
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGold nanoparticles conjugated with anti-CD133 monoclonal antibody and 5-fluorouracil chemotherapeutic agent as nanocarriers for cancer cell targeting
Manali Haniti Mohd-Zahid a,
Siti Nadiah Zulkiflib,
Che Azurahanim Che Abdullahbc,
JitKang Limd,
Sharida Fakurazie,
Kah Keng Wongf,
Andee Dzulkarnaen Zakariag,
Norzila Ismailh,
Vuk Uskokovići,
Rohimah Mohamudf and
Iskandar Z. A*a
a,
Siti Nadiah Zulkiflib,
Che Azurahanim Che Abdullahbc,
JitKang Limd,
Sharida Fakurazie,
Kah Keng Wongf,
Andee Dzulkarnaen Zakariag,
Norzila Ismailh,
Vuk Uskokovići,
Rohimah Mohamudf and
Iskandar Z. A*a
aDepartment of Chemical Pathology, School of Medical Sciences, Universiti Sains Malaysia, 16150 Kubang Kerian, Kelantan, Malaysia. E-mail: iskandarza@usm.my
bMaterial Synthesis and Characterization Laboratory, Institute of Advanced Technology, Universiti Putra Malaysia, Serdang 43400, Selangor, Malaysia
cDepartment of Physics, Faculty of Science, Universiti Putra Malaysia, Serdang 43400, Selangor, Malaysia
dSchool of Chemical Engineering, Universiti Sains Malaysia, 14300 Nibong Tebal, Penang, Malaysia
eDepartment of Human Anatomy, Faculty of Medicine and Health Sciences, Universiti Putra Malaysia, Serdang 43400, Selangor, Malaysia
fDepartment of Immunology, School of Medical Sciences, Universiti Sains Malaysia, 16150 Kubang Kerian, Kelantan, Malaysia
gDepartment of Surgery, School of Medical Sciences, Universiti Sains Malaysia, 16150 Kubang Kerian, Kelantan, Malaysia
hDepartment of Pharmacology, School of Medical Sciences, Universiti Sains Malaysia, 16150 Kubang Kerian, Kelantan, Malaysia
iAdvanced Materials and Nanobiotechnology Laboratory, TardigradeNano LLC, Irvine, CA 92604, USA
First published on 30th April 2021
Abstract
The enhanced permeability and retention effect allows for passive targeting of solid tumours by nanoparticles carrying anticancer drugs. However, active targeting by incorporation of various ligands onto nanoparticles can provide for a more selective and enhanced chemotherapeutic effect and complement the deficiencies of the passive targeting approach. Here we report on the design of the carboxyl-terminated PEGylated gold nanoparticles (AuNPs), their functionalization with anti-CD133 monoclonal antibody (mAb) via a crosslinking reaction, and subsequent 5-fluorouracil (5-FU) drug loading. The synthesized products in the form of stable colloids were characterised using a range of physicochemical techniques, including X-ray diffraction (XRD), UV-Vis spectroscopy, transmission electron microscopy (TEM), and dynamic light scattering (DLS). Conjugation of anti-CD133 mAb onto PEGylated AuNPs was confirmed with the use of UV-Vis, BCA protein assay and fluorescence microscopy. HCT116 colorectal cancer cells abundantly expressed CD133: 92.4 ± 1.3%, as measured by flow cytometry. Whereas PEGylated AuNPs not conjugated with anti-CD133 mAb accumulated mainly at the cellular membrane, nanoparticles conjugated with anti-CD133 mAb were contained within the nuclear region of the cells. Anti-CD133 mAb conjugation facilitated the specific intracellular uptake due to specific antigen–antibody binding interaction. In vitro cytotoxicity studies on HCT116 cells showed that PEGylated AuNPs and PEGylated AuNPs-CD133 did not elicit any toxicity at any of the tested concentrations. Meanwhile, 5-FU-PEGylated AuNPs-CD133 significantly reduced the cell viability relative to the treatment with 5-FU-PEGylated AuNPs without anti-CD133 mAb conjugates (p < 0.0001). This study shows that the conjugation of nanocarriers with the anti-CD133 antibody improves the specific targeting of 5-FU against colorectal cancer cells. These results demonstrate that simultaneous functionalisation of PEGylated AuNPs with antibodies and chemotherapeutic drugs is a viable strategy to combat cancer through targeted drug delivery.
1. Introduction
Selective targeting of cancer cells in the body with chemotherapeutic molecules presents one of the major challenges for medical researchers all across the globe. The enhanced permeability and retention (EPR) effect allows for passive targeting of solid tumours by nanoparticles carrying anticancer drugs.1 According to this effect, nanoparticles smaller than 100 nm but also sufficiently large to escape the renal clearance may pass through the 380–780 nm sized pores in the leaky neovasculature of tumours.2,3 However, the considerable disparity between the number of preclinical papers reporting on this effect and the number of corresponding products reaching the market has raised fundamental questions over its validity.4 Nowadays, it is known that the EPR effect is significantly more complex that its proposed mechanism of extravasation of nanoparticles through endothelial lining gaps due to leaky vasculature,5 with numerous factors affecting its dependency on the patient and the tumour type.6 Also, because of the low renal clearance and prolonged circulation times required for this effect to take place, types of treatment utilizing it are insufficiently selective and impose a considerable toxicity onto healthy cells in the vicinity of the tumour and elsewhere in the body. Active targeting by incorporation of various ligands onto nanoparticles can provide for a more selective and enhanced chemotherapeutic effect while avoiding the imposition of significant damage to the healthy cells.7 This approach builds upon the augmented expression of specific surface receptors in cancer cells compared to their healthy counterparts. Such surface markers can act as targets for molecular or nanoparticle therapies that differentiate between cancer and normal cells. In theory and in practice, such therapies deliver their chemotherapeutic payloads selectively into cancer cells, thus sparing the healthy cells from the adverse side effects of chemotherapy. This form of active targeting can complement the passive targeting mechanism and increase its efficiency in suppressing the tumour growth.Gold nanoparticles (AuNPs) are promising nanocarriers in drug delivery due to their unique physicochemical properties.8 In general, AuNPs with high surface-to-volume ratios avoid the circulation clearance via the reticuloendothelial system (RES) and can be used to achieve passive targeting of the tumour site through the EPR effect. In addition, such unique features, together with the ease of surface medication due to high surface reactivity of Au,9,10 also make AuNPs convenient for active targeting of tumours. As such, using any of the numerous surface functionalisation strategies, AuNPs can easily fulfill the role of cancer therapeutic agents.11 In addition to their use as drug delivery vehicles, AuNPs can be used as biomedical imaging contrast agents by relying on their unique surface plasmon resonance (SPR).12,13
Polyethylene glycol (PEG) is a nonionic hydrophilic polymer that provides steric stabilisation and biocompatibility to nanoparticles in biological media.14,15 Functionalisation of nanoparticles with PEG presents a standard approach to extend their circulation half-life and achieve a desired biodistribution.16 A preclinical study reported that PEGylated AuNPs (CYT-6091) escaped the immediate clearance by the RES, thereby improving the chemotherapeutic efficacy.17 In 2016, Suk et al.18 reported that AuNPs coated with PEG circumvented phagocytosis and hence increased the circulation time by promoting the stealth effect on the nanoparticle surface. This behavior confers PEG as a favorable surface modifier of AuNPs, providing that the functional groups in PEG assist the further covalent modification of nanoparticles for targeted drug delivery.
Amine-containing drug, 5-fluorouracil (5-FU), is an antineoplastic drug for the treatment of solid cancers such as CRC. It is a natural pyrimidine uracil analog and acts as a thymidylate synthase inhibitor to interfere with the deoxyribonucleic acid (DNA) and ribonucleic acid (RNA) synthesis, thereby interfering with their processing and functioning and through that inducing cell death.19–21 Despite the efficacy of 5-FU in chemotherapeutic treatments, short half-life, non-selectivity and poor biodistribution are included amongst its properties, limiting its therapeutic effects.22 Drug delivery systems using nanoparticles have emerged as a topic of interest to address these shortcomings of conventional chemotherapeutic agents, of which 5-FU is one member.
