
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnderstanding liquefaction in halide perovskites upon methylamine gas exposure†
Wencai Zhoua,
Zilong Zheng *a,
Yue Lu*a,
Manling Suia,
Jun Yinb and
Hui Yana
*a,
Yue Lu*a,
Manling Suia,
Jun Yinb and
Hui Yana
aFaculty of Materials and Manufacturing, Beijing University of Technology, Beijing 100124, China. E-mail: zilong.zheng@bjut.edu.cn; luyue@bjut.edu.cn
bDivision of Physical Science and Engineering, King Abdullah University of Science and Technology, Thuwal 23955-6900, Kingdom of Saudi Arabia
First published on 8th June 2021
Abstract
Methylamine (CH3NH2, MA) gas-induced fabrication of organometal CH3NH3PbI3 based perovskite thin films are promising photovoltaic materials that transform the energy from absorbed sunlight into electrical power. Unfortunately, the low stability of the perovskites poses a serious hindrance for further development, compared to conventional inorganic materials. The solid-state perovskites are liquefied and recrystallized from CH3NH2. However, the mechanism of this phase transformation is far from clear. Employing first principles calculations and ab initio molecular dynamics simulations, we investigated the formation energy of primary defects in perovskites and the liquefaction process in CH3NH2 vapor. The results indicated that defect-assisted surface dissolution leads to the liquefaction of perovskite thin films in CH3NH2 vapor. Two primary defects were studied: one is the Frenkel pair defect (including both negatively charged interstitial iodide ion (Ii−) and iodide vacancy (VI+) at the PbI2-termination surface, and the other is the Schottky defects (methylammonium vacancy, VMA) at the MAI-termination surface. Moreover, the defect-induced disorder in the microstructure reduces the degeneration of energy levels, which leads to a blue shift and broader absorption band gap, as compared to the clean perovskite surface. The mechanism of how defects impact the surface dissolution could be applied for the further design of high-stability perovskite solar cells.
I. Introduction
Halide perovskite solar cells (PSCs) have been regarded as one of the most promising materials for their exceptional optoelectronic properties, including high absorption coefficients, long charge carrier lifetime, and flexibility in material growth and architecture.1–5 The transition probability between the band edges is mainly derived from intra-atomic Pb s-orbitals to Pb p-orbitals in PSCs and results in a higher optical absorption coefficient, compared with GaAs-based solar cells.6 The band spitting induced by the strong spin–orbit coupling, leading to an indirect transition for non-radiative recombination from conduction band to valance band, responsible for long charge carrier lifetime in PSCs.7Although the solar to electrical power conversion efficiency (PCE) has improved remarkably from 3.8% (ref. 8) to 25.2% (ref. 9) during the past 10 years, the stability of perovskite materials is the most critical issue for commercial solar cells applications, for instance, instabilities of the crystal structure and photoelectric properties.10–15 Therefore, the defect-related stability generated in the fabrication process, and the nature of defects in organometal halide perovskites are the focus of extensive experimental and theoretical investigations.
