
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGround- and excited-state characteristics in photovoltaic polymer N2200†
Guanzhao Wen‡
a,
Xianshao Zou‡b,
Rong Hu c,
Jun Penga,
Zhifeng Chena,
Xiaochuan Hed,
Geng Dong*ef and
Wei Zhang*a
c,
Jun Penga,
Zhifeng Chena,
Xiaochuan Hed,
Geng Dong*ef and
Wei Zhang*a
aSchool of Physics and Materials Science, Guangzhou University, Guangzhou 510006, China. E-mail: wzhang@gzhu.edu.cn; Tel: +86-136-4279-2676
bDivision of Chemical Physics, Lund University, Lund 22100, Sweden
cSchool of Materials Science and Engineering, Chongqing University of Arts and Sciences, Chongqing 402160, China
dSongshan Lake Materials Laboratory, Dongguan 523808, China
eDepartment of Biochemistry and Molecular Biology, Shantou University Medical College, Shantou 515041, China. E-mail: gdong@stu.edu.cn; Tel: +86-187-3110-6711
fMedical Informatics Research Center, Shantou University Medical College, Shantou 515041, China
First published on 14th June 2021
Abstract
As a classical polymer acceptor material, N2200 has received extensive attention and research in the field of polymer solar cells (PSCs). However, the intrinsic properties of ground- and excited-states in N2200, which are critical for the application of N2200 in PSCs, remain poorly understood. In this work, the ground- and excited-state properties of N2200 solution and film were studied by steady-state and time-resolved spectroscopies as well as time-dependent density functional theory (TD-DFT) calculations. The transition mechanism of absorption peaks of N2200 was evaluated through the natural transition orbitals (NTOs) and hole–electron population analysis by TD-DFT. Time-resolved photoluminescence (TRPL) study shows that the lifetimes of singlet excitons in N2200 chlorobenzene solution and film are ∼90 ps and ∼60 ps, respectively. Considering the absolute quantum yield of N2200 film, we deduce that the intrinsic lifetime of singlet exciton can be as long as ∼20 ns. By comparing the TRPL and transient absorption (TA) kinetics, we find that the decay of singlet excitons in N2200 solution is dominated by a fast non-radiative decay process, and the component induced by intersystem crossing is less than 5%. Besides that, the annihilation radius, annihilation rate and diffusion length of singlet excitons in N2200 film were evaluated as 3.6 nm, 2.5 × 10−9 cm3 s−1 and 4.5 nm, respectively. Our work provides comprehensive information on the excited states of N2200, which is helpful for the application of N2200 in all-PSCs.
1. Introduction
Over the past few decades, polymer solar cells (PSCs) including p-type conjugated polymers and n-type organic semiconductors have been widely studied by researchers due to the outstanding advantages such as lightweight, flexibility, and low cost.1–4 Among the electron acceptor materials of PSCs, fullerenes are the most widely studied because of their good carrier mobility and electron capture ability in blend films.5–7 However, the weak light absorption of fullerene in the visible-near infrared region limits the further improvement of the power conversion efficiency (PCE) of PSCs. Therefore, alternative materials for fullerenes need to be developed. In recent years, the rapid development of non-fullerene acceptors, such as polymers and small organic molecules, has greatly promoted the improvement of PCE of PSCs.8–10 At present, the PCE of single-junction PSCs based on non-fullerene acceptors has reached 18%,11 which is suitable for commercialization in terms of PCE.Poly{[N,N′-bis(2-octyldodecyl)-naphthalene-1,4,5,8-bis(dicarboximide)-2,6-diyl]-alt-5,5'-(2,2′-bithiophene)} (known as Polyera ActivInk N2200, also named as P(NDI2OD-T2)), was first reported and successfully applied for transistors with a high electron mobility of 0.85 cm2 V−1 s−1 in 2009.12 Since then, N2200 is often employed as acceptor material in all-PSCs.13–15 In 2011, P3HT:N2200 photovoltaic system was constructed for the application of PSCs, and a PCE of 0.16% was achieved.16 After that, the PCE of N2200-based all-PSCs have made great progresses from the development of donor material design and device optimization.17–20 For instance, Wang et al. employed PBDTTS-FTAZ and N2200 as an absorption-complementary active layer to get a PCE of 4.8%.19 Li and co-workers selected a fluorine medium bandgap copolymer J51 as donor to match N2200, then acquiring a PCE of 8.27%.17 It is worth mentioning that the PTzBI-Si:N2200 photovoltaic system showed a milestone PCE of 11.76% after the morphology optimization.20
Time-resolved optical techniques are important tools for understanding photoelectrical conversion properties in n-type polymer-based PSCs.19,21–23 For example, Zhang et al. studied the carrier properties of PBDB-T:N2200 solar cells by using time-resolved microwave conductivity (TRMC), they found that the all-PSC (PBDB-T:N2200) has a longer TRMC lifetime compared with those of ITIC based (PBDB-T:ITIC) and PCBM based (PBDB-T:PCBM) devices.21 Kim et al. studied the photo-stability of PBDTTTPD:P(NDI2HD-T) under various light soaking conditions by using transient absorption, they found that the charge generation and transport dynamics were not changed under photo-stress, suggesting a superior photo-stability of n-type polymer (P(NDI2HD-T)) based devices under N2 conditions.23 Chen et al. studied exciton dissociation processes in all-PSCs by using time-resolved photoluminescence (TRPL), they found the exciton dissociation efficiency of solar cells can be enhanced by introducing a dye molecule into the n-type polymer N2200.22
A critical step of charge photogeneration processes is related to the migration of photogeneration excitons to the donor/acceptor interface where they can be dissociated into the free charge carriers.24,25 To realize high exciton dissociation efficiency, the excitons must diffuse and reach the interface within its lifetime.26 Therefore, exciton diffusion length of neat polymer is an essential parameter for high-efficient organic photovoltaics materials and devices. Moreover, exciton diffusion characteristics are closely related to the excited-state properties of organic semiconductor materials. Although the dissociation process of N2200 exciton in blend active layer has been studied by using TRPL,23 the excited-state properties of neat N2200 have not been systematically studied yet.
In this work, the ground- and excited-states properties of N2200 solution and film were studied by steady-state and time-resolved spectroscopies as well as time-dependent density functional theory (TD-DFT) calculations. First, hole–electron population analysis and natural transition orbitals (NTOs) by TD-DFT were employed to understand the transition mechanism of steady-state absorption peaks of N2200 solution. Then, TRPL and transient absorption (TA), were employed to understand the singlet exciton lifetime and the decay paths of the excited state in N2200 solution and film. Finally, the annihilation radius, annihilation rate and diffusion length of singlet excitons, which are closely correlated with charge photogeneration processes in N2200-based solar cells, were evaluated based on exciton annihilation dynamics of N2200 film under various excitation fluencies. Our work emphasizes the role of controlling the size of N2200 phase in active layers for developing high efficient N2200-based solar cells.