CD133 is a 97 kDa pentaspan transmembrane glycoprotein involved in membrane organisation and various signaling cascades.23 CD133 is known to be overexpressed in colorectal cancer (CRC) cells and has been proposed as a marker for the CRC stem cells that may predict the tumour progression, therapy prognosis and chemotherapeutic resistance.24,25 Cancer stem cell theory proposes the stemness of the tumour tissue as responsible for sustaining the tumour growth.26 Sharing the same major signaling pathways with the embryonic stem cell, cancer stem cells are believed to possess a cancer-initiating ability, thereby giving rise to the cancer recurrence.25,27 Given its prominence on the cell surface, CD133 presents a logical target for actively targeting nanomedicines. For this reason, CD133 has been investigated as a potential target for the CRC management via specific antibody-antigen targeting.28–30
In the current study, anti-CD133 monoclonal antibody (mAb) was utilised for targeted 5-FU delivery to CD133+ CRC cells. Carboxyl-terminated surface of PEGylated AuNPs enabled the conjugation of anti-CD133 via the amide linkage. Activation of the carboxyl terminus prior to anti-CD133 mAb conjugation was achieved using EDC/sulfo-NHS as the crosslinking agent. Here, the reaction of carboxylates with the EDC reagent produces an unstable reactive O-acylisourea ester, while subsequent reaction with sulfo-NHS enhances the coupling efficiency by producing semi-stable amine-reactive NHS-ester prior to the formation of the stable amide crosslinks with the amine groups of the protein.31–33 Meanwhile, noncovalent 5-FU loading via the electron cloud of π back-bonded carbonyl oxygen of PEGylated AuNPs allows sustained drug release to enhance chemotherapeutic effects.34
In order to reinvigorate the chemotherapeutic effects of 5-FU, it is pivotal to timely design the right drug carrier system with improved selectivity, effectiveness and controlled release of the drug into systemic circulation. Herein, we set out to investigate the cytotoxicity effects of carboxyl end-functionalised gold nanoparticles grafted with 5-FU in HCT116 colorectal cancer cells. The AuNPs and PEGylated AuNPs were prepared by the chemical reduction method and surface modification with thiolated PEG with carboxyl termination, respectively, under optimal synthesis conditions. The PEGylated AuNPs were further conjugated with anti-CD133 mAb via EDC/sulfo-NHS coupling reaction and functionalised with 5-FU, as illustrated in Fig. 1. The physical, chemical and cytotoxic properties of PEGylated AuNPs, PEGylated AuNPs-CD133, 5-FU-PEGylated AuNPs and 5-FU-PEGylated AuNPs-CD133 were investigated. Cellular internalisation of nanoparticles in HCT116 cells was also studied using fluorescence microscopy.
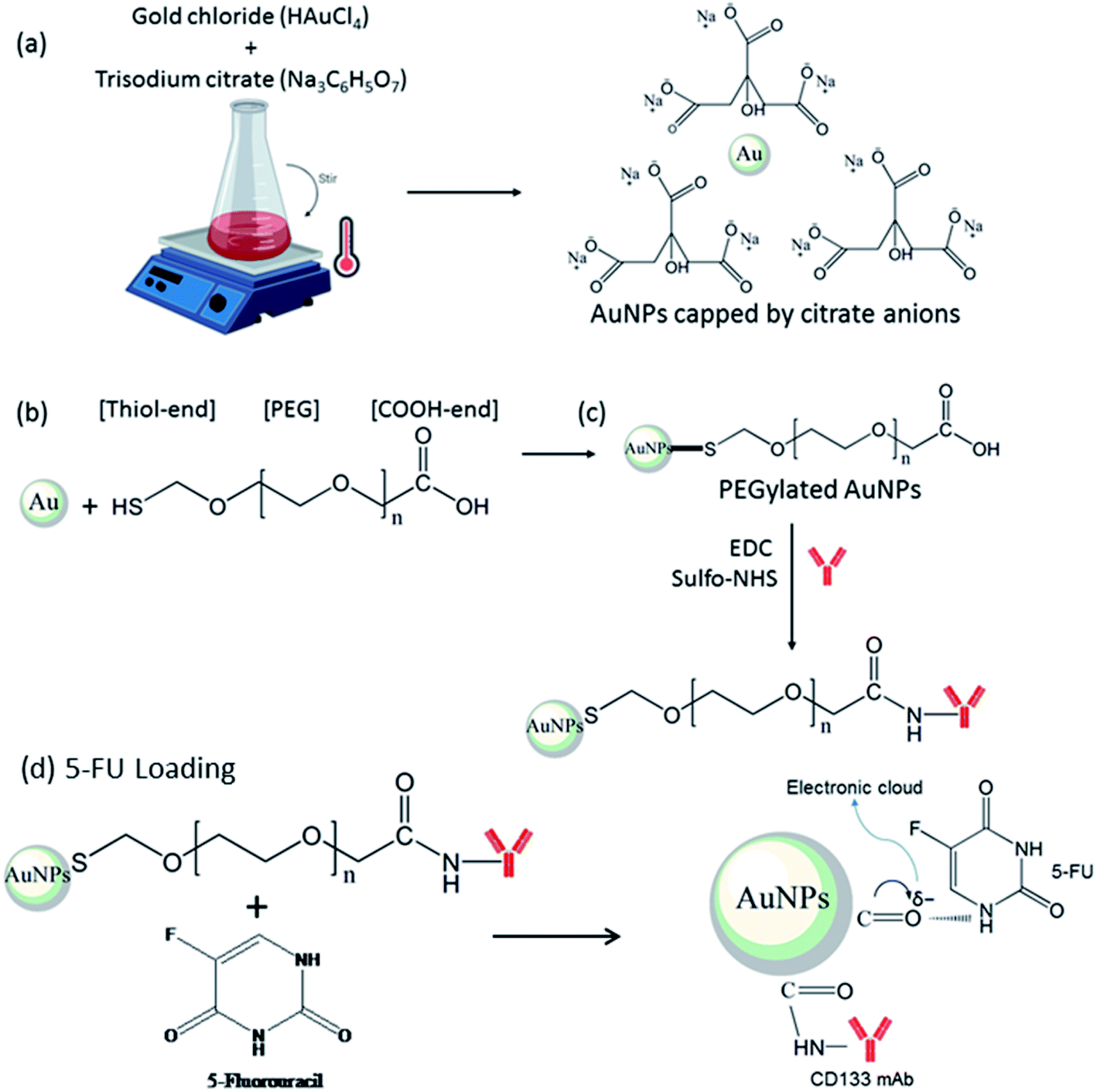 | ||
| Fig. 1 Schematic illustration of (a) AuNPs synthesis via chemical reduction of gold chloride to produce AuNPs with citrate anion capping, (b) AuNPs surface modification by thiolated PEG to displace citrate anions on the AuNPs surface and form PEGylated AuNPs with a COOH group on the other terminus for further functionalization, (c) PEGylated AuNPs functionalised with CD133 antibody. The carboxylates of PEGylated AuNPs were activated via EDC/sulfo-NHS coupling chemistry to form stable amide linkage with the amine group of mAb CD133. (d) 5-FU loading via the electron cloud of π back-bonded carbonyl oxygen of PEGylated AuNPs. | ||
2. Materials and methods
2.1. Materials
Gold chloride (HAuCl4), trisodium citrate (Na3C6H5O7), poly(ethylene glycol) 2-mercaptoethyl ether acetic acid (HS-PEG-COOH, Mw = 5 kDa), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC), and dimethyl sulfoxide (DMSO) were purchased from Sigma-Aldrich (St. Louis, MO, USA). N-hydroxysulfosuccinimide (NHS) was purchased from Cayman Chemical. Thiazolyl blue tetrazolium bromide (MTT) reagent was purchased from Nacalai Tesque (Kyoto, Japan). CD133 mAb conjugated with phycoerythrin (PE) was purchased from Thermo Fisher Scientific (Rockford, IL). 5-FU was purchased from Pfizer (New York, NY, USA) and used as received. Hoechst 33342 staining dye solution and the anti-fade fluorescence mounting medium were from Abcam (Cambridge, UK).2.2. Gold nanoparticle synthesis and PEGylation surface modification
Gold nanoparticles (AuNPs) were synthesised following a chemical reduction method.35 In brief, 2 mL of trisodium citrate (1%) were added into the boiling gold chloride solution (20 mL, 1 mM). The colour of the solution turned from slightly yellow to a deep ruby red colour, approximately after 2 minutes, indicating the formation of AuNPs. Surface modification of AuNPs was performed by the direct mixing of an HS-PEG-COOH solution with the AuNP colloid, as described by Hsiao et al.,36 with minor modifications. Briefly, 1 mL of 1 mM HS-PEG-COOH was added into 4 mL of the pristine AuNP colloid and incubated overnight at room temperature to allow for the ligand exchange to occur. Two washing cycles were conducted by collecting the suspended particles with centrifugation at 7500 × g for 60 minutes followed by resuspension back to 4 mL of distilled water to remove the excess thiolated PEG. The pH of the final pristine solution was found to be ∼6.9. For physical characterisation, we further monitored the (i) extinction profile of this suspension using UV-Vis spectroscopy (UV-1800 Shimadzu), (ii) the absolute size of the AuNPs formed using transmission electron microscopy (Carl Zeiss AG-LIBRA 120 kV) and (iii) the polydispersity index and hydrodynamic size of the AuNPs (Malvern Nanosizer). AuNPs and PEGylated AuNPs were kept at 4 °C until further use. The schematic illustration of AuNPs synthesis by chemical reduction and surface modification by thiolated PEG was shown in Fig. 1(a) and (b). The number density of the nanoparticles (N) was determined via UV-Vis spectroscopy.372.3. Conjugation of anti-CD133 antibody and 5-FU drug loading
Anti-CD133 mAb was covalently conjugated onto carboxyl end-PEGylated AuNPs by means of EDC/NHS chemistry. EDC/NHS is known as a crosslinker agent able to activate the carboxyl end of PEGylated AuNPs for functionalisation.38 The activation of the carboxyl end with EDC/sulfo-NHS according to the method by Parolo et al.39 was used in a slightly modified form. Briefly, 50 μL of EDC (10 mg mL−1) and 10 μL of sulfo-NHS (100 mg mL−1) were mixed and allowed to sit for 10 minutes at room temperature. The resulting solution was mixed with 5 mL PEGylated AuNPs, and PBS buffer was added up to a final volume of 10 mL prior to 30 minutes incubation at 24 °C. The excess EDC/sulfo-NHS was removed by centrifugation at 7500 × g for 60 minutes. Then, the pellet solution containing activated carboxyl groups of PEGylated AuNPs was quickly reacted with 10 μg of anti-CD133 PE-conjugated mAb, and the volume was diluted to the final volume of 2 mL in PBS buffer. The reaction was incubated for 2 hours at room temperature. Excess reagents were removed by washing with cold PBS at 4 °C for 60 minutes at 7500 × g. Supernatant was kept for the unconjugated protein concentration analysis in a bicinchoninic