Moreover, the defect states have a significant influence on the photoelectric performance,16–18 and it is necessary to improve the general understanding of fundamental characteristics of defects in PSCs. For instance, deep levels of intrinsic point defects in the band gap of PSCs can act as the centers of defect-assisted Shockley–Read–Hall (SRH) non-radiative recombination. Yin et al.19 found the dominant point defects in PSCs to be shallow level states, which have low formation energies and benign property, compared with deep levels. The defect clusters at the interfacial surface or grain boundaries in polycrystalline perovskite films were explored via experimental observations, and those defect-assisted trapping states could result in hysteresis in current–voltage characteristics.20–22 Following a defect-assisted model, Uratani et al.23 investigated the charge carrier trapping process with interface phase separation based on first principles calculations. Considering Lewis acids/bases have unoccupied orbitals/lone-pair electrons, which can passivate the undercoordinated I−/Pb2+ ion on the MAI/PbI2 terminated surface, which helps reduce the charge trapping and subsequent J–V hysteresis.24–26 For instance, the nitrogen-atoms in methylamine could provide a lone pair of electrons as a weak Lewis base, and Zhou et al.27 proposed the application of methylamine gas in the fabrication of perovskite films in 2015. During the methylamine gas conversion of the films, MAPbI3 perovskites generate a stable liquid-state intermediate, and then start to recrystallize following methylamine gas exposure. Methylamine-assisted (MAPbI3) perovskite films exhibit much low defect density, long carrier lifetime, excellent environmental stability, and much better photoelectric performance. The MA gas-induced liquefaction has been widely applied in numerous processes, such as device preparation28,29 and recycling.30,31 Although various reports have been proposed for the solid–liquid phase transformation progress, the intrinsic mechanism remains far from clear.27–33 It has been known that the MA molecules have interactions with the inorganic [PbI6]4− framework, given the coordination between lone pair electrons on nitrogen-atoms in methylamine and unoccupied orbitals in Pb2+ cations. Therefore, the MA-gas induced liquefaction was reported as a substitution of iodine with MA32 and NH3 molecules.30,31 It has been reported that the liquefaction process is associated with a MA–MA+ dimer-induced phase transition from three dimensional (3D) α phase to 1D δ phase, and the MA–MA+ dimer structure was also reported by He and Pan simultaneously.30 The interaction between MA molecules with MA+ cations indicates that the engineering of the A site of the halide perovskite is also important as the B site in the MA-gas induced fabrication.28 The experimental study provides a great contribution towards uncovering the PSC liquefaction mechanism, and could be widely applied for PSC fabrication. Moreover, it is significant for understanding the PSC liquefaction process in theory as well. Hence, it is urgent to determine the role of CH3NH2 vapor in the liquefaction process, and guide in designing better engineering for the perovskite film fabrication.
In this study, we investigated the defect formation energies and methylamine molecule adsorption energies on perovskite surfaces (including PbI2-termination and MAI-termination) based on the density functional theory (DFT). Then, considering the low defect formation energy of methylammonium vacancy (VMA), we studied defect-assisted surface dissolution combined with the interactions between methylamine gas and perovskite surfaces using ab initio molecular dynamics simulations (AIMD). Finally, we explored the disorder and energy-shift of the absorption band gap, resulting from perovskite surface dissolution.
II. Method
First principles calculations were performed with DFT methods as implemented in the Vienna Ab initio Simulation Package (VASP),34 and the long-range van der Waals interactions proposed by Grimme et al. (DFT-D3) to add a correction to the conventional Kohn–Sham DFT energy.35 DFT calculations were performed via standard Perdew–Burke–Ernzerhof (PBE) generalized gradient approximation (GGA),36 within the framework of the projector-augmented-wave (PAW) method.37 The cut-off energy was set to 500 eV, and we established 1 × 1 × 2 supercells with additional methylamine molecules to calculate the adsorption energies. In order to eliminate the properties of defects, we then increased the system to 2 × 2 × 2 supercells to calculate the defect formation energies, activation potential barriers of transition state, and defect-assisted methylamine penetrating dynamic progress. The modeling MAPbI3 clean surfaces were constructed using a thickness of three-layer MAPbI3 slabs with an additional 15 Å vacuum region (Fig. 1(a)). The atomic positions were fully relaxed with the conjugate gradient method until the atomic forces were less than 0.01 eV Å−1. The adsorption energy (Eads) of the equilibrium structures was calculated using the following equation,where ECH3NH2, EMAPbI3, and ECH3NH2+MAPbI3 are the total energies of a free methylamine molecule (CH3NH2), clean MAPbI3 surface, and MAPbI3 surface with adsorbed CH3NH2 molecules via the converged self-consistent field (SCF) method, respectively. N is the total number of the adsorbed CH3NH2 molecules on the clean MAPbI3 surface in the system.

 | ||
| Fig. 1 (a) Additional methylamine molecules (green color) absorbing (red region) and penetrating (yellow region) on the PbI2-termination MAPbI3 (001) clean surface. (b) Adsorption energy (per methylamine) as a function of the adsorbed methylamine numbers (N from 1 to 4) based on each (c) equilibrium structure. | ||
The transition state calculation was carried out via the climbing nudged elastic band (CINEB) method.38 More than 5 configurations were applied between the initial state and the final state, and the force threshold was set to 0.05 eV Å−1.