2. Materials and methods
2.1 Sample information
The polymer N2200 (Mw: 100–150 kDa; PDI: 3.0–4.5) was used as purchased from Solarmer Materials Inc. (Beijing) without further processing. For N2200 solution preparation, N2200 was dissolved in chlorobenzene (CB) solution and was put into a 1 mm cuvette for spectrum measurements. For the fabrication of N2200 film, N2200 were dissolved in CB to obtain a solution of 8 mg mL−1. The glass substrates were treated by ultrasonication in detergent and washed with deionized water, acetone, ethanol, and isopropyl alcohol. Then, the dissolved N2200 solution was dropped on the glass substrate and spin-coated at 1000 rpm for 50 s to fabricate the N2200 film with a thickness of ∼105 nm that measured by a step profiler (Alpha-Step D-100). Sample preparation, dissolution, and spin-coating were all performed in a nitrogen-filled glove box.2.2 Steady-state absorption and photoluminescence (PL) spectra measurements
Absorption spectra of N2200 solution and film were recorded on the UV-visible spectrometer Agilent 8453. For fluorescence measurements, the sample surface was at an angle of 45° with the excitation light. The excitation wavelength was 532 nm for N2200 solution and film. Photoinduced fluorescence was collected by an optical fiber spectrometer (AvaSpec-ULS2048CL-RS-EVO-UA) through a series of collection lenses. The collected fluorescence signals were all spectrally corrected by a standard light source (Ocean Optics, LS-1-CAL). All the measurements were conducted at room temperature in air.2.3 Transient absorption (TA) measurements
TA experiments were performed on a HARPIA-TA system (Light Conversion). A 1030 nm pulsed laser with a pulse duration of 190 fs and repetition rate of 100 kHz, was used as the fundamental light source of the HARPIA-TA system. The 1030 nm laser was divided into two parts by a beam splitter. One beam was employed to pump an optical parametric amplifier system (OPA, Light Conversion) system, and the output at 720 nm and 750 nm was used for exciting N2200 solution and films, respectively. The other beam was employed to generate the probe light in the range of 500–950 nm by exciting a sapphire plate. The delay between pump and probe was regulated by a mechanical delay stage. All the measurements were conducted at room temperature in air.2.4 Time-resolved photoluminescence (TRPL) and absolute photoluminescence quantum yield (PLQY) measurements
TRPL was measured in a setup described in the previous works.27,28 A fs pulsed laser (Tsunami, Spectra-Physical) with wavelength of 800 nm, pulse duration of ∼100 fs and repetitive rate of 80 MHz, was employed as the laser source. The frequency-doubled laser (400 nm) was employed as exciting light. PL signal of the sample was collected and focused into the input slit of the spectrometer (Chromex). The output PL from the spectrometer was directed into and measured by a streak camera system (Hamamatsu C6860). TRPL images have been corrected by a reference white light source (LS-1-CAL, Ocean Optics). The absolute PL quantum yield (QY) of the N2200 solution was measured by an integration sphere method. The sample was placed into the integrating sphere (F3029, Quanta-φ, HORIBA), and was excited by a CW laser with an output wavelength of 660 nm. An optical fiber spectrometer (Avaspec-ULS2048CL-RS-EVO-UA, Avantes) was used to measure the output light from the integrating sphere. All measurements were performed at room temperature in air.3. Results and discussion
3.1 Molecular structure and frontier orbitals
The TD-DFT calculations at the B3LYP/6-311G(d,p)29,30 level were carried out with Gaussian 16 software package31 for understanding the molecular orbitals properties of N2200 polymer. Considering the chain length would affect the lowest unoccupied molecular orbital (LUMO) and the highest occupied molecular orbital (HOMO) energy levels, N2200 oligomers from one to seven repeating units have been calculated. Since the alkyl branches (R = C8H17 and C10H21) on naphthalene diimide (NDI) unit of oligomers typically have a weak influence on the molecular orbital properties of polymers,32 alkyl branches were substituted by methyl groups during the calculations.The calculated