acid (BCA) assay. The conjugates were then added with 1 μL of 2% bovine serum albumin (BSA) in PBS buffer (10 mM, pH 7.4) to stabilize the conjugated-AuNPs solution. The resulting solution was then incubated for 60 minutes at room temperature. The final conjugates were resuspended in 2 mL distilled water and assigned as PEGylated AuNPs-CD133.The schematic illustration of the conjugation of anti-CD133 mAb to the PEGylated AuNPs by EDC/sulfo-NHS chemistry is shown in Fig. 1(c). Anti-CD133 mAb conjugation was indirectly measured by BCA assay using a commercial kit following the manufacturer's protocols (Pierce™ BCA Protein Assay Kit, Thermo Scientific). Briefly, 25 μL of conjugates supernatant or BSA standard was mixed with 200 μL working reagent in a 96-well plate. The mixture was then mixed for 30 seconds using plate shaker, incubated for 30 minutes at 37 °C in dark and read for absorbance at 562 nm. The loading of 5-FU onto PEGylated AuNPs and PEGylated AuNPs-CD133 were via non-covalent loading by the π back bond carbonyl group of PEG,34 as illustrated in Fig. 1(d). In brief, 10 μL 5-FU (1.28 mM) were added to 3 mL of PEGylated AuNPs and PEGylated AuNPs-CD133 solution. The mixture was incubated overnight at room temperature by constant stirring. Any unloaded 5-FU was separated by 60 minutes centrifugation at 7500 × g at 24 °C.
2.4. Nanoparticle characterisations
The synthesized AuNPs were characterised by X-ray diffraction (XRD) to identify their crystalline form. The sample for XRD was prepared by collecting the AuNPs pellet after centrifuging at 7500 × g for 1 hour, keeping at −80 °C overnight and freeze-drying to get the powdered sample. UV-Vis spectroscopy (UV-1800 spectrophotometer, Shimadzu) was used to monitor the absorbance spectrum of the AuNPs within the wavelength range of 400–600 nm. The size and the morphology of the particles were measured using transmission electron microscopy (TEM, Carl Zeiss AG-LIBRA 120) operating at 120 kV. Hydrodynamic sizes of nanoparticles were measured using the dynamic light scattering (DLS) technique on a Malvern Nanosizer.2.5. Cell culture
HCT116 cells was obtained from Universiti Kebangsaan Malaysia Medical Molecular Biology Institute (UMBI) and cultured in DMEM (Nacalai Tesque, Kyoto, Japan) enriched with 10% fetal bovine serum (FBS; Tico Europe). Cells were grown in T75 cm2 tissue culture flask and kept at 37 °C with 5% CO2. The medium was changed every 2 days and cells were passaged once they reached ∼80% confluence. The cells were used throughout the study between passages 3 and 9.2.6. Flow cytometry analysis
The percentage of HCT116 cells expressing CD133 was analyzed using flow cytometry. After cell harvesting, the suspended cells were incubated with PE-conjugated anti-CD133 antibody (Thermo Fisher Scientific, Rockford, IL), according to the manufacturer's recommendations. The cells were then analyzed on a BD FACSCanto II flow cytometer (Becton Dickinson, Franklin Lakes, NJ). The flow cytometric analysis was performed using FlowJo Software (TreeStar Inc., USA).2.7. Cellular imaging uptake of FITC-labeled gold nanoparticles
To study the internalisation of nanocarrier, FITC dye was conjugated with nanoparticles via hydroxyl group of FITC and carbonyl group of PEGylated AuNPs. Briefly, 50 μL (1.2 mg mL−1) FITC was added into 1 mL nanoparticles and incubated overnight at room temperature by constant stirring. The solution was centrifuged at 8182 × g for 20 minutes to discard unconjugated FITC and the pellet was dispersed in 1 mL PBS. FITC-labeled nanoparticles were stored in dark until further use. For fluorescence cell imaging, cells were cultured on square coverslips and treated with 100 μL of either PEGylated AuNPs, PEGylated AuNPs-CD133 or 5-FU-PEGylated AuNPs-CD133 in 2 mL of culture media. After 24 hours, cells were fixed with 4% paraformaldehyde prior to Hoechst staining for nuclei. Cells were then mounted on slide with mounting medium and observed under Olympus BX41 Microscope and images were further analysed by ImageJ software.2.8. Cytotoxicity effects using MTT reagent
The viability assay was measured in a 96-well plate using MTT reagent according to manufacturer's instructions (Nacalai Tesque, Kyoto, Japan). The nanoparticles were left under the UV light in Biological Safety Cabinet Class II (BSC II) for 3 hours for sterilisation prior to the cell treatments. HCT116 cells were seeded (2 × 104 cells per well) in complete medium and left to grow overnight. For cytotoxicity activity of free 5-FU and 5-FU in PEGylated AuNPs and CD133 mAb-conjugated form, HCT116 colorectal cancer cells were treated with various concentrations of 5-FU and (i) 5-FU-PEGylated AuNPs and (ii) 5-FU-PEGylated AuNPs-CD133 for 24 hours. After treatment, culture media were discarded and replaced by serum-free media. MTT reagent (5 mg mL−1) was then added into each well and incubated for 4 hours at 37 °C. Then, culture medium containing MTT was discarded and DMSO was added to solubilise the purple formazan. After 15 min of incubation, the absorbance was measured at 517 nm using ELISA microplate reader. Cell viability was expressed as the percentage relative to untreated cells.2.9. Statistical analysis
In all figures, error bars are the standard error of the mean (n = 3). GraphPad Prism 6 software was used for all the statistical analyses. Student's t-test was used to compare the statistical significance of the treatment groups relative to the control group. Two-tailed p-value below 0.05 was considered as statistically significant.3. Results and discussion
3.1. Synthesis of gold nanoparticles and PEGylated gold nanoparticles
Gold nanoparticles (AuNPs) have displayed a promising potential as nanocarriers to specifically target tumour cells.40,41 In this present study, AuNPs were synthesised based on the Turkevich method, also known as chemical reduction method. Citrate ions acted as reducing agents to reduce gold ions (Au3+) into zero-valent gold atoms (Au0) in a boiling solution.42 In addition, citrate anions adsorbed onto AuNPs surface also served as a capping agent,43 which contributed to the colloidal stability and maintained monodispersity by imposing electrostatic repulsion between the particles.Direct loading of 5-FU onto the citrate-stabilized AuNPs surface is not possible because of the hydrophobic barrier posed by the citrate capping before hydrophilic 5-FU. Thus, the AuNPs surface needs to be modified for 5-FU functionalisation. Here, poly(ethylene glycol)-2-mercaptoethyl ether acetic acid, HS-PEG-COOH, a heterobifunctional PEG with a thiol at one end and a carboxyl group at the other end was used as a linker and steric stabiliser. The high binding affinity of gold (Au) towards thiol (–SH) facilitated the ligand exchange between citrate anions and thiolated PEG. Under constant stirring, the ligand exchange resulted in colloidal gold known to be very stable and well-defined in suspension. In this regard, thiolated PEG has successfully replaced citrate anions through the chemical adsorption or chemisorption via the extremely stable S–Au bonds,44 which provided steric stabilisation to the AuNPs.