The AIMD simulations were performed using the CP2K/Quickstep package.39 MD simulations were performed with NVT ensemble at room temperature of 300 K. The total simulation time is 30 picoseconds with a time step of 1 femtosecond. The wave functions of the valence electrons were expanded in terms of Gaussian functions with molecularly optimized double ζ polarized basis sets (m-DZVP),40 which ensure a small basis set superposition error, and core electrons were described with norm-conserving Goedecker, Teter, and Hutter (GTH) pseudopotentials.41
III. Results and discussion
Two typical crystal structures of the MAPbI3 perovskite could exist at room temperature, namely tetragonal phase and cubic phase. The former one is more stable than the latter.42 Therefore, in this study, we only focused on the surface dissolution in the tetragonal phase. From previous work, the (001) surface is one of the most common surfaces in perovskites,43 and our results indicated the optimized lattice parameters are a = 8.09 Å, b = 8.73 Å, and c = 12.86 Å, which are in good agreement with the experimental measurement.44–46 To have a better illustration of the diffusion progress, we took the PbI2-termination (001) surface as an example, and marked the area of the vacuum region and inner structure, as shown in Fig. 1(a).As the initial configurations, we made one CH3NH2 molecule over 4 Å above on both PbI2-termination surface and MAI-termination surface, respectively. Then, we obtained the equilibrium adsorbing structures based on DFT energy minimization, as shown in adsorption energy and bond lengths in Table 1. On the PbI2-termination surface, CH3NH2 has a strong interaction with Pb2+ via a covalent bond, which is lower in energy (−1.06 eV), and CH3NH2 kept located in the vacuum region at the equilibrium configuration for adsorption. Considering the high-concentration methylamine gas, we established the modeling system by increasing numbers up to four adsorbed methylamine molecules (Fig. 1(b)). It should be noted that four is the maximum number of adsorbed CH3NH2 on the 1 × 1 slab because of steric effects. The adsorption energy (assigned in each CH3NH2) increases to −0.71 eV. The configuration of the Pb2+ ion is 5d86s0, which can accept four lone pair electrons, while two can fill up the 5d-orbital and the other two fill up the 6s-orbital. Alternatively, the maximum bond number between each Pb atom and adsorbed CH3NH2 is less than two, considering one nitrogen-atom of adsorbed CH3NH2 can provide two lone pair electrons. When more than two CH3NH2 molecules absorb on one Pb atom, there is no bond connection between them, additional CH3NH2 was stabilized via intermolecular hydrogen bonds among CH3NH2. Therefore, the absolute value of adsorption energy, |Eads|, decreases from 1.06 eV to 0.71 eV, when more CH3NH2 adsorbed. Therefore, in the preparation of perovskites, the high-concentration methylamine treatment process may not be applicable for liquefaction on the PbI2-termination surface.
| Eads (eV) | Distance (Å) | |
|---|---|---|
| PbI2-termination | −1.06 | 2.52 |
| MAI-termination | −0.51 | 1.65 |
Moreover, numerous lone pair electrons filling up Pb2+ unoccupied orbitals result in the reduction of the dipole interaction and bridging the polar ionic Pb–I bond (Fig. 1(c)). The generation of the Frenkel pair (interstitial iodide ion (Ii−) and iodide vacancy (VI+)) is more favorable via the free I− ion.
On the other hand, we investigated the adsorption on a MAI-termination perovskite surface, and Eads is −0.51 eV, which is mainly induced by intermolecular hydrogen bond interactions between CH3NH2 molecules, wherein one is outside the perovskite surface, and the other is inside, as shown in the configuration in Fig. 2 on the left. The stable dimer structure agrees well with the recent experimental reports.28,30,31 The absolute value of Eads is actually the same as the energy of the N⋯H bond (around 0.5 eV),47 where the molecules are not constrained by the lattice structure.