electron density distributions of LUMO and HOMO for NDI, bithiophene (T2) units and N2200 oligomers are shown in Fig. 1. For the NDI and T2 units, we find both the electron densities of the LUMO and HOMO wave functions are distributed over the whole units. For the N2200-1 oligomer, the electron density of the LUMO wave function tends to be localized on the NDI unit, while the electron density of the HOMO wave function is delocalized over both the NDI and T2 units. By examining the electron densities of HOMO and LUMO at various repeating units (n), we find that the electron densities of LUMO and HOMO are distributed on each repeating unit evenly when n < 4, while for longer oligomers (n > 4), the electron densities are localized on several repeating units. Moreover, we note that the LUMO energy level becomes lower, while the HOMO energy level becomes higher (as shown in Table 1), resulting in a reduced band-gap with the increase of repeating units. This result shows that when the number of repeating units increases to 6, the LUMO and HOMO energy levels of N2200 oligomers change very little.
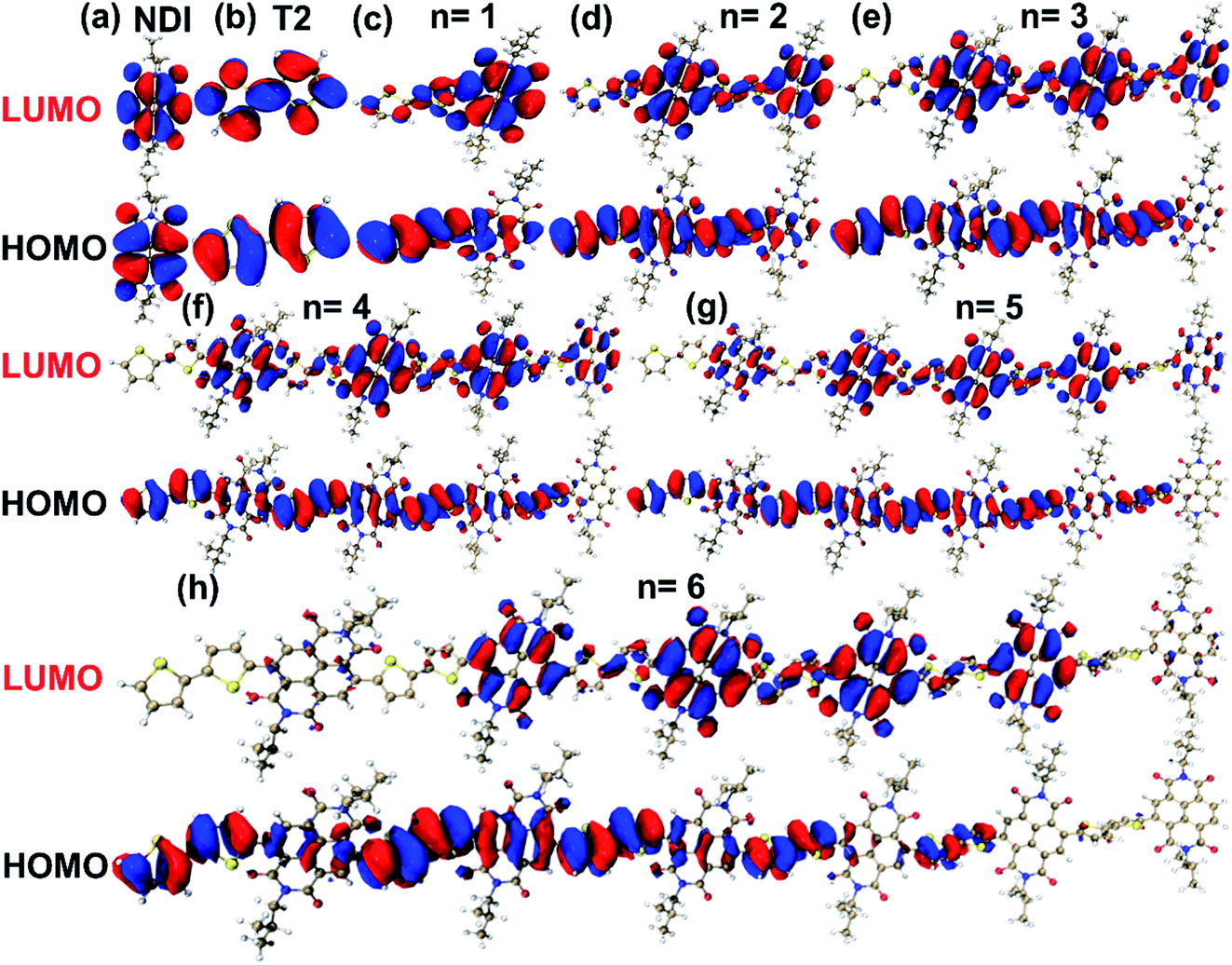 | ||
| Fig. 1 The electron density distribution of LUMO (up) and HOMO (down) energy levels for NDI, T2 units and N2200 oligomers with various repeating units by TD-DFT at the level of B3LYP/6-311G(d,p). n is the number of repeating units. The isosurface value was set at 0.01 a. u. | ||
| N2200-n | HOMO (eV) | LUMO (eV) | Band gap (eV) |
|---|---|---|---|
| N2200-1 | −5.862 | −3.569 | 2.293 |
| N2200-2 | −5.757 | −3.644 | 2.113 |
| N2200-3 | −5.730 | −3.677 | 2.053 |
| N2200-4 | −5.704 | −3.693 | 2.011 |
| N2200-5 | −5.670 | −3.703 | 1.997 |
| N2200-6 | −5.678 | −3.708 | 1.970 |
| N2200-7 | −5.680 | −3.706 | 1.974 |
3.2 The steady-state optical properties
Fig. 2 shows the normalized UV-vis absorption and PL spectra of N2200 in CB solution and thin-film. It can be seen that the absorption spectrum exhibits two distinct absorption peaks at around 390 and 700 nm in both solution and film. To understand the transition mechanism of these absorption peaks, the absorption spectra of N2200 oligomers with a dielectric constant of 5.6968 (CB solution) were simulated by TD-DFT at the level of B3LYP/6-311G(d,p), as shown in Fig. 2(a) and ESI Fig. S1.† It can be seen that the main absorption peaks are red-shifted with increasing the oligomer length. TD-DFT calculations show that, with the increase of oligomer length, LUMO energy level becomes lower and HOMO energy level becomes higher (as shown in Table 1), which will result in a reduced band-gap with the increase of repeating units. The reduced band-gap induces the redshift of the main absorption peaks with the increase of oligomer length. We also noted that the simulated UV-vis absorption spectra are red-shifted compared with the experimental data. This could origin from the rough description of charge transfer and Rydberg states presented by B3LYP.33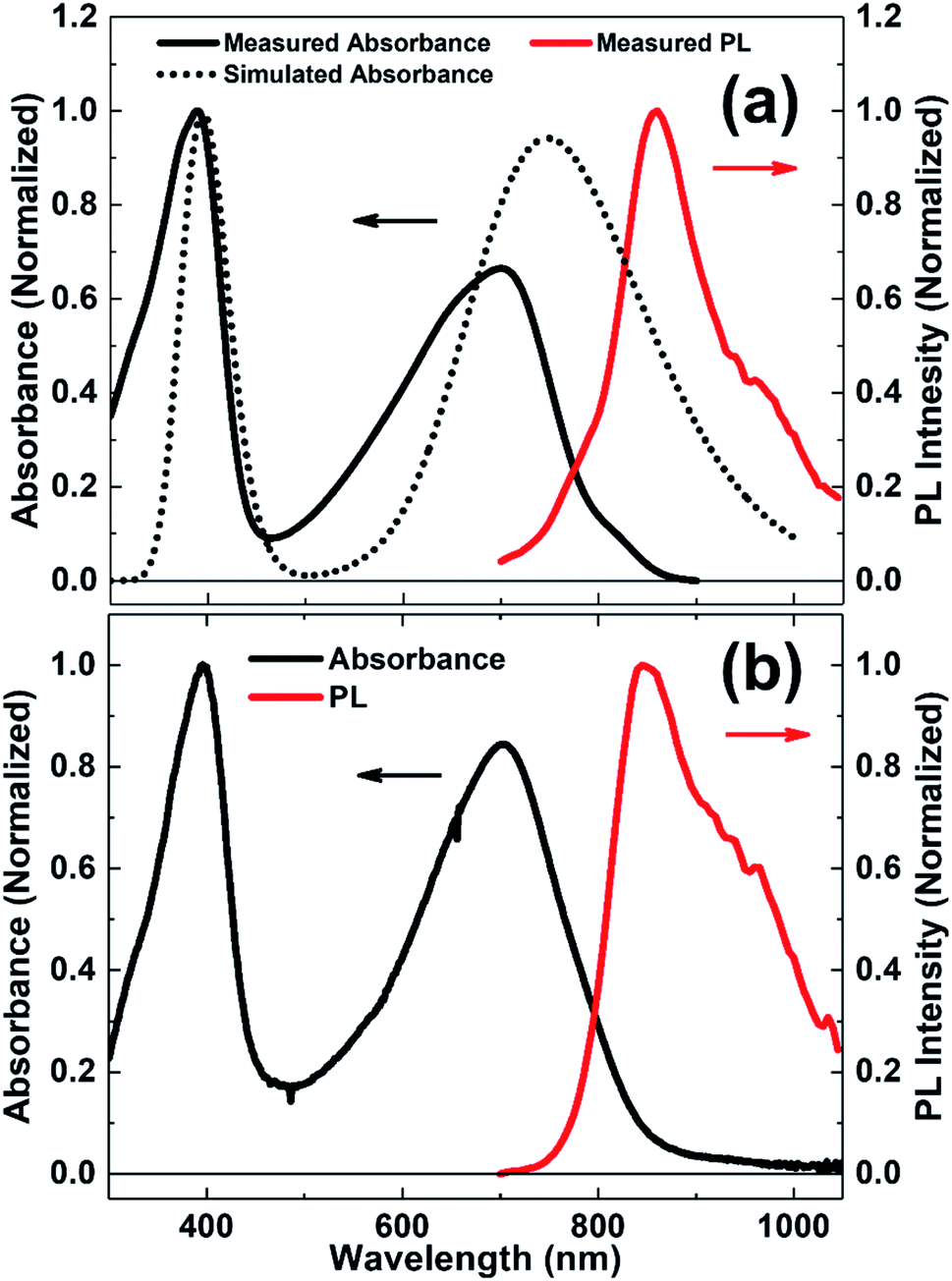 | ||
| Fig. 2 (a) The experimental (solid lines) and simulated (dot line) absorption and PL spectra of N2200 in chlorobenzene solution; the simulation was performed by TD-DFT at the level of B3LYP/6-311G(d,p) and a Gaussian function with a full width at half-maximum of 0.45 eV was used. (b) Steady-state absorption (black line) and PL (red line) spectra of N2200 thin film. The excitation wavelength for PL measurements of N2200 solution and film is 532 nm. | ||
By examining the calculation of N2200 oligomer with three repeating units, the absorption characteristics are assigned. The absorption peak of 700 nm (experimental value) in N2200 solution is characterized by S0 → S1 transition mainly contributed by HOMO (H) → LUMO (L) (83.9%) and H−1 → L+1 (6.9%) molecular orbital transitions. In addition, the absorption band at around 390 nm (experimental value) is predicted by the transition from the S0 to S16 excited state, that mainly contributed by H−3 → L+1 (14.6%), H → L+4 (14.5%), H−11 → L (10.9%) and H → L+3 (10.5%). To get a better insight into the electronic excitation process of these two absorption peaks in the N2200 solution, the NTOs34 and hole–electron population analysis35 were calculated in Multiwfn (version 3.7)36 and VMD (version 1.9.4)37 software, as shown in ESI Fig. S2 and S3.