3.2. Characterisations of gold nanoparticles and PEGylated gold nanoparticles
The XRD pattern of AuNPs shown in Fig. 2 exhibited distinctive peaks at 38.14°, 44.31°, 64.58° and 77.53° in the 2-theta range of 20°–80°, which were indexed as (111), (200), (220) and (311) crystallographic planes, respectively. Comparison of the peaks in the XRD pattern with the standard database (ICDD PDF card number 00-004-0784) confirmed that the synthesised product comprised the face-centred cubic lattice of Au. The relative sharpness of the diffraction peaks also suggested that the synthesised AuNPs are highly crystalline for their size.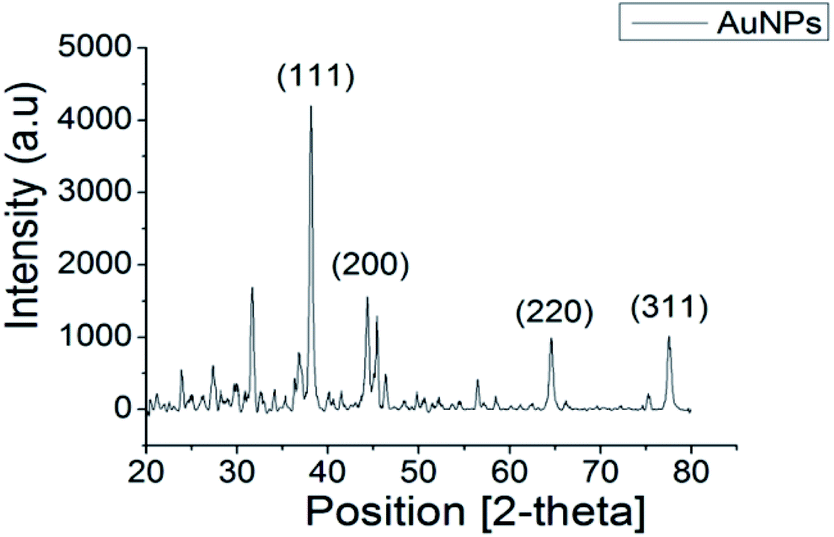 | ||
| Fig. 2 XRD pattern of synthesised AuNPs showing peaks of (111), (200), (220), (311) confirming the crystalline phases of as-synthesised material is gold. | ||
The optical properties of AuNPs and PEGylated AuNPs were measured using UV-Vis spectroscopy to confirm their formation. The SPR peaks of AuNPs and PEGylated AuNPs were detected at 522 nm and 525.5 nm, respectively [Fig. 3(A)]. This red shift of the absorption maximum suggests that PEGylation alters the dielectric properties at the surface of AuNPs. As shown in Fig. 3, AuNPs colloids were ruby red in colour, while the PEGylated AuNPs exhibited a more purplish colour, conforming to this red shift in the SPR due to PEGylation, which can be associated with the minimum size increment.45 The classical Mie theory predicts practically no changes to the peak wavelength of the SPR in the extinction spectra, but experimentally, the particle size increase by ∼5 nm usually shifts this peak to higher wavelengths by ∼2–3 nm in the 10–20 nm particle size range.46 Meanwhile, TEM images of AuNPs showed that the majority of AuNPs were spherical in shape, with a mean size of 13.0 ± 1.6 nm and low polydispersity [Fig. 3(B)]. Following PEGylation, AuNPs displayed signs of hydration layer assembly and aggregation occurring with low dispersed nanoparticles [Fig. 3(C)]. However, the PEGylated AuNPs nanoparticles remained in a single particle suggesting the preservation of the short-range repulsive hydration layer around the nanoparticles. Both pure and PEGylated systems were typified by pronounced colloidal stability [Fig. 3(B) and (C)], which is a prerequisite for their parenteral administration. The size of AuNPs increased from 13.0 ± 1.6 nm to 19.0 ± 4.6 nm after the modification with PEG, as in good agreement with the UV-Vis spectrometry characteristics might be from the agglomerates formation. Because this particle size increase is observed at the level of primary particles [Fig. 3(B) and (C)], this excludes particle aggregation due to PEGylation as a reason. More probably, the introduction of PEG in a non-buffered system modified the redox potential of the citrate anions and shifted the reaction balance back to the reactants, thereby inducing a late particle growth. Buffering with the combination of citric acid and sodium citrate and reversing the reagent addition may be a way to prevent this particle growth in the PEGylation stage.47
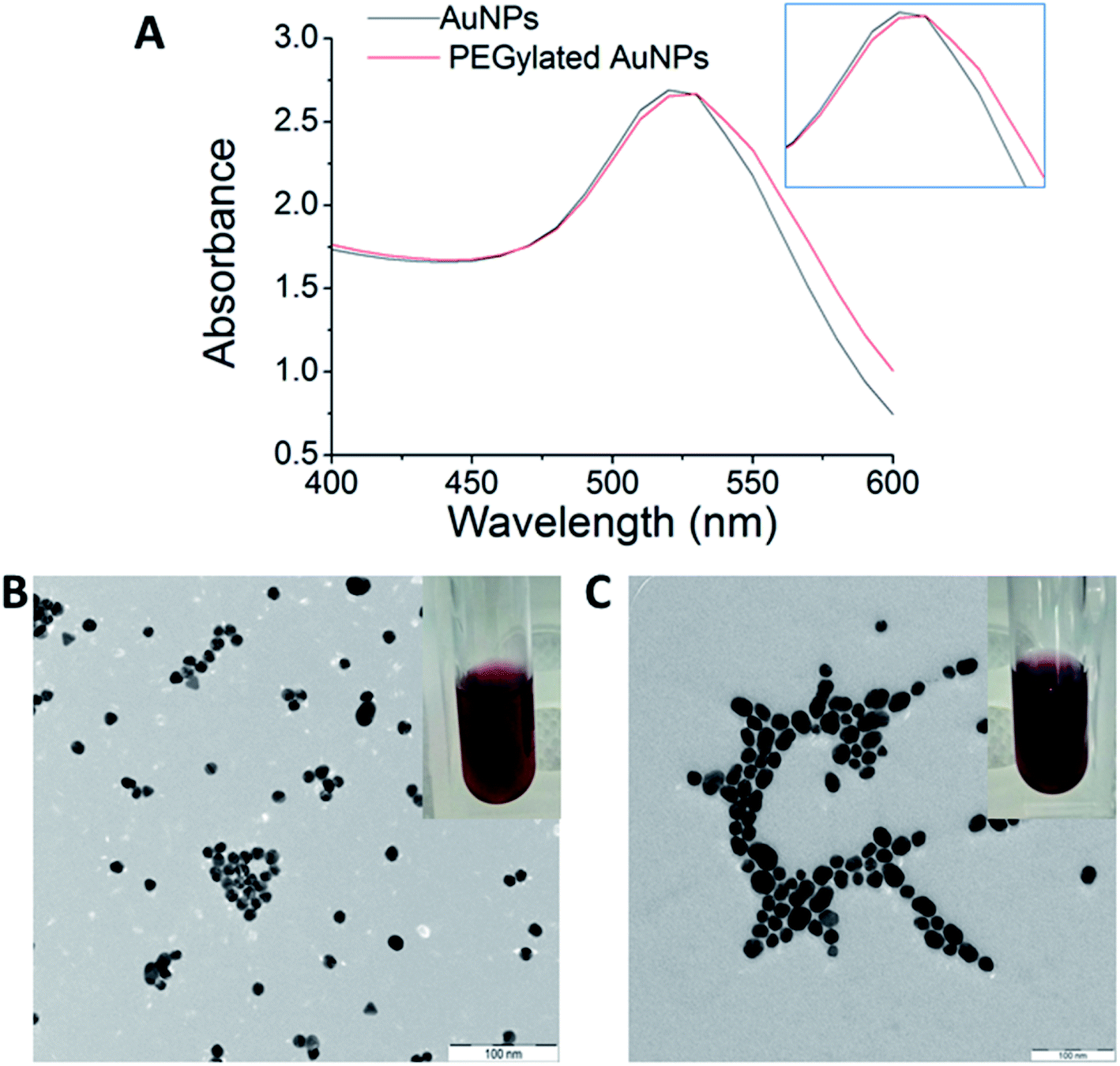 | ||
Fig. 3 (A) UV-Vis spectra of colloidal AuNPs and PEGylated AuNPs. Transmission electron microscopy images of (B) AuNPs and (C) PEGylated AuNPs. Magnification: 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000×, scale bar: 100 nm. Colloidal AuNPs stabilized by citrate displayed ruby red color (B), while PEGylated AuNPs displayed a more purple-like color (C). 000×, scale bar: 100 nm. Colloidal AuNPs stabilized by citrate displayed ruby red color (B), while PEGylated AuNPs displayed a more purple-like color (C). | ||