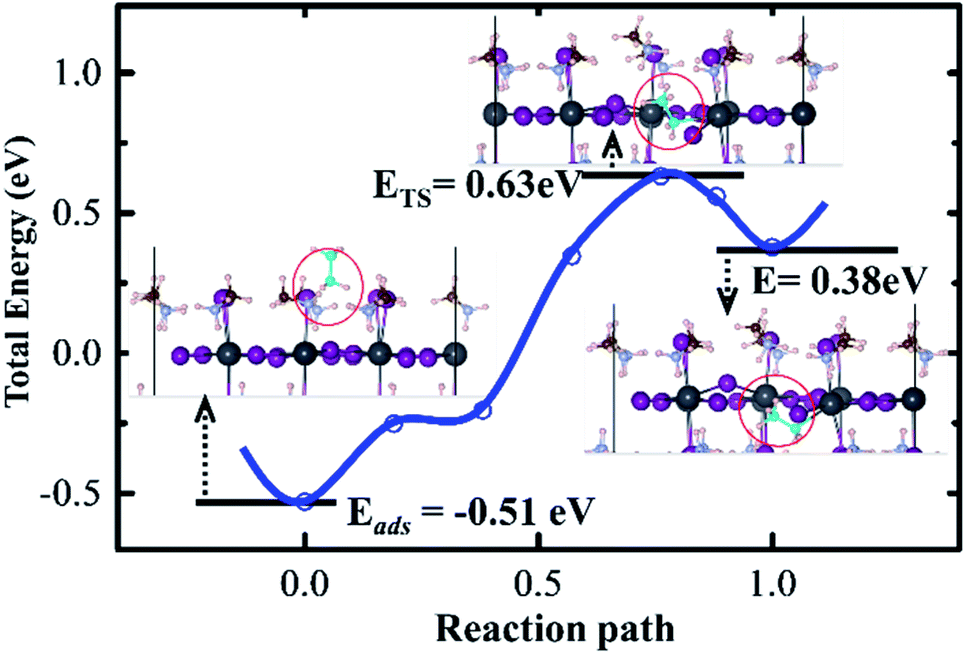 | ||
| Fig. 2 Energies and configurations along with the reaction path from the reactant equilibrium structure (CH3NH2 adsorbing on the perovskite surface), transition state (TS) structure to the product equilibrium structure (CH3NH2 penetrating into the surface). Zero point of total energy defined by the configuration, in which there is no interaction between adsorbed CH3NH2 and perovskite surface. | ||
In order to understand the mechanism of the CH3NH2 penetration process on the MAI-termination surface, the transition states between the two equilibrium configurations, adsorbed and penetrated structures, were obtained (Fig. 2). The activation potential barrier is as high as 1.14 eV (Eads + ETS), indicating that the CH3NH2 penetration process is hardly possible, and methylamine treatment should be avoided on the clean MAI-termination surface.
Given the defects on the perovskite surface are unavoidable, the mechanism of defect-assisted CH3NH2 gas penetration into the perovskite could be available. To verify this hypothesis, initially, we obtained the defect formation on both the PbI2-termination and MAI-termination surfaces via the DFT method. Employing the point defect model, the MA, Pb, and I vacancies (VMA, VPb, and VI) are included, and the values of their formation energies are 0.51 eV (of VMA), 0.73 eV (of VPb) and 0.90 eV (of VI) from the small to the large. The lowest defect formation energy of VMA implies that it is highly likely to exist on the MAI-termination perovskite surface (Table 2).
| Defect | Formation energy (eV) | |||
|---|---|---|---|---|
| I-rich | Pb-rich | |||
| PbI2-termination | VPb | 0.73 | 2.56 | |
| VI | 2.71 | 0.90 | ||
| MAI-termination | VMA | 0.51 | 1.44 | |
| VI | 2.63 | 1.71 | ||
Therefore, we further performed AIMD simulations on the MAI-termination surface with CH3NH2 gas exposure to compare with the CH3NH2 dissolution process, (i) on the clean surface and (ii) on MA-vacancy defective surface, as shown in the model configuration in Fig. 3(a).
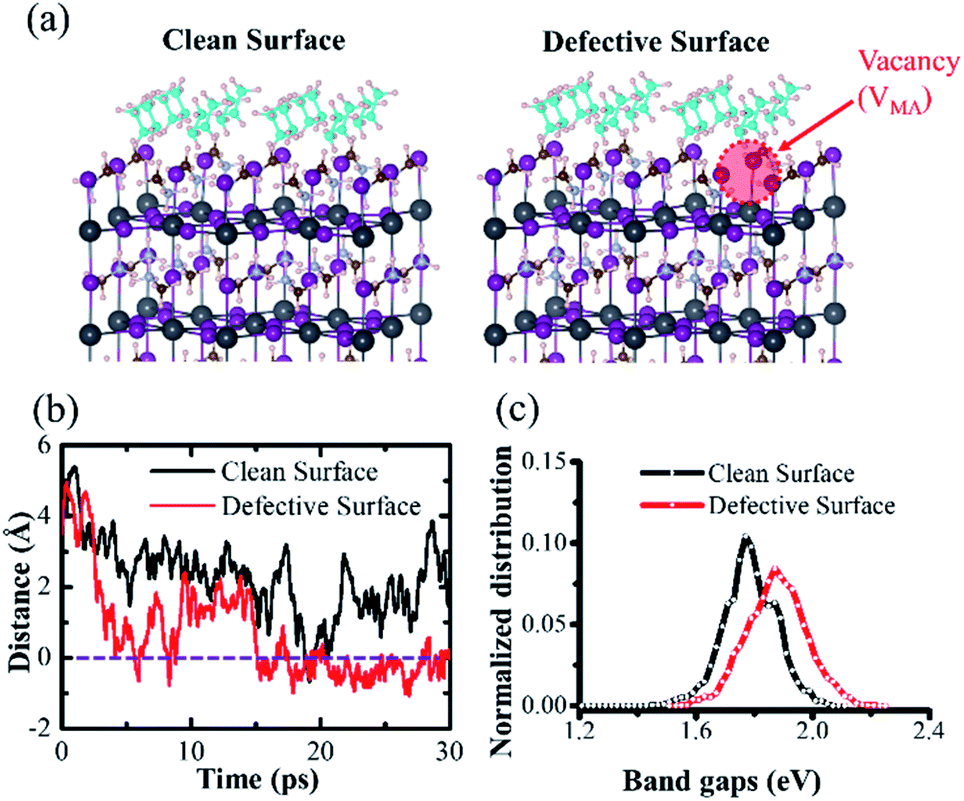 | ||
| Fig. 3 Model configures (a) of both the clean perovskite surface and MA-vacancy defective surface in AIMD simulations. (b) Distances between adsorbed CH3NH2 (green color) and perovskite surfaces as a function of time (negative values represent the CH3NH2 molecule penetrating into the perovskite surface). The energy distributions of optical band gaps (c) obtained from AIMD simulations. | ||
A 2 × 2 × 2 supercell for perovskites was established, which contained 16 adsorbed CH3NH2 molecules on the perovskite surface, as the model for high-concentration CH3NH2. 16 is the maximum number of CH3NH2 molecules, which covered the entire model 2 × 2 surface. For better illustration, the clean surface denotes the MA gas adsorbed on the pristine MAI-terminated surface, and the defective surface indicates the MAI-terminated surface with additional VMA defect. Due to defect, the absorbed CH3NH2 molecule started diffusing into the inner structure in 5 picosecond; however, it was pushed out immediately. It then penetrated into the surface in the 16 ps region, and remained localized in the inner structure. The dynamic distance between adsorbed CH3NH2 and MA-vacancy defective surface confirmed the process, (Fig. 3(b)), while the negative values of distances indicate CH3NH2 penetration into the surface. Furthermore, we calculated the statistical distribution of the band edge level and the bandgap variation and fit the status with the Gaussian function. The structural disorder on the defective perovskite surface is large, which reduces the degeneration of the conduction band (CB) level and valence band level (VB). As a result, the optical band gaps become broad. Therefore, the AIMD simulation presented a blue-shift and broader absorption band gap for MA-vacancy defective perovskites, given the enhanced disorder character of the inorganic framework.
On the clean surface, the difference is the adsorbed CH3NH2 barely diffuse into the inner structure, and remain located outside the perovskite surface. The small structural disorder leads to the optical band gaps becoming sharper, see Fig. 3(c).
Moreover, considering the strong absorption between CH3NH2 and the clean PbI2-termination surface, see Table 1, we explored the CH3NH2 diffusion process via AIMD simulations with at room temperature of 300 K, while the snapshots were exported every 10 ps, and configurations represented in Fig. 4(b).
 | ||
| Fig. 4 (a) Radical distribution functions of Pb–I bonds and (b) configurations obtained from AIMD simulations every 10 picoseconds (0 ps, 10 ps, 20 ps, and 30 ps), respectively. The green molecules are the adsorbed methylamine molecules, and the bottom panel (c) was exported in 30 ps AIMD simulations, where one adsorbed methylamine molecule was penetrating into the perovskite surface, following vacancy defect generation. | ||
Induced by the thermal fluctuations and adsorption dynamics, the PbI2-termination surface underwent slight distortion, as shown in the results of AIMD simulations. The breaking Pb–I bonds enable I− ion to move slightly from the ideal position outside the perovskite surface, and generate a positive charge iodine vacancy (VI+) at the original site, resulted in the formation of a Frenkel pair defect. However, unlike defect-assisted MAI-termination surface dissolution in CH3NH2 gas exposure, our simulation implied that the adsorbed CH3NH2 molecule cannot penetrate into the inner structure from the PbI2-termination surface even with defect assistance. This could be because of the large binding interaction between the surface Pb2+ ion with diffused CH3NH2 molecules. To further investigate the ordered/disordered character in materials, we obtained the radical distribution function (RDF) of Pb–I bonds, as shown in Fig. 4(a). The calculated RDF did not change significantly in AIMD simulations, which indicated that the amount of generated Frenkel pair defects (Ii−/VI+) on the PbI2-termination surface was much less, and insufficient to assist CH3NH2 diffusion into the perovskite surface. In CH3NH2 gas exposure, the dissolution on the PbI2-termination surface hardly occurred.
IV. Conclusions
In summary, we investigated the methylamine gas adsorption and diffusion process on organometal Halide (MAPbI3) perovskite surfaces based on the density functional theory and ab initio molecular dynamics simulations. The plumbum atom (Pb) has a strong covalent bond with the methylamine molecule, and the binding energy is as strong as 1.1 eV, which prevents MA gas from diffusing through the perovskite surface or dissolving on the PbI2-termination surface when exposed to MA gas. On the MAI-termination surface, the defect formation energy of MA vacancy is as low as 0.5 eV, which implies it is likely to exist in perovskites. Our simulations implied the adsorbed methylamines could be encouraged to penetrate into the defective MAI-termination surface, rather than from the clean surface, which means that the defect-assisted surface dissolution may lead to the liquefaction of perovskite thin films in methylamine vapor. The energies of adsorption and defect formation indicated a high-concentration methylamine treatment process may be applied for defective perovskite surface. Moreover, following methylamine adsorption, defect-assisted surface dissolution result in a blue-shift and broader absorption band gap. The mechanism of defect-assisted perovskite surface dissolution could be applied for further defects engineering in high-stability perovskite solar cells.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 62034001, 22033006, 52073005). We also acknowledge for the financial support from Key Laboratory of Advanced Functional Materials, Education Ministry of China, and supercomputer center of Institute of Advanced Energy Materials and Devices in BJUT.References
- J. Huang, Y. Yuan, Y. Shao and Y. Yan, Nat. Rev. Mater., 2017, 2, 17042 CrossRef CAS.
- X. Li, D. Bi, C. Yi, J.-D. Décoppet, J. Luo, S. M. Zakeeruddin, A. Hagfeldt and M. Grätzel, Science, 2016, 353, 58–62 CrossRef CAS PubMed.
- Y. Wu, X. Yang, W. Chen, Y. Yue, M. Cai, F. Xie, E. Bi, A. Islam and L. Han, Nat. Energy, 2016, 1, 16148 CrossRef CAS.
- H. Tsai, W. Nie, J.-C. Blancon, C. C. Stoumpos, R. Asadpour, B. Harutyunyan, A. J. Neukirch, R. Verduzco, J. J. Crochet, S. Tretiak, L. Pedesseau, J. Even, M. A. Alam, G. Gupta, J. Lou, P. M. Ajayan, M. J. Bedzyk, M. G. Kanatzidis and A. D. Mohite, Nature, 2016, 536, 312–316 CrossRef CAS PubMed.
- T. Leijtens, G. E. Eperon, A. J. Barker, G. Grancini, W. Zhang, J. M. Ball, A. R. S. Kandada, H. J. Snaith and A. Petrozza, Energy Environ. Sci., 2016, 9, 3472–3481 RSC.
- W.-J. Yin, J.-H. Yang, J. Kang, Y. Yan and S.-H. Wei, J. Mater. Chem. A, 2015, 3, 8926–8942 RSC.
- F. Zheng, L. Z. Tan, S. Liu and A. M. Rappe, Nano Lett., 2015, 15, 7794–7800 CrossRef CAS PubMed.
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050 CrossRef CAS PubMed.
- NREL, https://www.nrel.gov/pv/assets/pdfs/best-research-cell-efficiencies.20200925.pdf, 2020.
- B. W. Park, N. Kedem, M. Kulbak, D. Y. Lee, W. S. Yang, N. J. Jeon, J. Seo, G. Kim, K. J. Kim, T. J. Shin, G. Hodes, D. Cahen and S. I. Seok, Nat. Commun., 2018, 9, 3301 CrossRef PubMed.
- B. Conings, J. Drijkoningen, N. Gauquelin, A. Babayigit, J. D’Haen, L. D’Olieslaeger, A. Ethirajan, J. Verbeeck, J. Manca, E. Mosconi, F. D. Angelis and H.-G. Boyen, Adv. Energy Mater., 2015, 5, 6160–6164 Search PubMed.
- E. Tenuta, C. Zheng and O. Rubel, Sci. Rep., 2016, 6, 37654 CrossRef CAS PubMed.
- S. N. Habisreutinger, D. P. McMeekin, H. J. Snaith and R. J. Nicholas, APL Mater., 2016, 4, 643 Search PubMed.
- T. A. Berhe, W.-N. Su, C.-H. Chen, C.-J. Pan, J.-H. Cheng, H.-M. Chen, M.-C. Tsai, L.-Y. Chen, A. A. Dubale and B.-J. Hwang, Energy Environ. Sci., 2016, 9, 323–356 RSC.
- N.-G. Park, M. Grätzel, T. Miyasaka, K. Zhu and K. Emery, Nat. Energy, 2016, 1, 16152 CrossRef CAS.
- M. V. Kovalenko, L. Protesescu and M. I. Bodnarchuk, Science, 2017, 358, 745–750 CrossRef CAS PubMed.
- H. Huang, M. I. Bodnarchuk, S. V. Kershaw, M. V. Kovalenko and A. L. Rogach, ACS Energy Lett., 2017, 2, 2071–2083 CrossRef CAS PubMed.
- X. Zheng, B. Chen, J. Dai, Y. Fang, Y. Bai, Y. Lin, H. Wei, X. C. Zeng and J. Huang, Nat. Energy, 2017, 2, 17102 CrossRef CAS.
- W. J. Yin, T. Shi and Y. Yan, Adv. Mater., 2014, 26, 4653–4658 CrossRef CAS PubMed.
- H. J. Snaith, A. Abate, J. M. Ball, G. E. Eperon, T. Leijtens, N. K. Noel, S. D. Stranks, J. T.-W. Wang, K. Wojciechowski and W. Zhang, J. Phys. Chem. Lett., 2014, 5, 1511–1515 CrossRef CAS PubMed.
- B. Chen, M. Yang, S. Priya and K. Zhu, J. Phys. Chem. Lett., 2016, 7, 905–917 CrossRef CAS PubMed.
- Q. Chen, H. Zhou, T.-B. Song, S. Luo, Z. Hong, H.-S. Duan, L. Dou, Y. Liu and Y. Yang, Nano Lett., 2014, 14, 4158–4163 CrossRef CAS PubMed.
- H. Uratani and K. Yamashita, J. Phys. Chem. Lett., 2017, 8, 742–746 CrossRef CAS PubMed.
- P. L. Qin, G. Yang, Z. W. Ren, S. H. Cheung, S. K. So, L. Chen, J. Hao, J. Hou and G. Li, Adv. Mater., 2018, 30, e1706126 CrossRef PubMed.