† For the S0 → S1 transition, the hole distribution of the NTO and overlap electron–hole density map are more clearly localized on the T2 donor unit, while the electron tends to distribute on the NDI acceptor unit, due to the large twists between the T2 and NDI units in N2200 monomer hindering the delocalization along the backbone.38 From the analysis of the overlap of electron–hole density, we find both the absorption peaks at around 390 and 700 nm exhibit the characteristics of the charge transfer transitions. Comparing with the absorption in solution, the absorption maximum of N2200 film slightly redshifts (∼5 nm). This can be attributed to stronger intermolecular interactions and better coplanar structures in the solid-state.39,40 Meanwhile, we observe a pronounced Stokes shift of the emission spectrum in both solution and film (∼160 nm for the solution, ∼145 nm for the film), which is due to energy losses between excitation and emission.41,42
3.3 Excited-state properties of N2200 solution and film
In PSCs, the exciton dissociation efficiency of the blended active layer is closely related to the lifetime of exciton in the neat donor (or neat acceptor) film. Assuming phase separation and exciton diffusion coefficient are the same for the blended active layers, a longer the exciton lifetime of the neat electron-donor (or neat electron-acceptor) film would have a longer exciton diffusion length and thus a higher exciton dissociation efficiency in the blend film, which could increase the PCE of PSCs.19 The TRPL measurements were first conducted to study the lifetime of singlet emissive excitons in N2200 solution and film, as shown in Fig. 3(a). We find that the kinetics of the N2200 solution and film can be fitted by single exponential decay functions. This suggests the kinetics are dominated by the first-order reaction process, and the higher-order reaction processes such as exciton annihilation can be neglected under the applied excitation fluence.43,44 It is shown that the PL lifetimes of N2200 solution and film are ∼90 ps and ∼60 ps, respectively. We note the lifetime of N2200 film is much shorter than some classic donor polymers, such as P3HT (∼470 ps),45 MEH-PPV (∼450 ps),46 and APFO-3 (∼500 ps).47 The fast decay rate of film could be dominated by either radiative recombination rate of N2200, non-radiative decay rate, or both. In the previous studies, the absolute PLQY of N2200 film is found to be ∼0.31%,19 which is much lower than regular polymer donors. Considering the PLQY (0.3%) and TRPL lifetime (∼60 ps) of N2200 film, the intrinsic exciton lifetime of N2200 film is deduced as 20 ns. Low PLQY of N2200, suggests the measured PL lifetime of N2200 film (∼60 ps) is dominated by non-radiative channels, for example, inter-system crossover processes, charge separation in the neat polymer film, etc.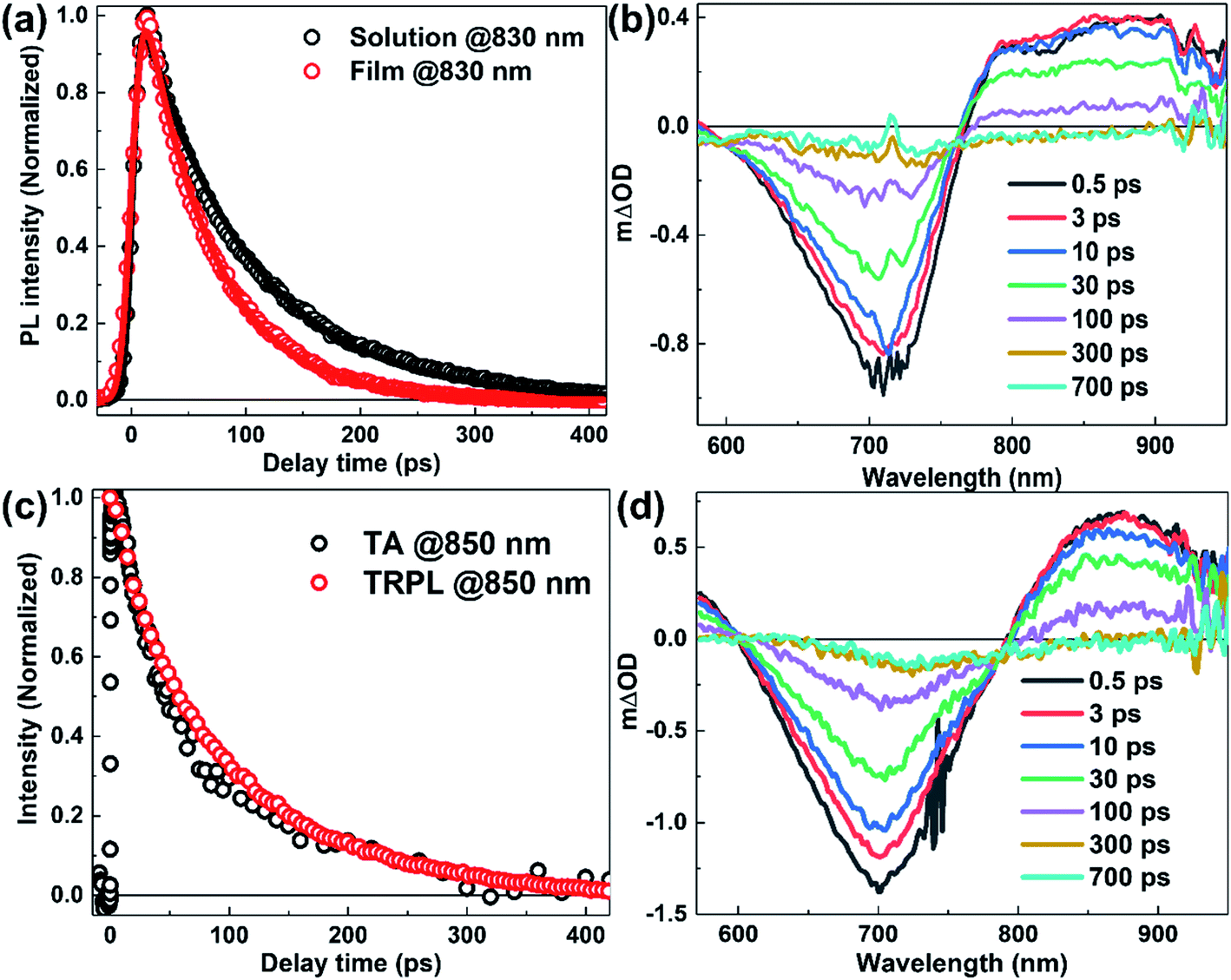 | ||
| Fig. 3 (a) TRPL kinetics of N2200 solution and film at indicated probe wavelength. The excited wavelength was 780 and 400 nm for N2200 solution and film, respectively. The solid lines are fitting curves based on single exponential decay functions. (b) TA spectrum of N2200 in CB solution at indicated delay time after photoexcitation at 720 nm with an excitation fluency of 8.49 × 1013 photons per cm2 per pulse. (c) Comparison of the TRPL and TA kinetics in the N2200 solution. (d) TA spectrum of N2200 film at indicated delay time after photoexcitation at 750 nm with an excitation fluency of 3.16 × 1013 photons per cm2 per pulse. | ||
