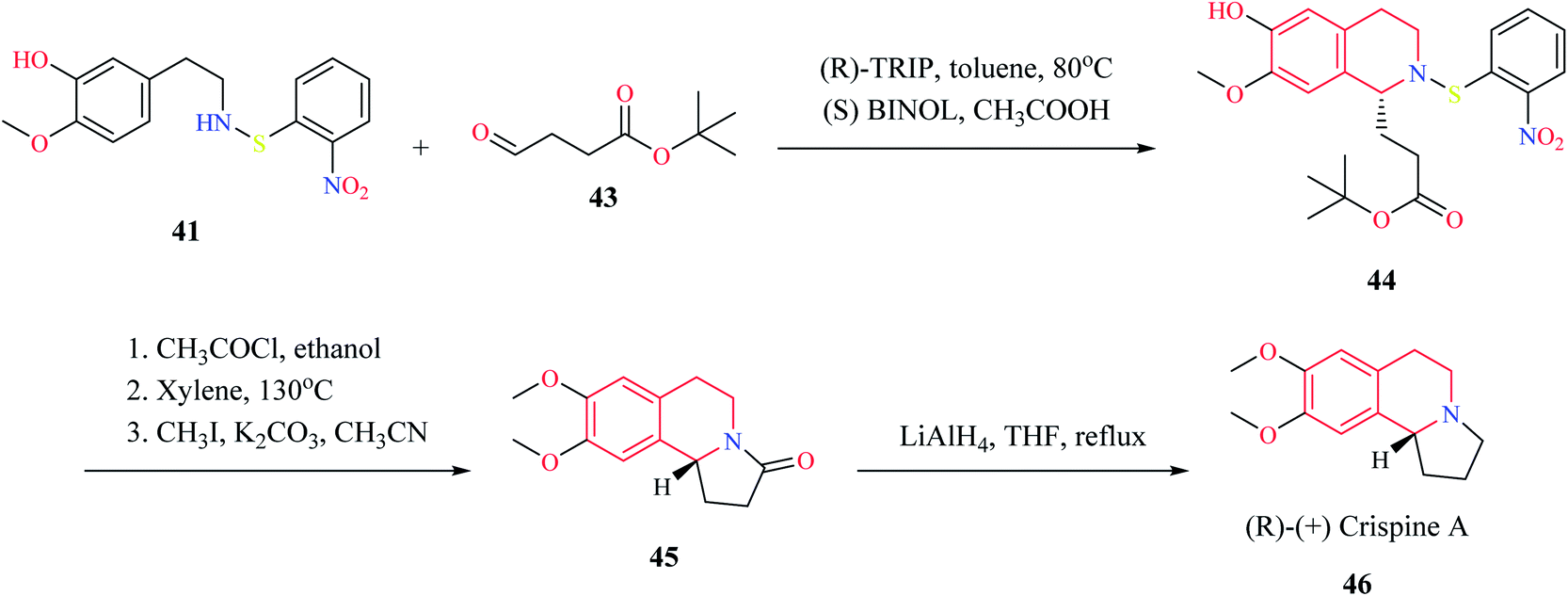 | ||
| Scheme 6 Synthesis of (R)-(+) crispine A 46. | ||
Microwave-assisted Pictet–Spengler reaction has also been used for the synthesis of 1-substituted THIQs.14 In such a strategy (Scheme 7), cyclization of one representative compound 2-(3,4-dimethoxyphenyl) ethylamine 25 with benzaldehyde 47 in the presence of trifluoroacetic acid (TFA) under microwave irradiation for 15 minutes afforded the title compound 48 in 98% yield.
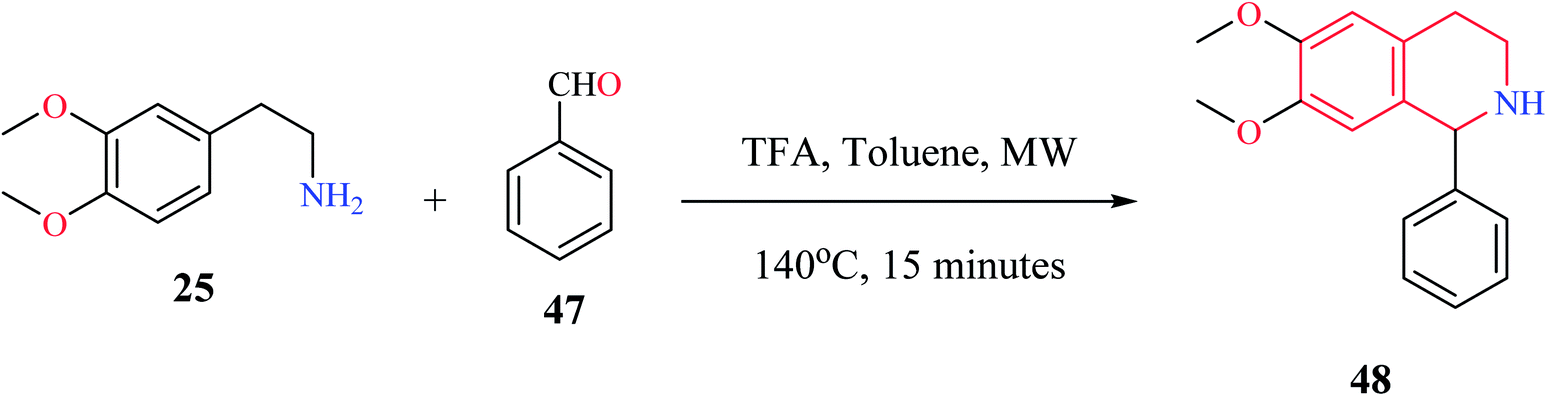 | ||
| Scheme 7 Microwave-assisted synthesis of compound 48. | ||
2.2 Bischler–Nepieralski reaction
Bischler–Nepieralski reaction is another commonly used strategy for the synthesis of isoquinoline derivatives. The reaction involves the cyclization of an N-acyl derivative of β-phenylethylamine 49 in the presence of a dehydrating agent such as POCl3, P2O5, ZnCl2 to generate 3,4-dihydro isoquinoline derivatives 50 (Scheme 8). The resulting dihydro isoquinoline 50 is then reduced to THIQs 51 using reducing agents like sodium borohydride, sodium cyanoborohydride, or catalytic hydrogenation process. Mechanistically, the reaction is said to proceed via intramolecular electrophilic aromatic substitution reaction.15,16 The presence of an electron-donating group like hydroxy or methoxy favors the cyclization step.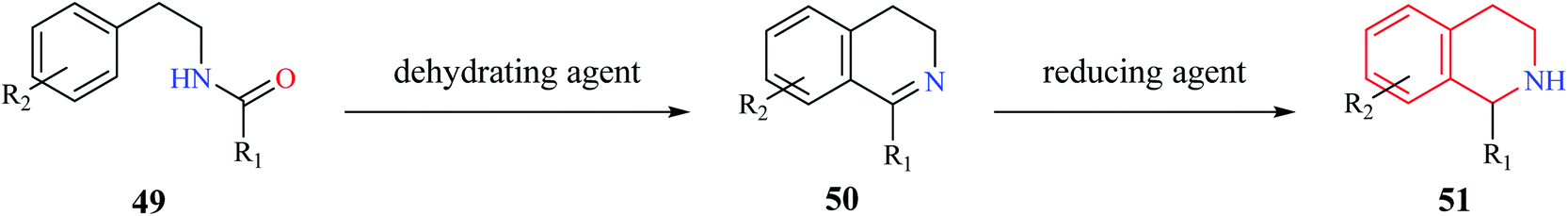 | ||
| Scheme 8 Bischler–Nepieralski synthesis of THIQ. | ||
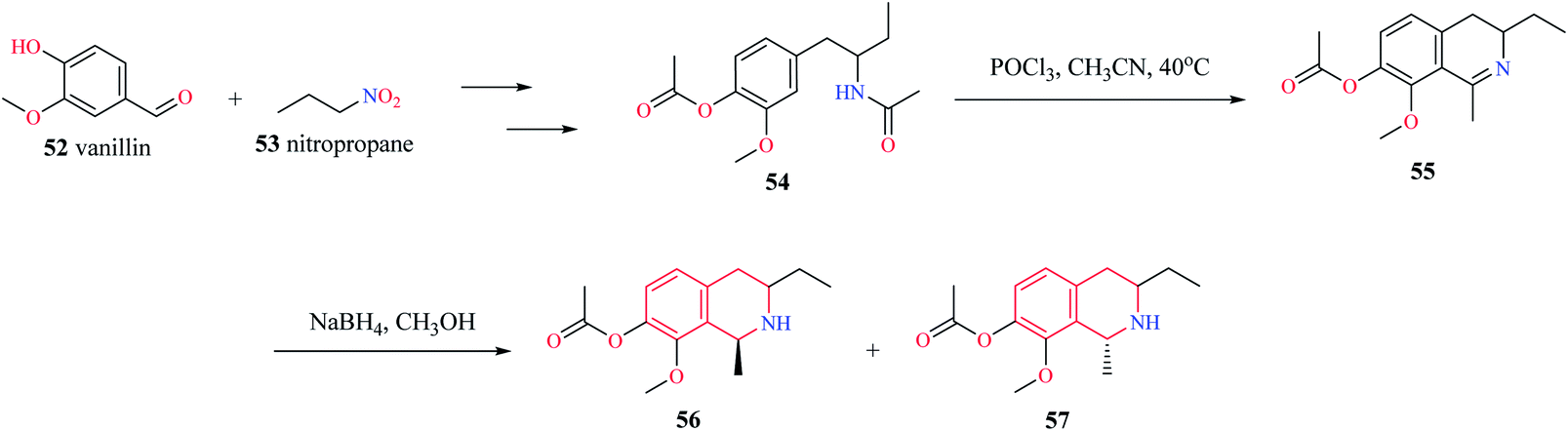
Mihoubi and co-workers synthesized THIQ derivatives 56–57 via Bischler–Nepieralski cyclization (Scheme 9).17 Initially, compound 54 was obtained from vanillin 52 and nitropropane 53. The compound 54 was then subjected to Bischler–Nepieralski cyclization using POCl3 to give the dihydroisoquinoline moiety 55 which was then reduced using NaBH4 to give the titled compounds 56–57.
 | ||
| Scheme 9 Synthesis of compounds 56–57. | ||
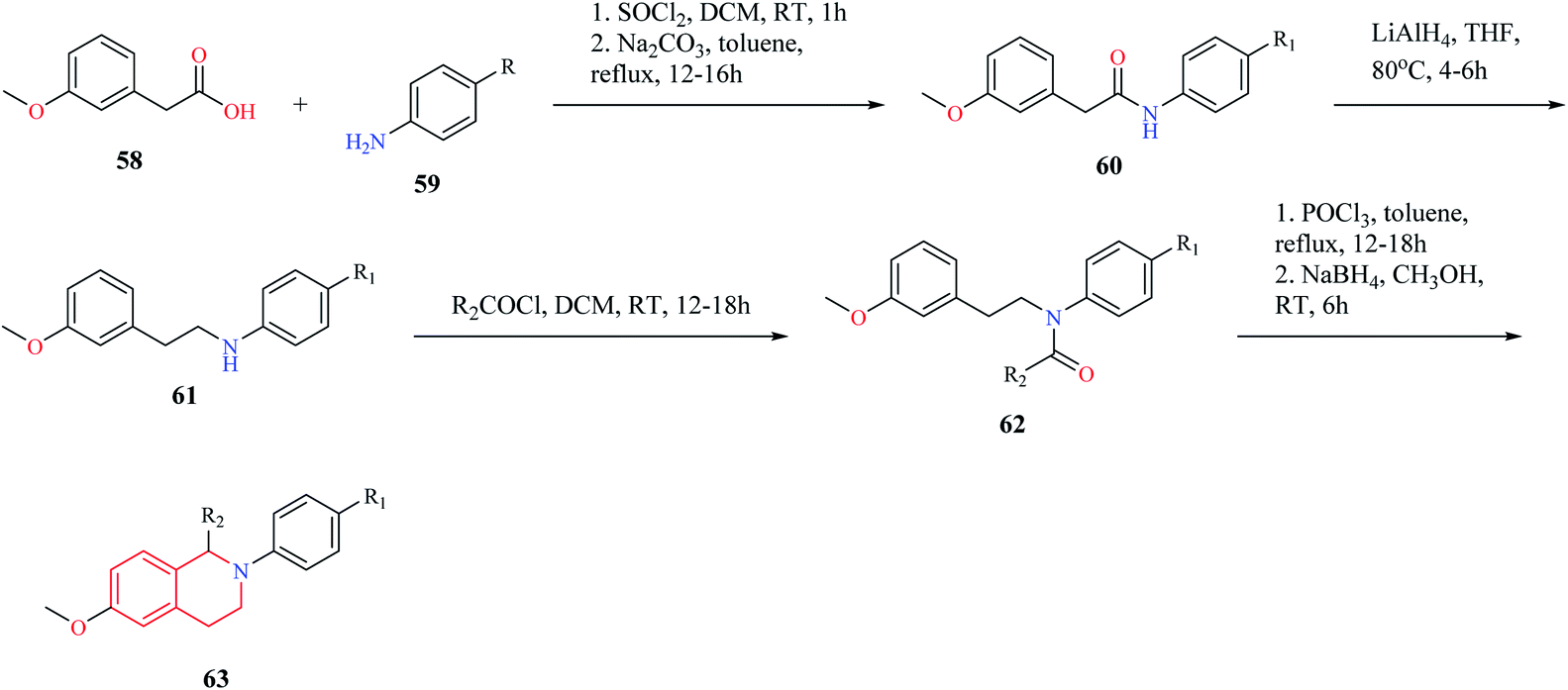
Mottinelli and co-workers synthesized N-substituted THIQs via Bischler–Nepieralski cyclization approach.18 Initially, 3-methoxy-phenyl acetic acid 58 was converted to its amide 60, which was then reduced to amine 61 using LiAlH4. The amine 61 was then converted to N-acyl derivative 62 using acyl chloride. Compound 62 was then subjected to Bischler–Nepieralski cyclization, followed by reduction with NaBH4 to give the N-substituted titled THIQ derivative 63 (Scheme 10).
 | ||
| Scheme 10 Synthesis of compound 63. | ||
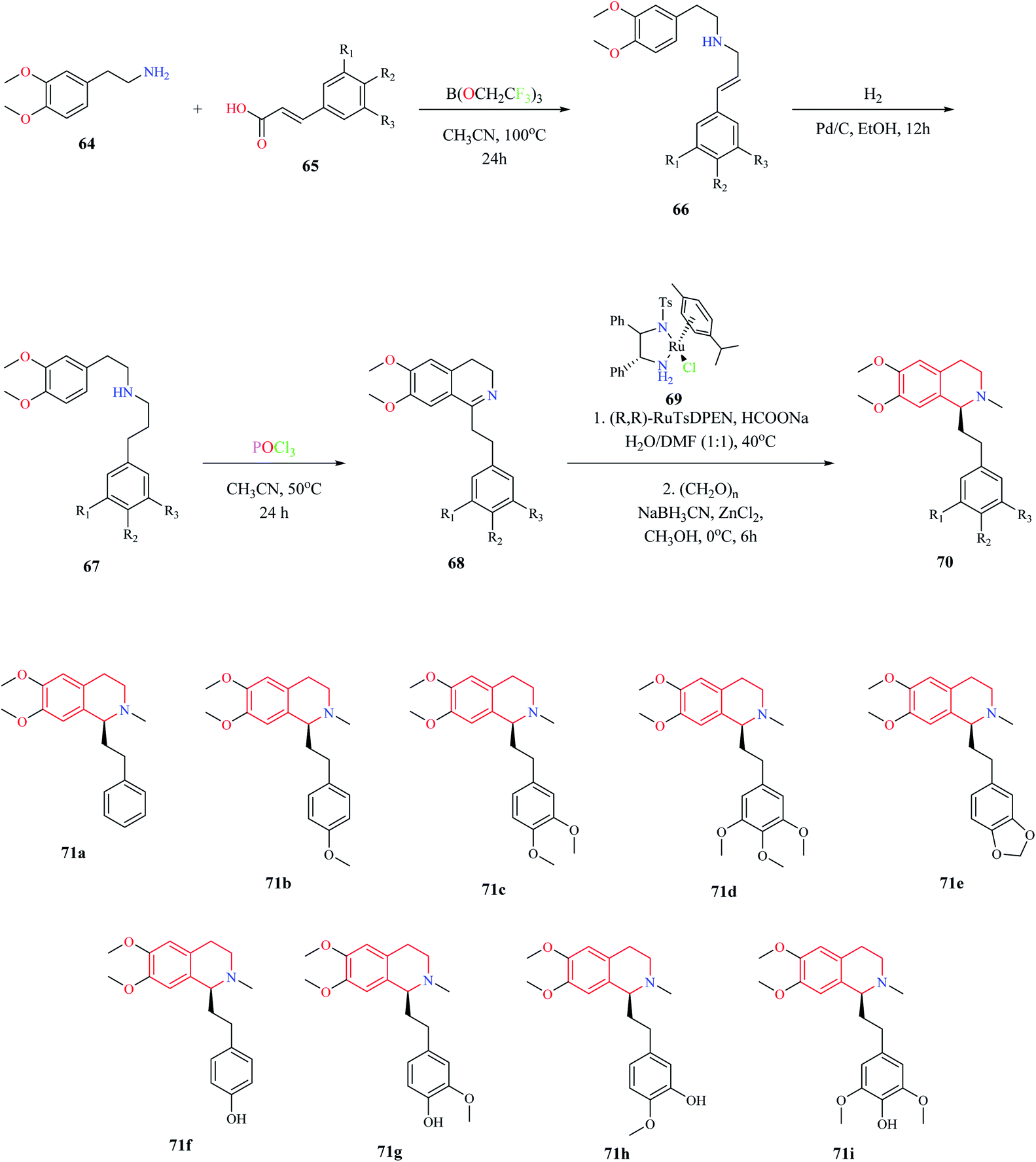
A biomimetic synthesis approach comprising of Bischler–Nepieralski cyclization and Noyori Asymmetric Transfer Hydrogenation (ATH) was used for the total synthesis of Dysoxylum alkaloids.19 The total synthesis consists of 5 steps. The first step involved the formation of zanthoxylamide protoalkaloid 66 from 2-(3,4-dimethoxyphenyl)-ethylamine 64 and (2-)3-phenylprop-2-enoic acid 65. Intermediate compound 66 was then subjected to catalytic hydrogenation, which furnished second intermediate compound 67. Intermediate compound 67 was then converted to the isoquinoline moiety 68 via Bischler–Nepieralski reaction. Compound 68 was then subjected to ATH reaction using a chiral catalyst (R,R)-RuTsDPEN 69 followed by reductive amination to obtain the final product 70 (Scheme 11). This approach was used for the total synthesis of Dysoxylum alkaloids 71a–i.
 | ||
| Scheme 11 Biomimetic approach for the synthesis of Dysoxylum alkaloids. | ||
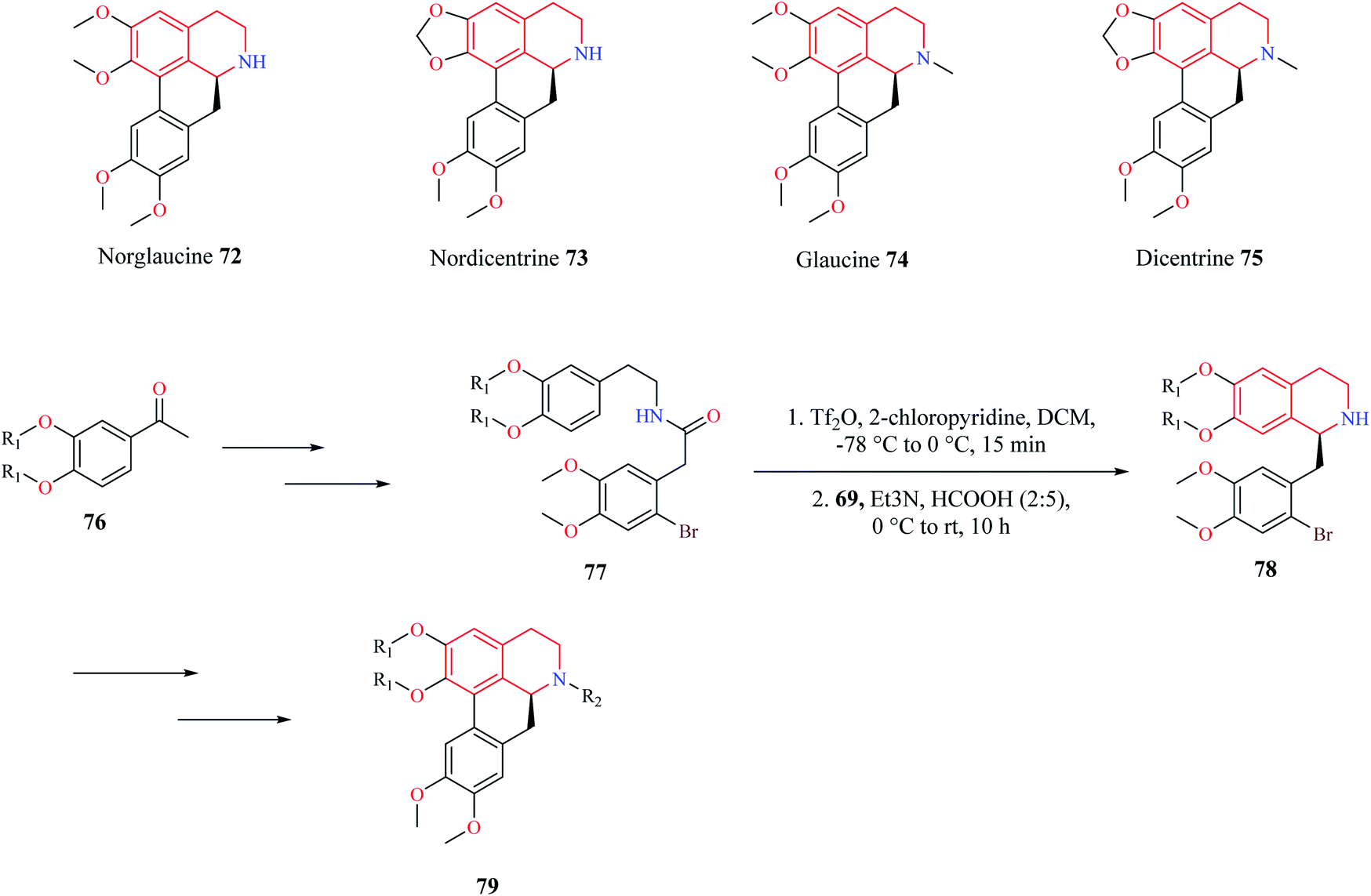
A similar synthetic approach comprising of Bischler–Nepieralski cyclization and Noyori ATH was used by Pieper and co-workers for the synthesis of enantioselective aporphine natural products 72–75 (Scheme 12).20 The precursor compound 77 required for Bischler–Nepieralski cyclization was obtained from 3,4-disubstituted acetophenone 76 using a sequence of reactions. Then intermediate compound 77 was treated with trifluoromethanesulfonic anhydride in the presence of 2-chloropyridine at low temperature to obtain the dihydro isoquinoline, which was then reduced using Noyori ATH to obtain the 1-substituted THIQ compound 78. The THIQ compound 78 was then converted into the final compound 79.
 | ||
| Scheme 12 Synthesis of aporphine natural products. | ||
2.3 Multicomponent reactions (MCR)
Multicomponent reactions (MCR) is a powerful strategy that has been routinely used for the construction of structurally diverse chemical compounds. MCR reactions have been used for the synthesis of compounds that have been bestowed with biological activity such as anticancer, antimicrobial, among others.21–23Rong et al. utilized a MCR comprising of aromatic aldehydes 80, N-methyl piperidin-4-one 81, and malononitrile 82 for the synthesis of 6-amino-8-aryl-2-methyl-1,2,3,4-tetrahydroisoquinoline-5,7-dicarbonitriles 83 under solvent-free conditions (Scheme 13).24 Almost 10 THIQ derivatives were synthesized with yields ranging from 85–97%.
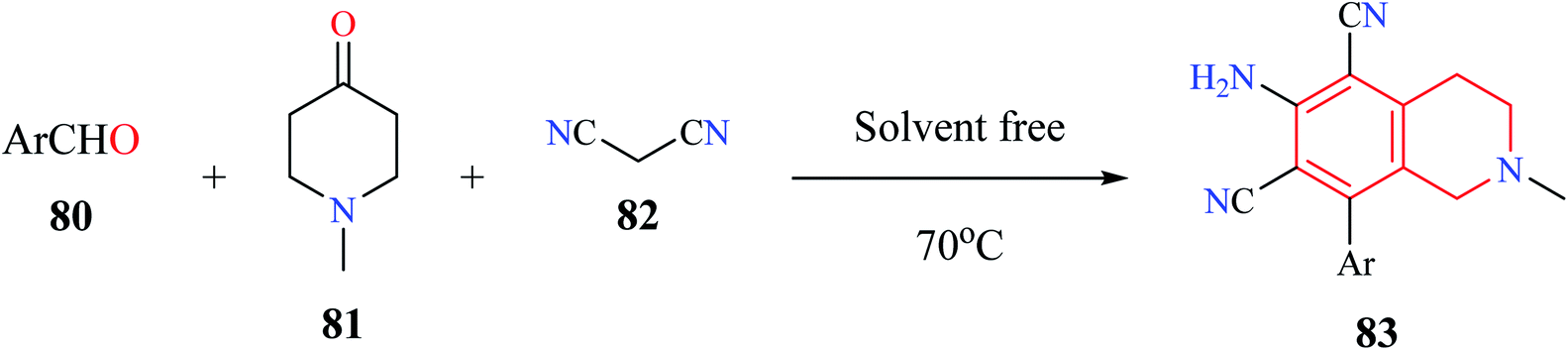 | ||
| Scheme 13 Synthesis of compound 83. | ||
A simple and efficient one-pot synthesis of novel N-alkyl substituted-6-amino-7-nitro-8-aryl-1,2,3,4-tetrahydroisoquinoline-5-carbonitrile derivatives 87 were synthesized using a MCR comprising of 1-alkylpiperidin-4-one 84, malononitrile 85, and β-nitro styrene 86 (Scheme 14).25 The reactions were hypothesized to proceed sequentially via Knoevenagel condensation, Michael addition, Thorpe–Ziegler cyclization, and air-promoted dehydrogenation processes.