- Y. Lin, L. Shen, J. Dai, Y. Deng, Y. Wu, Y. Bai, X. Zheng, J. Wang, Y. Fang, H. Wei, W. Ma, X. C. Zeng, X. Zhan and J. Huang, Adv. Mater., 2017, 29, 1604545 CrossRef PubMed.
- L. Liu, W.-H. Fang, R. Long and O. V. Prezhdo, J. Phys. Chem. Lett., 2018, 9, 1164–1171 CrossRef CAS PubMed.
- Z. Zhou, Z. Wang, Y. Zhou, S. Pang, D. Wang, H. Xu, Z. Liu, N. P. Padture and G. Cui, Angew. Chem., Int. Ed. Engl., 2015, 54, 9705–9709 CrossRef CAS PubMed.
- X. Huang, R. Chen, G. Deng, F. Han, P. Ruan, F. Cheng, J. Yin, B. Wu and N. Zheng, J. Am. Chem. Soc., 2020, 142, 6149–6157 CrossRef CAS PubMed.
- D.-N. Jeong, D.-K. Lee, S. Seo, S. Y. Lim, Y. Zhang, H. Shin, H. Cheong and N.-G. Park, ACS Energy Lett., 2019, 4, 1189–1195 CrossRef CAS.
- X. Y. Feng, K. W. Ng, S. P. Wang, W. Z. Chen, Z. Z. Zhang, W. Chen, Y. Y. Zhao, B. Tu, Z. K. Tang, H. Pan and Z. B. He, J. Mater. Chem. A, 2020, 8, 13585–13593 RSC.
- X. Feng, Q. Guo, J. Xiu, Z. Ying, K. W. Ng, L. Huang, S. Wang, H. Pan, Z. Tang and Z. He, Cell Rep. Phys. Sci., 2021, 2, 100341 CrossRef.
- D. Bogachuk, L. Wagner, S. Mastroianni, M. Daub, H. Hillebrecht and A. Hinsch, J. Mater. Chem. A, 2020, 8, 9788–9796 RSC.
- Y. Jiang, E. J. Juarez-Perez, Q. Ge, S. Wang, M. R. Leyden, L. K. Ono, S. R. Raga, J. Hu and Y. Qi, Mater. Horiz., 2016, 3, 548–555 RSC.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem, Phys., 2000, 113, 9901–9904 CrossRef CAS.
- J. VandeVondele, M. Krack, F. Mohamed, M. Parrinello, T. Chassaing and J. Hutter, Comput. Phys. Commun., 2005, 167, 103–128 CrossRef CAS.
- J. VandeVondele and J. Hutter, Comput. Phys. Commun., 2007, 127, 114105 Search PubMed.
- S. Goedecker, M. Teter and J. Hutter, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 1703–1710 CrossRef CAS PubMed.
- Y. Kawamura, H. Mashiyama and K. Hasebe, J. Phys. Soc. Jpn., 2002, 71, 1694 CrossRef CAS.
- X. Liu and K. Sohlberg, Complex Met., 2014, 1, 103–121 CrossRef.
- T. Baikie, Y. Fang, J. M. Kadro, M. Schreyer, F. Wei, S. G. Mhaisalkar, M. Graetzel and T. J. White, J. Mater. Chem. A, 2013, 1, 5628–5641 RSC.
- S. Piskunov, E. A. Kotomin, E. Heifets, J. Maier, R. I. Eglitis and G. Borstel, Surf. Sci., 2005, 575, 75–88 CrossRef CAS.
- S. Colella, E. Mosconi, P. Fedeli, A. Listorti, F. Gazza, F. Orlandi, P. Ferro, T. Besagni, A. Rizzo, G. Calestani, G. Gigli, F. De Angelis and R. Mosca, Chem. Mater., 2013, 25, 4613–4618 CrossRef CAS.
- B. A. Gong, B. Jing, Q. Z. Li, Z. B. Liu, W. Z. Li, J. B. Cheng, Q. C. Zheng and J. Z. Sun, Theor. Chem. Acc., 2010, 127, 303–309 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra01458g |
| This journal is © The Royal Society of Chemistry 2021 |