TA measurements of N2200 solution and film were conducted for a better understanding of the excited-state decay processes. Fig. 3(b) shows the TA spectrum of N2200 solution at various delay times. It can be seen that a broad positive absorption band and a negative bleaching band appear immediately in the wavelength range of 600–950 nm after photoexcitation, and then the bands decay with time. To assign the absorption band, TA kinetics at 850 nm is compared with TRPL kinetics, which is induced by the emission of singlet excitons, as shown in Fig. 3(c). We find that the TA kinetics at 850 nm is similar to the TRPL kinetics in N2200 solution, strongly supporting that the absorption band in the wavelength range of >760 nm origin from the absorption of S1 excitons. Since TA kinetics at 850 nm can be fitted by the single exponential decay function, the exciton annihilation process can be also neglected for the measurement under the employed excitation fluency. By comparing TRPL kinetics with TA bleaching kinetics at 700 nm, we find that there is a long-lived component of TA kinetics at 700 nm besides the fast decay component corresponding to the decay component of TRPL kinetics, as shown in ESI Fig. S5.† The long-lived component is most likely induced by the formation of triplet excitons. It should be noted that the proportion of the long-lived component is less than 5%, indicating the intersystem crossing induced S1 exciton loss is less than 5%. In other words, the PLQY could achieve >95% if the intersystem crossing is the only loss channel of S1 exciton. However, the PLQY of N2200 solution is measured as 0.68% after photoexcitation at 660 nm by using integration sphere methods. This suggest inter-system crossing is not the dominant loss channel, decay of TRPL kinetics is dominated by other non-radiative recombination processes, such as internal-conversion induced fast non-radiative decay.
The TA spectrum of N2200 film (Fig. 3(d)) exhibits a bleaching band at ∼700 nm and an absorption band at ∼850 nm that can be attributed to the absorption of singlet excitons (see ESI Fig. S6†). By examining the kinetics at 700 nm under various excitation fluencies (Fig. 4(a)), we find the decay becomes faster with increasing excitation power at an early timescale, which can be attributed to the exciton–exciton annihilation under high excitation power.
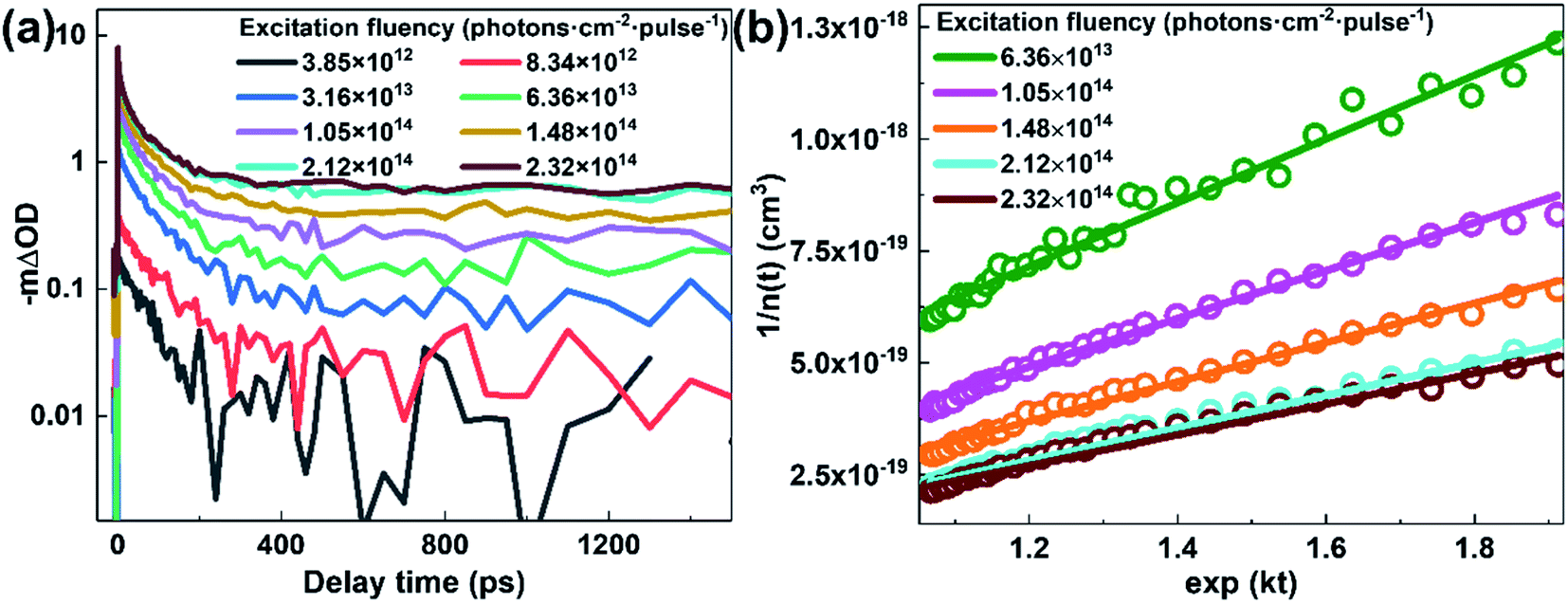 | ||
| Fig. 4 (a) TA kinetics of N2200 film at a probe wavelength of 700 nm after photoexcitation at 750 nm under various excitation fluencies. (b) Reciprocal of exciton density vs. exp(kt) of N2200 film under various excitation fluencies. The solid lines are linear fitting curves. | ||
3.4 Exciton diffusion in N2200 film
After photoexcitation, singlet excitons generated in the polymer films. The photogenerated excitons can diffuse along the single conjugated chain or among different chains within the exciton lifetime. The exciton diffusion processes are influenced by many exciton decay processes, such as recombine back to the ground state through radiative or nonradiative channels, transit into triplet states through intersystem crossing. When the two excitons encounter each other in the polymer films, exciton–exciton annihilation can happen. As a result, the energy of one exciton transfers to the other and recombine back to the ground state, meanwhile, the other exciton acquires the energy and transits into a higher excited state. Thus, half of the amounts of excitons will be lost during exciton–exciton annihilation processes. Apparently, exciton–exciton annihilation is a second-order processes, and a higher exciton concentration could lead to a more pronounced annihilation phenomenon.43,44In conjugated polymers, exciton–exciton annihilation can be employed to estimate the diffusion length of singlet exciton.26,44,48–51 The singlet exciton dynamics can be described as44
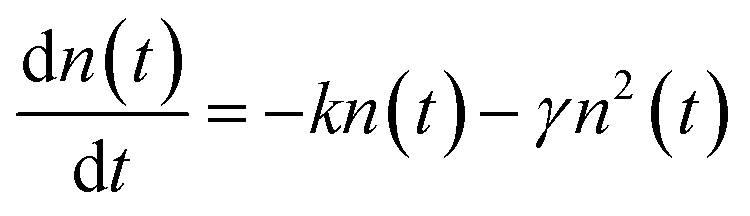 | (1) |
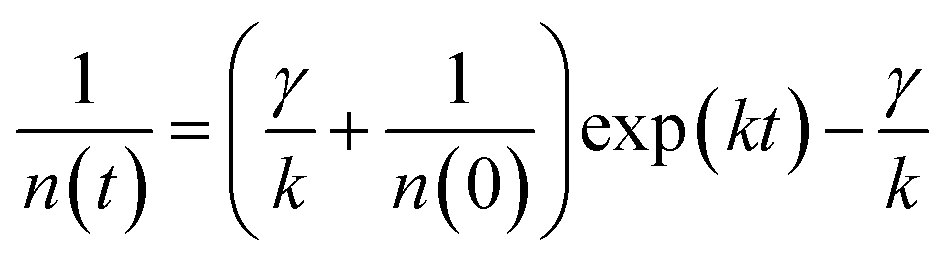 | (2) |
Apparently, the time-independent annihilation rate γ can be determined from the linear fit when n(0) and n(t) are known. In this work, exciton dynamics that measured by transient absorption were employed to analyze the exciton–exciton annihilation properties of N2200 film. Fig. 4(a) shows the TA kinetics of the N2200 film under various excitation fluencies ranging from 3.85 × 1012 to 2.32 × 1014 photons per cm2 per pulse probing at 700 nm. As the excitation fluency is increasing above 1013 photons per cm2 per pulse, it can be seen that the TA kinetics decays faster with increasing excitation fluency in the time range of <200 ps, due to more pronounced exciton–exciton annihilation under higher excitation power. Meanwhile, no pronounced differences were observed for the kinetics in the time range of >200 ps among the different excitation fluence.