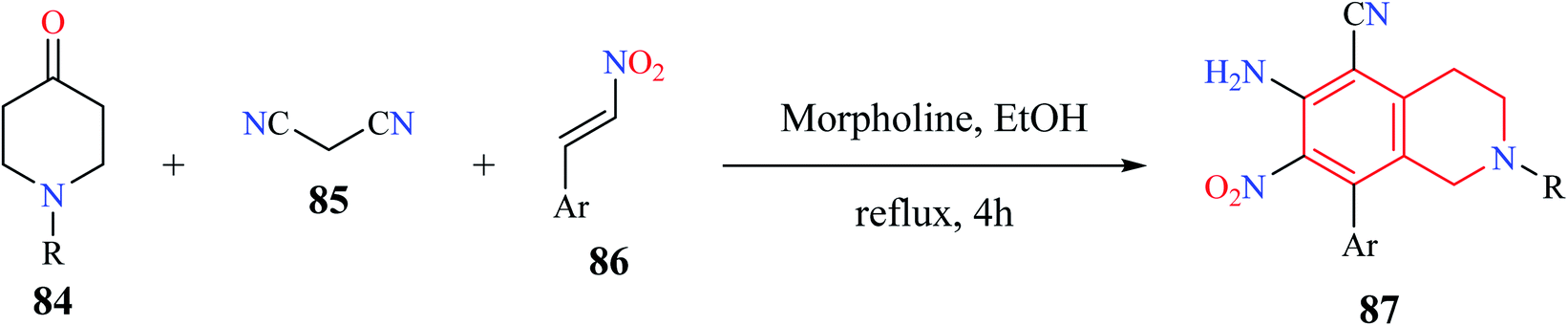 | ||
| Scheme 14 Synthesis of compound 87. | ||
A one-pot, three-component synthesis comprising of 2-bromophenethylsulfonamide 88, acryloyl chloride 89, and primary or secondary amine 90 was used for the synthesis of functionalized THIQ derivatives 91 via domino Heck–aza-Michael reactions (Scheme 15).26 Highly functionalized derivatives of THIQ were synthesized using this domino reaction with moderate to excellent yields (28–97%).
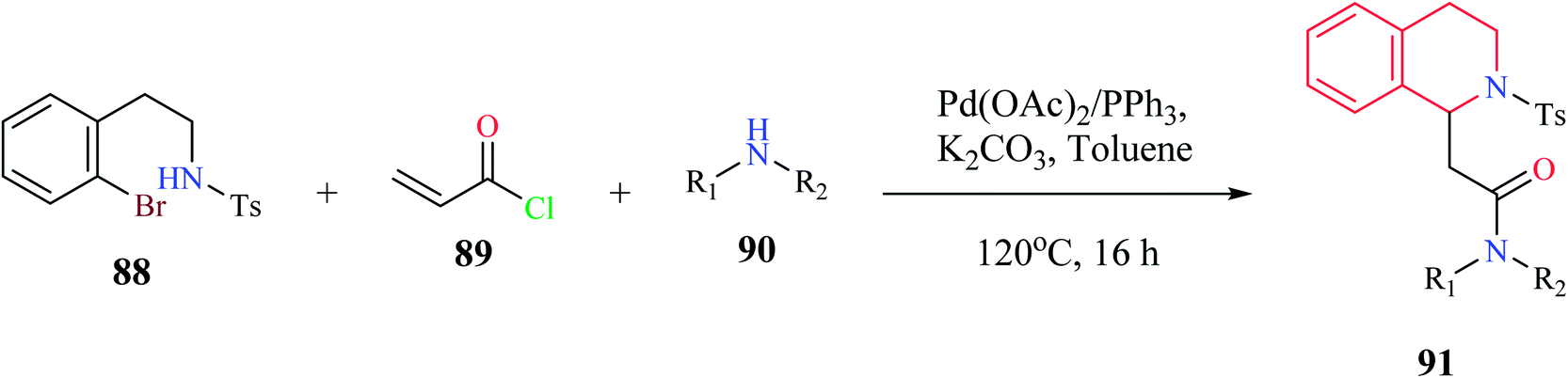 | ||
| Scheme 15 Synthesis of compound 91. | ||
A one-pot, four-component (92–95) enantioselective synthesis of 1,3,4-substituted THIQ analogs 97 was reported by Jiang and co-workers (Scheme 16).27 The cascade reaction comprising of Mannich–aza-Michael reaction was developed using a synergistic catalytic system consisting of ruthenium complex with a chiral Brønsted acid 96. Almost 15 THIQ derivatives were synthesized using the optimized procedure with good yields and enantiomeric purity.
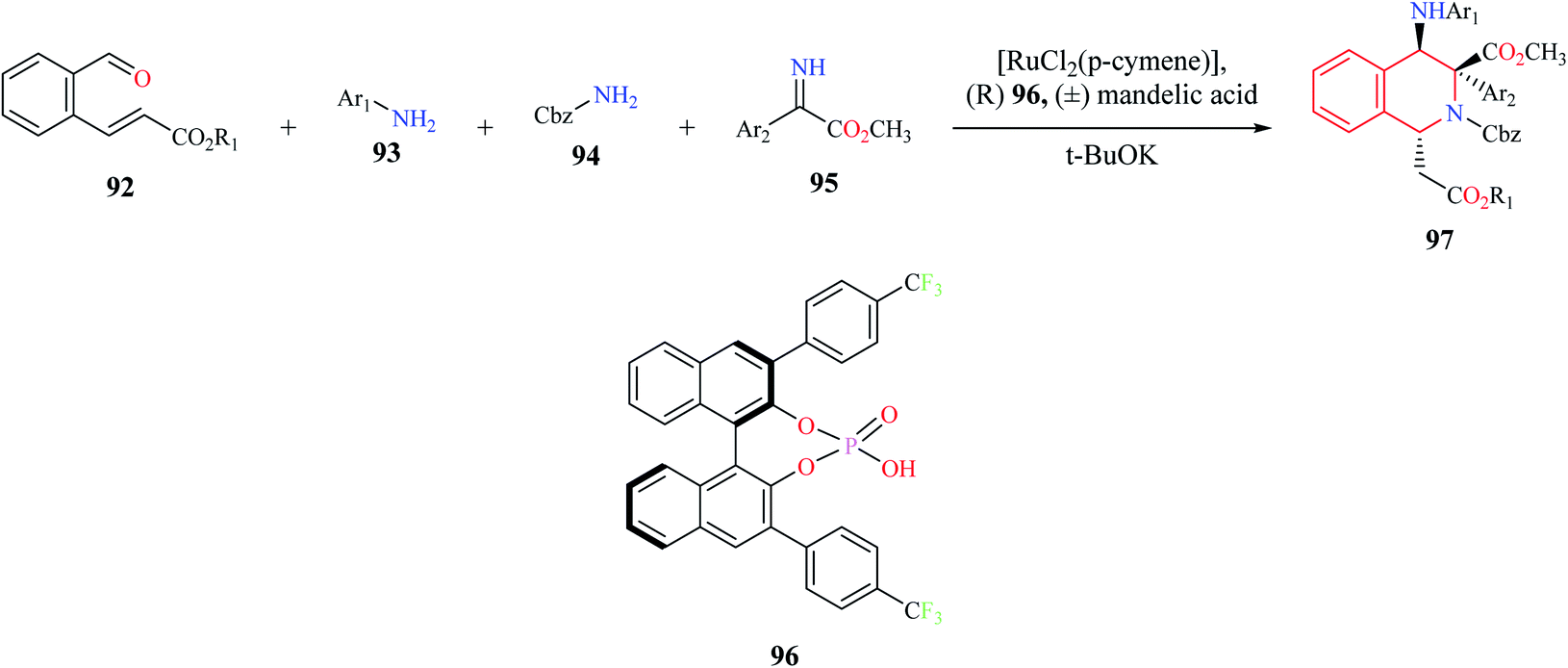 | ||
| Scheme 16 Enantioselective synthesis of compound 97. | ||
A one-pot, MCR comprising of 2-(2-bromoethyl)-benzaldehyde 98, isocyanide 99, amine 100, and azide 101 was used for the synthesis of tetrazolyl-THIQ derivatives 102 under catalyst-free conditions (Scheme 17).28 The optimized synthetic methodology exhibited a wide substrate scope with excellent yields (up to 99%). The reaction was postulated to proceed sequentially via an intramolecular cyclization/Ugi-azide reaction.
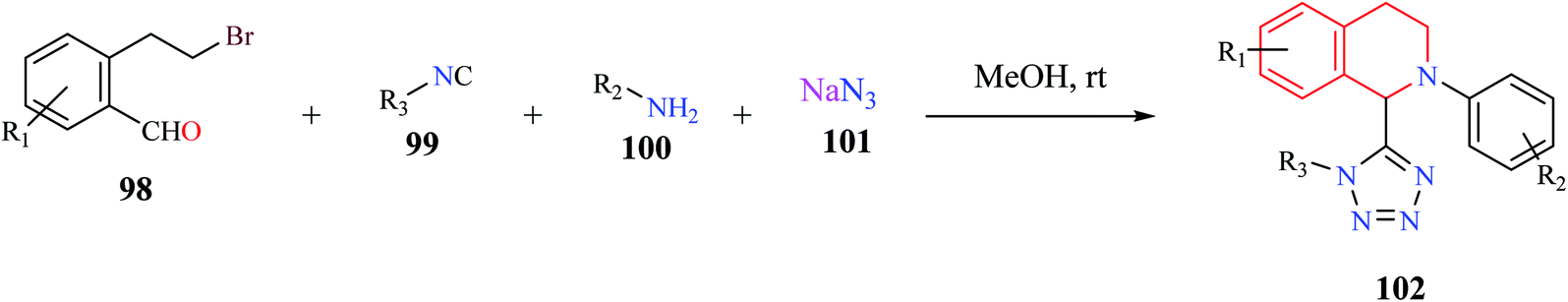 | ||
| Scheme 17 Synthesis of tetrazolyl-THIQ analogs. | ||
2.4 Intramolecular hydroamination reaction
Henderson et al. reported the synthesis of THIQ derivatives via acid-catalyzed intramolecular hydroamination reaction from 2-aminoethyl styrene derivatives 103 which afforded good to excellent yields of the corresponding tetrahydroisoquinolines 104 (Scheme 18).29 This approach represents an alternative strategy to the classical Pictet–Spengler method for the syntheses of THIQ compounds.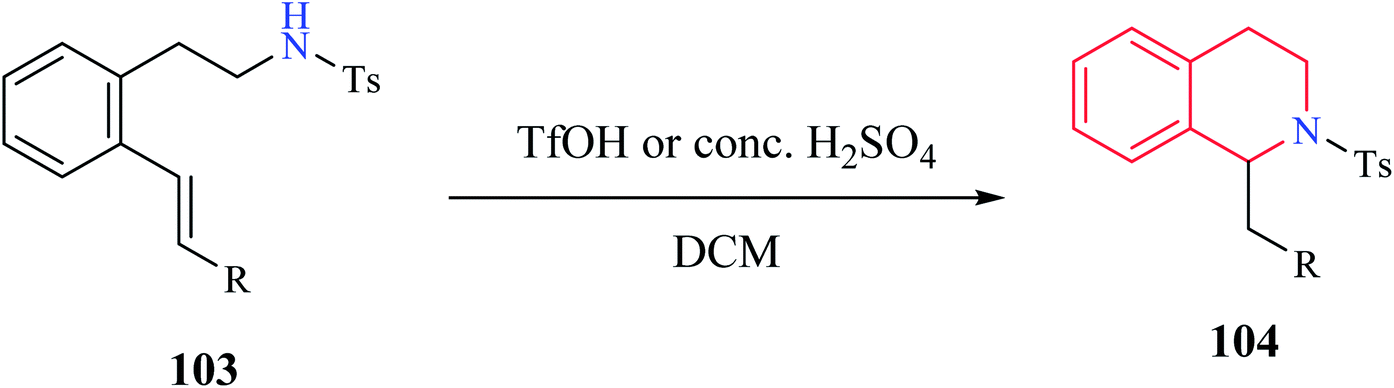 | ||
| Scheme 18 Synthesis of compound 104. | ||
Different catalytic systems have been reported for the conversion of amino alkenes to corresponding THIQs via intramolecular hydroamination reaction. Ogata et al. reported the total synthesis of (s)-laudanosine which involved a key asymmetric intramolecular hydroamination reaction (Scheme 19).30 The key intermediate 107 was obtained from 2-bromo-4,5-dimethoxy benzaldehyde 105 and 4-ethenyl-1,2-dimethoxy benzene 106 using a sequence of reactions. The intermediate compound 107 was then subjected to chiral bisoxazoline 108–lithium diisopropylamide (LDA)-catalyzed asymmetric intramolecular hydroamination to obtain (s)-laudanosine 109.
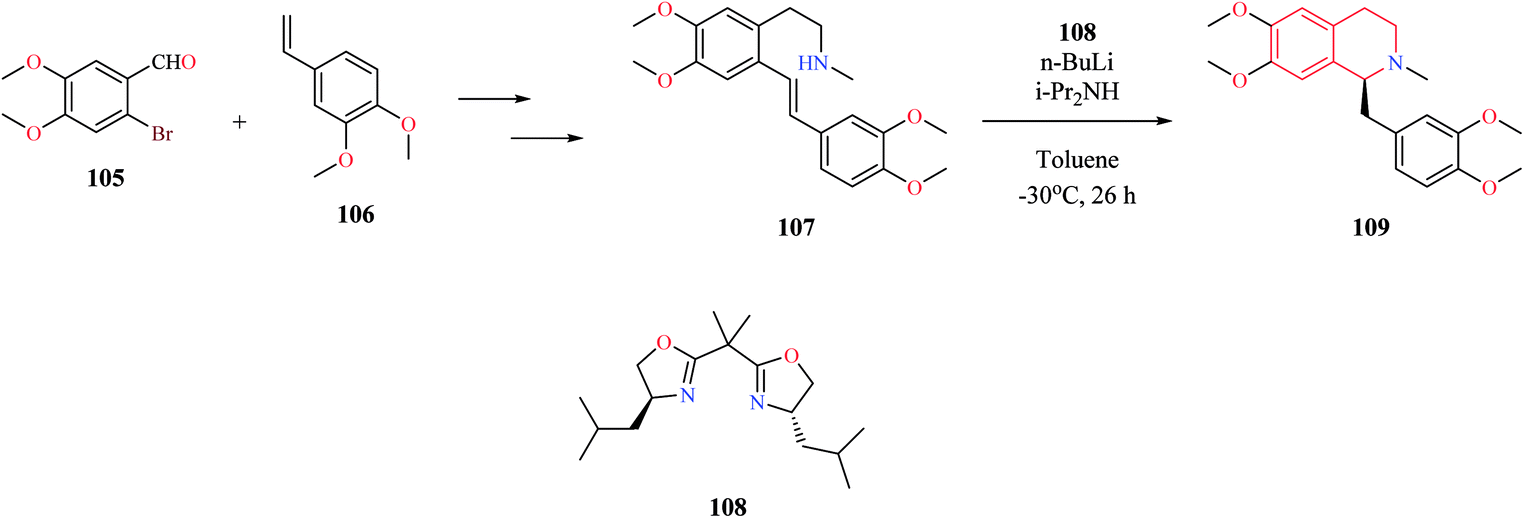 | ||
| Scheme 19 Synthesis of (s)-laudanosine 109. | ||
Tussing and co-workers reported a borane-catalyzed THIQ synthesis via intramolecular hydroamination (Scheme 20).31 A sequential reaction comprising of tris-(pentafluorophenyl)-borane 110 catalyzed hydroamination and hydrogenation reaction was used for the conversion of amino alkyne 111 to the corresponding THIQ compound 112 in 61% yield.
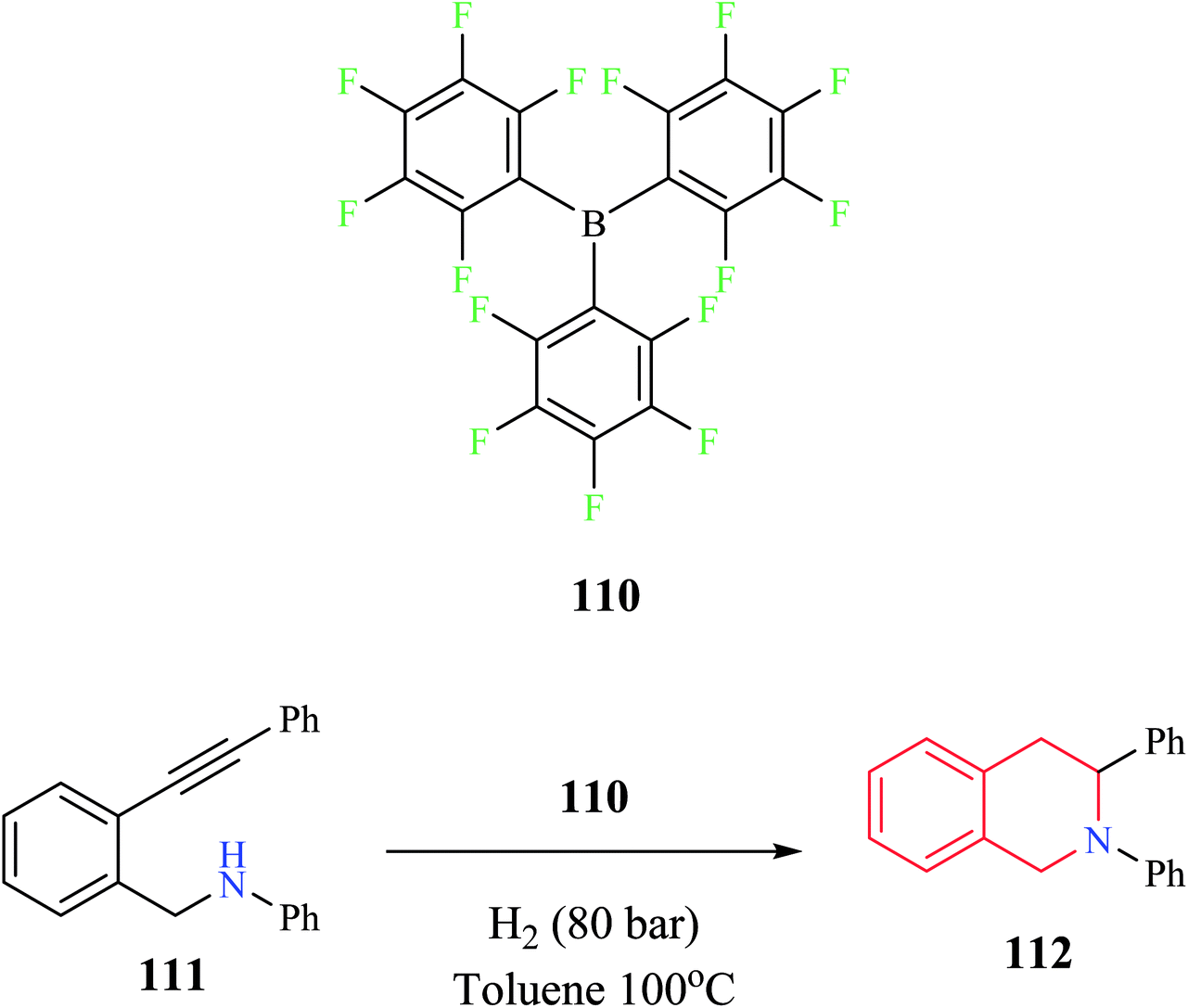 | ||
| Scheme 20 Borane-catalyzed intramolecular hydroamination reaction for the synthesis of compound 112. | ||
A convenient gold(III) chloride catalyzed intramolecular hydroamination for the synthesis of 1-alkyl-3-diethoxyphosphoryl-THIQ analog was reported by Murashkina and co-workers (Scheme 21).32 Alkynyl substituted α-aminophosphonates 113 was first subjected to AuCl3 catalyzed hydroamination reaction, which was then followed by reduction with NaBH4 to give the THIQ analogs 114 with good to excellent yields.
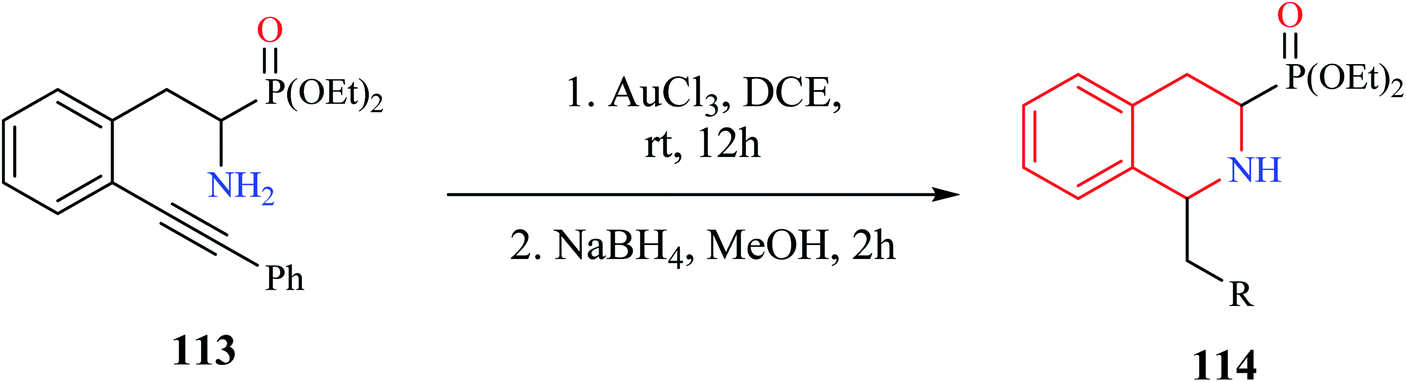 | ||
| Scheme 21 Gold catalyzed intramolecular hydroamination reaction for the synthesis of compound 114. | ||
Dai and co-workers reported an enantioselective copper-catalyzed intramolecular hydroamination reaction for the preparation of THIQ analog (Scheme 22).33 The amino alkene 115 was converted to enantiomerically pure compound 117 with the aid of Cu(OAc)2 and a chiral catalyst (R,R)-Ph-BPE 116.
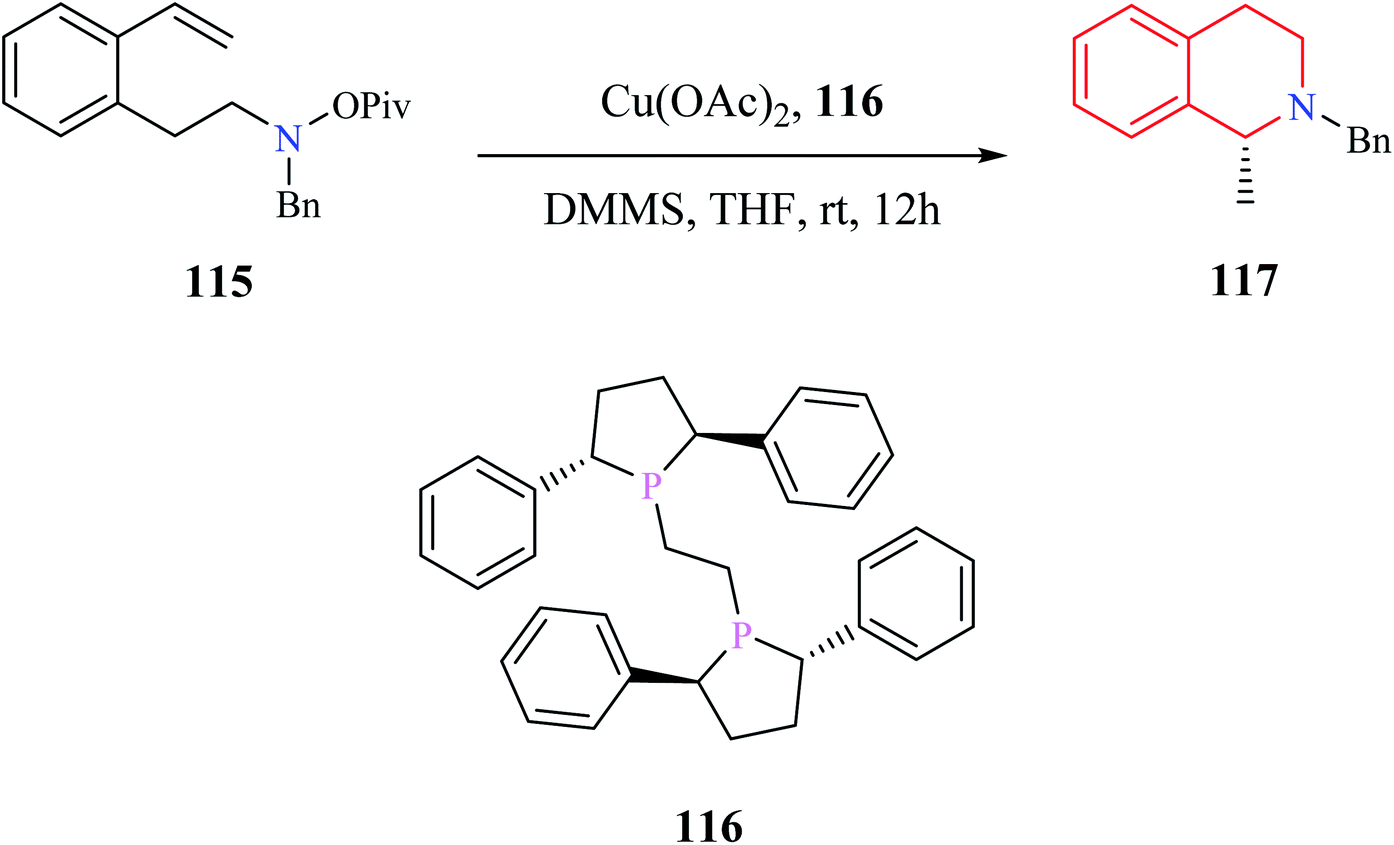 | ||
| Scheme 22 Enantioselective copper-catalyzed intramolecular hydroamination reaction for the synthesis of compound 117. | ||
Pictet–Spengler condensation and Bischler–Nepieralski reaction are some of the routinely used synthetic strategies for the construction of THIQ core. However, they often involve multiple steps and are restricted to substitution at the 1st position of the THIQ nucleus. On the other hand, MCR provides an exciting opportunity to generate diverse libraries of THIQs in one-pot and atom economic synthetic protocols.