Z-Average and the polydispersity index (PDI) of AuNPs and PEGylated AuNPs are tabulated in Table 1. The average hydrodynamic size of AuNPs and PEGylated AuNPs were 27.88 nm and 50.18 nm, respectively, and the PDI values were 0.408 and 0.521, respectively. As expected, the hydrodynamic diameters of AuNPs and PEGylated AuNPs were larger compared to the core nanoparticle sizes measured under TEM. This increase in the hydrodynamic diameters measured by DLS compared to values measured by TEM is mostly caused by particle aggregation evident under TEM [Fig. 3(B) and (C)] and only to a minor degree by the contribution of the hydration layer surrounding the nanoparticles in an aqueous medium. Further, an increase in the particle size by 20–40 nm has been typically observed as the result of PEGylation.48–50
| λ (nm) | Peak absorption (OD) | Size (nm) | Z-Average (nm) | PDI | Number density of nanoparticles, N (particles per mL) | Concentration (μg mL−1) | |
|---|---|---|---|---|---|---|---|
| AuNPs | 522 | 2.698 | 13 | 27.88 | 0.408 | 7.249 × 1012 | 161.172 |
| PEGylated AuNPs | 525.5 | 2.678 | 19 | 50.18 | 0.521 | 2.212 × 1012 | 153.542 |
Based on UV-Vis and TEM results, the estimated number density of the nanoparticles (N) was calculated using the following equation:
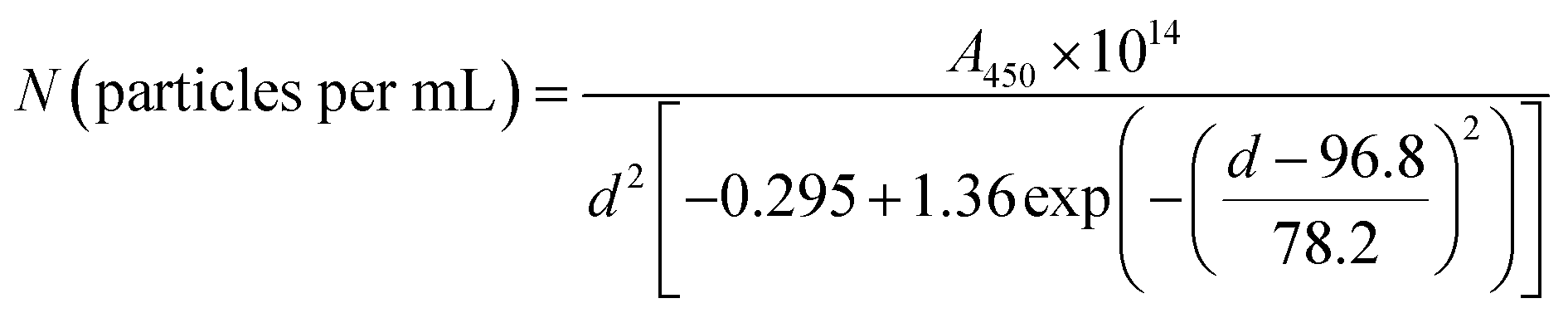 | (1) |
| C (μg mL−1) = N × gold density × (1/6)(22/7) × d3 × 10−21 × 106. | (2) |
3.3. Conjugation of anti-CD133 mAb
In the present study, PEGylated AuNPs were conjugated with anti-CD133 mAb via the EDC/sulfo-NHS reaction. Conjugation with mAb was entailed by the red shift of the maximum absorbance by PEGylated AuNPs, as visible from UV-Vis spectra [Fig. 4(A)]. The wavelength for the maximum absorbance of PEGylated AuNPs-CD133 increased from 525.5 nm to 527 nm, indicating the physicochemical interaction between the AuNPs and the protein along their interface.51 The estimated amount of covalently conjugated anti-CD133 mAb onto PEGylated AuNPs could be determined from the difference between the total amount of the initial antibody added and the amount of unconjugated antibody recovered following the separation via centrifugation. A calibration curve was generated using the BCA protein assay across a concentration range of 0–2000 μg mL−1 [Fig. 4(B)]. A linear relationship was observed with R2 value > 0.997. Based on the BSA standard curve, the efficiency of the uptake of anti-CD133 mAb by PEGylated AuNPs via EDC/sulfo-NHS crosslinking was 82.35% relative to the total amount of the protein added.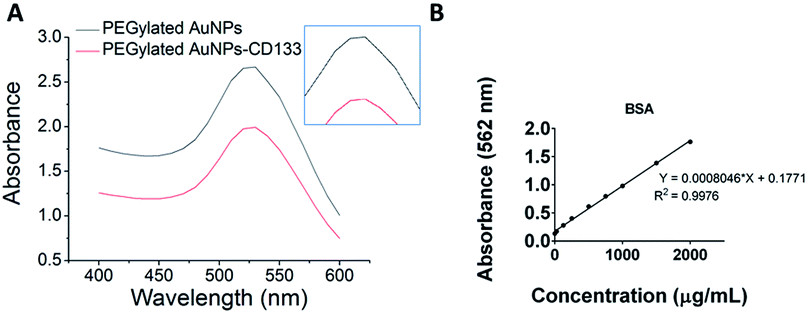 | ||
| Fig. 4 Functionalization of PEGylated AuNPs with anti-CD133 monoclonal antibody conjugated via EDC/sulfo-NHS chemistry, as measured by (A) UV-Vis spectra and (B) BCA assay using BSA standard protein analysis. | ||
3.4. CD133 expression and localization in HCT116 cells
Flow cytometry was used to confirm the expression of CD133 in HCT116 cells. Meanwhile, fluorescence microscopy demonstrated the localisation of CD133 cell marker. As illustrated in Fig. 5(A), 92.4 ± 1.3% of HCT116 cells expressed CD133. CD133 is a transmembrane protein, preferentially located in the plasma membrane protrusions and has been associated with poor prognosis in CRC,52 high resistance towards chemoradiotherapy.53 It was also proposed as the surface marker for CRC stem cells.24 Moreover, immunohistochemical staining performed on colon cancer samples of patients has demonstrated a strong correlation between CD133 expression and liver metastasis.54 To confirm the localisation of CD133 in HCT116 cells, cellular nuclei were counterstained with DAPI, while the cells were stained with anti-CD133 mAb conjugated with PE. As shown by the merged image in Fig. 5(B), CD133 was highly expressed at the cellular membrane.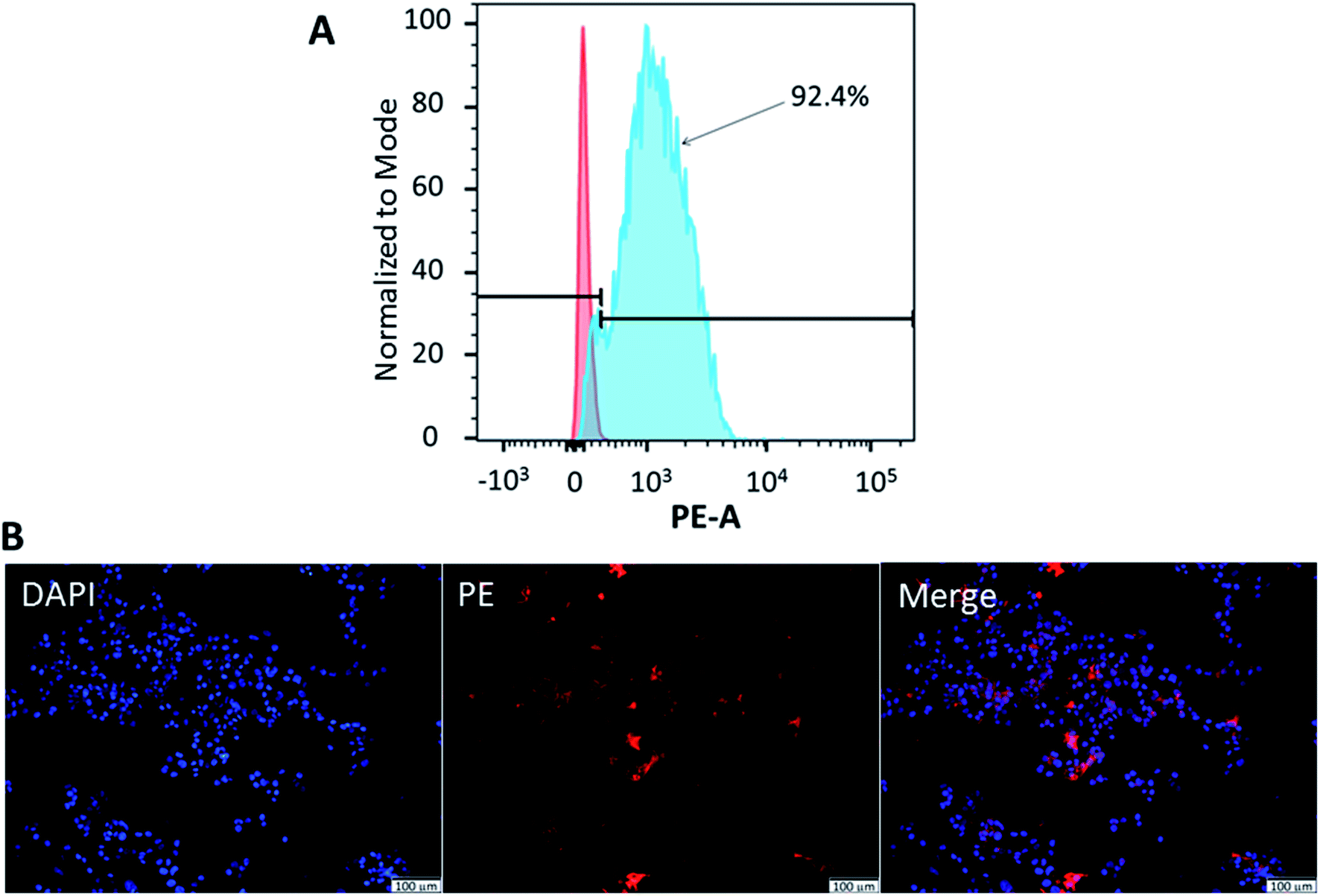 | ||