The exciton density n(t) can be determined from the ΔOD(t) in TA kinetics using the known initial singlet exciton concentration n(0). Then, TA kinetics in the time range of <200 ps in Fig. 4(a) can be rearranged as Fig. 4(b) and ESI Fig. S9.† The rate constant of annihilation γ is determined as (2.4 ± 0.2) × 10−9 cm3 s−1 for N2200 film by fitting the kinetics in Fig. 4(b) with eqn (2). The figure including the y-intercept at the exp(kt) = 0 can be found in ESI Fig. S9.† According to eqn (2), y-intercept at the exp(kt) = 0 represents the −γ/k, where γ represents the rate constant of annihilation, k is the first-order exciton decay rate. The annihilation rate can be correlated with diffusion coefficient as51
| γ = 4πDRa | (3) |
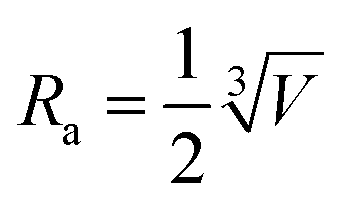 , which is ∼3.6 nm under the photoexcitation at 750 nm. By substituting parameters γ and Ra into eqn (3), the diffusion constant for the singlet exciton of N2200 film was estimated as D = (5.3 ± 0.4) × 10−4 cm2 s−1. This allows us to estimate the three-dimensional exciton diffusion length by using Ld = (6Dτ)1/2, which characterizes the mean effective distance for the exciton diffusion within its lifetime,49 that is 4.3 ± 0.2 nm in terms of the exciton lifetime of 60 ps.
, which is ∼3.6 nm under the photoexcitation at 750 nm. By substituting parameters γ and Ra into eqn (3), the diffusion constant for the singlet exciton of N2200 film was estimated as D = (5.3 ± 0.4) × 10−4 cm2 s−1. This allows us to estimate the three-dimensional exciton diffusion length by using Ld = (6Dτ)1/2, which characterizes the mean effective distance for the exciton diffusion within its lifetime,49 that is 4.3 ± 0.2 nm in terms of the exciton lifetime of 60 ps.
In PSCs, exciton dissociation is a critical process determining the PCE of devices. In order to make the device have high energy conversion efficiency, the exciton dissociation efficiency should be as high as possible. In other words, the excitons generated in N2200 phase of the blend film need to reach the interface within its diffusion length to ensure high exciton dissociation efficiency. The diffusion length of 4.3 ± 0.2 nm in N2200 film suggest the size of N2200 phase should be controlled within 4.3 ± 0.2 nm when optimizing the morphology of N2200 based devices. Many methods, such as thermal annealing,53 adding the additive of DIO,54 can be employed to for regulating the scale of donor (or acceptor) phase in the blended active layers. We note that exciton diffusion coefficient of N2200 can be comparable with some polymer semiconductors, such as POMeOPT (6.0 × 10−4 cm2 s−1)55 and PCE11 (1.0 × 10−3 cm2 s−1).56 Although the diffusion coefficient of N2200 film can be compared with these two polymers, the diffusion length of N2200 exciton is much smaller due to a shorter exciton lifetime in N2200 film. At the same time, we note that the exciton diffusion length of N2200 is much smaller than that of classical non-fullerene small molecule acceptors, such as ITIC (18.2 nm)57 and IT-4F (18.8 nm).57 Therefore, compared with PSCs based on small molecule acceptors, N2200 based solar cells should strictly control the size of acceptor phase in active layer.
4. Conclusion
We have studied the excited-states properties in N2200 solution and film by the optical spectroscopies as well as TD-DFT calculations. The transition mechanism of the absorption peaks of N2200 was evaluated through the NTOs and hole–electron population analysis by TD-DFT. The lifetime of singlet exciton in N2200 solution and film are found to be ∼90 ps and ∼60 ps, respectively. The short lifetime of N2200 is due to the fast non-radiative decay process. The annihilation radius, annihilation rate and diffusion length of singlet exciton in N2200 film were evaluated as 3.6 nm, 2.5 × 10−9 cm3 s−1 and 4.5 nm, respectively. Our work emphasizes the role of controlling the size of N2200 phase in active layers for developing high efficient N2200 based solar cells.Author contributions
Guanzhao Wen: software, investigation, data curation, funding acquisition, writing – original draft. Xianshao Zou: investigation, data curation, writing – review & editing. Rong Hu: investigation, data curation, funding acquisition, writing – review &editing. Jun Peng: methodology, writing – review & editing. Zhifeng Chen: methodology, funding acquisition. Xiaochuan He: investigation. Geng Dong: supervision, software, writing – review & editing. Wei Zhang: conceptualization, writing – review & editing, supervision, funding acquisition.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was supported by the funding from Natural Science Foundation of China (Grant No. 21603020, 21903017), Natural Science Foundation of Guangdong Province (Grant No. 2020A1515010411, 2019A1515010783), Key Research Project of Guangzhou University (Grant No. YK2020003), Young Talents Program of Guangzhou University (Grant No. RQ2020080), Guangzhou Municipal Science and Technology Project (Grant No. 202001010002), Science and Technology Research Program of Chongqing Municipal Education Commission (Grant No. KJQN201901319, KJQN202001323) and Innovation Research for the Postgraduates of Guangzhou University (Grant No. 2020GDJC-M50).References
- C. W. Tang, Two-layer organic photovoltaic cell, Appl. Phys. Lett., 1986, 48(2), 183–185 CrossRef CAS.