3. Biological activity
3.1 Anti-bacterial activity
Guzman et al. synthesized three series (A, B, C) of THIQ analogs to elucidate a common anti-TB pharmacophore.34 The synthesized analogs were evaluated against two mycobacterium species – Mycobacterium bovis BCG and M. tuberculosis H37Rv. Most of the compounds in the A-series were found to be inactive except compound 118 which exhibited moderate potency against both the mycobacterium species. Compounds 119 and 120 were found to be the most potent from the B series. The third series of compounds were designed and synthesized based on the structure of active compound 120. 121–123 were some of the compounds that were found to be active in this series. Moreover, the active compounds from each of the three series were evaluated for the inhibitory property against MurE synthetase, a crucial enzyme in peptidoglycan biosynthesis. Compounds 119, 120, 122 exhibited effective MurE inhibition. However, the correlation between the MurE inhibition and phenotypic inhibitory effects was low, indicating another possible mechanism of action. The SAR is discussed in Fig. 2.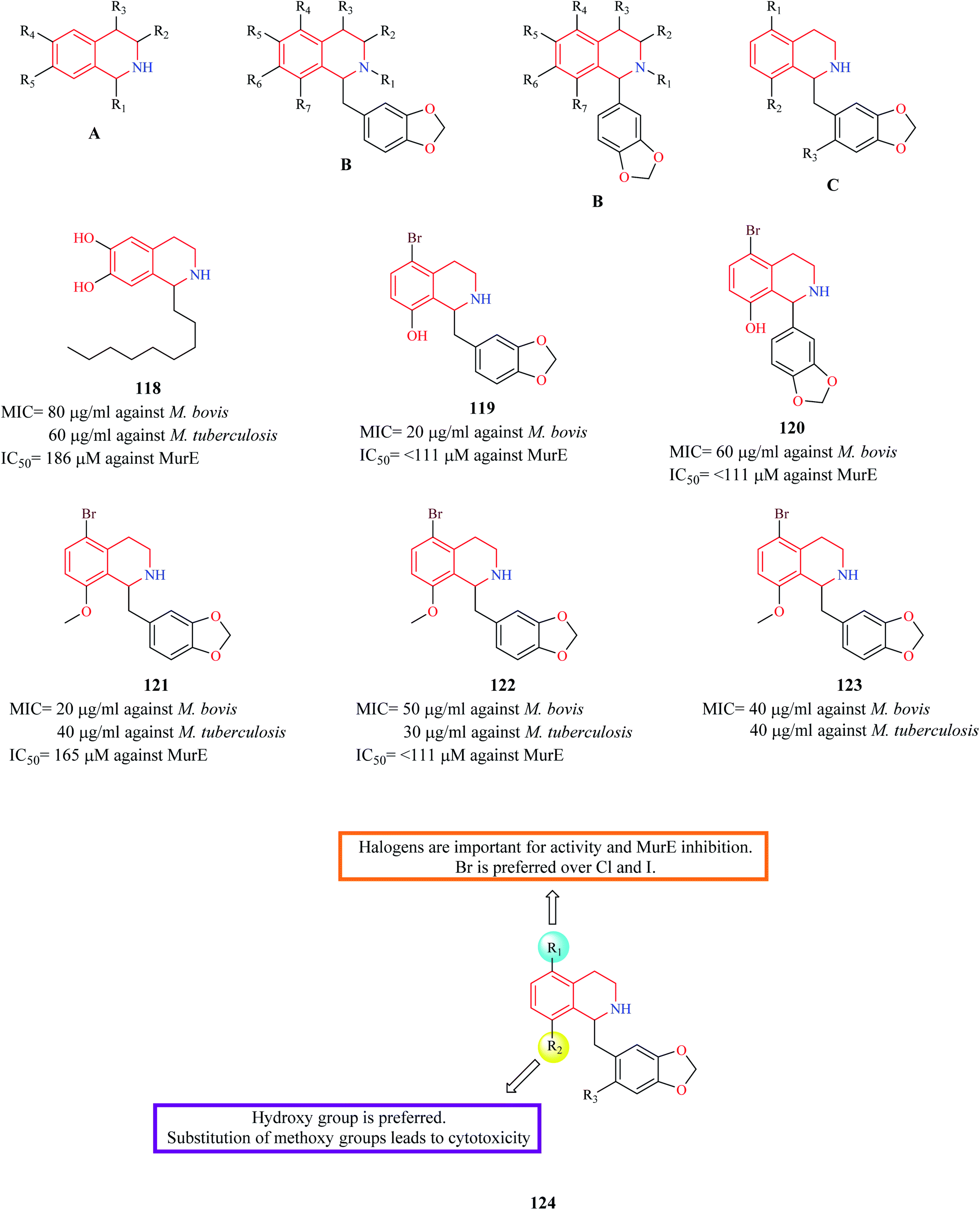 | ||
| Fig. 2 THIQ analogs 118–123 as anti-bacterial agents. | ||
Farha et al. designed and synthesized analogs of ticlopidine 125 and clopidogrel 126 (Fig. 3), anti-platelet drugs used for cardiac disorders.35 Anti-platelet drugs ticlopidine and clopidogrel have been shown to potentiate the activity of beta-lactam antibiotics against methicillin-resistant Staphylococcus aureus (MRSA) by inhibiting TarO, the first enzyme in the synthesis of wall teichoic acids (WTA). Ticlopidine being a prodrug is extensively metabolized to its anti-platelet metabolite leaving only traces of the intact drug in the plasma. Hence, the thiophene ring of ticlopidine as wells as its analog clopidogrel was replaced with a phenyl ring to generate compounds 127 and 128 containing the THIQ nucleus. Several derivatives were designed and synthesized based on compounds 127 and 128 and they were evaluated for their synergism with cefuroxime against MRSA. Compound 129 exhibited maximum synergism with cefuroxime.
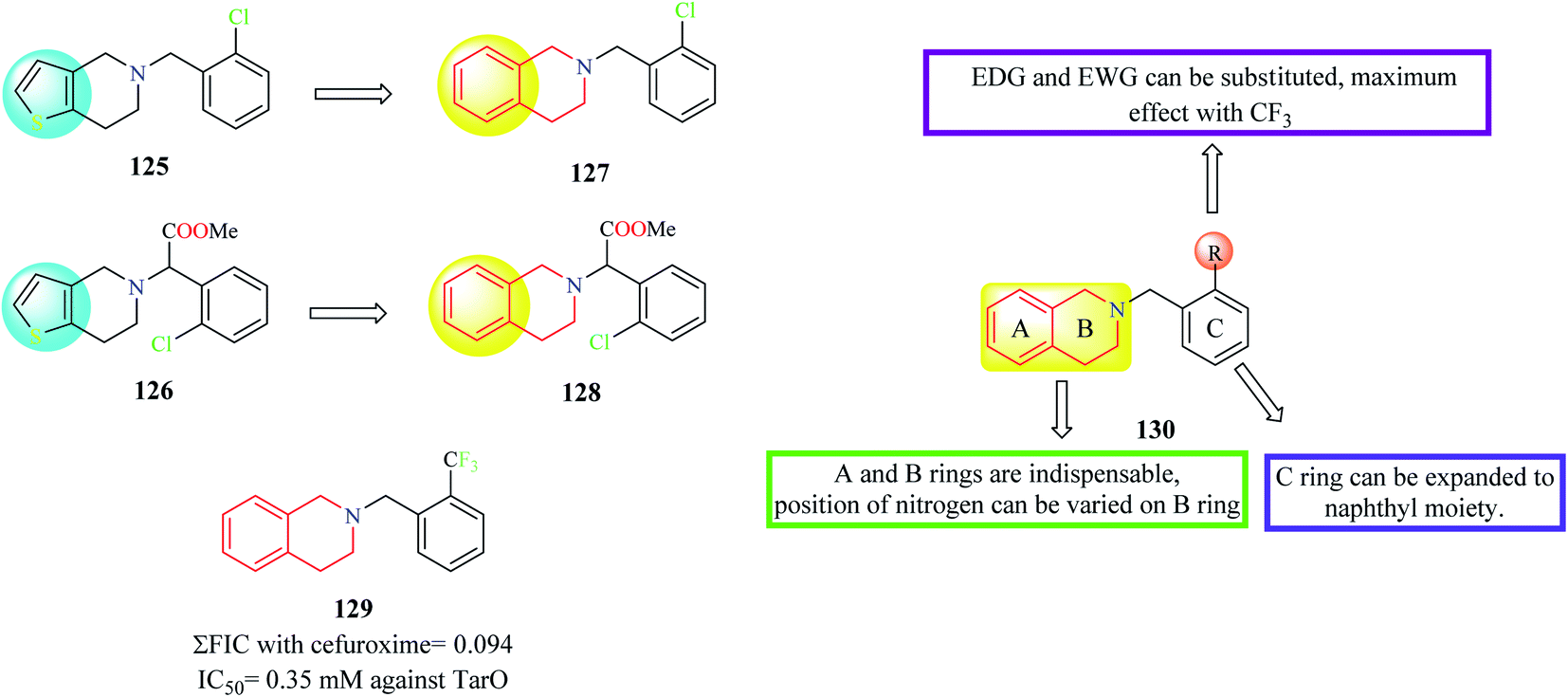 | ||
| Fig. 3 SAR profile of ticlopidine and clopidogrel analogs. | ||
A novel compound (E)-2-benzyl-3-(furan-3-yl)-6,7-dimethoxy-4-(2-phenyl-1H-inden-1-ylidene)-1,2,3,4-tetrahydroisoquinoline 131 (Fig. 4) was synthesized and evaluated for its antibacterial property against eight pathogenic bacterial strains.36 The strains that were most susceptible to the action of compound 131 at 25 μg ml−1 were Staphylococcus epidermidis and Klebsiella pneumonia.
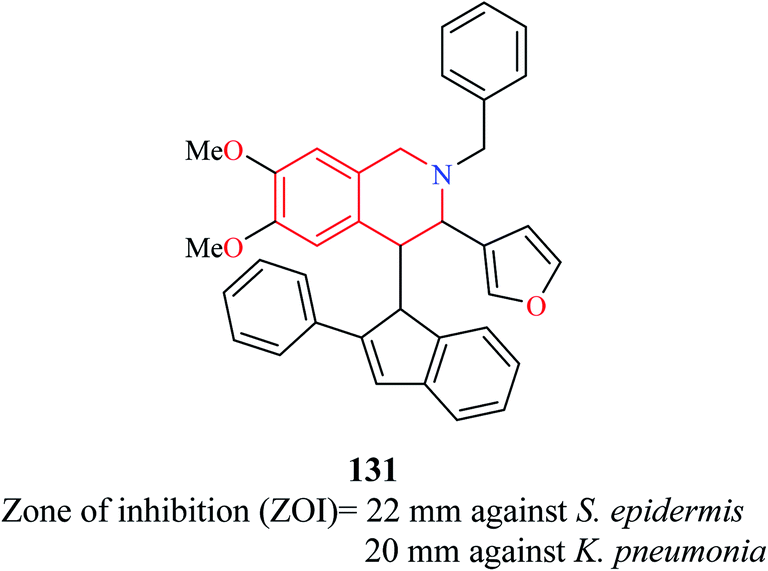 | ||
| Fig. 4 Structure of compound 131. | ||
Novel derivatives of THIQ: chiral quaternary N-spiro ammonium bromides containing 3′,4′-dihydro-1′H-spiro-[isoindoline-2,2′-isoquinoline] were evaluated for their bacteriostatic and bactericidal property against nine pathogenic bacterial strains.37 Most of the analogs were more potent against Gram-negative bacterial strains. Compounds 132–135 (Fig. 5) were found to be more potent than control norfloxacin against Campylobacter jejuni. Compound 136 was the most promising compound as it possessed more potent antibacterial property than ciprofloxacin against Streptococcus mutans and Bacillus subtilis.
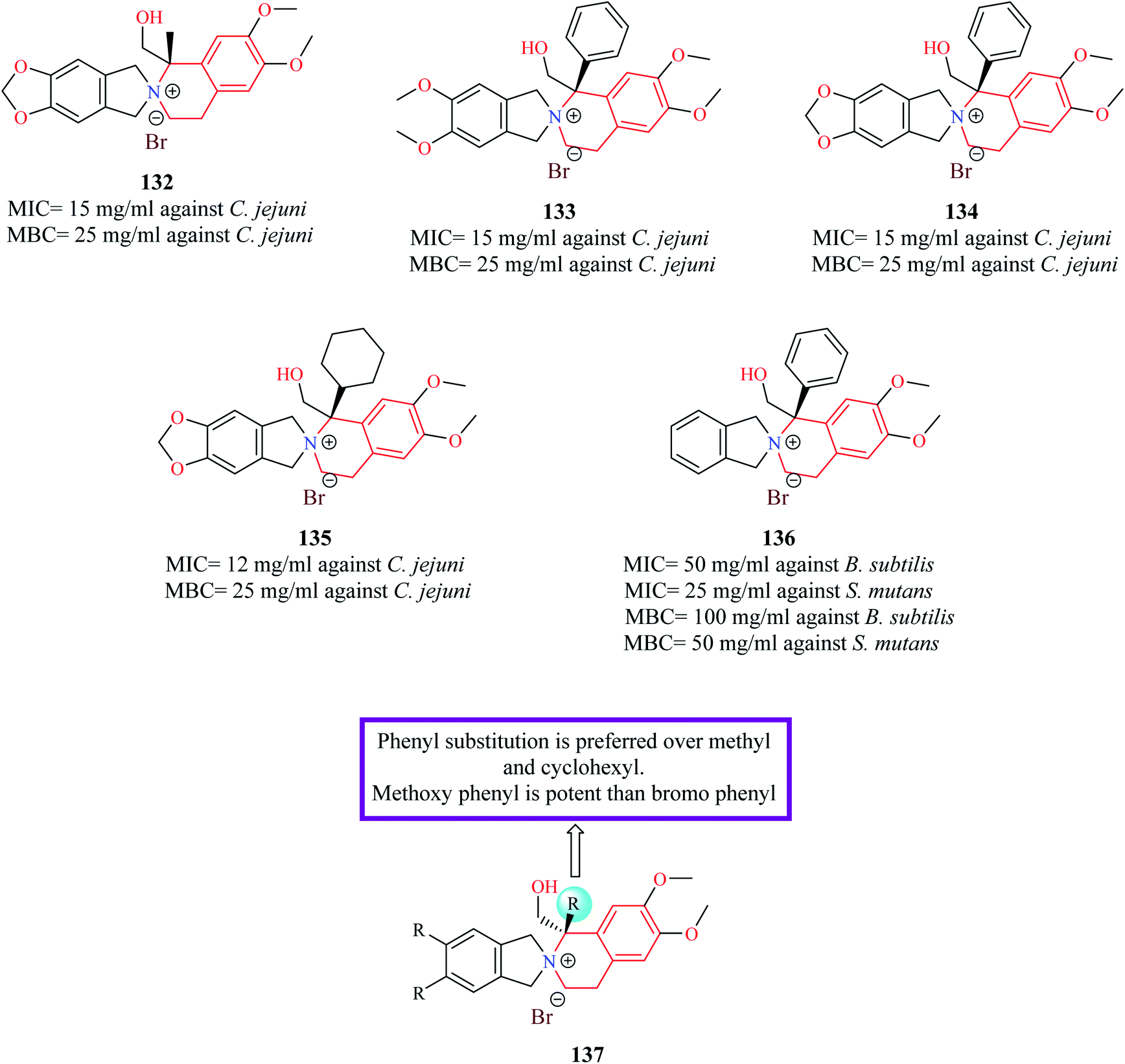 | ||
| Fig. 5 Structure of THIQ analogs 132–136 as anti-bacterial agents. | ||
Novel THIQ analogs containing lipid-like choline moiety were synthesized and evaluated for their antibacterial properties by Zablotskaya et al. Incorporation of these lipid-like substituents may improve the absorption properties of the compounds. Compounds 138 and 139 (Fig. 6) were some of the analogs that exhibited good antibacterial activity. Compound 138 was more active against Gram-positive bacteria's whereas compound 139 was superior against Gram-negative bacterial species. Moreover, these compounds also possessed inhibitory property against DNA gyrase, a vital enzyme that is involved in DNA topology.38 Similarly, organosilicon lipid-like derivatives of THIQ analogs were also reported by Zablotskaya et al. Compounds 140 and 141 were found to exhibit good antibacterial activity along with DNA gyrase inhibitory action.39
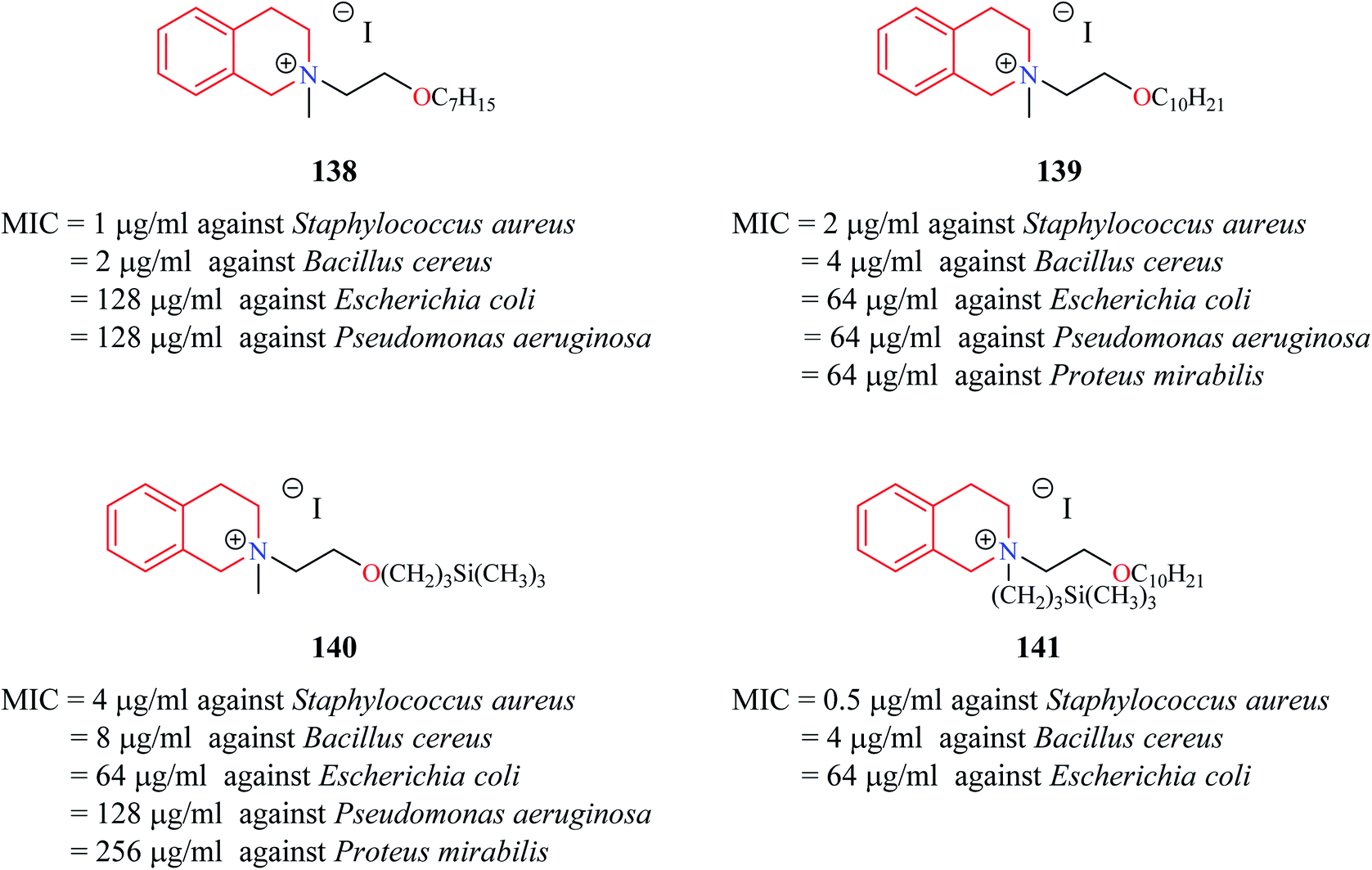 | ||
| Fig. 6 Structure of compounds 138–141 as anti-bacterial agents. | ||
A series of 5,8-disubstituted THIQ analogs were synthesized by Lu et al. The synthesized analogs were evaluated for their anti-mycobacterial property.40 Compounds 142 and 143 (Fig. 7) were some of the potent analogs against Mycobacterium tuberculosis. Compound 143 also exhibited good clearance property. Moreover, it was also found to exhibit potent inhibitory activity against mycobacterial (M. smegmatis) ATP synthetase enzyme with an IC50 value of 1.8 μg ml−1 with about 9-fold selectivity over the human counterpart. The SAR of these groups of analogs is summarized in Fig. 7.
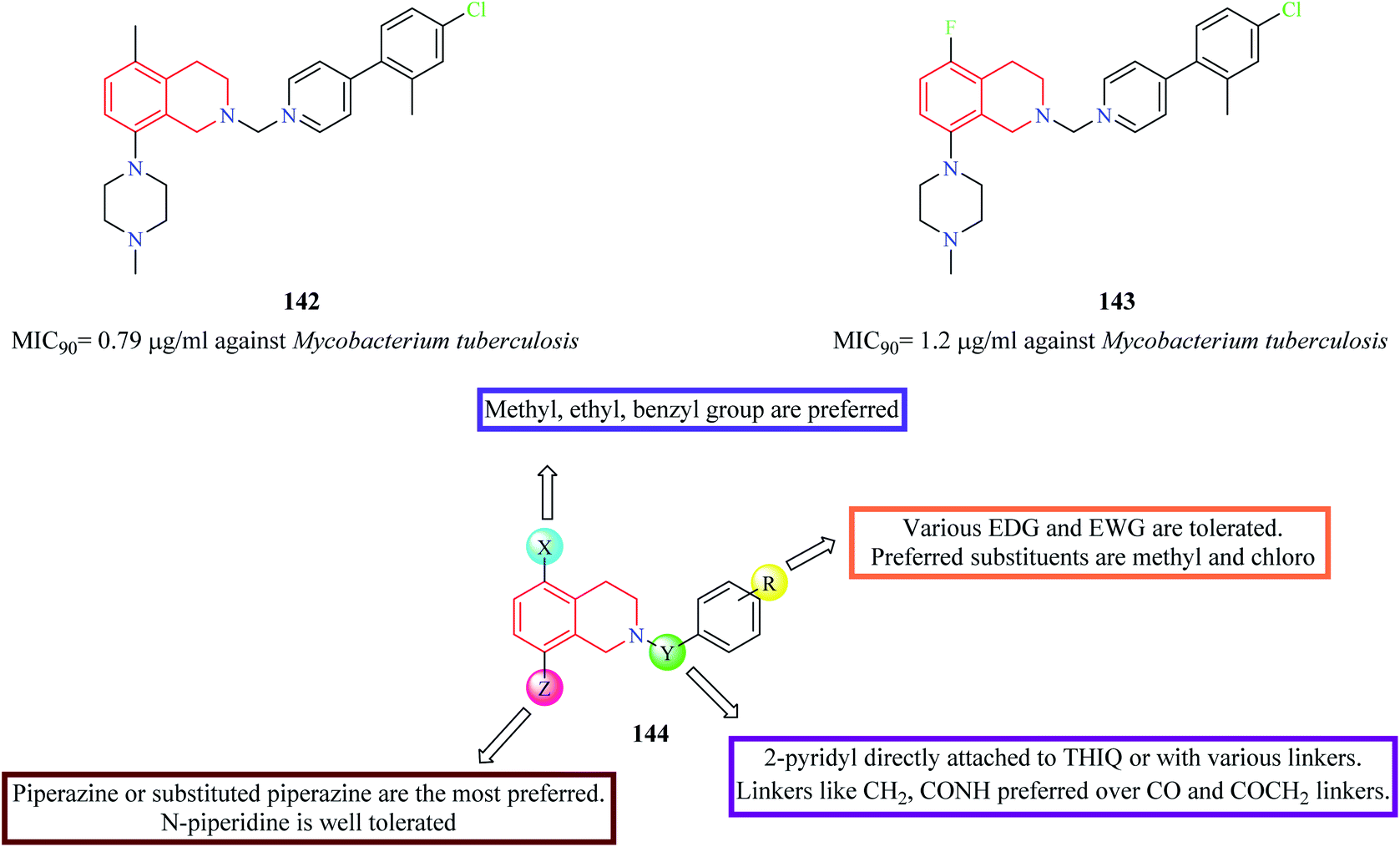 | ||
| Fig. 7 SAR profile of 5,8-disubstituted THIQ analogs. | ||
3.2 Anti-fungal activity
Novel N-substituted THIQ analogs were synthesized and evaluated for their antifungal activity.41 Initially, these analogs were evaluated against four fungi species and their zone of inhibition was determined. Compounds 145 and 146 (Fig. 8) produced a comparable zone of inhibition against Candida glabrata as that of the standard drug clotrimazole. Their MIC values were determined against different fungal species. Compound 145 exhibited the most potent activity against Saccharomyces cerevisiae (MIC = 1 μg ml−1) whereas compound 146 was more potent against Yarrowia lipolytica with a MIC value of 2.5 μg ml−1. Furthermore, these compounds were shown to interfere with the ergosterol biosynthetic pathway. Specifically, these compounds inhibited delta-8,7-isomerase.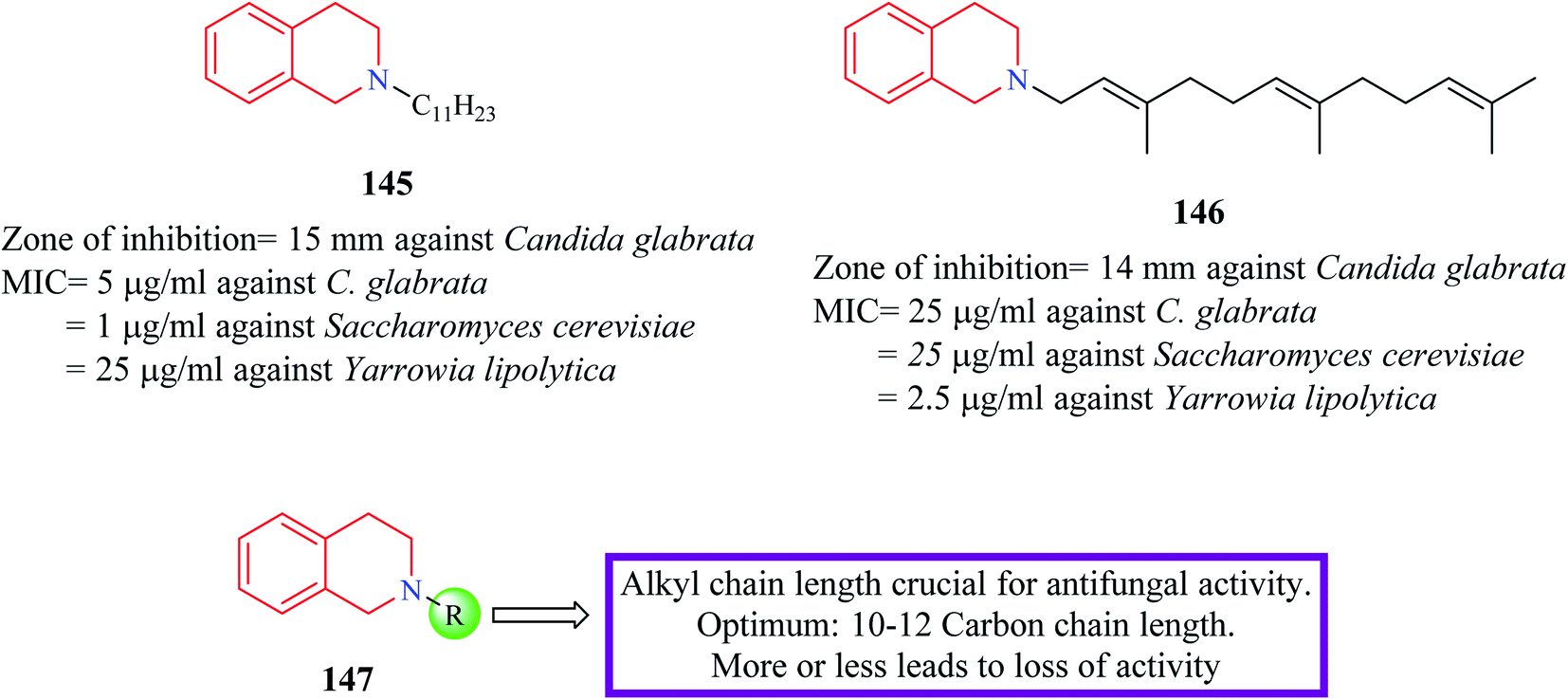 | ||
| Fig. 8 SAR profile of N-substituted THIQ analogs along with their anti-fungal activity. | ||
The antifungal potency of novel analogs of the pyrrolo-THIQ fused system was reported by Sutariya and co-workers.42 Compound 148 and its stereoisomer compound 149 (Fig. 9) exhibited equipotent activity as that of nystatin against pathogenic fungi C. albicans. Two bis (THIQ) derivatives of undecane 150 and 151 were evaluated for their antifungal activity against C. albicans.43 Compound 150 (zone of inhibition = 20 mm) exhibited better antifungal activity than nystatin (zone of inhibition = 15 mm). Zablotskaya and co-workers also evaluated compounds 138–141 for their antifungal property.38,39 They exhibited potent antifungal activity as that of the standard drug fluconazole.
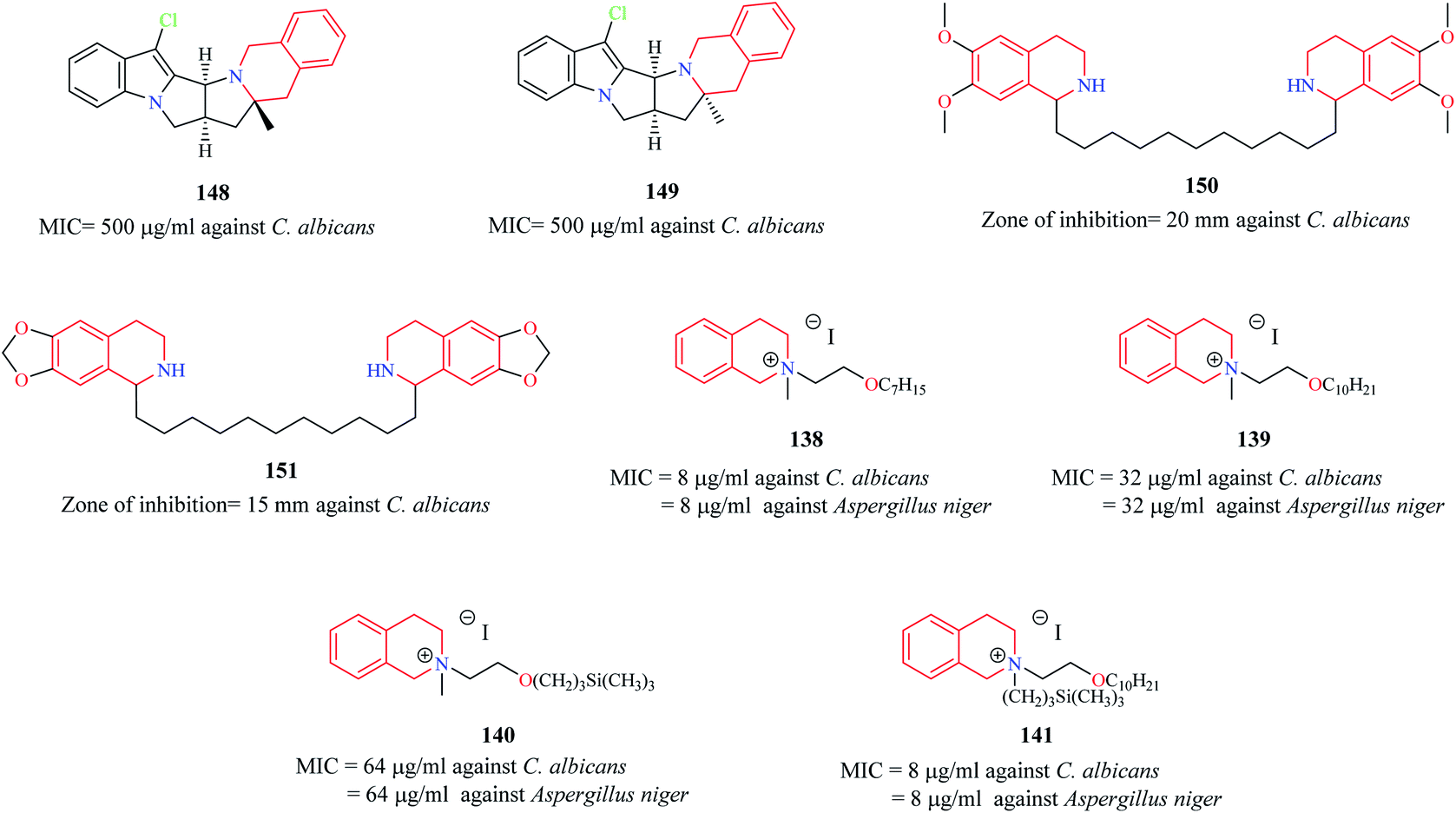 | ||
| Fig. 9 Structure of anti-fungal THIQ analogs 148–151. | ||
3.3 Anti-viral activity
Murugesan et al. synthesized novel THIQ analogs and evaluated their Human immunodeficiency virus-1 (HIV-1) reverse transcriptase (RT) inhibitory property.44 Compounds 152 and 153 exhibited moderate activity against HIV-1 RT. Molecular docking studies were performed to predict the binding pose of compounds 152 and 153 (Fig. 10). Docking studies indicated that these compounds adopted “butterfly-like” conformation like non-nucleoside reverse transcriptase inhibitors (NNRTIs) (e.g. nevirapine, efavirenz) within the binding pocket of RT. Three novel derivatives of THIQ analogs 154–156 as NNRTIs were shown to exhibit potent activity against the HIV RT.45 Both these analogs inhibited the polymerase activity of the RT in the nanomolar range. The interactions of compound 156 in the binding pocket of the RT are depicted in Fig. 11. Moreover, both the compounds also prevented HIV-induced cell death in vitro. Continuing their studies on identifying potent anti-HIV derivatives, Zhan and others synthesized 2-(3-(2-chlorophenyl)pyrazin-2-ylthio)-N-arylacetamide analogs via a structure-based bioisosterism approach and evaluated their anti-HIV activity.46 One of the derivatives 157 containing THIQ scaffold exhibited potent anti-HIV activity (IC50 = 4.10 μm).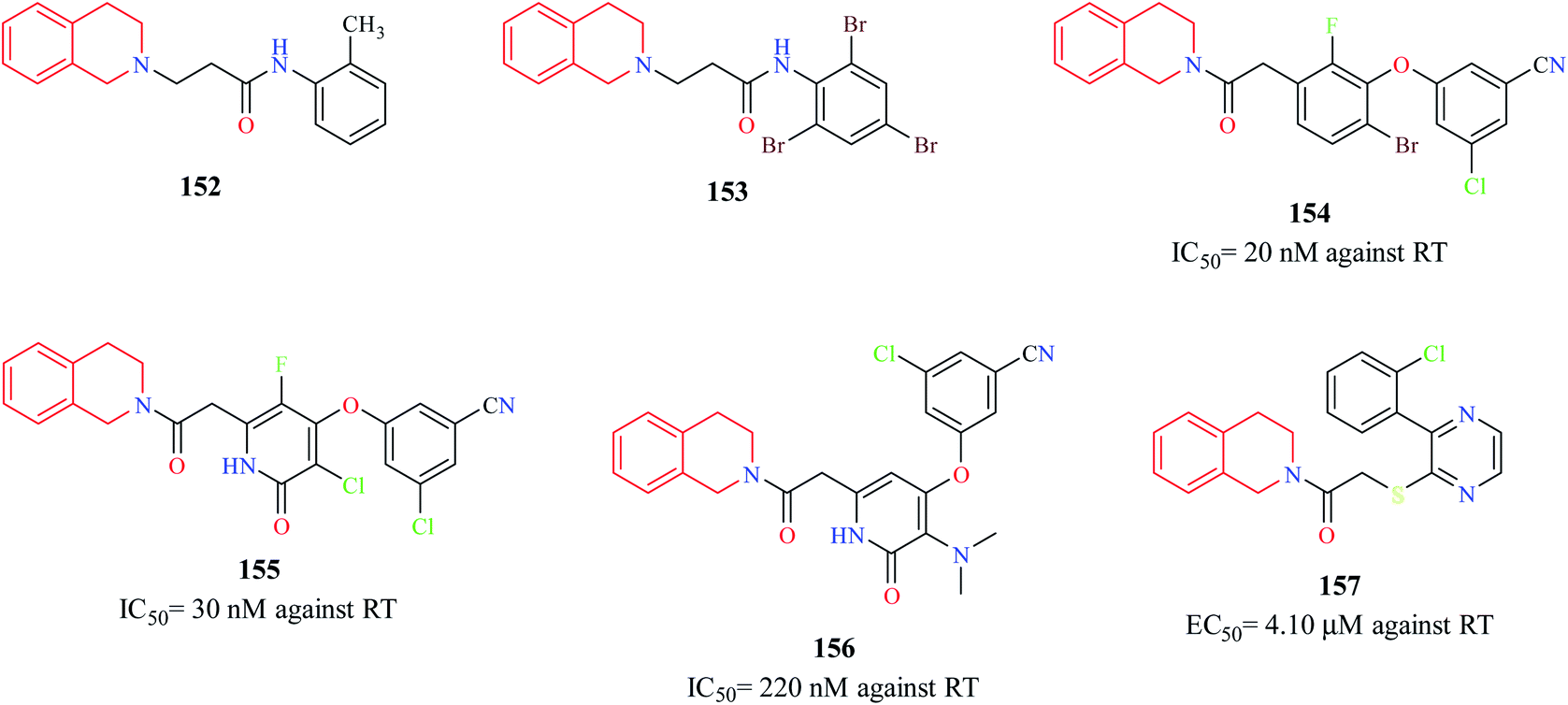 | ||
| Fig. 10 Structure of THIQ analogs 152–157 as NNRTIs. | ||
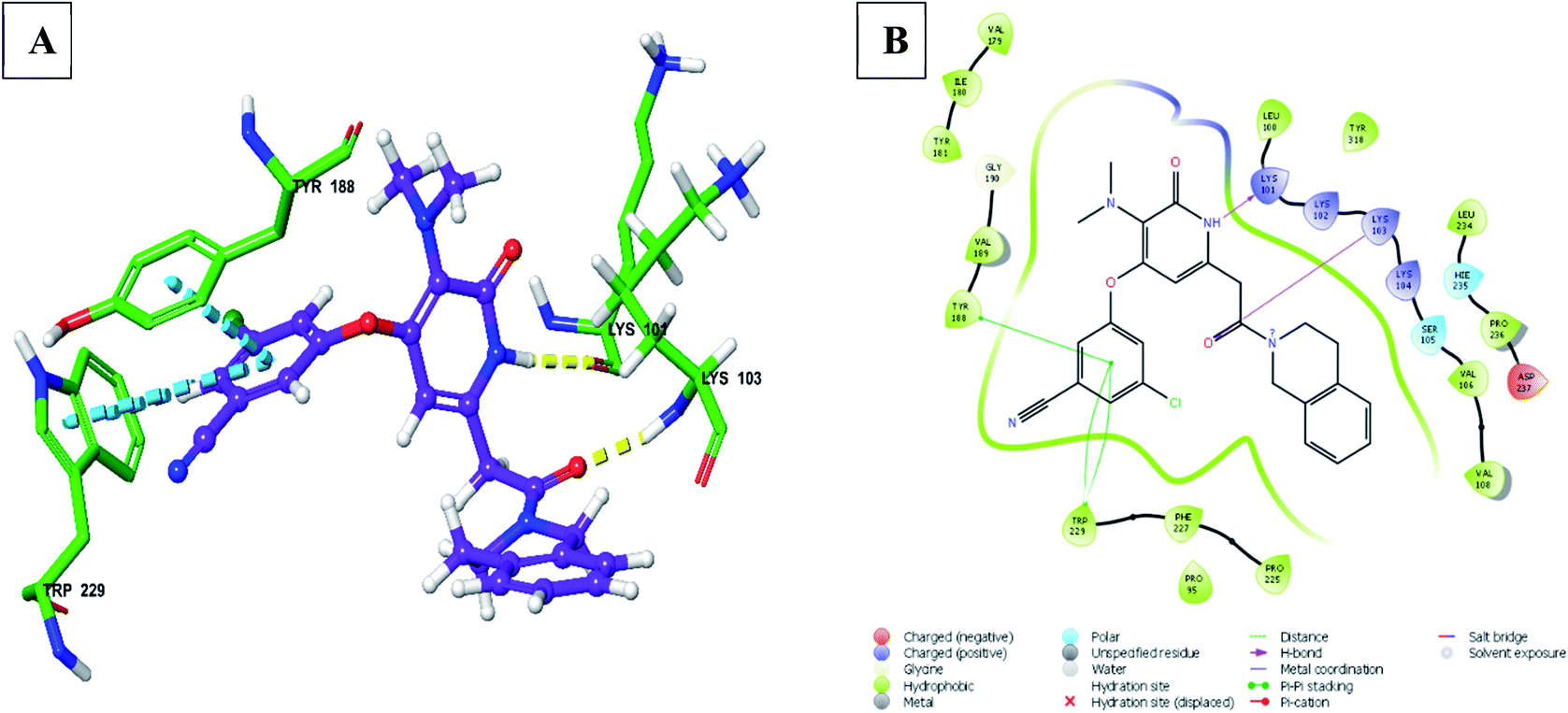 | ||
| Fig. 11 (A) 3D docked pose of compound 156 in the binding pocket of RT (yellow – hydrogen bond, blue – pi–pi interaction), (B) interaction of compound 156 with surrounding amino acid residues in 2D (PDB ID – 3FFI) (visualized using Maestro visualizer).47 | ||
Chander et al. designed and synthesized two series of THIQ analogs 158 and 159 (Fig. 12) as HIV-RT inhibitors.48 Thirty derivatives were synthesized based on compounds 158 and 159 and about eight analogs exhibited more than 50% inhibition at 100 μM. Compounds 160 and 61 were the most promising of them all with % RT inhibition values of 74.82% and 72.58% respectively. Molecular docking studies were also performed to shed light on the binding pattern of compounds 160 and 161 at the active site of the target HIV-1 RT. 6,7-Dimethoxy THIQ of both the analogs were found to exhibit hydrophobic contacts with Tyr-188, Tyr-181, and Trp-229. The nitrogen of acetamide exhibited hydrogen bond interaction with Lys-101, an active residue of RT. These interactions may be the reason for their significant in vitro activity.
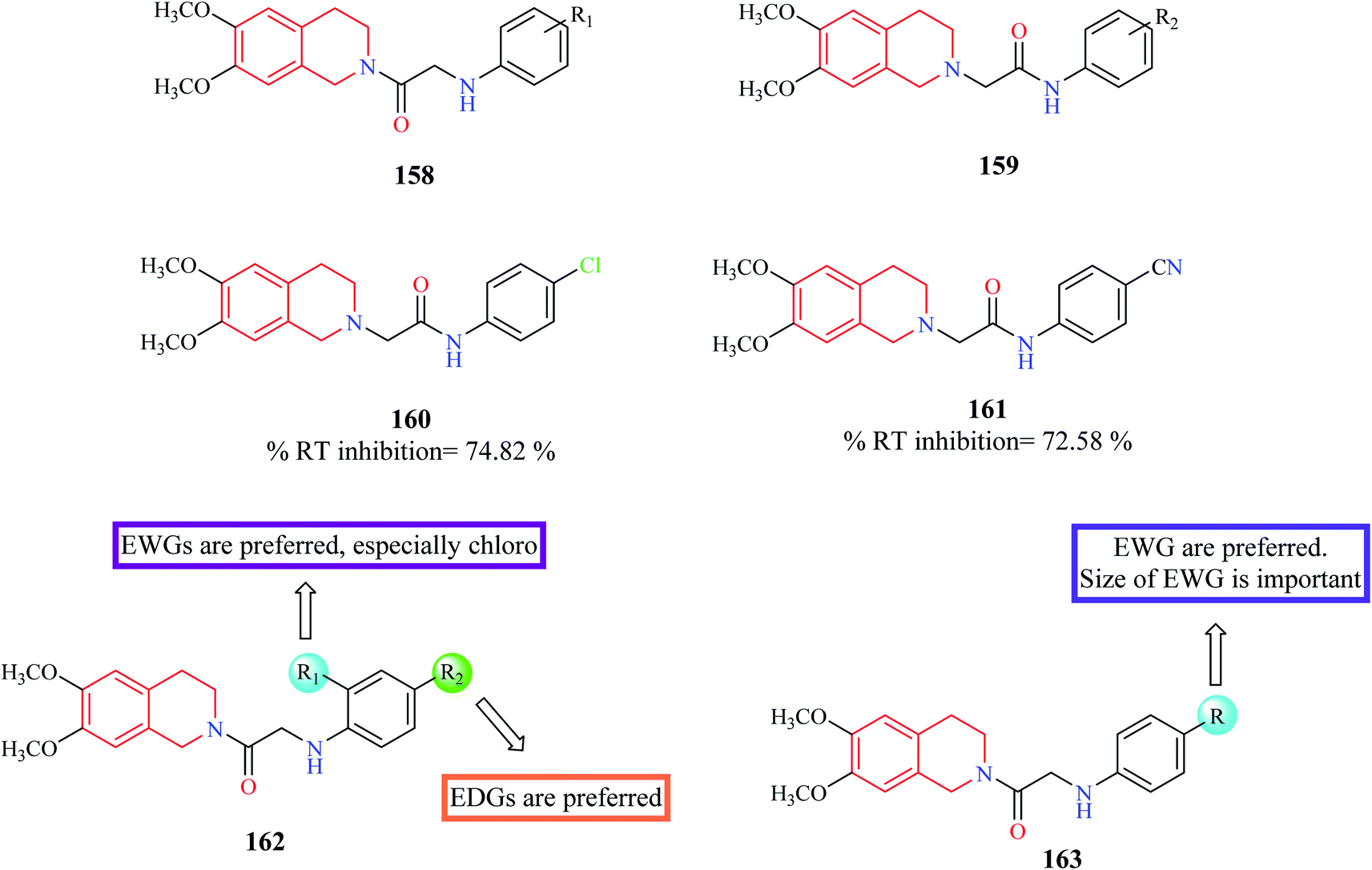 | ||
| Fig. 12 SAR profile of N-substituted THIQ analogs as RT inhibitors. | ||
Compound 164 (BMS-626529) (Fig. 13) is a potent HIV-1 attachment inhibitor. The prodrug form of compound 164, Fostemsavir 165 is FDA approved for the treatment of HIV. The benzamide moiety in the compound 164 was susceptible to in vivo metabolism. Therefore, Swidorski and co-workers made efforts to replace the piperazine benzamide moiety with THIQ scaffold in order to improve the metabolic stability and solubility to preclude the use of prodrug.6 Initial efforts lead to the development of compounds 166 and 167 with enhanced potency against HIV-1 when compared with compound 164. Compound 166 also exhibited similar solubility (19 μg ml−1) like that of compound 164 (22 μg ml−1). However, compounds 166 and 167 had a very low metabolic stability when compared with compound 164. Various analogs were synthesized based on compound 167 in order to improve the metabolic stability without comprising solubility. Though the potency of compounds was found to increase, none of the synthesized analogs were found to exhibit better properties than compound 167. Further studies identified 1st position of the THIQ scaffold as a soft spot for metabolization. The 1st position of the THIQ in compound 167 was then functionalized to generate compounds 168 and 169. The potency of both the compounds decreased when compared with compound 167. Moreover, aqueous solubility decreased, and cytotoxicity enhanced. This study demonstrates the immense potential of the THIQ scaffold against HIV.
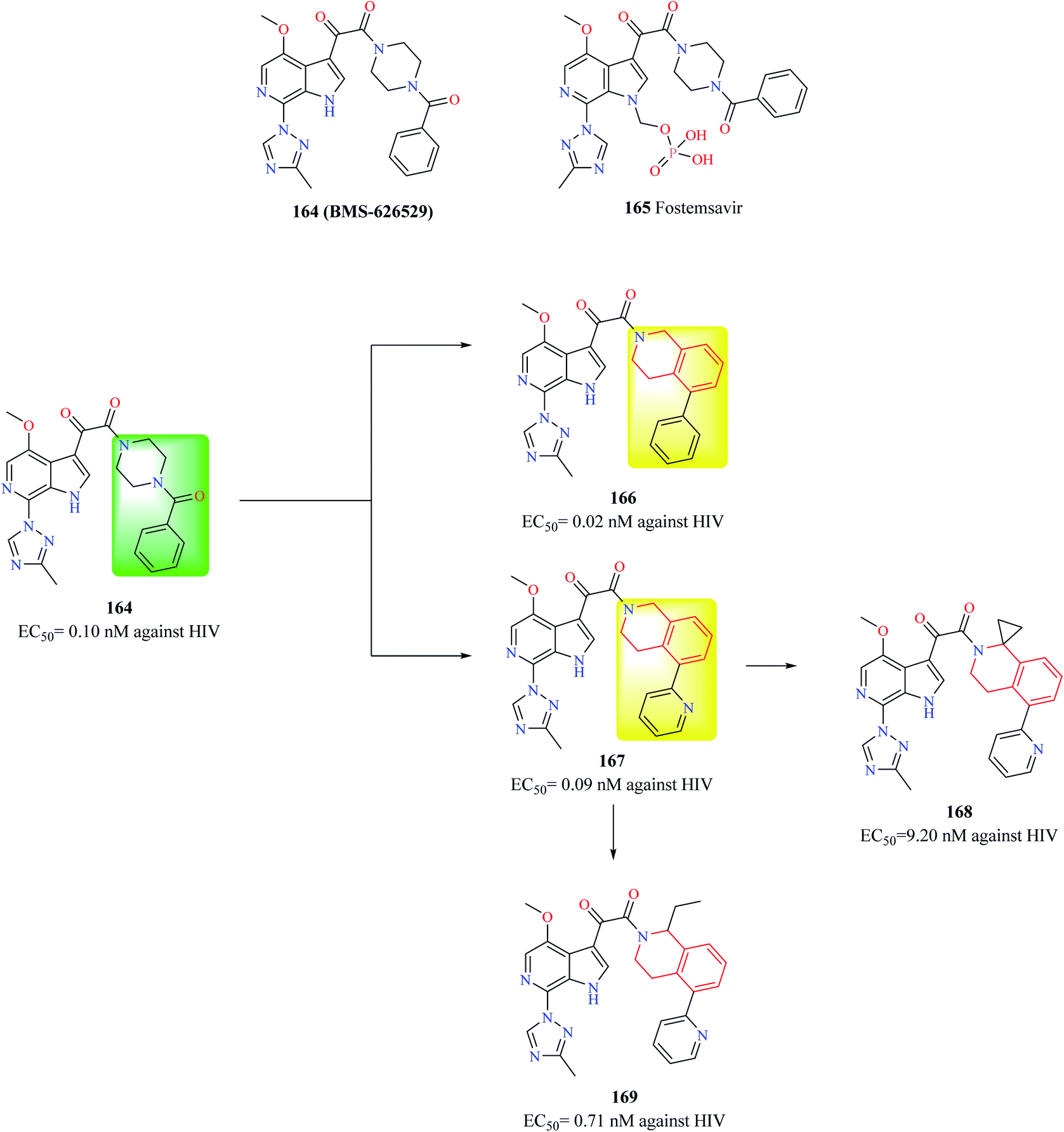 | ||
| Fig. 13 Design of THIQ analogs as anti-HIV agents. | ||
PAN (N-terminus of polymeric acidic protein) endonuclease is considered an ideal target for the development of anti-influenza agents. They are highly conserved across different influenza strains and more importantly, have no human counterpart. Liao et al. modified dopamine 170 (Fig. 14) to develop novel THIQ analogs.49 Compounds 171 and 172 exhibited potent activity against influenza virus A in vitro. Compound 172 exhibited concentration-dependent inhibition of PAN endonuclease with an EC50 value of 489.39 nM and a KD value of 94.19 nM. Molecular docking studies revealed that compound 172 was able to occupy the active site in the target protein. Compound 172 was able to form hydrophobic contacts with the active site residue His 41 as well as chelate the Mn metal ion in the active site.
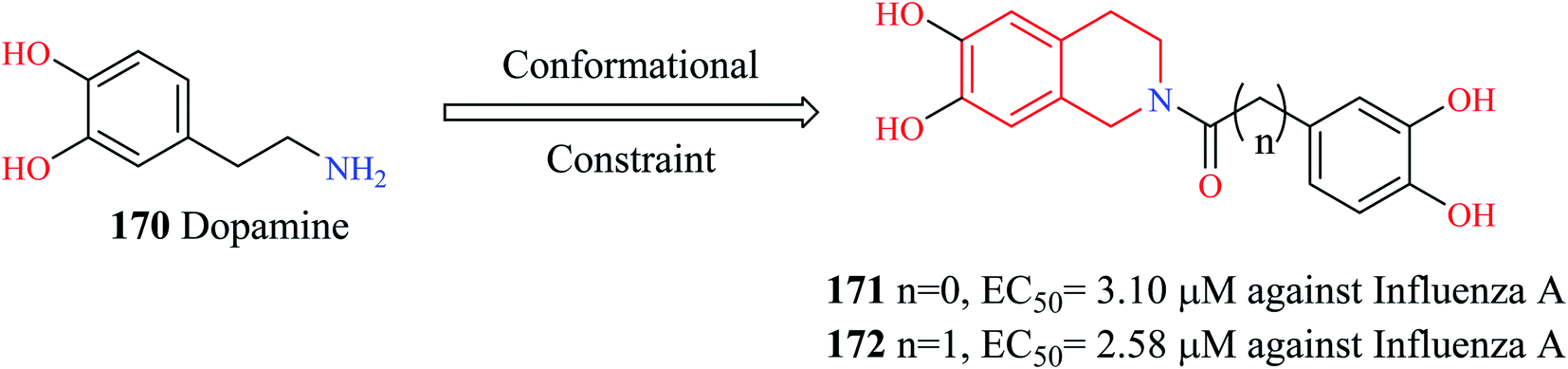 | ||
| Fig. 14 Design of THIQ analogs as anti-influenza A agents. | ||
3.4 Anti-malarial activity
High-throughput phenotypic screening of over 300![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 compounds was performed against the 3D7 strain of Plasmodium falciparum to discover novel hits for pre-clinical development.50 Phenotypic screening resulted in the identification of about 1300 validated hits. Most of the potent hits belonged to three different scaffolds: THIQ, diaminonaphthoquinones, and dihydropyridine. Over 40 analogs belonged to the THIQ core with most of the compounds exhibiting potent activity against drug-resistant strains of malaria. Compound 173 (Fig. 15) was identified as the most active of them all with EC50 in the sub-nanomolar range against all the tested malarial strains. Compound 173 also exhibited acceptable pharmacokinetic properties.50 However, the compounds were found to exhibit poor solubility and metabolic vulnerability. Further hit-to-lead studies were conducted on compound 173 to generate pre-clinical candidates for development.51 Different groups were substituted on N-2, C-3, and C-4 positions of compound 173 to address potency, solubility, and metabolic stability. Phenyl group was the most preferred substituent on the amide nitrogen. Substitution of aliphatic or other aromatic systems did not improve the potency. In general, EDG groups were preferred on the para position (the only exception being fluoro), and EWG groups were preferred on the meta-position in the phenyl ring. However, none of the substituents improved solubility. Hence, different substitutions were made on C-3 to address this problem. Although potency improved with a 5-membered heterocyclic ring system, no improvement in solubility was observed. A consensus was obtained with pyridine or pyrazole substitution. There was a slight decrease in potency upon pyridine substitution, but solubility was enhanced. The next substitution on N-2 was done to address the problem related to metabolic vulnerability as well as solubility. Both the lipophilicity as well as the shape of the substituent on N-2 had a profound effect on the potency. Though better potency was observed with iso-butyl substitution, they are metabolically labile. Therefore, N-2,2,2-trifluoroethyl substituent was substituted to afford stability. After extensive studies, two compounds 174 and 175 (also known as (+)-SJ733) were identified as ideal lead candidates.51 Both the compounds exhibited good solubility and metabolic stability, and potent antimalarial activity in vitro and in vivo. Compound 175 was then subjected to intense pre-clinical studies.52 The studies demonstrated that compound 175 possessed potent in vivo activity, high oral bioavailability, and good safety margins. Further, mechanistic studies indicated that compound 175 exhibits its antimalarial activity by inhibiting Plasmodium falciparum ATPase 4 (PfATP4).52 Compound 175 was also shown to have a favorable pharmacokinetic and safety profile in phase 1 clinical trials in humans.53
000 compounds was performed against the 3D7 strain of Plasmodium falciparum to discover novel hits for pre-clinical development.50 Phenotypic screening resulted in the identification of about 1300 validated hits. Most of the potent hits belonged to three different scaffolds: THIQ, diaminonaphthoquinones, and dihydropyridine. Over 40 analogs belonged to the THIQ core with most of the compounds exhibiting potent activity against drug-resistant strains of malaria. Compound 173 (Fig. 15) was identified as the most active of them all with EC50 in the sub-nanomolar range against all the tested malarial strains. Compound 173 also exhibited acceptable pharmacokinetic properties.50 However, the compounds were found to exhibit poor solubility and metabolic vulnerability. Further hit-to-lead studies were conducted on compound 173 to generate pre-clinical candidates for development.51 Different groups were substituted on N-2, C-3, and C-4 positions of compound 173 to address potency, solubility, and metabolic stability. Phenyl group was the most preferred substituent on the amide nitrogen. Substitution of aliphatic or other aromatic systems did not improve the potency. In general, EDG groups were preferred on the para position (the only exception being fluoro), and EWG groups were preferred on the meta-position in the phenyl ring. However, none of the substituents improved solubility. Hence, different substitutions were made on C-3 to address this problem. Although potency improved with a 5-membered heterocyclic ring system, no improvement in solubility was observed. A consensus was obtained with pyridine or pyrazole substitution. There was a slight decrease in potency upon pyridine substitution, but solubility was enhanced. The next substitution on N-2 was done to address the problem related to metabolic vulnerability as well as solubility. Both the lipophilicity as well as the shape of the substituent on N-2 had a profound effect on the potency. Though better potency was observed with iso-butyl substitution, they are metabolically labile. Therefore, N-2,2,2-trifluoroethyl substituent was substituted to afford stability. After extensive studies, two compounds 174 and 175 (also known as (+)-SJ733) were identified as ideal lead candidates.51 Both the compounds exhibited good solubility and metabolic stability, and potent antimalarial activity in vitro and in vivo. Compound 175 was then subjected to intense pre-clinical studies.52 The studies demonstrated that compound 175 possessed potent in vivo activity, high oral bioavailability, and good safety margins. Further, mechanistic studies indicated that compound 175 exhibits its antimalarial activity by inhibiting Plasmodium falciparum ATPase 4 (PfATP4).52 Compound 175 was also shown to have a favorable pharmacokinetic and safety profile in phase 1 clinical trials in humans.53
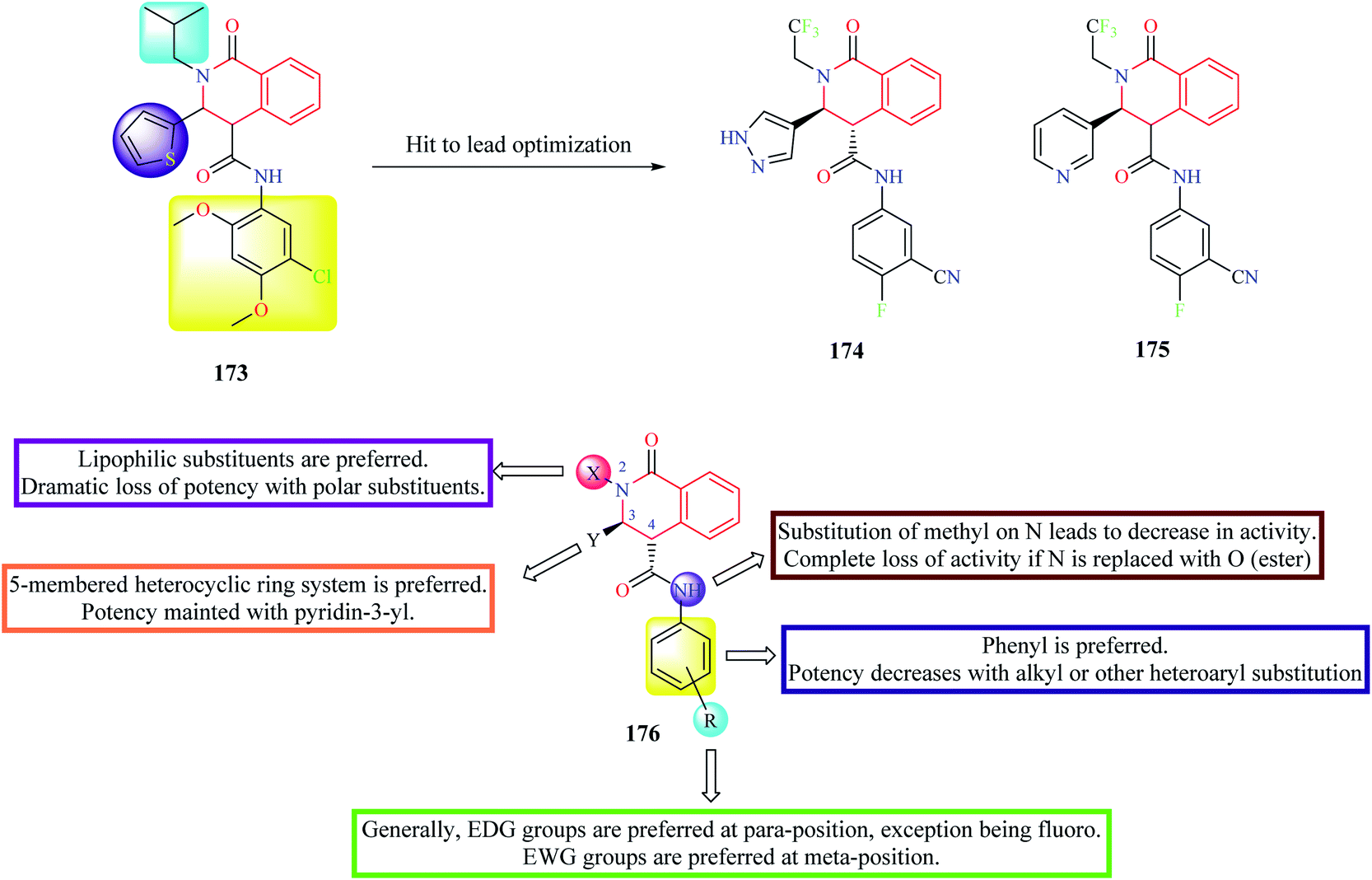 | ||
| Fig. 15 Hit to lead optimization of compound 173 along with SAR profile. | ||
21 novel derivatives of 1-aryl-6-hydroxy-THIQ analogs were designed and synthesized by Hanna et al.54 The synthesized analogs were evaluated for their antiplasmodial activity against P. falciparum. More than 15 compounds were found to exhibit moderate antimalarial activity. Compounds 177–179 (Fig. 16) exhibited potent antimalarial activity comparable to that of the standard drug chloroquine. They also possessed good selectivity indices and were also predicted to possess good pharmacokinetic properties. Further mechanistic and in vivo studies are required to further their stance as prospective antimalarial drug candidates.
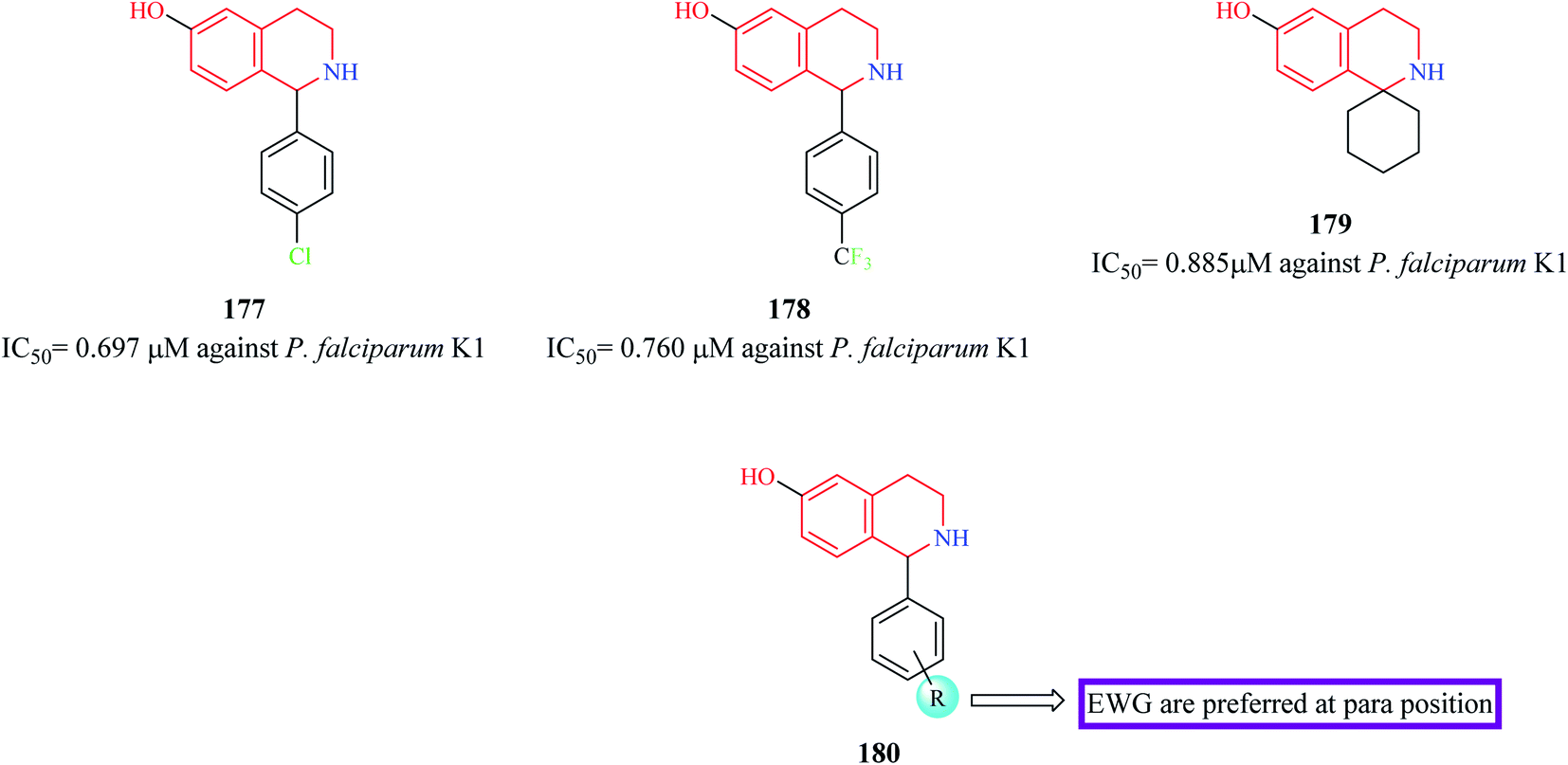 | ||
| Fig. 16 Structure of THIQ analogs 177–179 as anti-malarial agents. | ||
Three THIQ analogs 181–183 (Fig. 17) from Medicines for Malaria Venture (MMV) Malaria Box were assessed for their antiplasmodial activity against P. falciparum in different stages of its life cycle: asexual blood stages, mature gametocyte stages, and early sporogonic stages.55 All three THIQ analogs exhibited potent activity against the asexual blood stage of P. falciparum. They were also highly potent against chloroquine-resistant P. falciparum W2 strain with IC50 values between 0.070-0.133 μM. They also exhibited good P. falciparum gametocidal activity. They were also found to have a profound effect on the early sporogonic stage.
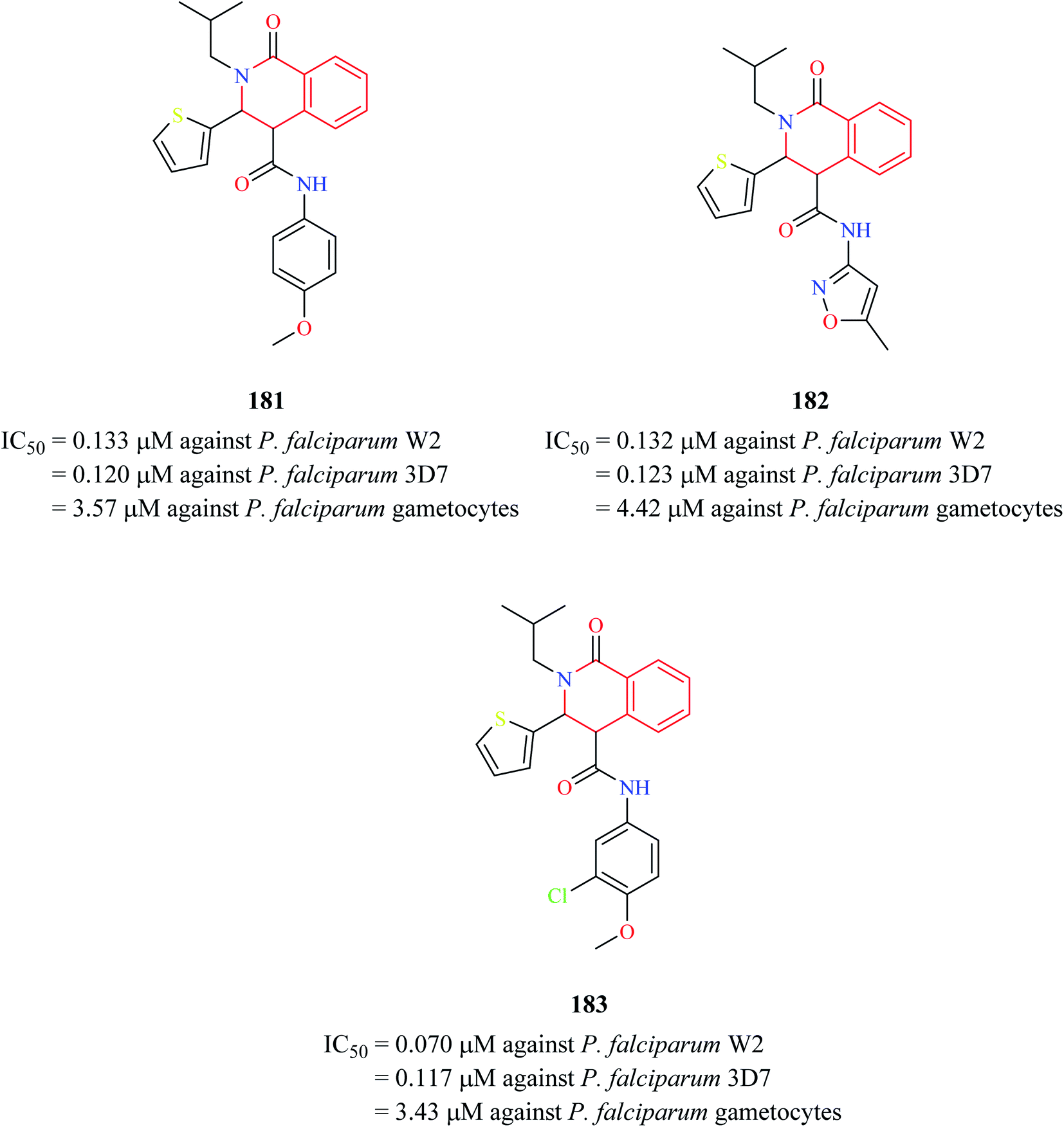 | ||
| Fig. 17 Structure of anti-malarial compounds 181–183. | ||
3.5 Anti-leishmanial activity
Chauhan et al. synthesized a series of triazine dimers as potential antileishmanial agents.56 Triazine-THIQ hybrid 184 (Fig. 18) exhibited good antileishmanial activity (IC50 = 7.62 μM) against the amastigotes form of Leishmania donovani with a good selectivity index.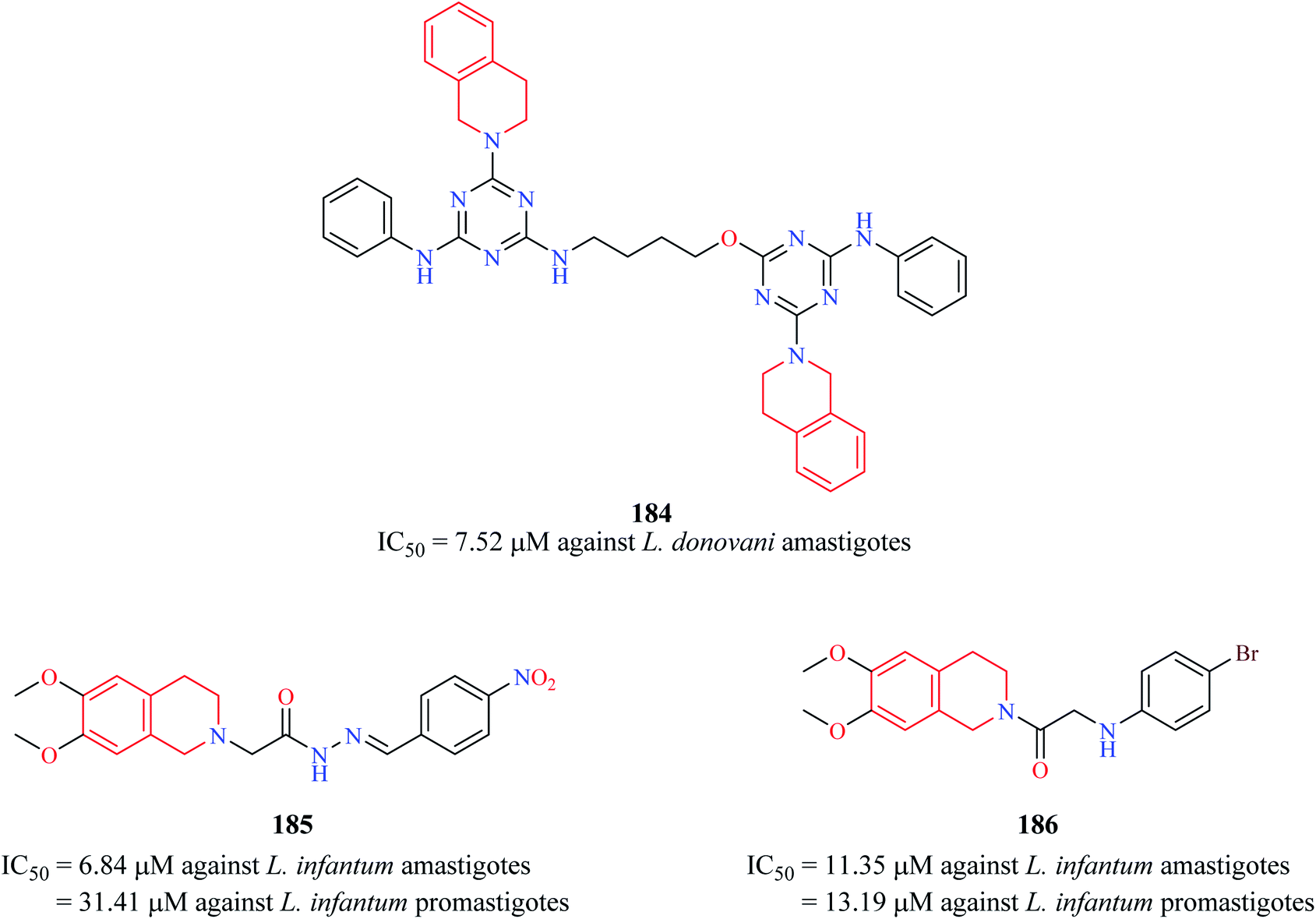 | ||
| Fig. 18 Structure of anti-leishmanial THIQ analogs 184–186. | ||
A series of novel THIQ analogs were synthesized and screened against the promastigote and amastigote forms of L. infantum.57 The THIQ analogs exhibited weak to potent activity against both forms of the leishmanial parasite. Compounds 185 and 186 exhibited potent activity against the amastigotes with IC50 values of 6.84 μM and 11.35 μM, respectively.
3.6 Anti-trypanosomal activity
Cullen et al. synthesized novel THIQ analogs by derivatizing the core scaffold 187 (Fig. 19).58 The synthesized analogs were screened for their inhibitory action against Trypanosoma brucei rhodesiense. Compounds 188–190 were found to exhibit potent activity antitrypanosomal activity. Preliminary SAR analysis revealed that compounds containing biphenyl methyl substitution at the phenolic OH displayed more potency and substitution at 1-position of THIQ nucleus had no effect on the antitrypanosomal activity.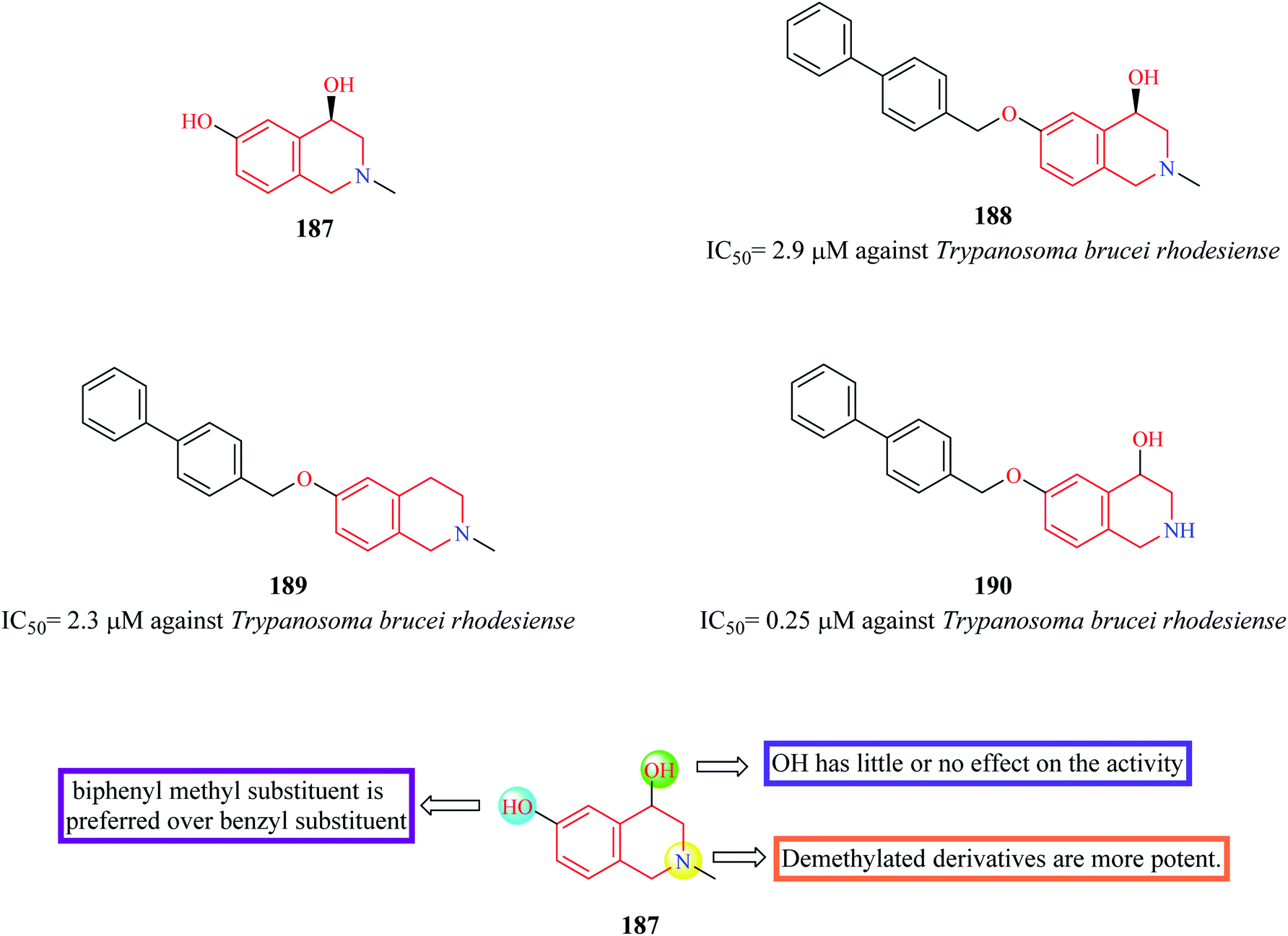 | ||
| Fig. 19 SAR profile of 4,6-dihydroxy THIQ analogs. | ||
3.7 Anti-schistosomal activity
Sadhu et al. synthesized novel praziquantel (PZQ) analogs with diversification at the aromatic and piperazine nucleus.59 Most of the synthesized compounds lacked activity, however, compounds 191 and 192 (Fig. 20) were found to exhibit potent activity against Schistosoma mansoni with 90% lethal concentration (LC90) values of 10 and 25 μM, respectively.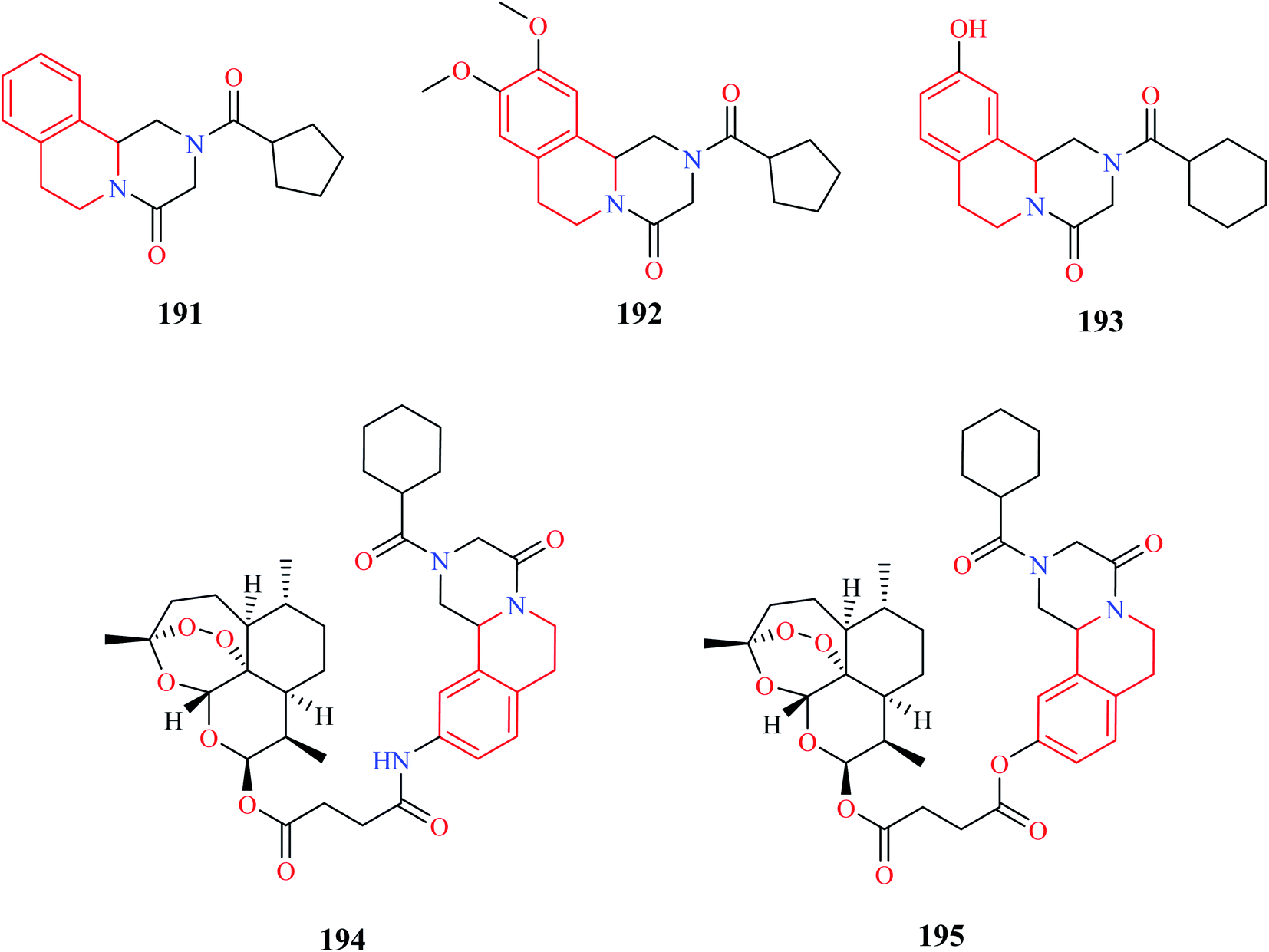 | ||
| Fig. 20 Structure of anti-schistosomal PZQ derivatives. | ||
Duan and co-workers designed novel PZQ analogs by hybridizing PZQ with another established antischistosomal drug, artemether.60 Initially a simple derivative of PZQ, 193 was synthesized, after which the hybrid compounds 194 and 195 were synthesized. All three compounds were evaluated for their antischistosomal activity against both the juvenile and adult forms of S. japonicum. Compounds 193–195 exhibited potent activity against the adult form of the parasite, much better than the standard drug PZQ. They were also found to be equally effective against the juvenile form, reducing the worm vitality to 100% in 72 hours at 15 μM. PZQ on the other hand wasn't effective at all. Compounds 193–195 were also evaluated for their worm reduction activity in vivo. All three compounds were moderately effective in reducing the worm load in mice. Importantly, compound 195 effectively reduced the worm load of both the adult and juvenile form of the parasite in mice, with a total worm reduction rate of 56.2% and 70.3%, respectively. Further in vivo studies demonstrated that compound 195 reduced the worm load by 60–85% in different developmental stages of the worm. Further dive into its mechanism of action revealed that compound 195 exhibited its activity by damaging the inner and outer walls of the worm tegument.61 Further in-depth studies were also conducted to prove its prowess as an antischistosomal agent.62 These compelling studies made compound 195 a possible drug candidate that can be explored further for the treatment of schistosomiasis.
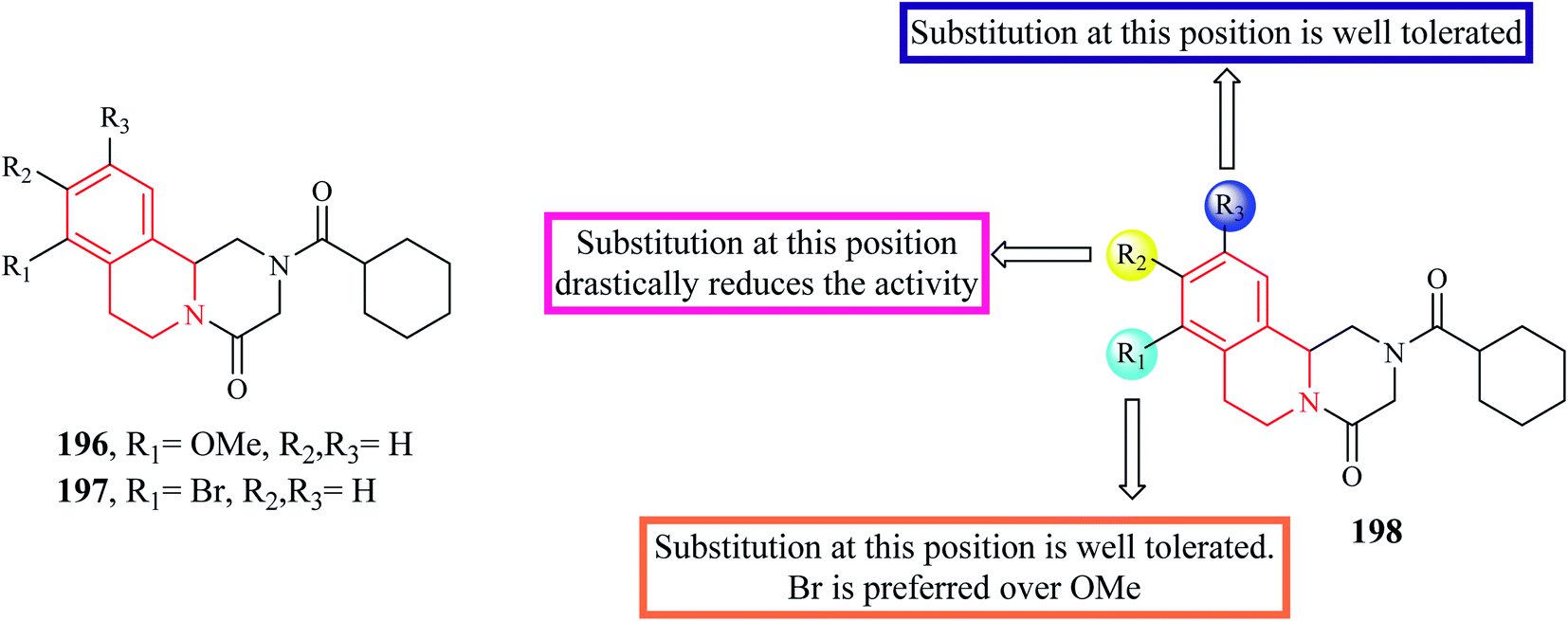
A series of novel PZQ derivatives were designed and synthesized by substituting different groups on the aromatic ring of PZQ.63 The synthesized compounds were evaluated against adult and juvenile forms of S. japonicum in vitro. A couple of compounds 196 and 197 (Fig. 21) exhibited potent antischistosomal activity. Compound 197 in particular, was equipotent as the standard drug PZQ. Compounds 196 and 197 were also effective against the juvenile form and compound 197 reduced the worm vitality to less than 10% at a concentration of 25 μM at the end of 72 hours. Compounds 196 and 197 were also tested for their efficacy in mice harboring 42 day old S. japonicum. Compounds 196 and 197 reduced the worm count by 44.3% and 54.3% respectively.
 | ||
| Fig. 21 SAR profile of PZQ derivatives (substitutions on phenyl group). | ||
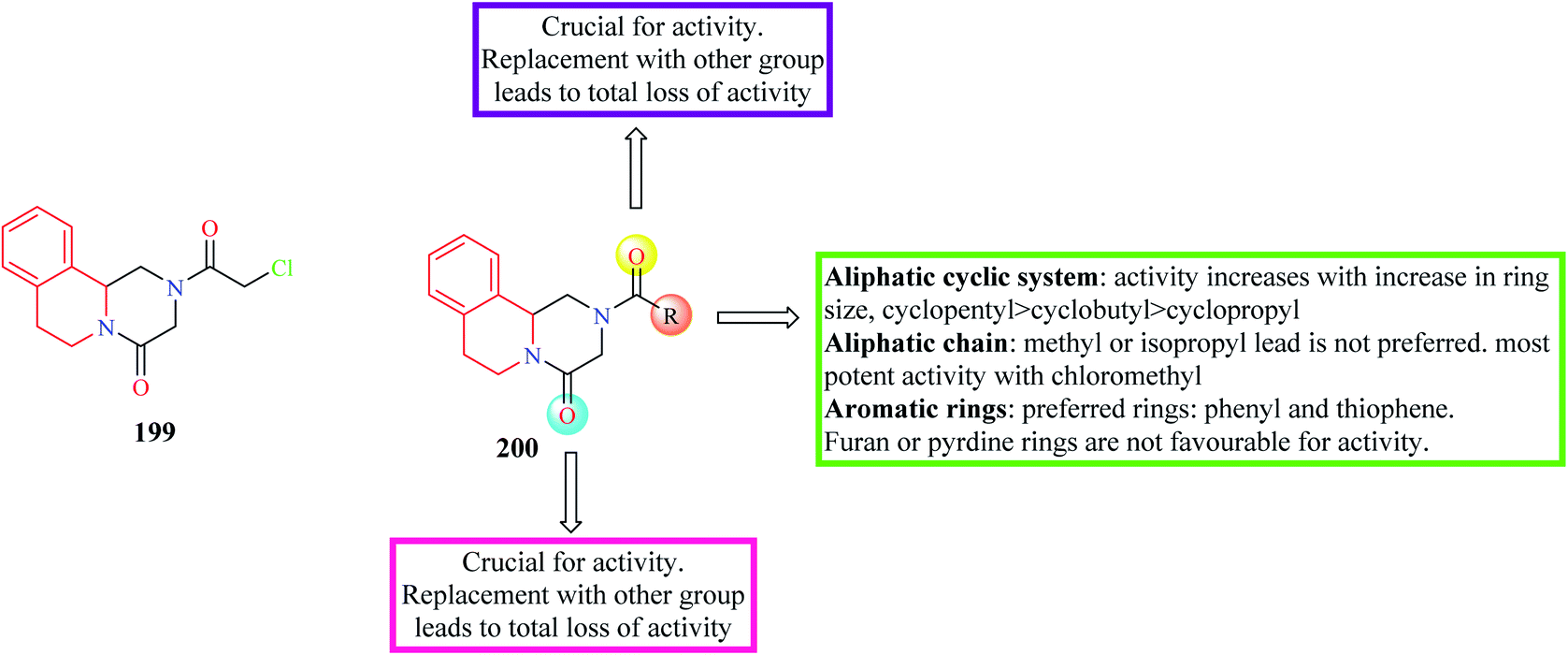
Wang et al. designed and synthesized novel PZQ derivatives and explored their antischistosomal activity.64 The compounds were designed by replacing the cyclohexane ring with different groups like an aliphatic chain, aromatic ring systems, aliphatic cyclic rings. The effect of the amide groups at positions 2 and 4 was also determined. Compound 199 (Fig. 22) containing a chloromethyl substituent was found to more potency than PZQ. Compound 199 was able to completely kill the worms at a concentration of 5 μM. The SAR is discussed in Fig. 22.
 | ||
| Fig. 22 SAR profile of PZQ derivatives (replacement of cyclohexane group). | ||
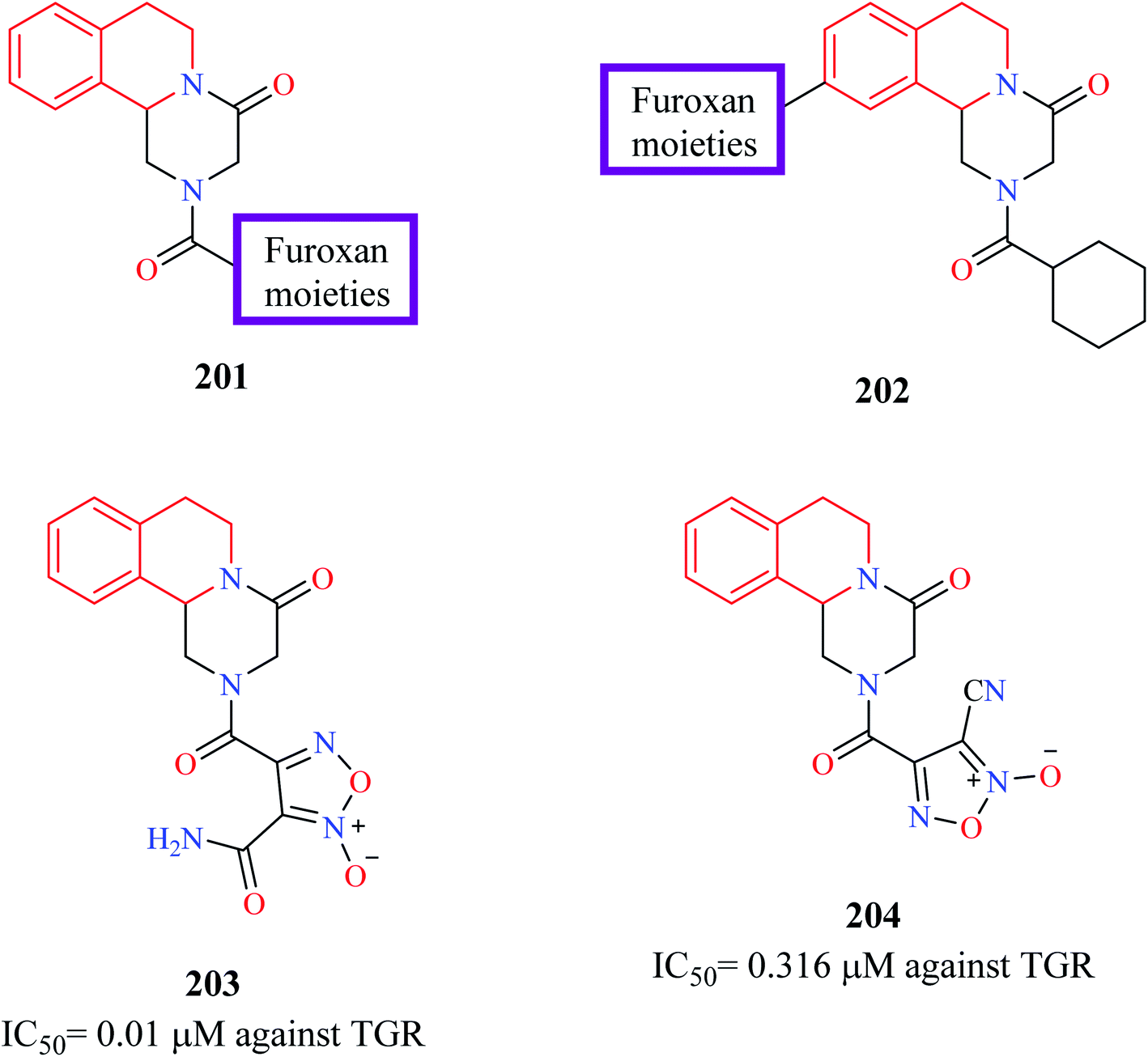
Guglielmo and others employed molecular hybridization technique to design novel PZQ-furoxans as potential antischistosomal agents.65 Furoxans are 1,2,5-oxadiazole-2-oxides that have been previously shown to be effective against schistosomiasis due to their NO donating capacity. Furoxan derivatives inhibit thioredoxin glutathione reductase (TGR), a multifunctional protein in the worms.66–69 Therefore, at least theoretically, hybridizing the furoxan scaffold with PZQ may lead to the development of potent antischistosomal agents. In the first series of hybrid compounds 201 (Fig. 23), the cyclohexyl group of PZQ was replaced with furoxans. In the second hybrid series 202, the furoxan moiety was bridged to the 10th position of PZQ using appropriate linkers. The synthesized compounds were evaluated for their ability to inhibit S. mansoni (TGR) and for their worm-killing ability. Compounds 203 and 204 were found to exhibit potent activity against TGR (IC50 = 0.01 μM and 0.316 μM, respectively). Both compounds 203 and 204 were also endowed with good worm-killing potency, in which compound 203 induced 80% of worm death in144 h while compound 204 induced 100% worm death in 72 h. In comparison, PZQ exhibited only 40% worm death at the end of 144 h.
 | ||
| Fig. 23 Structure of anti-schistosomal PZQ-furoxan hybrid analogs. | ||
3.8 Anti-depressant activity
The antidepressant potential of 1-methyl THIQ 205 (Fig. 24) in animal models of depression was demonstrated by Antkiewicz-Michaluk and co-workers.70 1-Methyl THIQ administered at a dose of 10, 25, 50 mg kg−1 i.p. caused a significant reduction of the immobility time in the forced swimming test (FST). 1-Methyl THIQ at a dose of 50 mg kg−1 i.p. produced better activity than the standard drug imipramine (dose = 30 mg kg−1 i.p.). The 1-methyl THIQ also significantly increased the swimming time (dose = 25, 50 mg kg−1 i.p.) and climbing activity (dose = 50 mg kg−1 i.p.) of the rats. 1-Methyl THIQ at the highest dose reduced the locomotor activity of rats like imipramine, thus ruling any psychostimulating activity. The neurochemical analysis revealed that 1-methyl THIQ produced a significant elevation in the serotonin levels with a simultaneous reduction in its metabolite 5-hydroxy indole acetic acid (5-HIAA). Moreover, it also affected the dopamine metabolism; causing a decrease in the levels of 3,4-dihydroxyphenylacetic acid (DOPAC) with a significant increase in the levels of 3-methoxytyramine (3-MT). These effects of 1-methyl THIQ are attributed to the fact that it is a well-characterized inhibitor of monoamine oxidase (MAO).71 Inhibition of MAO by 1-methyl THIQ causes elevation of serotonin in the rat striatum.72 1-Methyl THIQ has also been shown to inhibit the MAO-dependent oxidation of dopamine and shift its catabolism towards Catechol O-methyltransferase (COMT) dependent pathway. Such a shift in dopamine metabolism also reduces oxidative stress in the brain (due to MAO-dependent dopamine oxidation), which is also ascribed to be involved in the pathophysiology of depression.73,74 Moreover, 1-methyl THIQ has also been demonstrated to be a scavenger of free radicals.75 A Similar antidepressant activity was also observed for the THIQ. THIQ administered at a dose of 25, 50 mg kg−1 was found to significantly decrease the immobility time and increase the swimming activity of the rats in FST. Besides exhibiting its antidepressant activity in the FST model, administration of THIQ for about 3 weeks completely restored normal sucrose consumption in the rats that were subjected to the chronic mild stress (CMS) test.76 Further studies also corroborated the antidepressant potential of THIQ and 1-methyl THIQ.77,78 Additionally, 1-methyl THIQ was subjected to intense in vitro, in vivo, and in silico toxicity studies.79 No significant toxicity was observed for 1-methyl THIQ in the in vitro and in vivo toxicity studies. The unique mechanism of action(s) of 1-methyl THIQ in depression supplemented with a lack of significant toxicity makes it an interesting antidepressant drug candidate for further studies.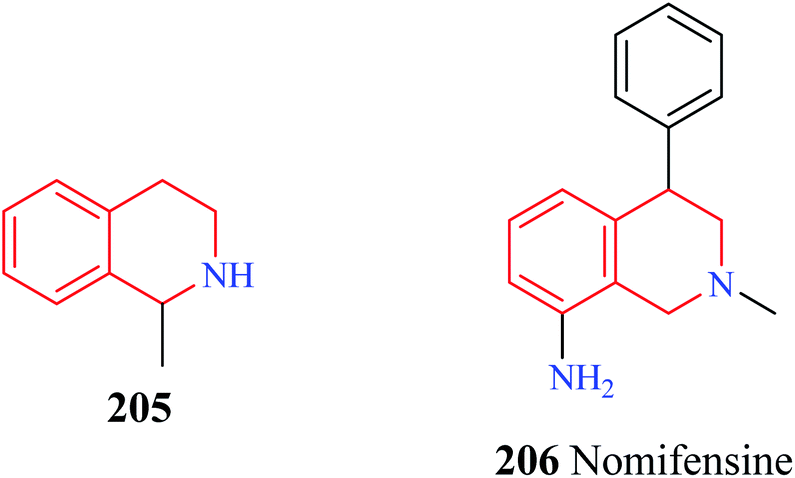 | ||
| Fig. 24 Structure of 1-methyl THIQ 205 and nomifensine 206. | ||
Nomifensine 206 is a norepinephrine and dopamine reuptake inhibitor that was used as an antidepressant in the 1970s. However, it was withdrawn from the market in the 1980s due to its toxicity. The aniline moiety in the nomifensine was attributed to the generation of toxic metabolites.80–82 Molino and co-workers designed a series of THIQ analogs based on the structure of nomifensine as dual norepinephrine and dopamine reuptake inhibitors.83 Removal of the amino group and optimization of the compounds around the THIQ core and 4-phenyl moiety resulted in the generation of compounds 207–209 (Fig. 25) with good potency against norepinephrine transporter (NET) and dopamine transporter (DAT). Compound 209 displayed significant NET occupancy (49%) in the ex vivo binding assay. Compound 209 was subjected to further studies wherein it was found to be selective for NET and DAT without any off-target activity. However, compound 209 was found to inhibit the CYP2D6 with moderate potency (IC50 = 0.48 μM), which might result in unwanted drug–drug interactions.
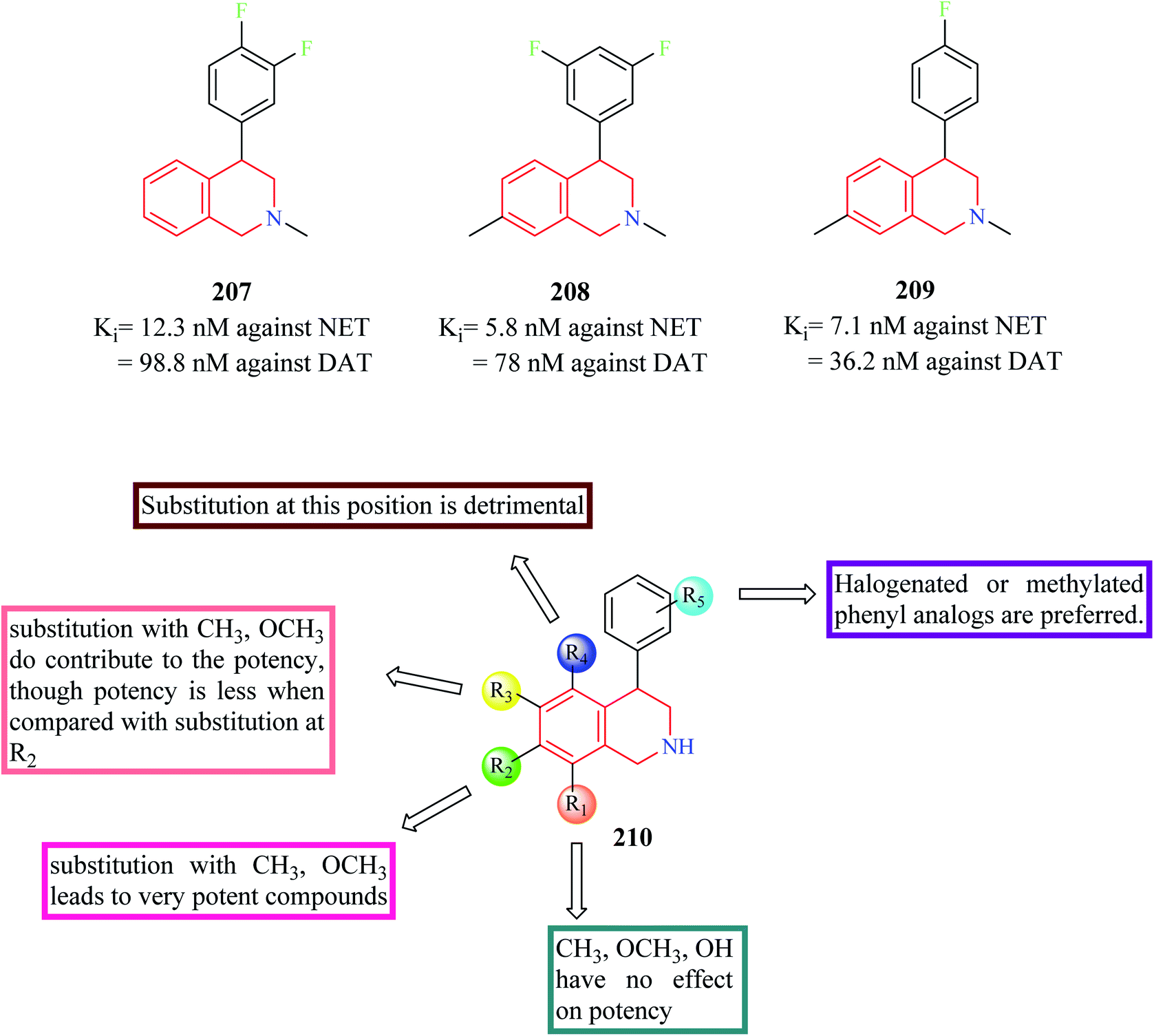 | ||
| Fig. 25 SAR profile of dual reuptake inhibitory THIQ derivatives. | ||
In subsequent studies, novel THIQ analogs were designed by taking cues from compound 209.84 This series of compounds explored the substitution of different heterobicyclic groups on the 4-position of the THIQ nucleus. Substitution with heterobicyclic groups gave rise to compounds that were capable of inhibiting NET, DAT, and Serotonin transporter (SERT), thus giving rise to triple reuptake inhibitors. In general, the (+) enantiomers were found to be more potent than their counterpart. In general, THIQs substituted with indole, benzofuran, and benzothiophene were found to be superior triple reuptake inhibitors with compound 211 (Fig. 26) being the most potent of them all. It was also found to exhibit significant potency against CYP2D6 (IC50 = 0.8 μM). Different analogs of compound 211 were then synthesized by substituting different groups on the 7-position of the THIQ core. Optimization of compound 211 led to the development of compound 212 with potent activity against all three transporters with low activity against CYP2D6 (IC50 = 11 μM). Compound 212 (dose = 3 mg kg−1 p/o) was also found to have good occupancy at SERT (95%), DAT (77%), NET (98%) in ex vivo binding assays. Compound 212 was found to possess significant antidepressant activity in FST and Tail suspension models, reducing the immobility time in both models. However, metabolic profiling of compound 212 lead to the discovery of a significant metabolite compound 213 that was found to possess equal potency against the three transporters as that of compound 212. Further studies corroborated that compound 213 might contribute to the in vivo pharmacology of compound 212. However, compound 213 was found to have an affinity for CYP2D6 (IC50 = 1 μM), therefore susceptible to drug–drug interactions. The SAR of these groups of compounds has been summarized in Fig. 26.
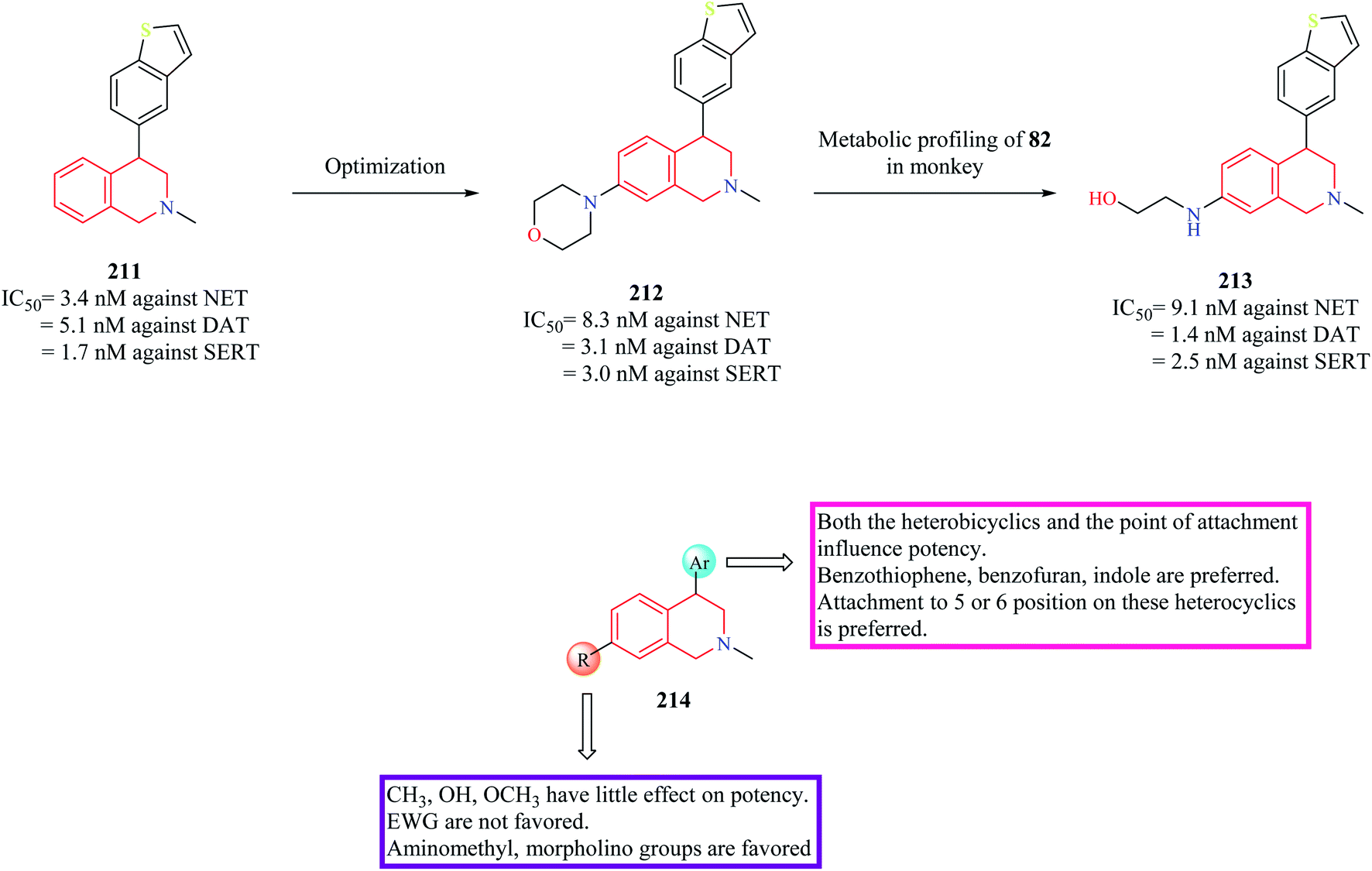 | ||
| Fig. 26 SAR profile of triple reuptake inhibitors. | ||
3.9 Anti-Alzheimer activity
Sukumarapillai and others designed a series of THIQ analogs and evaluated their inhibitory potential against cholinesterases: acetylcholinesterase (AChE) and butyrylcholinesterase (BChE), a widely explored target for the development of anti-Alzheimer agents.85 A couple of compounds 215 and 216 (Fig. 27) were found to exhibit good inhibitory potential against both the enzymes with their activity being more pronounced against BChE. Enzyme kinetic studies revealed that the compounds exhibited a mixed-mode of inhibition against both the enzymes. Molecular docking studies were performed to determine the probable binding mode of the compounds. Compound 215 was found to exhibit hydrophobic and hydrogen bond interactions with the active site residues of the enzymes. Sudhapriya et al. synthesized triazolo-benzodiazepino-THIQ fused analogs as potential AChE inhibitors. Compound 217 was found to potently inhibit AChE with an IC50 value of 0.25 μM.86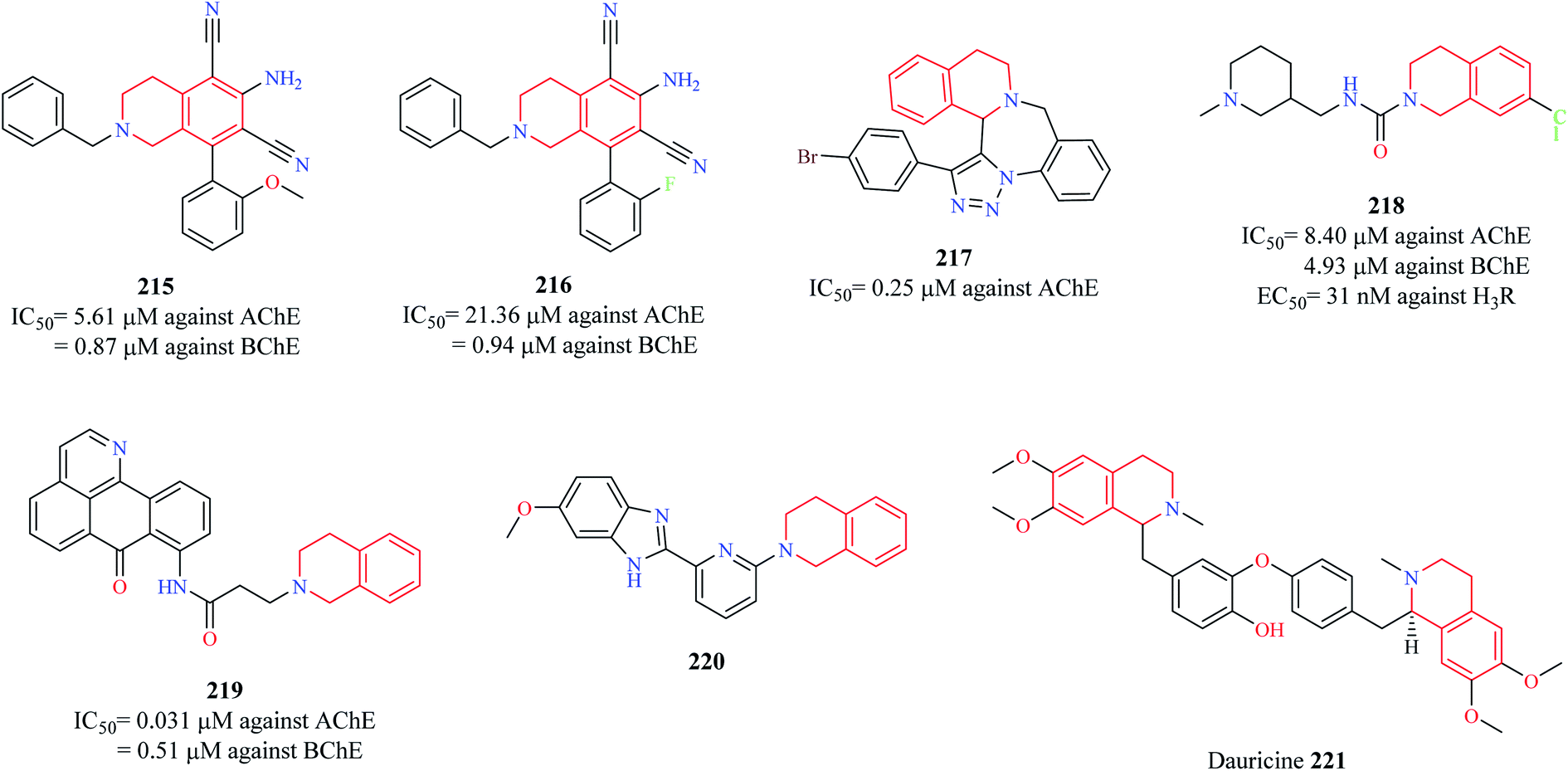 | ||
| Fig. 27 Structure of anti-Alzheimer THIQ analogs 215–221. | ||
Since Alzheimer's disease (AD) is a multifactorial disease involving intricate targets and molecular pathways, the development of ligands that can engage more than one target seems an interesting approach to combat AD. A similar strategy was taken up by Ghamari and co-workers87,88 wherein they tested the potential of compound 218 to act as cholinesterase inhibitor and Histamine 3 receptor (H3R) antagonist, an emerging target for the management of AD.89,90 Compound 218 was found to exhibit nanomolar potency against the H3R (EC50 = 31 nM) and micromolar potencies against AChE (IC50 = 8.40 μM) and BChE (IC50 = 4.93 μM) with no significant impact on the viability of CHO-K1 cells. Molecular docking studies revealed that compound 218 had a similar binding pattern as that of donepezil in the active site of AChE with THIQ ring exhibiting a π–π stacking and hydrophobic interaction with Trp286 and Try341 respectively. Additionally, the nitrogen of the THIQ ring system was found to have a hydrogen bond interaction with Tyr124. Chen and co-workers designed a new series of oxoisoaporphine–tetrahydroisoquinoline analogs based on the concept of molecular hybridization as multi-target directed ligands (MTDLs) in AD.91 Compound 219 exhibited the following properties: (i) strongly suppressed the production of NO-induced by LPS in BV-2 microglia and RAW 264.7 macrophage (ii) compound 219 also suppressed the expression of iNOS in RAW 264.7 macrophage cells (iii) compound 219 was also shown to suppress the LPS-induced production of pro-inflammatory cytokines such as TNF-α and IL-β (iv) compound 219 was able to reduce Aβ42 levels by 37.6% (v) exhibited potent inhibitory activity against AChE (IC50 = 0.031 μM) and BChE (IC50 = 0.51 μM) (vi) caused a significant delay in paralysis (due to Aβ1–42 toxicity) in C. elegans GMC101 in vivo model. These results suggest that compound 219 may be a promising anti-Alzheimer agent that needs further exploration. A similar strategy was conceptualized by Fang et al. wherein they developed THIQ–benzimidazole hybrids as MTDLs as a potential treatment strategy for AD.92 Among them, compound 220 was found to possess good anti-neuroinflammatory property (IC50 = 5.07 μM against NO production) by inhibiting the production of iNOS and pro-inflammatory cytokines (TNF-α and IL-1β) in BV-2 cells. It was also shown to possess moderate inhibitory activity against β-amyloid precursor protein cleaving enzyme 1 (BACE1) and potent neuroprotective activity against glutamate-induced cell death in HT22 cells by inhibiting the production of reactive oxygen species (ROS) and increasing the levels of glutathione. It possessed good blood–brain barrier (BBB) penetration ability, making it an interesting candidate for further development.
Dauricine 221 is a benzyl THIQ alkaloid that has been conferred with neuroprotective effects in AD. Different mechanisms have been proposed for its neuroprotective effects. It was found to reduce Aβ accumulation by activating the X-box binding protein 1 (XBP-1) and eukaryotic translation initiation factor (eIF2α) arms of the unfolded protein responses (UPR), which results in the clearance of Aβ oligomers and their associated toxicities.93 The anti-apoptotic and strong antioxidant properties of dauricine also accounted for its neuroprotective effects in AD.94 Further studies indicated that it reduced tau phosphorylation along with Aβ plaque formation by altering UPR, mitochondrial function, and ROS clearance.95
3.10 Anti-Parkinson activity
The endogenous THIQs – 1-benzyl THIQ 222 (Fig. 28) and 1-methyl THIQ 205 have contrasting effects in Parkinson's disease (PD). 1-Benzyl THIQ 222 potentiates the metabolism of dopamine, increases the production of free radicals by activating the dopamine oxidation pathway and inhibiting dopamine metabolism via the COMT pathway, and induces cell death via apoptotic mechanisms.96–98 It is also regarded as an etiological factor in idiopathic PD. The levels of 1-benzyl THIQ in cerebrospinal fluid (CSF) were found to be significantly higher (about three times) in PD patients than in control patients.99 Studies have demonstrated that chronic administration of 1-benzyl THIQ produces parkinsonian-like symptoms in primates and rodents highlighting its neurotoxic effects.99–101 In contrast, 1-methyl THIQ, another endogenous THIQ, has been demonstrated to translate its neuroprotective role in animal models of PD as well. Studies have demonstrated that acute and chronic administration of 1-methyl THIQ completely antagonizes the neurotoxic effect of 1-benzyl THIQ on dopaminergic neurons. A similar neuroprotective activity was also observed for THIQ.102,103 Furthermore, chronic administration of both THIQ and 1-methyl THIQ have been shown to offer protection against 6-hydroxy dopamine-induced disturbances in the dopaminergic neurons.104,105 Several other synthetic THIQs have also been shown to exert neuroprotective effects in animal models of PD. Taguchi and co-workers evaluated the neuroprotective effects of 1,3-dimethyl THIQ 223 and 1,3-dimethyl-N-propargyl THIQ 224 on 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and methyl-4-phenylpyridinium ion (MPP+), an active metabolite of MPTP, induced PD like symptoms in mice.106 Though 1,3-dimethyl THIQ did not offer much neuroprotective effects, 1,3-dimethyl-N-propargyl THIQ was found to prevent the adverse events induced by MPTP and MPP+. Stereoselectivity had an important role to play as cis and trans isomers of 1,3-dimethyl-N-propargyl THIQ was found to have different neuroprotective functions. Both the isomers were found to prevent MPTP-induced bradykinesia, tyrosine hydroxylase (TH) positive cell loss, and decreased dopamine levels with the trans isomer of 1,3-dimethyl-N-propargyl THIQ exhibiting superior activity than its counterpart. However, the cis isomer was found to afford superior protection against MPP+ induced cell death than the trans isomer. In a similar study, 3-methyl-N-propargyl-THIQ 225 prevented MPTP induced adverse events in mice while a THIQ analog without the propargyl group, 3-methyl THIQ 226 was found to have only weak activity against MPTP induced PD like symptoms.107 These studies shed light on the importance of the propargyl group in the neuroprotective activity. It is speculated that compounds like 224 and 225 exhibit its anti-parkinsonism action by inhibiting the neurotoxicity caused by MPP+ by competing with MPP+ in the catecholamine uptake sites.107 However, further studies are required to elucidate the exact role of the propargyl group in neuroprotection. Son and others disclosed the promising potential of PTIQ 227 in PD.108 Compound 227 suppressed the production of matrix metalloproteinase-3 (MMP-3, which plays an important role in dopaminergic neurodegeneration) in the CATH.a cell and also offered cytoprotection. Compound 227 also downregulated MMP-3, COX-2, and other proinflammatory cytokines like IL-1β, TNF-α in BV-2 microglial cells activated with Lipopolysaccharides (LPS). Compound 227 also attenuated the adverse events induced by MPTP in mice. Moreover, compound 227 was also bestowed with the following pharmacokinetic properties – (i) compound 227 was stable against liver microsomal enzymes (ii) exhibited high IC50 values against CYP450 enzymes (iii) no side effect against hERG channel (iv) no cytotoxicity towards liver cells (v) no lethality to animals at 1000 mg kg−1 i.p. (vi) readily penetrates BBB. Similar pharmacological effects were observed for AMTIQ 228, a derivative of PTIQ. Compound 228 suppressed inflammatory responses in activated microglial BV-2 cells and attenuated the effects of MPTP (motor deficits and dopamine neurodegeneration) in mice. Compound 228 was stable against liver microsomes, lacked CYP450 inhibitory effect, and was also demonstrated to possess better BBB penetration than PTIQ.109 Oleracein E 229 a naturally occurring THIQ analog was demonstrated to provide neuroprotection against rotenone-induced PD in cell and animal models.110 Oleracein E was found to improve the motor functions in a PD mouse model, preserved and maintained TH positive neurons, and density of dopaminergic fibers in the substantia nigra pars compacta. Further mechanistic studies revealed that oleracein E exhibits its neuroprotective effects in cell and animal models via diverse mechanisms like scavenging free radicals and reducing oxidative stress, downregulating the phosphorylation of extracellular regulated kinases (ERKs), and inhibiting neuronal apoptosis.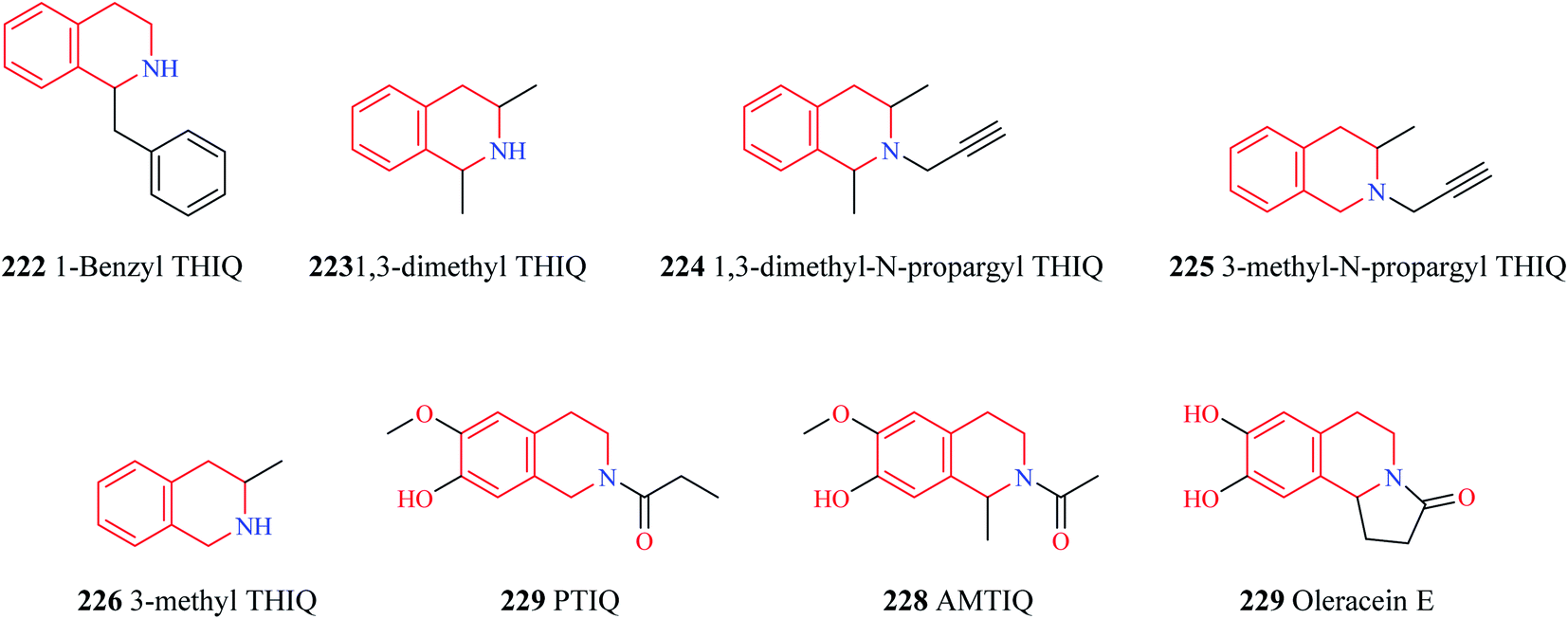 | ||
| Fig. 28 Structure of THIQ analogs 222–229. | ||
4. Conclusion
THIQ is a privileged scaffold present in a number of clinically used drug molecules. The information amassed in this review article sheds light on the biological potential and SAR of THIQ based analogs across different facets of diseases. Various strategies for the construction of the THIQ core scaffold with special emphasis on multi-component reactions (MCR) are discussed.Though Pictet–Spengler condensation and Bischler–Nepieralski reaction are the most commonly used synthesis strategies for constructing the THIQ core, MCR provides an opportunity to explore the chemical space to synthesize diverse THIQ analogs. Mechanistically, THIQ analogs exhibit their pharmacological properties by diverse mechanisms as described in Fig. 29.
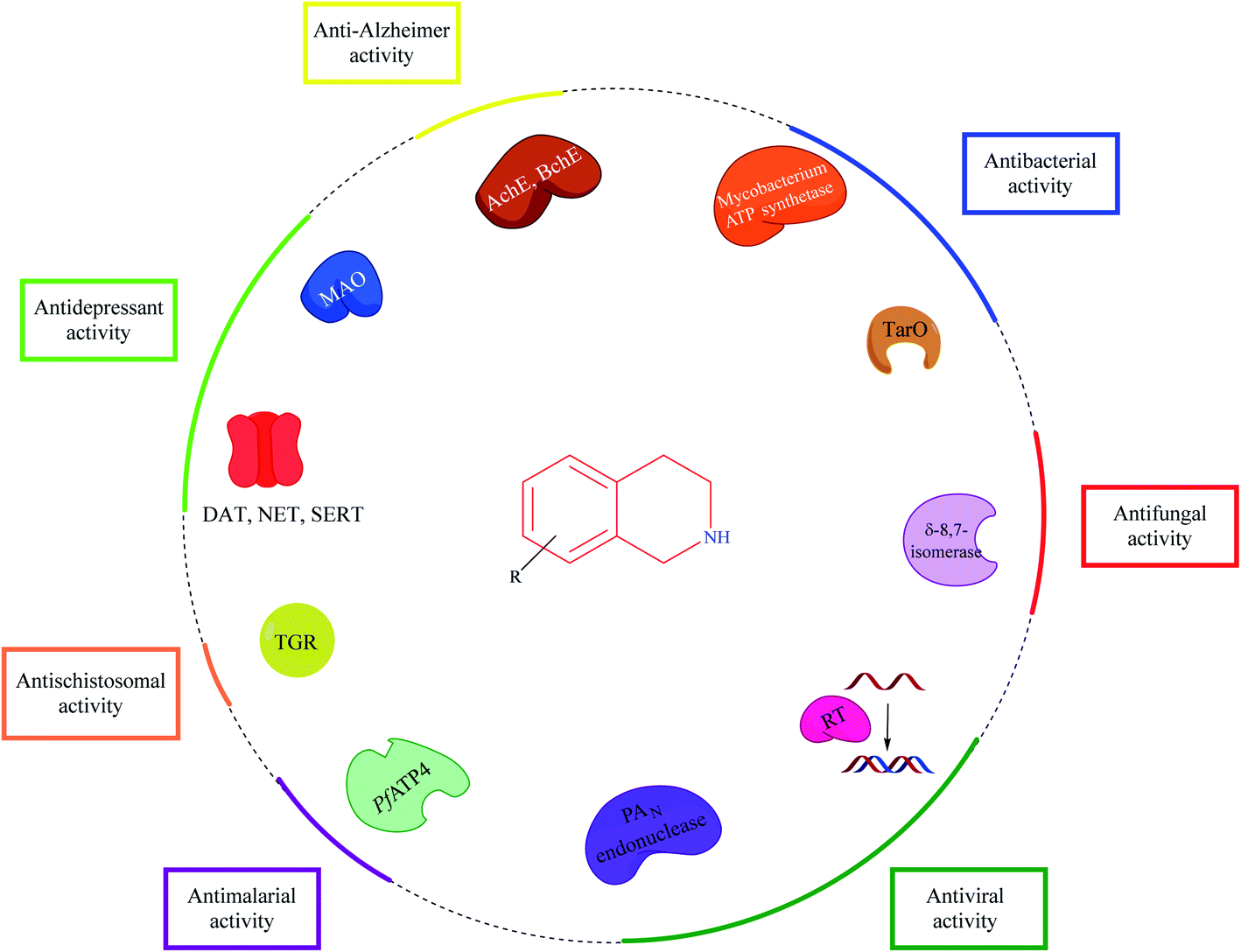 | ||
| Fig. 29 Potential targets and their associated activities for THIQ based analogs. | ||
Importantly, THIQs showed a profound effect on the central nervous system. Endogenous amines like 1-methyl THIQ and other synthetic THIQ analogs have been demonstrated to possess good neuroprotective properties, which have been shown to alleviate symptoms of depression and Parkinson's disease in animal models. However, there is still a lot to explore about these derivatives like delineating the importance of propargyl group in neuroprotection. Emerging drug discovery paradigm like MTDLs, consisting of THIQ based hybrid analogs, is a promising strategy for the development of novel anti-Alzheimer agents that needs further exploration. Drug design strategies like structure-based drug design, ligand-based drug design, fragment-based drug design, and molecular hybridization could be employed for the design of novel THIQ analogs against various ailments.
Future medicinal chemistry efforts could be targeted to improve the potency and pharmacokinetic properties of existing THIQ analogs and unearth the mechanistic basis of activity at the molecular level. Medicinal chemistry strategies like bioisosteric replacement, scaffold hopping, SAR among others could be employed for the optimization of identified lead compounds in order to enter the clinical trial phases of drug discovery. The synthetic ease combined with the nucleophilicity of the secondary nitrogen makes it possible for hybridizing the THIQ core with other privileged heterocyclic scaffolds for developing potent and effective hybrid analogs.
Conflicts of interest
The authors have no conflicts of interest to declare.Acknowledgements
SM and KVGC thank the Department of Biotechnology for the research grant. This work was carried under the grant of the Department of Biotechnology, Indo-Spain, New Delhi. (Ref. No. BT/IN/Spain/39/SM/2017-2018). BKK is thankful to the Ministry of Tribal Affairs, Government of India for providing financial assistance (Award no. 201920-NFST-TEL-01497). SM and BKK thank BITS-Pilani, Pilani campus for providing adequate facilities like access to scientific journals to do this review study.References
- J. D. Scott and R. M. Williams, Chem. Rev., 2002, 102, 1669–1730 CrossRef CAS PubMed.
- R. K. Tiwari, D. Singh, J. Singh, A. K. Chhillar, R. Chandra and A. K. Verma, Eur. J. Med. Chem., 2006, 41, 40–49 CrossRef CAS PubMed.
- J. Zhu, J. Lu, Y. Zhou, Y. Li, J. Cheng and C. Zheng, Bioorg. Med. Chem. Lett., 2006, 16, 5285–5289 CrossRef CAS PubMed.
- A. Kumar, S. B. Katiyar, S. Gupta and P. M. S. Chauhan, Eur. J. Med. Chem., 2006, 41, 106–113 CrossRef CAS PubMed.
- X. H. Liu, J. Zhu, A. na Zhou, B. A. Song, H. L. Zhu, L. S. Bai, P. S. Bhadury and C. X. Pan, Bioorg. Med. Chem., 2009, 17, 1207–1213 CrossRef CAS PubMed.
- J. J. Swidorski, Z. Liu, Z. Yin, T. Wang, D. J. Carini, S. Rahematpura, M. Zheng, K. Johnson, S. Zhang, P. F. Lin, D. D. Parker, W. Li, N. A. Meanwell, L. G. Hamann and A. Regueiro-Ren, Bioorg. Med. Chem. Lett., 2016, 26, 160–167 CrossRef CAS PubMed.
- M. E. Zarranz De Ysern and L. A. Ordoñez, Prog. Neuro-Psychopharmacol., 1981, 5, 343–355 CrossRef CAS PubMed.
- I. P. Singh and P. Shah, Expert Opin. Ther. Pat., 2017, 27, 17–36 CrossRef CAS PubMed.
- A. Pictet and T. Spengler, Ber. Dtsch. Chem. Ges., 1911, 44, 2030–2036 CrossRef CAS.
- W. M. Whaley and T. R. Govindachari, in Organic Reactions, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2011, pp. 151–190 Search PubMed.
- C. Kuhakarn, N. Panyachariwat and S. Ruchirawat, Tetrahedron Lett., 2007, 48, 8182–8184 CrossRef CAS.
- C. Gremmen, M. J. Wanner and G. J. Koomen, Tetrahedron Lett., 2001, 42, 8885–8888 CrossRef CAS.
- E. Mons, M. J. Wanner, S. Ingemann, J. H. Van Maarseveen and H. Hiemstra, J. Org. Chem., 2014, 79, 7380–7390 CrossRef CAS PubMed.
- E. Awuah and A. Capretta, J. Org. Chem., 2010, 75, 5627–5634 CrossRef CAS PubMed.
- S. Nagubandi and G. Fodor, J. Heterocycl. Chem., 1980, 17, 1457–1463 CrossRef CAS.
- M. Nicoletti, D. O'Hagan and A. M. Z. Slawin, J. Chem. Soc., Perkin Trans. 1, 2002, 2, 116–121 Search PubMed.
- M. Mihoubi, N. Micale, A. Scala, R. M. Jarraya, A. Bouaziz, T. Schirmeister, F. Risitano, A. Piperno and G. Grassi, Molecules, 2015, 20, 14902–14914 CrossRef CAS PubMed.
- M. Mottinelli, M. Sinreih, T. L. Rižner, M. P. Leese and B. V. L. Potter, ChemMedChem, 2020, 1–34 Search PubMed.
- C. E. Puerto Galvis and V. V. Kouznetsov, J. Org. Chem., 2019, 84, 15294–15308 CrossRef CAS PubMed.
- P. Pieper, E. McHugh, M. Amaral, A. G. Tempone and E. A. Anderson, Tetrahedron, 2020, 76, 130814 CrossRef CAS.
- L. Weber, Curr. Med. Chem., 2002, 9, 2085–2093 CrossRef CAS PubMed.
- P. Slobbe, E. Ruijter and R. V. A. Orru, MedChemComm, 2012, 3, 1189–1218 RSC.
- D. Insuasty, J. Castillo, D. Becerra, H. Rojas and R. Abonia, Molecules, 2020, 25, 1–71 CrossRef PubMed.
- L. Rong, L. Gao, H. Han, H. Jiang, Y. Dai and S. Tu, Synth. Commun., 2010, 40, 289–294 CrossRef CAS.
- K. Balamurugan, V. Jeyachandran, S. Perumal and J. C. Menéndez, Tetrahedron, 2011, 67, 1432–1437 CrossRef CAS.
- D. L. Priebbenow, F. M. Pfeffer and S. G. Stewart, Eur. J. Org. Chem., 2011, 1632–1635 CrossRef CAS.
- J. Jiang, X. Ma, C. Ji, Z. Guo, T. Shi, S. Liu and W. Hu, Chem.–Eur. J., 2014, 20, 1505–1509 CrossRef CAS PubMed.
- A. H. Shinde, N. Archith, S. Malipatel and D. S. Sharada, Tetrahedron Lett., 2014, 55, 6821–6826 CrossRef CAS.
- L. Henderson, D. W. Knight and A. C. Williams, Tetrahedron Lett., 2012, 53, 4657–4660 CrossRef CAS.
- T. Ogata, T. Kimachi, K. I. Yamada, Y. Yamamoto and K. Tomioka, Heterocycles, 2012, 86, 469–485 CrossRef CAS.
- S. Tussing, M. Ohland, G. Wicker, U. Flörke and J. Paradies, Dalton Trans., 2017, 46, 1539–1545 RSC.
- A. V. Murashkina, A. Y. Mitrofanov, Y. K. Grishin, V. B. Rybakov and I. P. Beletskaya, ChemistrySelect, 2018, 3, 6810–6813 CrossRef CAS.
- X.-J. Dai, O. D. Engl, T. León and S. L. Buchwald, Angew. Chem., 2019, 131, 3445–3449 CrossRef.
- J. D. Guzman, T. Pesnot, D. A. Barrera, H. M. Davies, E. McMahon, D. Evangelopoulos, P. N. Mortazavi, T. Munshi, A. Maitra, E. D. Lamming, R. Angell, M. C. Gershater, J. M. Redmond, D. Needham, J. M. Ward, L. E. Cuca, H. C. Hailes and S. Bhakta, J. Antimicrob. Chemother., 2014, 70, 1691–1703 CrossRef PubMed.
- M. A. Farha, K. Koteva, R. T. Gale, E. W. Sewell, G. D. Wright and E. D. Brown, Bioorg. Med. Chem. Lett., 2014, 24, 905–910 CrossRef CAS PubMed.
- S. Murugavel, N. Manikandan, D. Lakshmanan, K. Naveen and P. T. Perumal, J. Chil. Chem. Soc., 2015, 60, 3015–3020 CrossRef CAS.
- K. Bielawski, K. Leszczyńska, Z. Kałuża, A. Bielawska, O. Michalak, T. Daniluk, O. Staszewska-Krajewska, A. Czajkowska, N. Pawłowska and A. Gornowicz, Drug Des., Dev. Ther., 2017, 11, 2015–2028 CrossRef CAS PubMed.
- A. Zablotskaya, I. Segal, A. Geronikaki, I. Shestakova, V. Nikolajeva and G. Makarenkova, Pharmacol. Rep., 2017, 69, 575–581 CrossRef CAS PubMed.
- A. Zablotskaya, I. Segal, G. Kazachonokh, Y. Popelis, I. Shestakova and V. Nikolajeva, Silicon, 2018, 10, 1129–1138 CrossRef CAS.
- G. L. Lu, A. S. T. Tong, D. Conole, H. S. Sutherland, P. J. Choi, S. G. Franzblau, A. M. Upton, M. U. Lotlikar, C. B. Cooper, W. A. Denny and B. D. Palmer, Bioorg. Med. Chem., 2020, 28, 115784 CrossRef CAS PubMed.
- J. Krauss, C. Müller, J. Kießling, S. Richter, V. Staudacher and F. Bracher, Arch. Pharm., 2014, 347, 283–290 CrossRef CAS PubMed.
- T. R. Sutariya, B. M. Labana, N. J. Parmar, R. Kant, V. K. Gupta, G. B. Plata and J. M. Padrón, New J. Chem., 2015, 39, 2657–2668 RSC.
- E. O. Terent′eva, A. S. Saidov, Z. S. Khashimova, N. E. Tseomashko, S. A. Sasmakov, D. M. Abdurakhmanov, V. I. Vinogradova and S. S. Azimova, Chem. Nat. Compd., 2017, 53, 328–332 CrossRef.
- S. Murugesan, S. Ganguly and G. Maga, J. Chem. Sci., 2010, 122, 169–176 CrossRef CAS.
- J. J. Kennedy-Smith, N. Arora, J. R. Billedeau, J. Fretland, J. Q. Hang, G. M. Heilek, S. F. Harris, D. Hirschfeld, H. Javanbakht, Y. Li, W. Liang, R. Roetz, M. Smith, G. Su, J. M. Suh, A. G. Villaseñor, J. Wu, D. Yasuda, K. Klumpp and Z. K. Sweeney, MedChemComm, 2010, 1, 79–83 RSC.
- P. Zhan, W. Chen, Z. Li, X. Li, X. Chen, Y. Tian, C. Pannecouque, E. De Clercq and X. Liu, Bioorg. Med. Chem., 2012, 20, 6795–6802 CrossRef CAS PubMed.
- Schrödinger, Schrödinger Release 2018-1, 2018 Search PubMed.
- S. Chander, P. Ashok, A. Singh and S. Murugesan, Chem. Cent. J., 2015, 9, 1–13 CrossRef CAS PubMed.
- Y. Liao, Y. Ye, S. Li, Y. Zhuang, L. Chen, J. Chen, Z. Cui, L. Huo, S. Liu and G. Song, Eur. J. Med. Chem., 2020, 189, 112048 CrossRef CAS PubMed.
- W. A. Guiguemde, A. A. Shelat, D. Bouck, S. Duffy, G. J. Crowther, P. H. Davis, D. C. Smithson, M. Connelly, J. Clark, F. Zhu, M. B. Jiménez-Díaz, M. S. Martinez, E. B. Wilson, A. K. Tripathi, J. Gut, E. R. Sharlow, I. Bathurst, F. El Mazouni, J. W. Fowble, I. Forquer, P. L. McGinley, S. Castro, I. Angulo-Barturen, S. Ferrer, P. J. Rosenthal, J. L. Derisi, D. J. Sullivan, J. S. Lazo, D. S. Roos, M. K. Riscoe, M. A. Phillips, P. K. Rathod, W. C. Van Voorhis, V. M. Avery and R. K. Guy, Nature, 2010, 465, 311–315 CrossRef CAS PubMed.
- D. M. Floyd, P. Stein, Z. Wang, J. Liu, S. Castro, J. A. Clark, M. Connelly, F. Zhu, G. Holbrook, A. Matheny, M. S. Sigal, J. Min, R. Dhinakaran, S. Krishnan, S. Bashyum, S. Knapp and R. K. Guy, J. Med. Chem., 2016, 59, 7950–7962 CrossRef CAS PubMed.
- M. B. Jiménez-Díaz, D. Ebert, Y. Salinas, A. Pradhan, A. M. Lehane, M. E. Myrand-Lapierre, K. G. O'Loughlin, D. M. Shackleford, M. J. De Almeida, A. K. Carrillo, J. A. Clark, A. S. M. Dennis, J. Diep, X. Deng, S. Duffy, A. N. Endsley, G. Fedewa, W. A. Guiguemde, M. G. Gómez, G. Holbrook, J. Horst, C. C. Kim, J. Liu, M. C. S. Lee, A. Matheny, M. S. Martínez, G. Miller, A. Rodríguez-Alejandre, L. Sanz, M. Sigal, N. J. Spillman, P. D. Stein, Z. Wang, F. Zhu, D. Waterson, S. Knapp, A. Shelat, V. M. Avery, D. A. Fidock, F. J. Gamo, S. A. Charman, J. C. Mirsalis, H. Ma, S. Ferrer, K. Kirk, I. Angulo-Barturen, D. E. Kyle, J. L. Derisi, D. M. Floyd and R. K. Guy, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E5455–E5462 CrossRef PubMed.
- A. H. Gaur, J. S. McCarthy, J. C. Panetta, R. H. Dallas, J. Woodford, L. Tang, A. M. Smith, T. B. Stewart, K. C. Branum, B. B. Freeman, N. D. Patel, E. John, S. Chalon, S. Ost, R. N. Heine, J. L. Richardson, R. Christensen, P. M. Flynn, Y. Van Gessel, B. Mitasev, J. J. Möhrle, F. Gusovsky, L. Bebrevska and R. K. Guy, Lancet Infect. Dis., 2020, 20, 964–975 CrossRef CAS.
- J. N. Hanna, F. Ntie-Kang, M. Kaiser, R. Brun and S. M. N. Efange, RSC Adv., 2014, 4, 22856–22865 RSC.
- H. M. Malebo, S. D'alessandro, Y. A. Ebstie, H. Sorè, A. R. T. Guedoung, S. J. Katani, S. Parapini, D. Taramelli and A. Habluetzel, Drug Des., Dev. Ther., 2020, 14, 1593–1607 CrossRef CAS PubMed.
- K. Chauhan, M. Sharma, R. Shivahare, U. Debnath, S. Gupta, Y. S. Prabhakar and P. M. S. Chauhan, ACS Med. Chem. Lett., 2013, 4, 1108–1113 CrossRef CAS PubMed.
- S. Chander, P. Ashok, R. M. Reguera, M. Y. Perez-Pertejo, R. Carbajo-Andres, R. Balana-Fouce, K. V. Gowri Chandra Sekhar and M. Sankaranarayanan, Exp. Parasitol., 2018, 189, 49–60 CrossRef CAS PubMed.
- D. R. Cullen, J. Pengon, R. Rattanajak, J. Chaplin, S. Kamchonwongpaisan and M. Mocerino, ChemistrySelect, 2016, 1, 4533–4538 CrossRef CAS.
- P. S. Sadhu, S. N. Kumar, M. Chandrasekharam, L. Pica-Mattoccia, D. Cioli and V. J. Rao, Bioorg. Med. Chem. Lett., 2012, 22, 1103–1106 CrossRef CAS PubMed.
- W. W. Duan, S. J. Qiu, Y. Zhao, H. Sun, C. Qiao and C. M. Xia, Bioorg. Med. Chem. Lett., 2012, 22, 1587–1590 CrossRef CAS PubMed.
- L. Dong, W. Duan, J. Chen, H. Sun, C. Qiao and C. M. Xia, PLoS One, 2014, 9, e112163 CrossRef.
- X. Wang, D. Yu, C. Li, T. Zhan, T. Zhang, H. Ma, J. Xu and C. Xia, Parasites Vectors, 2019, 12, 1–13 CrossRef PubMed.
- Z. X. Wang, J. L. Chen and C. Qiao, Chem. Biol. Drug Des., 2013, 82, 216–225 CrossRef CAS PubMed.
- W. L. Wang, L. J. Song, X. Chen, X. R. Yin, W. H. Fan, G. P. Wang, C. X. Yu and B. Feng, Molecules, 2013, 18, 9163–9178 CrossRef CAS PubMed.
- S. Guglielmo, D. Cortese, F. Vottero, B. Rolando, V. P. Kommer, D. L. Williams, R. Fruttero and A. Gasco, Eur. J. Med. Chem., 2014, 84, 135–145 CrossRef CAS PubMed.
- G. Rai, C. J. Thomas, W. Leister and D. J. Maloney, Tetrahedron Lett., 2009, 50, 1710–1713 CrossRef CAS PubMed.
- H. M. Alger, A. A. Sayed, M. J. Stadecker and D. L. Williams, Int. J. Parasitol., 2002, 32, 1285–1292 CrossRef CAS.
- A. Gasco and K. Schoenafinger, in Nitric Oxide Donors, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, FRG, 2005, pp. 131–175 Search PubMed.
- A. A. Sayed, A. Simeonov, C. J. Thomas, J. Inglese, C. P. Austin and D. L. Williams, Nat. Med., 2008, 14, 407–412 CrossRef CAS PubMed.
- A. Wąsik, E. Mozdzeń, I. Romańska, J. Michaluk and L. Antkiewicz-Michaluk, Eur. J. Pharmacol., 2013, 700, 110–117 CrossRef PubMed.
- A. Patsenka and L. Antkiewicz-Michaluk, Pol. J. Pharmacol., 2004, 56, 727–734 CAS.
- L. Antkiewicz-Michaluk, A. Wąsik and J. Michaluk, Neurotoxic. Res., 2014, 25, 1–12 CrossRef CAS PubMed.
- L. Antkiewicz-Michaluk, J. Michaluk, M. Mokrosz, I. Romanska, E. Lorenc-Koci, S. Ohta and J. Vetulani, J. Neurochem., 2001, 78, 100–108 CrossRef CAS PubMed.
- M. Maes, R. Yirmyia, J. Noraberg, S. Brene, J. Hibbeln, G. Perini, M. Kubera, P. Bob, B. Lerer and M. Maj, Metab. Brain Dis., 2009, 24, 27–53 CrossRef CAS PubMed.
- L. Antkiewicz-Michaluk, J. W. Lazarewicz, A. Patsenka, M. Kajta, E. Zieminska, E. Salinska, A. Wasik, K. Golembiowska and J. Vetulani, J. Neurochem., 2006, 97, 846–856 CrossRef CAS PubMed.
- E. Mozdzeń, M. Papp, P. Gruca, A. Wąsik, I. Romańska, J. Michaluk and L. Antkiewicz-Michaluk, Eur. J. Pharmacol., 2014, 729, 107–115 CrossRef PubMed.
- E. Możdżeń, A. Wąsik, I. Romańska, J. Michaluk and L. Antkiewicz-Michaluk, Pharmacol. Rep., 2017, 69, 566–574 CrossRef PubMed.
- A. Wąsik, E. Możdżeń, J. Michaluk, I. Romańska and L. Antkiewicz-Michaluk, Neurotoxic. Res., 2014, 25, 323–334 CrossRef PubMed.
- E. Możdżeń, I. Babińska, J. Wójcikowski and L. Antkiewicz -Michaluk, Pharmacol. Rep., 2019, 71, 1140–1146 CrossRef PubMed.
- B. Habibi, J. P. Cartron, M. Bretagne, P. Rouger and C. Salmon, Vox Sang., 1981, 40, 79–84 CrossRef CAS PubMed.
- J. H. Harrison and D. J. Jollow, J. Pharmacol. Exp. Ther., 1986, 238, 1045–1054 CAS.
- J. Yu, D. G. Brown and D. Burdette, Drug Metab. Dispos., 2010, 38, 1767–1778 CrossRef CAS PubMed.
- A. D. Pechulis, J. P. Beck, M. A. Curry, M. A. Wolf, A. E. Harms, N. Xi, C. Opalka, M. P. Sweet, Z. Yang, A. S. Vellekoop, A. M. Klos, P. J. Crocker, C. Hassler, M. Laws, D. B. Kitchen, M. A. Smith, R. E. Olson, S. Liu and B. F. Molino, Bioorg. Med. Chem. Lett., 2012, 22, 7219–7222 CrossRef CAS PubMed.
- S. Liu, C. Zha, K. Nacro, M. Hu, W. Cui, Y. L. Yang, U. Bhatt, A. Sambandam, M. Isherwood, L. Yet, M. T. Herr, S. Ebeltoft, C. Hassler, L. Fleming, A. D. Pechulis, A. Payen-Fornicola, N. Holman, D. Milanowski, I. Cotterill, V. Mozhaev, Y. Khmelnitsky, P. R. Guzzo, B. J. Sargent, B. F. Molino, R. Olson, D. King, S. Lelas, Y. W. Li, J. Kim, T. Molski, A. Orie, A. Ng, R. Haskell, W. Clarke, R. Bertekap, J. Oconnell, N. Lodge, M. Sinz, S. Adams, R. Zaczek and J. E. Macor, ACS Med. Chem. Lett., 2014, 5, 760–765 CrossRef CAS PubMed.
- D. K. Sukumarapillai, K. Kooi-Yeong, Y. Kia, V. Murugaiyah and S. K. Iyer, Med. Chem. Res., 2016, 25, 1705–1715 CrossRef CAS.
- N. Sudhapriya, A. Manikandan, M. R. Kumar and P. T. Perumal, Bioorg. Med. Chem. Lett., 2019, 29, 1308–1312 CrossRef CAS.
- N. Ghamari, O. Zarei, D. Reiner, S. Dastmalchi, H. Stark and M. Hamzeh-Mivehroud, Chem. Biol. Drug Des., 2019, 93, 832–843 CrossRef CAS PubMed.
- N. Ghamari, S. Dastmalchi, O. Zarei, J. A. Arias-Montaño, D. Reiner, F. Ustun-Alkan, H. Stark and M. Hamzeh-Mivehroud, Chem. Biol. Drug Des., 2020, 95, 279–290 CrossRef CAS PubMed.
- A. D. Medhurst, A. R. Atkins, I. J. Beresford, K. Brackenborough, M. A. Briggs, A. R. Calver, J. Cilia, J. E. Cluderay, B. Crook, J. B. Davis, R. K. Davis, R. P. Davis, L. A. Dawson, A. G. Foley, J. Gartlon, M. I. Gonzalez, T. Heslop, W. D. Hirst, C. Jennings, D. N. C. Jones, L. P. Lacroix, A. Martyn, S. Ociepka, A. Ray, C. M. Regan, J. C. Roberts, J. Schogger, E. Southam, T. O. Stean, B. K. Trail, N. Upton, G. Wadsworth, J. A. Wald, T. White, J. Witherington, M. L. Woolley, A. Worby and D. M. Wilson, J. Pharmacol. Exp. Ther., 2007, 321, 1032–1045 CrossRef CAS PubMed.
- D. Vohora and M. Bhowmik, Front. Syst. Neurosci., 2012, 1–27 Search PubMed.
- Y. Chen, C. Su, L. Wang, J. Qin, S. Wei and H. Tang, Mol. Diversity, 2019, 23, 709–722 CrossRef CAS.
- Y. Fang, H. Zhou, Q. Gu and J. Xu, Eur. J. Med. Chem., 2019, 167, 133–145 CrossRef CAS PubMed.
- Z. Pu, S. Ma, L. Wang, M. Li, L. Shang, Y. Luo and W. Chen, Amyloid-beta Degradation and Neuroprotection of Dauricine Mediated by Unfolded Protein Response in a Caenorhabditis elegans Model of Alzheimer's disease, 2018, vol. 392 Search PubMed.
- L. Wang, Z. Pu, M. Li, K. Wang, L. Deng and W. Chen, Life Sci., 2020, 243, 1–8 Search PubMed.
- P. Liu, X. Chen, H. Zhou, L. Wang, Z. Zhang, X. Ren, F. Zhu, Y. Guo, X. Huang, J. Liu, P. S. Spencer and X. Yang, Oxid. Med. Cell. Longevity, 2018, 2018, 2025914 Search PubMed.
- L. Antkiewicz-Michaluk, J. Michaluk, M. Mokrosz, I. Romanska, E. Lorenc-Koci, S. Ohta and J. Vetulani, J. Neurochem., 2001, 78, 100–108 CrossRef CAS PubMed.
- A. Wąsik, I. Romańska and L. Antkiewicz-Michaluk, Neurotoxic. Res., 2009, 15, 15–23 CrossRef.
- S. Shavali and M. Ebadi, Neurotoxicology, 2003, 24, 417–424 CrossRef CAS PubMed.
- Y. Kotake, Y. Tasaki, Y. Makino, S. Ohta and M. Hirobe, J. Neurochem., 1995, 65, 2633–2638 CrossRef CAS PubMed.
- Y. Kotake, S. Ohta, I. Kanazawa and M. Sakurai, Neuroscience, 2003, 117, 63–70 CrossRef CAS PubMed.
- R. Kohta, Y. Kotake, T. Hosoya, T. Hiramatsu, Y. Otsubo, H. Koyama, Y. Hirokane, Y. Yokoyama, H. Ikeshoji, K. Oofusa, M. Suzuki and S. Ohta, J. Neurochem., 2010, 114, 1291–1301 CAS.
- A. Wąsik, I. Romańska, J. Michaluk, A. Zelek-Molik, I. Nalepa and L. Antkiewicz-Michaluk, Neurotoxic. Res., 2016, 29, 351–363 CrossRef PubMed.
- A. Wąsik, I. Romańska and L. Antkiewicz-Michaluk, Neurotoxic. Res., 2016, 30, 648–657 CrossRef PubMed.
- A. Wąsik, D. Polak, I. Romańska, J. Michaluk and L. Antkiewicz-Michaluk, Pharmacol. Rep., 2016, 68, 1205–1213 CrossRef PubMed.
- A. Wąsik, I. Romańska, A. Zelek-Molik and L. Antkiewicz-Michaluk, Neurotoxic. Res., 2018, 33, 523–531 CrossRef PubMed.
- N. Katagiri, S. Chida, K. Abe, H. Nojima, M. Kitabatake, K. Hoshi, Y. Horiguchi and K. Taguchi, Brain Res., 2010, 1321, 133–142 CrossRef CAS PubMed.
- K. Saitoh, K. Abe, T. Chiba, N. Katagiri, T. Saitoh, Y. Horiguchi, H. Nojima and K. Taguchi, Pharmacol. Rep., 2013, 65, 1204–1212 CrossRef CAS PubMed.
- H. J. Son, J. A. Lee, N. Shin, J. H. Choi, J. W. Seo, D. Y. Chi, C. S. Lee, E. M. Kim, H. Choe and O. Hwang, Br. J. Pharmacol., 2012, 165, 2213–2227 CrossRef CAS PubMed.
- H. J. Son, S. H. Han, J. A. Lee, C. S. Lee, J. W. Seo, D. Y. Chi and O. Hwang, Eur. J. Pharmacol., 2016, 771, 152–161 CrossRef CAS PubMed.
- H. Sun, X. He, C. Liu, L. Li, R. Zhou, T. Jin, S. Yue, D. Feng, J. Gong, J. Sun, J. Ji and L. Xiang, ACS Chem. Neurosci., 2017, 8, 155–164 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2021 |