| Fig. 5 CD133 expression and localisation in HCT116 cells, as analyzed by (A) flow cytometry and (B) immunocytochemical analysis using fluorescence microscopy. Red staining: CD133 expression on plasma membrane; blue staining: nucleus. Magnification: 20×, scale bar: 100 μm. | ||
3.5. Cellular imaging of PEGylated AuNPs and 5-FU-PEGylated AuNPs-CD133
Next, we investigated the cellular internalisation of nanoparticles with and without the anti-CD133 mAb in HCT116 cells, which abundantly expressed CD133 on their membranes. Fluorescence microscopy provides a direct and simple approach to monitor intracellular trafficking pathways as well as the cellular fate of nanoparticles. The uptake of the nanoparticles labeled with fluorophores by HCT116 cells was visualised using a single or combination of three different markers: DAPI, FITC and PE. DAPI (blue) was used to counterstain nuclei whereas PEGylated AuNPs were visualised using the FITC (green) signal. PE (red) signal was used to visualise the localisation of PEGylated AuNPs conjugated with anti-CD133 mAb. The isothiocyanate group in FITC facilitates the attachment of FITC molecules onto the PEGylated AuNPs surface.55 The presence of nanoparticles in HCT116 cells was indicated by the images obtained from the FITC channel. Meanwhile, the PE channel represented the images of nanoparticles conjugated with anti-CD133 mAb.As shown in Fig. 6, in vitro intracellular colocalisation of FITC-labeled PEGylated AuNPs with DAPI as the nuclear dye in HCT116 cells demonstrated that the accumulation of PEGylated AuNPs was mainly at the cellular membrane. In contrast, interestingly, nanoparticles conjugated with anti-CD133 mAb appeared to be contained within the nuclear regions of the cells. The evident the PE expression in HCT116 cells also acts as an indirect indicator of successful conjugation of PEGylated AuNPs with anti-CD133 mAb via EDC/sulfo-NHS reaction (Fig. 6). This result supports our nation that anti-CD133 mAb conjugation facilitates the specific intracellular uptake. This effect was likely due to the specific antigen–antibody binding interaction. These results confirm that anti-CD133 mAb can be used as a targeting ligand to match the CD133 antigen receptors present on the CRC cell surface. The given mAb can act as a guidance missile to help the nanoparticle carrier target specific antigenic sites on the tumour surface. Other than antibodies, folate has been utilized earlier as a ligand to target overexpressed folic acid on the surface of solid tumours.56,57
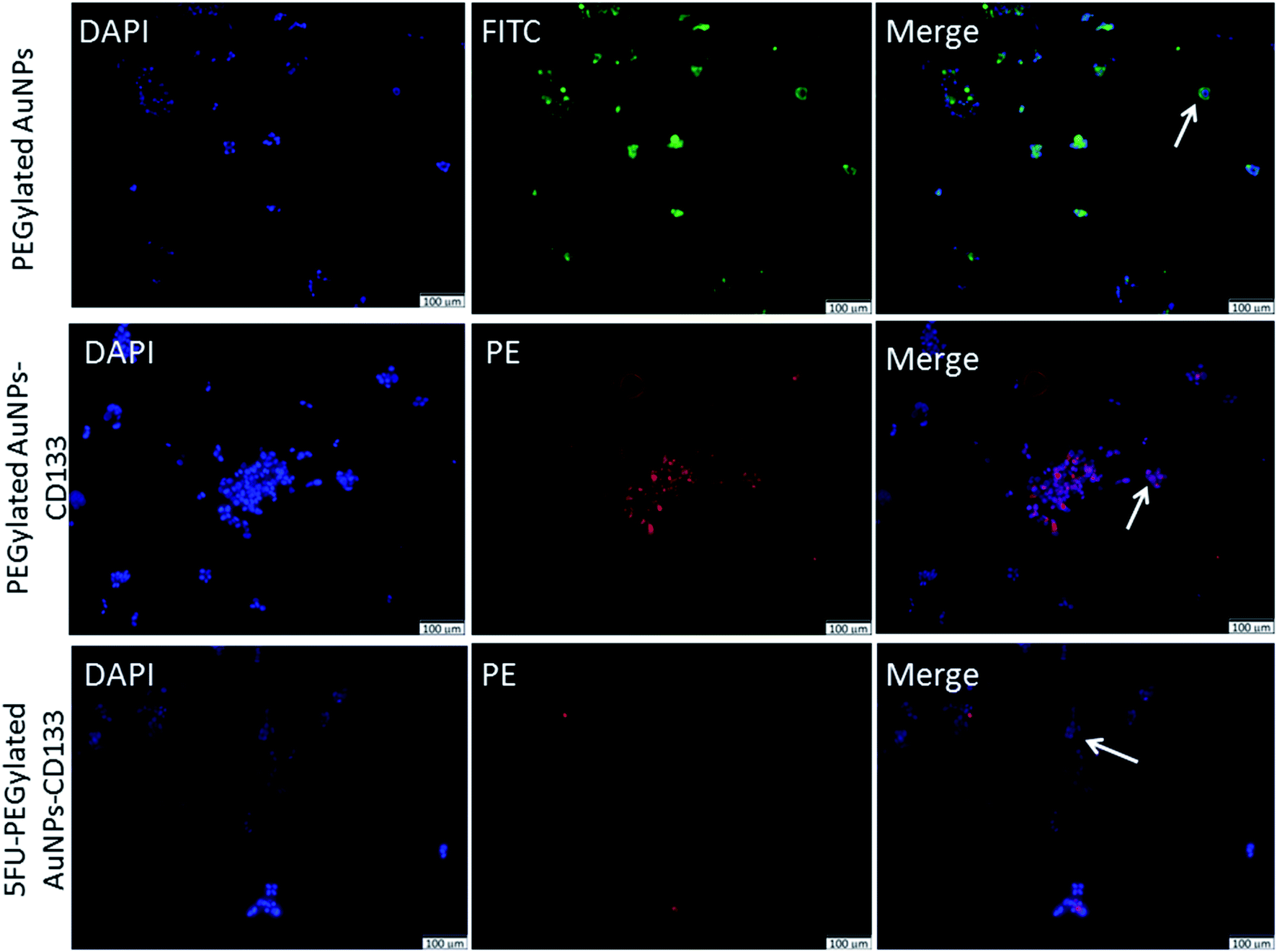 | ||
| Fig. 6 Intracellular localisation of nanoparticles incubated for 24 hours at a 5% dilution of the stock solution (7.7–8.0 μg mL−1). Arrows point at examples of localization of PEGylated AuNPs without or with anti-CD133 mAb conjugates. Blue staining: nucleus, Green staining: PEGylated AuNPs, red staining: CD133 expression. Magnification: 20×, scale bar: 100 μm. | ||
However, as reports from previous studies suggest, the uptake of nanoparticles occurs in a time- and concentration-dependent manner.58 Thus, the recommended future studies include investigation of different incubation times in order to obtain the optimum cell entry as well as optimum chemotherapeutic effects of 5-FU-PEGylated AuNPs-CD133 against CRC cells.
3.6. Cytotoxicity of 5-fluorouracil and nanoparticles
To assess the toxicity of 5-FU and the synthesized nanoparticles against HCT116 cells, the MTT viability assay was carried out. The 24 hours exposure of HCT116 cells against 5-FU showed dose–response relationship with IC50 of 57.29 μg mL−1 [Fig. 7(A)]. 5-FU is an antineoplastic agent involving in thymidylate synthase inhibition to subsequently induce DNA cell damage.22 Meanwhile, after exposing HCT116 cells to the PEGylated AuNPs and PEGylated AuNPs-CD133 for 24 hours, no toxicities were observed even up to the 10% dilution of the pristine stock solution (15.4–16.1 μg mL−1) [Fig. 7(B)]. These findings suggest that PEGylated AuNPs and PEGylated AuNPs conjugated with CD133 mAb were nontoxic and suitable to be used as nanocarrier, given the relative cell survival of 95% and higher. In contrast, 5-FU PEGylated AuNPs and 5-FU PEGylated AuNPs-CD133 conjugates significantly reduced cell viabilities at all the tested concentrations. Still, the cytotoxic effect was far more pronounced for the 5-FU-PEGylated AuNPs-CD133 conjugates than for 5-FU-PEGylated AuNPs, reaching p < 0.0001 for most concentration. These results unequivocally indicate the enhanced CRC cell targeting using anti-CD133 mAb and its ability to facilitate the delivery of 5-FU to specific sites in cancerous cells. 5-FU is a genotoxic drug; due to its identical metabolic pathway with the natural pyrimidine uracil, 5-FU is readily incorporated into nucleic acids, where it interferes with RNA and DNA processing, leading ultimately to cell death.20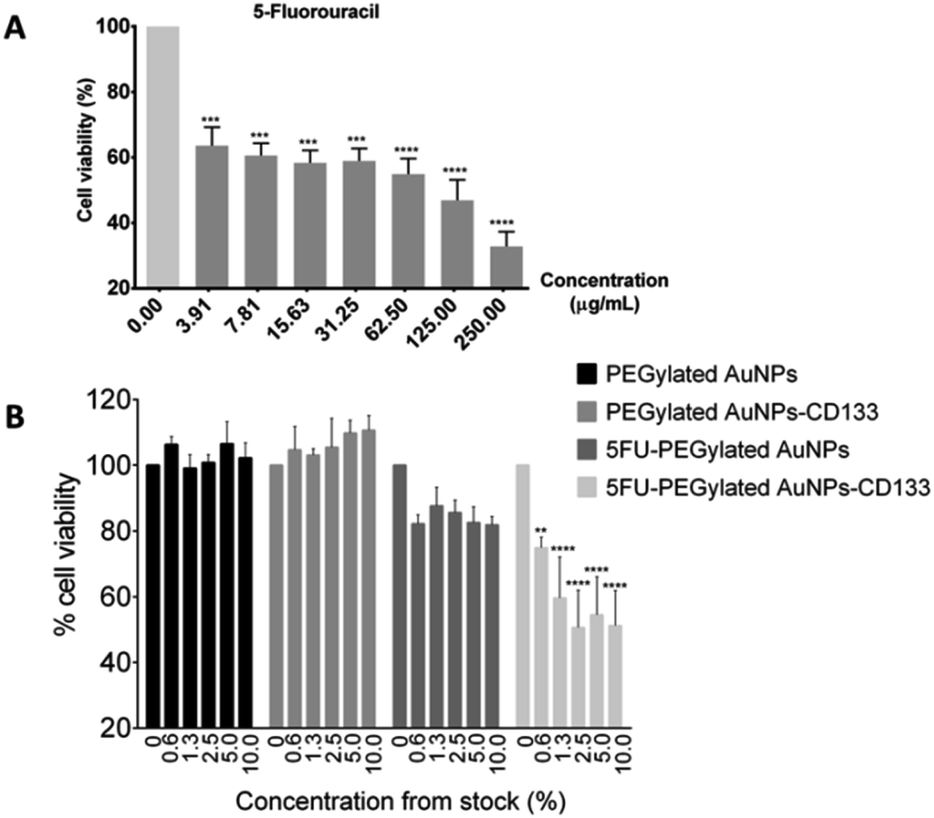 | ||
| Fig. 7 Cell viability on the HCT116 cells of (A) free 5-fluorouracil and (B) as-synthesised nanoparticles, as measured by MTT assay. Stock dilutions of nanoparticles in the 0.6–10% range correspond to nanoparticle concentration range of 1.0–16.1 μg mL−1 in the cell culture medium. Data are presented as mean ± SEM (n = 3). Asterisks denote the levels of confidence with respect to the statistical difference between 5-FU PEGylated AuNPs-CD133 and 5-FU PEGylated AuNPs sample groups: ****p < 0.0001, ***p < 0.05, **p < 0.01 vs. 0% concentration. | ||
Undoubtedly, the medical applications using the nanoparticles particularly AuNPs require inevitably many future research and assessment since there are complex interactions of nanodrugs with cellular and humoral constituents of the immune system. Thus, in vitro studies may not translated well to the effects in medical application. However, preclinical trials data provide valuable information to allow more rapid development of new treatments and technologies due to their efficacy and cost-effectiveness. Nanoparticles with stealth engineering designs may offer immune evasion of the host by considering the specific cells to be targeted. In pharmacology, the natural nano-sized particles such as viruses demonstrate ways on escaping the host immune system to gain access for intracellular target which may be a valuable understanding for the current researchers of targeted drug delivery.59
4. Conclusions
In conclusion, we have successfully modified the surface of AuNPs with heterobifunctional PEG having a carboxyl end for anti-CD133 mAb conjugation to facilitate specific targeting of cancer cells and the direct delivery of chemotherapeutic 5-FU. The antibody was successfully conjugated via EDC/sulfo-NHS coupling reaction used to activate the carboxyl termini of PEGylated AuNPs. The results from fluorescence imaging and the viability assay suggested that 5-FU-PEGylated AuNPs conjugated with anti-CD133 mAb facilitated cellular internalisation and the release of the 5-FU payload at specific sites that triggered cancer cell death. Abundant CD133 expression on the surface of CRC cells confers anti-CD133 mAb conjugates as an excellent candidate for the targeted drug delivery systems to enhance the current CRC treatment and management. However, additional studies investigating the pathways of internalization by CRC cells are recommended for better insight into conditions governing the nanoparticle uptake as grounds for the further optimization of the chemotherapeutic effects of 5-FU.Author contributions
IZA conceived the original idea. IZA, RM and CACA designed the study and supervised the work. MHMZ prepared and characterised the 5-FU-PEGylated AuNPs-CD133 synthesis, analysed the results and drafted the article. SNZ prepared and characterised the AuNPs and PEGylated AuNPs. WKK gave advices regarding mAb purchasing, reviewed and edited the article. JKL and SF gave advices regarding characterisations of the sample, reviewed and edited the article. ADZ, NI and VU reviewed and edited the article. All authors have read and approved the article submission.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors would like to acknowledge Nanotedd Group at Faculty of Physics, Universiti Putra Malaysia for assistance with synthesis and characterisation studies, Department of Immunology, Universiti Sains Malaysia for the flow cytometry facility and Universiti Sains Malaysia under Research University Individual (RUI Grant No. 1001/PPSP/8012296) and Graduate Research Assistant (GRA) Assist for the financial support.Notes and references
- E. Blanco, H. Shen and M. Ferrari, Nat. Nanotechnol., 2015, 33, 941–951 CAS.
- Y. Nakamura, A. Mochida, P. L. Choyke and H. Kobayashi, Bioconjug. Chem., 2016, 27, 2225–2238 CrossRef CAS PubMed.
- R. Ngoune, A. Peters, D. V. Elverfeldt, K. Winkler and G. Pütz, J. Control. Release, 2016, 238, 58–70 CrossRef CAS PubMed.
- Y. Shi, R. van der Meel, X. Chen and T. Lammers, Theranostics, 2020, 10, 7921–7924 CrossRef PubMed.
- S. Sindhwani, A. M. Syed, J. Ngai, B. R. Kingston, L. Maiorino, J. Rothschild, P. MacMillan, Y. Zhang, N. U. Rajesh, T. Hoang, J. L. Y. Wu, S. Wilhelm, A. Zilman, S. Gadde, A. Sulaiman, B. Ouyang, Z. Lin, L. Wang, M. Egeblad and W. C. W. Chan, Nat. Mater., 2020, 19, 566–575 CrossRef CAS PubMed.
- H. Maeda, H. Nakamura and J. Fang, Adv. Drug Deliv. Rev., 2013, 65, 71–79 CrossRef CAS PubMed.
- V. Uskoković and D. P. Uskoković, Nanotechnologies for Preventive and Regenerative Medicine: An Emerging Big Picture, ed. V. Uskoković and D. Uskoković, Micro and Nano Technologies Volume Series, Elsevier, Oxford, UK, 2018, pp. 513–566 Search PubMed.
- H. Jahangirian, K. Kalantari, Z. Izadiyan, R. Rafiee-Moghaddam, K. Shameli and T. J. Webster, Int. J. Nanomedicine, 2019, 14, 1633–1657 CrossRef CAS PubMed.
- S. Ahmad, A. A. Zamry, H. T. T. Tan, K. K. Wong, J. K. Lim and R. Mohamud, Mol. Immunol., 2017, 91, 123–133 CrossRef CAS PubMed.
- J. Liu, R. Papadakis and H. Li, Appl. Phys. Lett., 2018, 113, 083108 CrossRef.
- Z. R. Goddard, M. J. Marín, D. A. Russell and M. Searcey, Chem. Soc. Rev., 2020, 49, 8774–8789 RSC.
- D. Xi, S. Dong, X. Meng, Q. Lu, L. Meng and J. Ye, RSC Adv., 2012, 2, 12515–12524 RSC.
- T. Ilovitsh, Y. Danan, R. Meir, A. Meiri and Z. Zalevsky, Sci. Rep., 2015, 5, 8244 CrossRef CAS PubMed.
- M. M. El-Hammadi, Á. V. Delgado, C. Melguizo, J. C. Prados and J. L. Arias, Int. J. Pharm., 2017, 516, 61–70 CrossRef CAS PubMed.
- L. Guerrini, R. A. Alvarez-Puebla and N. Pazos-Perez, Mater., 2018, 11, 1154 CrossRef PubMed.
- V. Uskoković, P. P. Lee, L. A. Walsh, K. E. Fischer and T. A. Desai, Biomaterials, 2012, 33, 1663–1672 CrossRef PubMed.
- S. K. Libutti, G. F. Paciotti, A. A. Bymes, H. R. Alexander, W. E. Gannon, M. Walker, G. D. Seidel, N. Yuldasheva and N. Tamarkin, Clin. Cancer Res., 2010, 16, 6139–6149 CrossRef CAS PubMed.
- J. S. Suk, Q. Xu, N. Kim, J. Hanes and L. M. Ensign, Adv. Drug Deliv. Rev., 2016, 99, 28–51 CrossRef CAS PubMed.
- D. B. Longley, D. P. Harkin and P. G. Johnston, Nature, 2003, 3, 330–338 CAS.
- M. W. Saif, K. N. Syrigos and N. A. Katirtzoglou, Expert Opin. Investig. Drugs, 2009, 18, 335–348 CrossRef CAS PubMed.
- T. A. Baudino, Curr. Drug Discov. Technol., 2015, 12, 3–20 CrossRef CAS PubMed.
- M. H. Mohd-Zahid, R. Mohamud, C. A. C. Abdullah, J. K. Lim, H. Alem, W. N. W. Hanaffi and Z. A. Iskandar, RSC Adv., 2020, 10, 973–985 RSC.
- P. M. Glumac and A. M. LeBeau, Clin. Transl. Med., 2018, 7, 18 CrossRef PubMed.
- F. Ren, W. Q. Sheng and X. Du, World J. Gastroenterol., 2013, 19, 2603–2611 CrossRef CAS PubMed.
- D. Abetov, Z. Mustapova, T. Saliev and D. Bulanin, Tumour Biol., 2015, 36, 1339–1353 CrossRef CAS PubMed.
- K. F. Hung, T. Yang and S. Y. Kao, J. Chin. Med. Assoc., 2019, 82, 814–818 CrossRef PubMed.
- C. L. Zhang, T. Huang, B. L. Wu, W. X. He and D. Liu, Oncotarget, 2017, 8, 75756–75766 CrossRef PubMed.
- S. T. Ning, S. Y. Lee, M. F. Wei, C. L. Peng, S. Y. F. Lin and M. H. Tsai, ACS Appl. Mater. Interfaces, 2015, 8, 17793–17804 CrossRef PubMed.
- M. Bostad, C. E. Olsen, Q. Peng, K. Berg, A. Høgset and P. K. Selbo, J. Control. Release, 2015, 206, 37–48 CrossRef CAS PubMed.
- M. Akbari, N. Shomali, A. Faraji, D. Shanehbandi, M. Asadi and A. Mokhtarzadeh, Cell Biol. Int., 2019, 44, 368–380 CrossRef PubMed.
- A. A. Bogdanov, A. L. Klibanov and V. P. Torchilin, FEBS Lett., 1988, 1988(231), 381–384 CrossRef.
- Q. Yan, H. N. Zheng, C. Jiang, K. Li and S. J. Xiao, RSC Adv., 2015, 5, 69939–69947 RSC.
- P. Carl, I. I. Ramos, M. A. Segundo and R. J. Schneider, PLoS One, 2019, 14, e0218686 CrossRef CAS PubMed.
- M. Gajendiran, H. Jo, K. Kim and S. Balasubramaniam, Int. J. Nanomedicine, 2019, 14, 819–834 CrossRef CAS PubMed.
- M. Tejaswi, M. C. Rao, P. V. D. Prasad, G. Giridhar, V. G. K. M. Pisipti and R. K. N. R. Manepalli, Rasayan J. Chem., 2016, 9, 697–705 CAS.
- P. F. Hsiao, S. Peng, T. C. Tang, S. Y. Lin and H. C. Tsai, Int. J. Nanomedicine, 2016, 11, 1867–1878 CAS.
- W. Haiss, N. T. K. Thanh, J. Aveyard and D. G. Fernig, Anal. Chem., 2007, 79, 4215–4221 CrossRef CAS PubMed.
- S. Zhang, M. Gong, D. Zhang, H. Yang, F. Gao and L. Zou, Eur. J. Radiol., 2014, 83, 1190–1198 CrossRef PubMed.
- C. Parolo, A. de la Escosura-Muñi, E. Polo, V. Grazú, J. M. de la Fuente and A. Merkoçi, ACS Appl. Mater. Interfaces, 2013, 5, 10753–10759 CrossRef CAS PubMed.
- Z. Z. J. Lim, J. E. J. Li, C. T. Ng, L. Y. L. Yung and B. H. Bay, Acta Pharmacol. Sin., 2011, 32, 983–990 CrossRef CAS PubMed.
- P. Singh, S. Pandit, V. R. S. S. Mokkapati, A. Garg, V. Ravikumar and I. Mijakovic, Int. J. Mol. Sci., 2018, 19, 1979 CrossRef PubMed.
- B. Contreras-Trigo, V. Díaz-García, E. Guzmán-Gutierrez, I. Sanhueza, P. Coelho, S. E. Godoy, S. Torres and P. Oyarzún, Sensors, 2018, 18, 2246 CrossRef PubMed.
- J. W. Park and J. S. Shumaker-Parry, J. Am. Chem. Soc., 2014, 136, 1907–1921 CrossRef CAS PubMed.
- S. H. Kim, E. M. Kim, C. M. Lee, D. W. Kim, S. T. Lim, M. H. Sohn and H. J. Jeong, J. Nanomater., 2012, 504026 Search PubMed.
- A. Qin, L. T. Fu, J. K. F. Wong, L. Y. Chau, S. P. Yip and T. M. H. Lee, ACS Appl. Mater. Interfaces, 2017, 9, 10472–10480 CrossRef CAS PubMed.
- J. Kimling, M. Maier, B. Okenve, V. Kotaidis, H. Ballot and A. Plech, J. Phys. Chem. B, 2006, 110, 15700–15707 CrossRef CAS PubMed.
- F. Schulz, T. Homolka, N. G. Bastús, V. Puntes, H. Weller and T. Vossmeyer, Langmuir, 2014, 30, 10779–10784 CrossRef CAS PubMed.
- J. Manson, D. Kumar, B. J. Meenan and D. Dixon, Gold Bull., 2011, 44, 99–105 CrossRef CAS.
- W. Xiao, J. Xiong, S. Zhang, Y. Xiong, H. Zhang and H. Gao, Int. J. Pharm., 2018, 538, 105–111 CrossRef CAS PubMed.
- Y. C. Dong, M. Hajfathalian, P. S. N. Maidment, J. C. Hsu, P. C. Naha, S. Si-Mohamed, M. Breuilly, J. Kim, P. Chhour, P. Douek, H. I. Litt and D. P. Cormode, Sci. Rep., 2019, 9, 14912 CrossRef PubMed.
- W. Hou, S. Wang, X. Wang, X. Han, H. Fan, S. Cao, J. Yue, Q. Wang, W. Jiang, C. Ding and S. Yu, PloS One, 2015, 10, e0122952 CrossRef PubMed.
- S. Chen, X. Song, Z. Chen, X. Li, M. Li, H. Liu and J. Li, PloS One, 2013, 8, e56380 CrossRef CAS PubMed.
- K. Hongo, S. Kazama, E. Sunami, N. H. Tsuno, K. Takahashi, H. Nagawa and J. Kitayama, Med. Oncol., 2012, 29, 2849–2857 CrossRef CAS PubMed.
- D. Horst, S. K. Scheel, S. Liebmann, J. Neumann, S. Maatz, T. Kirchner and A. Jung, J. Pathol., 2009, 219, 427–434 CrossRef CAS PubMed.
- L. Ao, F. Gao, B. Pan, R. He and D. Cui, Anal. Chem., 2006, 78, 1104–1106 CrossRef CAS PubMed.
- V. Dixit, J. Van den Bossche, D. M. Sherman, D. H. Thompson and R. P. Andres, Bioconjug. Chem., 2006, 17, 603–609 CrossRef CAS PubMed.
- S. Hossen, M. K. Hossain, M. K. Basher, M. N. H. Mia, M. T. Rahman and M. J. Uddin, J. Adv. Res., 2019, 15, 1–18 CrossRef CAS PubMed.
- X. F. Du, B. J. Zhu, Z. C. Cai, C. Wang and M. X. Zhao, ACS Omega, 2019, 4, 17850–17856 CrossRef CAS PubMed.
- B. Fadeel, Front. Immunol., 2019, 10, 1–10 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2021 |