- J. J. M. Halls, C. A. Walsh, N. C. Greenham, E. A. Marseglia, R. H. Friend and S. C. Moratti, et al., Efficient photodiodes from interpenetrating polymer networks, Nature, 1995, 376(6540), 498–500 CrossRef CAS.
- N. Zhang, Y. Xu, X. Zhou, W. Zhang, K. Zhou and L. Yu, et al., Synergistic effects of copolymerization and fluorination on acceptor polymers for efficient and stable all-polymer solar cells, J. Mater. Chem. C, 2019, 7(45), 14130–14140 RSC.
- S. Li, Z. Zhang, M. Shi, C. Z. Li and H. Chen, Molecular electron acceptors for efficient fullerene-free organic solar cells, Phys. Chem. Chem. Phys., 2017, 19(5), 3440–3458 RSC.
- Z. He, C. Zhong, S. Su, M. Xu, H. Wu and Y. Cao, Enhanced power-conversion efficiency in polymer solar cells using an inverted device structure, Nat. Photonics, 2012, 6(9), 591–595 CrossRef.
- B. C. Thompson and J. M. Fréchet, Polymer-fullerene composite solar cells, Angew. Chem. Int. Ed., 2008, 47(1), 58–77 CrossRef CAS PubMed.
- M. C. Scharber, D. Mühlbacher, M. Koppe, P. Denk, C. Waldauf and A. J. Heeger, et al., Design rules for donors in bulk-heterojunction solar cells-towards 10% energy-conversion efficiency, Adv. Mater., 2006, 18(6), 789–794 CrossRef CAS.
- J. Hou, O. Inganäs, R. H. Friend and F. Gao, Organic solar cells based on non-fullerene acceptors, Nat. Mater., 2018, 17(2), 119–128 CrossRef CAS PubMed.
- G. Zhang, J. Zhao, P. C. Y. Chow, K. Jiang, J. Zhang and Z. Zhu, et al., Nonfullerene acceptor molecules for bulk heterojunction organic solar cells, Chem. Rev., 2018, 118(7), 3447–3507 CrossRef CAS PubMed.
- Y. Cui, H. Yao, L. Hong, T. Zhang, Y. Xu and K. Xian, et al., Achieving over 15% efficiency in organic photovoltaic cells via copolymer design, Adv. Mater., 2019, 31(14), 1808356 CrossRef PubMed.
- Q. Liu, Y. Jiang, K. Jin, J. Qin, J. Xu and W. Li, et al., 18% Efficiency organic solar cells, Sci. Bull., 2020, 65(4), 272–275 CrossRef CAS.
- H. Yan, Z. Chen, Y. Zheng, C. Newman, J. R. Quinn and F. Dötz, et al., A high-mobility electron-transporting polymer for printed transistors, Nature, 2009, 457(7230), 679–686 CrossRef CAS PubMed.
- X. Liu, Y. Zou, H.-Q. Wang, L. Wang, J. Fang and C. Yang, High-performance all-polymer solar cells with a high fill factor and a broad tolerance to the donor/acceptor ratio, ACS Appl. Mater. Interfaces, 2018, 10(44), 38302–38309 CrossRef CAS PubMed.
- J. Liu, Y. Shi, J. Dong, M. I. Nugraha, X. Qiu and M. Su, et al., Overcoming coulomb interaction improves free-charge generation and thermoelectric properties for n-doped conjugated polymers, ACS Energy Lett., 2019, 4(7), 1556–1564 CrossRef CAS.
- R. Wang, Y. Yao, C. Zhang, Y. Zhang, H. Bin and L. Xue, et al., Ultrafast hole transfer mediated by polaron pairs in all-polymer photovoltaic blends, Nat. Commun., 2019, 10(1), 398 CrossRef CAS PubMed.
- S. Fabiano, Z. Chen, S. Vahedi, A. Facchetti, B. Pignataroc and M. A. Lo, Role of photoactive layer morphology in high fill factor all-polymer bulk heterojunction solar cells, J. Mater. Chem., 2011, 21(16), 5891–5896 RSC.
- L. Gao, Z. G. Zhang, L. Xue, J. Min, J. Zhang and Z. Wei, et al., All-polymer solar cells based on absorption-complementary polymer donor and acceptor with high power conversion efficiency of 8.27%, Adv. Mater., 2016, 28(9), 1884–1890 CrossRef CAS PubMed.
- Z. Li, L. Ying, P. Zhu, W. Zhong, N. Li and F. Liu, et al., A generic green solvent concept boosting the power conversion efficiency of all-polymer solar cells to 11%, Energy Environ. Sci., 2019, 12(1), 157–163 RSC.
- Z. Li, W. Zhang, X. Xu, Z. Genene, D. Di Carlo Rasi and W. Mammo, et al., High-performance and stable all-polymer solar cells using donor and acceptor polymers with complementary absorption, Adv. Energy Mater., 2017, 7(14), 1602722 CrossRef.
- L. Zhu, W. Zhong, C. Qiu, B. Lyu, Z. Zhou and M. Zhang, et al., Aggregation-induced multilength scaled morphology enabling 11.76% efficiency in all-polymer solar cells using printing fabrication, Adv. Mater., 2019, 31(41), 1902899 CrossRef CAS PubMed.
- Q. Zhang, Y. Xin, Y. Feng, B. W. Larson, G. M. Su and M. M. Yin, et al., Understanding the interplay of transport-morphology-performance in PBDB-T-based polymer solar cells, Sol. RRL, 2020, 4(4), 1900524 CrossRef CAS.
- D. Chen, J. Yao, L. Chen, J. Yin, R. Lv and B. Huang, et al., Dye-incorporated polynaphthalenediimide acceptor for additive-free high-performance all-polymer solar cells, Angew. Chem. Int. Ed., 2018, 57(17), 4580–4584 CrossRef CAS PubMed.
- T. Kim, R. Younts, W. Lee, S. Lee, K. Gundogdu and B. J. Kim, Impact of the photo-induced degradation of electron acceptors on the photophysics, charge transport and device performance of all-polymer and fullerene–polymer solar cells, J. Mater. Chem. A, 2017, 5(42), 22170–22179 RSC.
- T. M. Clarke and J. R. Durrant, Charge photogeneration in organic solar cells, Chem. Rev., 2010, 110(11), 3736–3767 CrossRef PubMed.
- Y. Huang, E. J. Kramer, A. J. Heeger and G. C. Bazan, Bulk heterojunction solar cells: morphology and performance relationships, Chem. Rev., 2014, 114(14), 7006–7043 CrossRef CAS PubMed.
- H. Y. Shin, J. H. Woo, M. J. Gwon, M. Barthelemy, M. Vomir and T. Muto, et al., Exciton diffusion in near-infrared absorbing solution-processed organic thin films, Phys. Chem. Chem. Phys., 2013, 15(8), 2867–2872 RSC.
- X. Su, X. Zeng, H. Němec, X. Zou, W. Zhang and M. T. Borgstrom, et al., Effect of hydrogen chloride etching on carrier recombination processes of indium phosphide nanowires, Nanoscale, 2019, 11(40), 18550–18558 RSC.
- X. Zou, C. Li, X. Su, Y. Liu, D. Finkelstein-Shapiro and W. Zhang, et al., Carrier recombination processes in gaas wafers passivated by wet nitridation, ACS Appl. Mater. Interfaces, 2020, 12(25), 28360–28367 CrossRef CAS PubMed.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields, J. Phys. Chem., 1994, 98(45), 11623–11627 CrossRef CAS.
- A. D. McLean and G. S. Chandler, Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z=11-18, J. Chem. Phys., 1980, 72(10), 5639–5648 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, et al., Gaussian 16. Revision B.01, Gaussian Inc., Wallingford CT, 2016 Search PubMed.
- L. M. Tolbert, Solitons in a box: The organic chemistry of electrically conducting polyenes, Acc. Chem. Res., 1992, 25(12), 561–568 CrossRef CAS.
- L. A. Leal, L. E. de Sousa, P. P. de Brito, B. G. E. Neto, A. M. Ceschin and W. F. da Cunha, et al., Optical properties of P3HT and N2200 polymers: A performance study of an optimally tuned DFT functional, J. Mol. Model., 2018, 24(1), 32 CrossRef PubMed.
- R. L. Martin, Natural transition orbitals, J. Chem. Phys., 2003, 118(11), 4775–4777 CrossRef CAS.
- Z. Liu, T. Lu and Q. Chen, An sp-hybridized all-carboatomic ring, cyclo[18]carbon: Electronic structure, electronic spectrum, and optical nonlinearity, Carbon, 2020, 165, 461–467 CrossRef CAS.
- T. Lu and F. Chen, Multiwfn: A multifunctional wavefunction analyzer, J. Comput. Chem., 2012, 33(5), 580–592 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, VMD: Visual molecular dynamics, J. Mol. Graph., 1996, 14(1), 33–38 CrossRef CAS PubMed.
- T. Kastinen and T. I. Hukka, Charge transfer characteristics of fullerene-free polymer solar cells via multi-state electronic coupling treatment, Sustainable Energy Fuels, 2020, 4(8), 4137–4157 RSC.
- Z. Genene, J. Wang, X. Meng, W. Ma, X. Xu and R. Yang, et al., High bandgap (1.9 eV) polymer with over 8% efficiency in bulk heterojunction solar cells, Adv. Electron. Mater., 2016, 2(7), 1600084 CrossRef.
- Z. Li, X. Xu, W. Zhang, Z. Genene, W. Mammo and A. Yartsev, et al., High-photovoltage all-polymer solar cells based on a diketopyrrolopyrrole–isoindigo acceptor polymer, J. Mater. Chem. A, 2017, 5(23), 11693–11700 RSC.
- X. Zou, G. Wen, R. Hu, G. Dong, C. Zhang and W. Zhang, et al., An insight into the excitation states of small molecular semiconductor Y6, Molecules, 2020, 25(18), 4118 CrossRef CAS PubMed.
- J. R. Lakowicz, Principles of fluorescence spectroscopy, Springer, New York, NY, USA, 3rd edn, 2006, pp. 27–61 Search PubMed.
- H. Wang, H. Y. Wang, B. R. Gao, L. Wang, Z. Y. Yang and X. B. Du, et al., Exciton diffusion and charge transfer dynamics in nano phase-separated P3HT/PCBM blend films, Nanoscale, 2011, 3(5), 2280–2285 RSC.
- A. J. Lewis, A. Ruseckas, O. P. M. Gaudin, G. R. Webster, P. L. Burn and I. D. W. Samuel, Singlet exciton diffusion in MEH-PPV films studied by exciton–exciton annihilation, Org. Electron., 2006, 7(6), 452–456 CrossRef CAS.
- N. Banerji, S. Cowan, E. Vauthey and A. J. Heeger, Ultrafast relaxation of the poly(3-hexylthiophene) emission spectrum, J. Phys. Chem. C, 2011, 115(19), 9726–9739 CrossRef CAS.
- D. E. Markov, C. Tanase, P. W. M. Blom and J. Wildeman, Simultaneous enhancement of charge transport and exciton diffusion in poly(p-phenylene vinylene) derivatives, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72(4), 045217 CrossRef.
- S. D. Dimitrov, B. C. Schroeder, C. B. Nielsen, H. Bronstein, Z. Fei and I. McCulloch, et al., Singlet exciton lifetimes in conjugated polymer films for organic solar cells, Polymers, 2016, 8(1), 14 CrossRef PubMed.
- Y. Firdaus, V. M. Le Corre, S. Karuthedath, W. Liu, A. Markina and W. Huang, et al., Long-range exciton diffusion in molecular non-fullerene acceptors, Nat. Commun., 2020, 11(1), 5220 CrossRef CAS PubMed.
- Y. Long, G. J. Hedley, A. Ruseckas, M. Chowdhury, T. Roland and L. A. Serrano, et al., Effect of annealing on exciton diffusion in a high performance small molecule organic photovoltaic material, ACS Appl. Mater. Interfaces, 2017, 9(17), 14945–14952 CrossRef CAS PubMed.
- P. E. Shaw, A. Ruseckas and I. D. W. Samuel, Exciton diffusion measurements in Poly(3-hexylthiophene), Adv. Mater., 2008, 20(18), 3516–3520 CrossRef CAS.
- G. J. Hedley, A. Ruseckas and I. D. W. Samuel, Light harvesting for organic photovoltaics, Chem. Rev., 2017, 117(2), 796–837 CrossRef CAS PubMed.
- W. Zhang, R. Hu, D. Li, M.-M. Huo, X.-C. Ai and J.-P. Zhang, Primary dynamics of exciton and charge photogeneration in solvent vapor annealed P3HT/PCBM films, J. Phys. Chem. C, 2012, 116(6), 4298–4310 CrossRef CAS.
- B. Fan, L. Ying, P. Zhu, F. Pan, F. Liu and J. Chen, et al., All-polymer solar cells based on a conjugated polymer containing siloxane-functionalized side chains with efficiency over 10%, Adv. Mater., 2017, 29(47), 1703906 CrossRef PubMed.
- A. Robitaille, S. A. Jenekhe and M. Leclerc, Poly(naphthalene diimide-alt-bithiophene) prepared by direct (hetero)arylation polymerization for efficient all-polymer solar cells, Chem. Mater., 2018, 30(15), 5353–5361 CrossRef CAS.
- Y. Zaushitsyn, K. G. Jespersen, L. Valkunas, V. Sundström and A. Yartsev, Ultrafast dynamics of singlet-singlet and singlet-triplet exciton annihilation in poly(3-2′-methoxy-5′octylphenyl)thiophene films, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75(19), 195201 CrossRef.
- Y. Zhang, M. T. Sajjad, O. Blaszczyk, A. J. Parnell, A. Ruseckas and L. A. Serrano, et al., Large crystalline domains and an enhanced exciton diffusion length enable efficient organic solar cells, Chem. Mater., 2019, 31(17), 6548–6557 CrossRef CAS.
- M. T. Sajjad, A. Ruseckas, L. K. Jagadamma, Y. Zhang and I. D. W. Samuel, Long-range exciton diffusion in non-fullerene acceptors and coarse bulk heterojunctions enable highly efficient organic photovoltaics, J. Mater. Chem. A, 2020, 8(31), 15687–15694 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra01474a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |