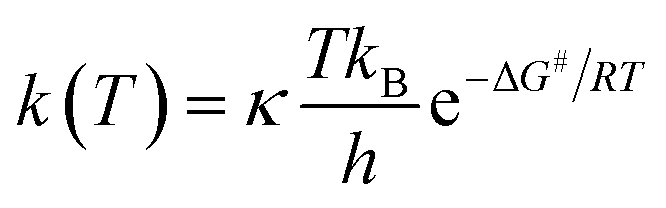DOI:
10.1039/D1RA01076J
(Paper)
RSC Adv., 2021,
11, 12971-12980
Benzofuran–stilbene hybrid compounds: an antioxidant assessment – a DFT study†
Received
8th February 2021
, Accepted 19th March 2021
First published on 6th April 2021
Abstract
The antioxidative activities of the original compound dehydro-δ-viniferin (1) and its three designated derivatives, 5-(2-(4-hydroxyphenyl)benzofuran-3-yl)benzene-1,3-diol (2), (E)-5-(2-(2-(4-hydroxyphenyl)benzofuran-5-yl)vinyl)benzene-1,3-diol (3), and (E)-5-(2-(3-(3,5-dihydroxyphenyl)benzofuran-5-yl)vinyl)benzene-1,3-diol (4), are extensively studied, mainly based on density functional theory (DFT) calculations using the hybrid functional B3LYP and the 6-311G(d,p) basis set. The O–H bond breakage is reasonably responsible for the antioxidative activity, whereas the environment greatly influences results. In solvents, namely water, methanol, and especially acetone, the SPL–ET mechanism (sequential proton loss–electron transfer) is mainly likely to be a antioxidative route for four studied compounds; however, the HAT mechanism (hydrogen atom transfer) is assigned to these compounds in the gaseous phase. Benzofuran–stilbene hybrid compound 3 with a planar structure is the best antioxidative agent; its 4′-OH induced the lowest PA values in liquid solvents as well as the lowest BDE value in the gas solvent. The kinetic investigation of the interactions of compounds 1–4 with HOO˙ radicals evidently provides the lowest Gibbs activation energy ΔG# = 4.7 kcal mol−1 and the highest rate constant K = 5.702 × 1010 L mol−1 s−1, which again are assigned to 3-4′-OH. At the theoretical level of B3LYP/6-311G(d,p)/LANL2DZ, the complex [La(compound 3)3] in gas induces a redshift in the UV-Vis spectrum.
Introduction
Oxygen radicals, such as hydroperoxyl (HOO˙), also known as reactive oxygen species (ROS), are unstable molecules that contain oxygen atoms that easily react with surrounding molecules.1 They are currently found in both living organisms and the atmosphere, and may cause DNA, protein, and cell damage.2 Hence, searching for new drugs that target these oxygen radicals has induced much interest.
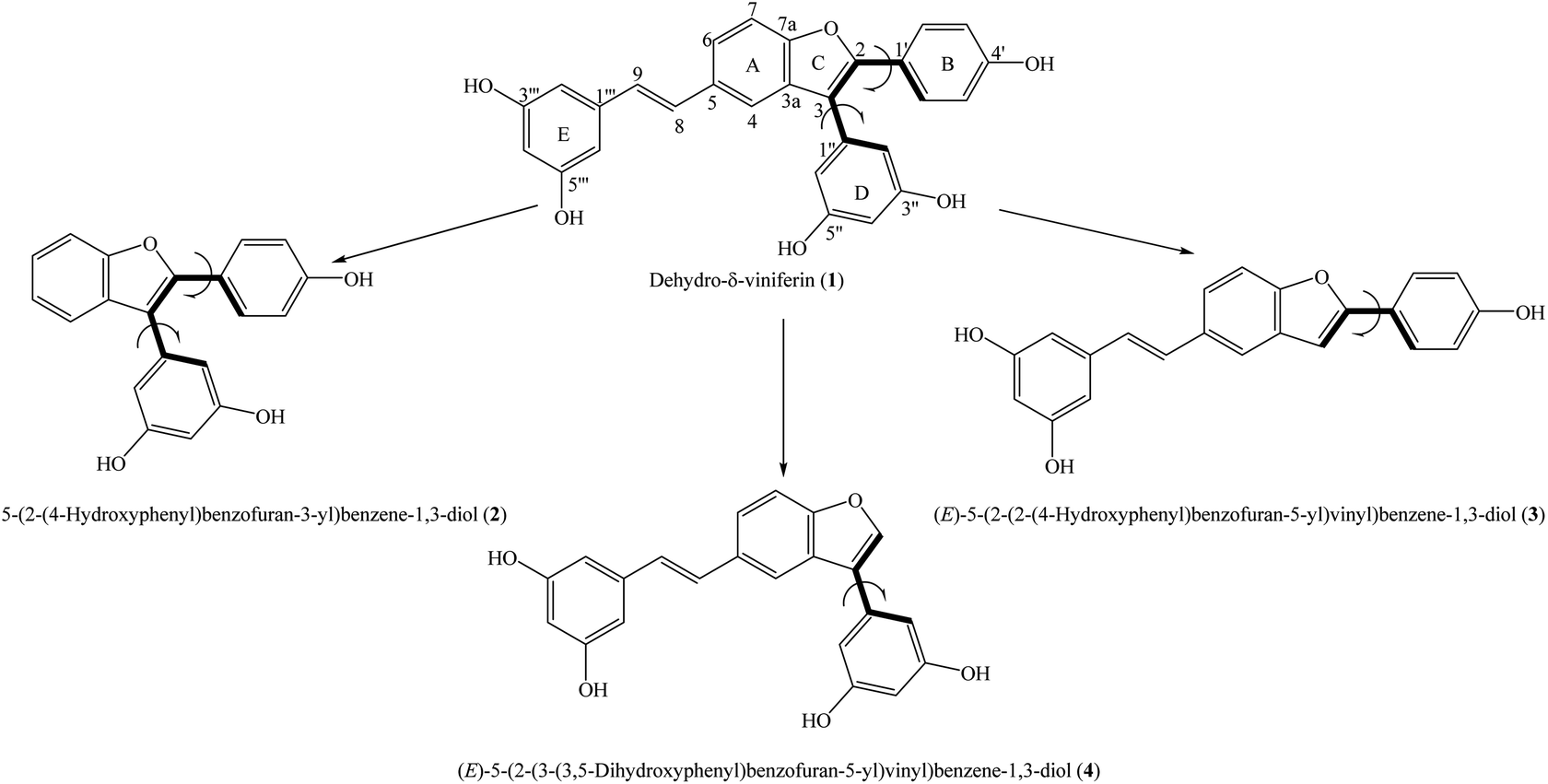
Total synthesis is a scientific-economic approach that involves using chemical reactions to create newly bioactive compounds; many useful medicines can be produced quickly by this approach. Total synthesis may help reduce inappropriate molecular sizes and toxic levels and can achieve enhanced bio-availability and solubility compared to natural constituents.3 Benzofurans and stilbenes are well-known derivatives used in antioxidative treatments.4,5 The synthesis of benzofuran–stilbene hybrid compounds has always been an interesting topic. Dehydro-δ-viniferin (1) is a representative example because this compound is made up of a combination of methyl 4-hydroxybenzoate and 3,5-dihydroxyacetophenone.6 5-(2-(4-Hydroxyphenyl)benzofuran-3-yl)benzene-1,3-diol (2), (E)-5-(2-(2-(4-hydroxyphenyl)benzofuran-5-yl)vinyl)benzene-1,3-diol (3), and (E)-5-(2-(3-(3,5-dihydroxyphenyl)benzofuran-5-yl)vinyl)benzene-1,3-diol (4) were successfully synthesized and can be seen to be the derivatives of compound 1 with the deletions of the functional moieties at carbons C-5, C-2 and C-3, respectively (Fig. 1).7 These agents have also shown antimicrobial activity. However, there have been no experimental or computational reports using these four compounds in antioxidative assessments to date. We now establish an antioxidative DFT-calculated investigation on derivatives 1–4, mostly based on the functional B3LYP/6-311G(d,p) basis set. The current paper considers thermal and kinetic analyses, with emphasis on highlighting the effects of structural–electronic properties and environmental factors on antioxidative outcomes.
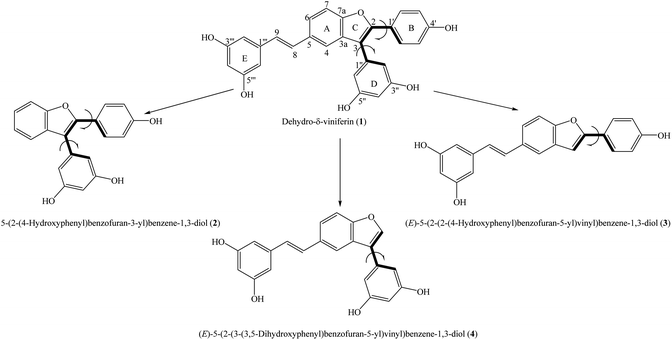 |
| | Fig. 1 General structures of compounds 1–4 with atom numbering. | |
Theoretical methodology
All DFT calculations were performed using the Gaussian 09 molecular package.8 The optimized procedure for the parent compound A-OH and its radicals, cationic radicals, and anions were computed in four mediums, namely gas, water, methanol and acetone, at the B3LYP/6-311G(d,p) level. Vibrational frequency calculations at the same level were applied to achieve accurate ZPE, and results also indicated that there is no imaginary frequency in the ground state. The IFE-PCM model was used to consider the effects of solvents.
According to many previous reports, the free radical HOO˙ capture of compound A-OH may be ruled by three well-known mechanisms.9–13
(1) The HAT route (hydrogen atom transfer)
This route is recognized as the splitting off of a proton from A-OH, which is intermediately transferred to the free radical HOO˙ (eqn (1)). This is the homolytic process, and it can be estimated by the BDE (bond dissociation enthalpy) (eqn (2)).
| | |
A-OH + HOO˙ → A-O˙ + HOOH
| (1) |
| | |
BDE = H(A-O˙) + H(H˙) − H(A-OH)
| (2) |
(2) The SET–PT route (the single electron transfer–proton transfer)
The SET–PT route consists of two stages (eqn (3)). The first stage involves the loss of an electron from A-OH to yield A-OH˙+, and it is characterized by the IP (ionization potential) (eqn (4)). A-OH+˙ is then deprotonated in the second stage; this step is the heterolytic process, and it is characterized by the PDE (proton dissociation enthalpy) (eqn (5)).
| | |
A-OH + HOO˙ → A-OH˙+ + HOO− → A-O˙ + HOOH
| (3) |
| | |
IP = H(A-OH+˙) + H(e−) − H(A-OH)
| (4) |
| | |
PDE = H(A-O˙) + H(H+) − H(A-OH˙+)
| (5) |
(3) The SPL–ET route (the sequential proton loss–electron transfer)
From eqn (6), the SPL–ET route involves two stages. In the first stage, A-OH is deprotonated to produce the anion A-O−. In the second stage, the anion A-O− is transferred an electron to generate the radical HOO˙. The PA (proton affinity) and the ETE (electron transfer enthalpy) are used to estimate energies for the first and second steps, respectively (eqn (7) and (8)).
| | |
A-OH → A-O− + H+; A-O− + HOO˙ → A-O˙ + HOO−; HOO− + H+ → HOOH
| (6) |
| | |
PA = H(A-O−) + H(H+) − H(A-OH)
| (7) |
| | |
ETE = H(A-O˙) + H(e−) − H(A-O−)
| (8) |
In the DFT method, H(M) is the total enthalpy of species M, and it can be expressed as in eqn (9).10
| | |
H(M) = E + ZPE + Htrans + Hrot + Hvib + RT
| (9) |
where
E and ZPE are the respective electronic and zero-point energies and
Htrans,
Hrot and
Hvib are the translational, rotational, and vibrational contributions to enthalpy, respectively. The
H(e
−) and
H(H
+) are extracted from the literature.
10
The antioxidative action of compound A-OH can also be estimated by using physicochemical parameters,9–13 comprising the EHOMO (the energy of the highest occupied molecular orbital), the ELUMO (the energy of the lowest unoccupied molecular orbital), the spin density, the MEP (molecular electrostatic potential), the chemical hardness η, the chemical potential μ, the electronegativity χ, the global electrophilicity index ω, and the condensed Fukui functions.
It relies on Koopman's rule and finite different approximation, and these physicochemical parameters can be written as below:
| | |
η ≈ −(ELUMO + EHOMO)/2
| (10) |
| | |
μ ≈ (EHOMO + ELUMO)/2 = −χ
| (11) |
| | |
ω− = −(3EHOMO + ELUMO)2/[16(EHOMO − ELUMO)]
| (13) |
| | |
ω+ = −(EHOMO + 3ELUMO)2/[16(EHOMO − ELUMO)]
| (14) |
Generally, the Fukui amounts are applied to justify the reactive sites in each molecule. Three Fukui indices, ft+, ft−, and fto, show the respective electrophilicity, nucleophilicity, and radical property, respectively (eqn (15)–(17)).
For nucleophilic attack,
| | |
ft+ = Qt(M + 1) − Qt(M)
| (15) |
For electrophilic attack
| | |
ft− = Qt(M) − Qt(M − 1)
| (16) |
For radical attack
| | |
fto = [Qt(M + 1) − Qt(M − 1)]/2
| (17) |
where
Q stands for the gross charge of atom t in a molecule.
Qt(M),
Qt(M + 1), and
Qt(M − 1) are the electronic population of atom t in neutral, anionic, and cationic molecules, respectively.
The B3LYP functional is also a usefully suitable approach for both thermodynamic and kinetic calculations.2,9,11,12,14,15 Of the conventional transition state theory (TST), at T = 298.0 K, the rate constant K in a radical reaction is related to the Gibbs activation energy ΔG#, the Wigner coefficient κ, the Plank constant h and the Boltzmann constant kB through eqn (18).
| |
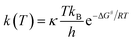 | (18) |
Results and discussion
Geometrical analysis
Structural analysis is one of the most effective methods of evaluating antioxidant activity, especially the behavior of hydroxy moieties.1 At the DFT/B3LYP/6-311G(d,p) level, the geometrical state of each compound did not show a difference in any of the four studied mediums (Fig. S1†). As can be seen, the π-electrons are distributed in the rings of all four compounds 1–4 and in the double bonds of compounds 1, 3, and 4. Compound 3 is completely planar [dihedral angle θ11(C3–C2–C1′–C2′) = 0°], whereas the planar property of the remaining compounds is lost, with θ11(C3–C2–C1′–C2′) = −21.4 to −24.7° and θ12(C2–C3–C1′′–C2′′) = −55.0 to −57.9° in compounds 1 and 2, respectively, and θ12(C2–C3–C1′′–C2′′) = −38.1 to −42.8° in compound 4 (Table S1†). Cai et al. (2014) suggested that planar molecules often display better antioxidative activity.16 Hence, compound 3 is expected to show better results. The O–H bond lengths in each compound fluctuate from 0.963 to 0.965 Å. Upon converting gases into liquids, the O–H bond distances in all four studied compounds 1–4 visibly increase by 0.001–0.002 Å. It may be expected that the solvent reduced the bond-breaking energies.
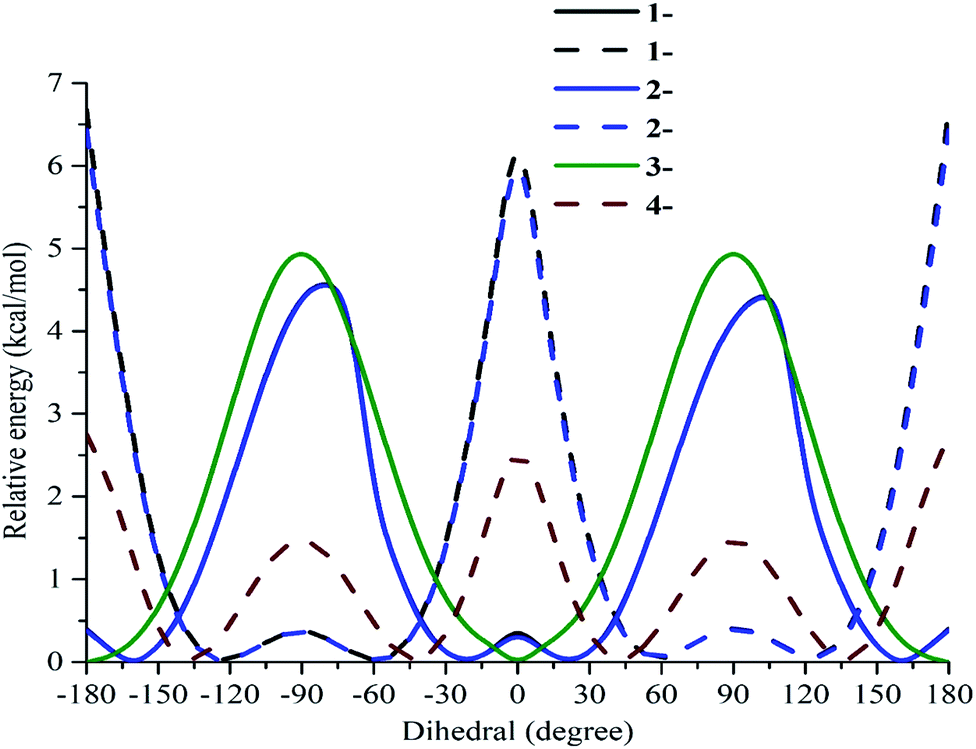
To confirm the relative orientations between phenyl units (rings B and D) and the benzofuran nucleus, the curves of the potential energies depending on dihedral angles θ were created in the gaseous phase at the B3LYP/6-311G(d) level. From −180 to 180°, the optimized processes resulted in a 10° change of θ11 in the first step and θ12 in the second step (Table S2†). All studied compounds 1–4 were further optimized around each minimal conformer for more correction.
As shown in Fig. 2, the phenyl unit and double bond at carbon C-5 of compounds 1 and 2 was not affected, because the curves of the two compounds are very similar to each other. Regarding the first scan of θ11, compounds 1 and 2 are composed of an absolute conformer at ±160° and ±20° and three interconvertible barriers at −80°, 0°, and 100°. In the second scan of θ12, the absolute conformer is located at ±120° and ±60°, whereas the three energy barriers peak at ±180°, ± 90° and 0°. Compound 3 appears to have an absolute conformer at ±180° and 0° as well as a barrier at ±90°, associated with ΔE = 5.02 kcal mol−1 relative to the absolute conformer when θ11 was scanned. Taking the scan of θ12 of compound 4 into account, an absolute conformer is found at ±140° and ±40°, while the three barriers are situated at ±180°, ±90° and 0° with ΔE = 1.51–2.75 kcal mol−1.
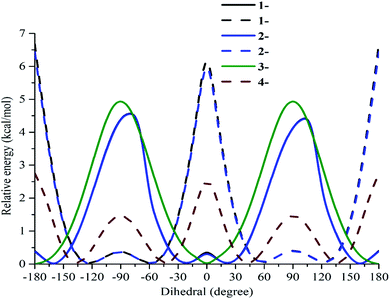 |
| | Fig. 2 Potential energy curves versus the dihedral angles θ11(C3–C2–C1′–C2′) and θ12(C2–C3–C1′′–C2′′) in the gaseous medium at the B3LYP/6-311G(d) level of theory. | |
Frontier molecular orbital theory (FMO), molecular electrostatic potential (MEP) and spin density
The FMO analysis evidently gives insight into the distribution of the electrons and predicts the antioxidative reaction sites because H-breakage is directly related to electron transfer.9–13 The FMO images of each compound are the same in gas and liquid solvents (Fig. S2†). The electrons have been spread over rings A–B–C–E and are few (or absent) in ring D of the HOMO-neutrals of all four compounds 1–4. In the LUMO-neutrals, the electrons are completely absent in rings B and D of compound 1, ring B of compound 3, and ring D of compound 4. In compounds 1 and 3, compared to the FMO neutrals, only the HOMO and LUMO pictures of the 4′-OH radical remain unchanged.9 It is possible to conclude that the 4′-OH of these two compounds is a good site for radical formation. HOMO and LUMO pictorial results of all radical cases of compound 2 and 4-5′′-OH radical resemble the neutral case. The electrons were found to be delocalized in the whole molecule in the HOMO of the 3′′′-OH and 5′′′-OH radicals of compound 4; however, they were absent in ring D. In another approach, a higher EHOMO and lower ELUMO induce a better Egap = EHOMO − ELUMO.9–13 The order of the EHOMO values of compounds 1–4 follows gas > acetone > methanol > water (Table S3†). In each compound, the Egap values slightly decreased when the gas was transformed into the solvents water, methanol, and acetone (Fig. S3†). Notably, compound 3 established the lowest Egap of an average of 3.955 eV in liquid. From these findings, the solvent is a main factor supporting ionization, and the energies for bond breakage of compound 3 appear to be lower.
MEP illustration, also known as molecular electrical potential pictorial surfaces, denotes the charges of a molecule in a 3D model. MEP basically relies on visual colors: red < orange < yellow < green < blue. Atoms colored red or yellow contain excess electrons (electrophilic attack), and the maximal positive charge is displayed by blue (nucleophilic attack).10 Green accompanies zero charge. As shown in Fig. S4,† the carbon and proton atoms of the four studied compounds 1–4 are green or blue, and yellow is assigned to oxygens. In this way, hydroxy groups act as nucleophiles.10
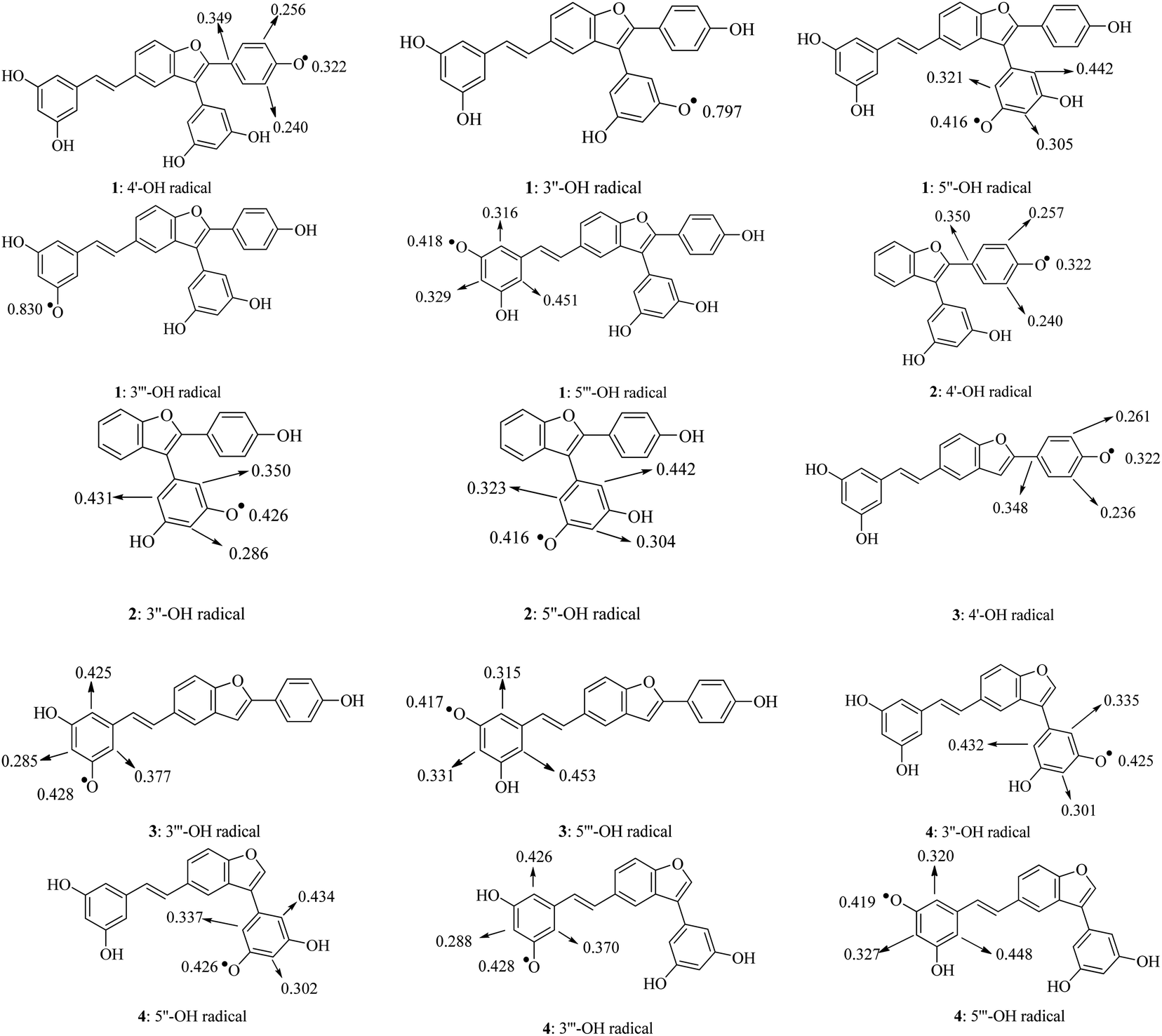
A good parameter to consider the speed of free radical quenching is the spin density. Greater delocalization of the spin density results in easier radical formation. Generally, the significant spin densities were symmetrically distributed in carbons C-2, C-4, and C-6 of the phenyl unit containing the O˙-radical (Fig. 3). It was found that radical formation in phenyl units is easier than in the benzofuran nucleus and double bonds. In particular, the gaseous spin density showed the same trend as the BDE, as an atom with a lower spin density will afford a lower BDE.17 The lowest value of 0.322 was found in the 4′-O˙ radicals of compounds 1–3; thereby, the HAT antioxidative mechanism is mainly due to 4′-OH bond breakage.
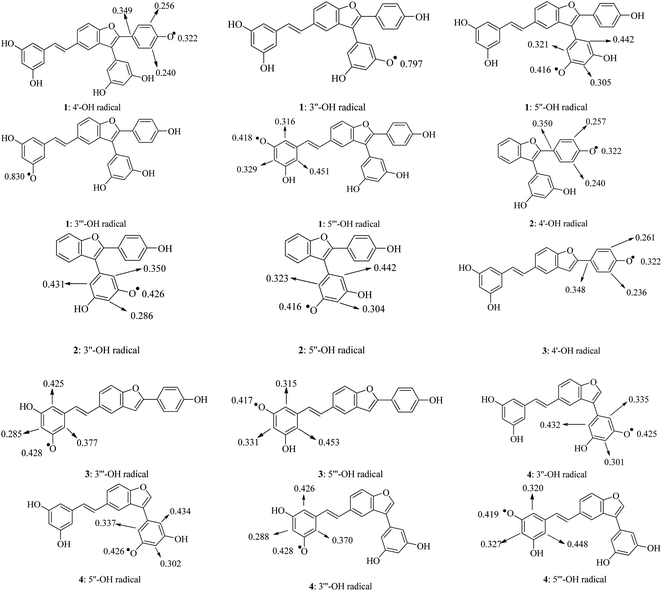 |
| | Fig. 3 Spin density distributions of structural radicals 1–4 in the gaseous medium at the B3LYP/6-311G(d,p) level of theory. | |
Electronic properties
The η parameter shows the charge transmission resistance of a molecule.1,2 The η values of naturally occurring benzofurans appear to be higher in solvents than in gas.11 However, in the current paper, the η values of each compound in the solvents are always lower than those in the gas (Table S3†). This may be due to the effect of rings D and E and the double bond. Table S3† also provides the results of the polarizability. The polar liquids, such as water and methanol, caused an increase in the polarizability in all four studied compounds. Based on the theoretical HSAB (hard and soft acids and bases) rule, it is possible that this is due to environmental responses for high oxidation/low polarizability to low oxidation/high polarizability.13 The χ parameter is opposite to μ, which demonstrates the tendency of an atom to receive a shared electron pair toward itself.9–13 A substance with a high χ promptly establishes equalization and also possesses low reactive energy.13 In all four studied compounds, the highest and lowest χ values were found in the polar solvent water and the non-polar medium gas, respectively. The χ values in methanol were found to be higher than those in acetone. Consequently, increasing the polarity of the environment is the first aim to increase the χ parameter. The ω parameter demonstrates the ability of a compound to accept an electron from its surroundings.9–13 The electrophilicity and nucleophilicity are in accordance with the high ω+ and ω− values, respectively. The ω, ω+ and ω− values of all four studied compounds increased when gas was converted into liquid (Table S3†). In each compound, the ω− values were ca. 3.3 times higher than the ω+ values. Resembling natural products, synthetic compounds 1–4 acted as electron donors instead of acceptors.9–13
The Fukui indices are also a fast approach to analyse the reactive efficacy of each atom. Evidence for the electrophilicity comes in Δft = ft+ − ft− > 0; the nucleophilicity is determined by Δft < 0.10–12 The carbons of compounds 1–4 play both roles. For instance, carbon C-2 of all the studied compounds serves as an electrophilic center (Δft > 0); however, carbon C-3 of compound 4, in contrast to that of compounds 1–3, acts as a nucleophilic site (Δft < 0). Except for 5′′-OH, the remaining OH groups display nucleophilicity (Table S4†). The efficiency of radical production can be determined by fto < 0.10–12 Most carbon and oxygen atoms are associated with fto > 0. However, carbon C-3 of all four studied compounds, especially the 4′-OH of compounds 1–3, induces fto < 0. Thus, 4′-OH is more likely to be the most active site in antiradical reactions.
Antioxidant mechanisms
HAT route. The calculated BDE values in the gaseous phase and liquids are provided in Table 1. In accordance with the above findings of the spin density, the 4′-OH of compounds 1–3 possesses the lowest BDE results. Particularly, 3-4′-OH was found to contain the lowest BDE values in each medium (78.6 kcal mol−1 in gas, 77.1 kcal mol−1 in water, 79.3 kcal mol−1 in methanol, and 76.6 kcal mol−1 in acetone) (Table 1). In most cases, the application of the polar solvent water or the weak polar solvent acetone resulted in a BDE decrease compared to the use of non-polar gas and medium polar methanol. The highest BDE values belonged to 1-3′′′-OH in both the gas and liquids, and there was a clear decrease in the cases of 3-3′′′-OH and 4-3′′′-OH. Similarly, 1-3′′-OH demonstrates higher BDE values than 2-3′′-OH and 4-3′′-OH. Hence, deletion of the functional groups at carbons C-3 and/or C-5 of compound 1 may be a good strategy for antioxidative activities. In general, the gaseous BDE values of the studied compounds 1–4 were comparable with those of flavonoids, which are well-known antioxidative agents.18–20
Table 1 Reaction enthalpies in the studied phases at 298 K for the radicals of the studied compounds 1–4 at the B3LYP/6-311G(d,p) level of theory (in kcal mol−1)
| Compounds |
BDE |
IP |
PDE |
| Gas |
Water |
Methanol |
Acetone |
Gas |
Water |
Methanol |
Acetone |
Gas |
Water |
Methanol |
Acetone |
| 1-4′-OH |
79.1 |
77.4 |
79.6 |
76.9 |
153.1 |
100.1 |
105.2 |
97.8 |
241.8 |
24.8 |
20.4 |
12.3 |
| 1-3′′-OH |
102.7 |
82.6 |
84.8 |
82.1 |
265.5 |
29.9 |
25.6 |
17.5 |
| 1-5′′-OH |
83.8 |
82.2 |
84.4 |
81.7 |
246.5 |
29.6 |
25.2 |
17.1 |
| 1-3′′′-OH |
103.2 |
100.4 |
102.8 |
100.4 |
265.9 |
47.8 |
43.7 |
35.7 |
| 1-5′′′-OH |
83.4 |
81.9 |
84.1 |
81.4 |
246.2 |
29.3 |
24.9 |
16.7 |
| 2-4′-OH |
78.9 |
77.4 |
79.6 |
76.9 |
157.3 |
100.3 |
105.4 |
98.1 |
237.4 |
24.6 |
20.2 |
11.9 |
| 2-3′′-OH |
84.2 |
82.5 |
84.7 |
82.0 |
242.8 |
29.7 |
25.3 |
17.1 |
| 2-5′′-OH |
83.7 |
82.2 |
84.4 |
81.7 |
242.2 |
29.4 |
25.0 |
16.8 |
| 3-4′-OH |
78.6 |
77.1 |
79.3 |
76.6 |
153.9 |
100.1 |
105.2 |
97.8 |
240.5 |
24.4 |
20.1 |
12.0 |
| 3-3′′′-OH |
83.7 |
82.3 |
84.5 |
81.8 |
245.6 |
29.7 |
25.4 |
17.2 |
| 3-5′′′-OH |
83.4 |
81.9 |
84.1 |
81.4 |
245.3 |
29.3 |
25.0 |
16.8 |
| 4-3′′-OH |
84.1 |
82.6 |
84.8 |
82.1 |
160.8 |
103.6 |
108.7 |
101.4 |
239.2 |
26.5 |
22.1 |
13.9 |
| 4-5′′-OH |
84.1 |
82.5 |
84.7 |
82.0 |
239.1 |
26.4 |
22.0 |
13.7 |
| 4-3′′′-OH |
83.9 |
82.4 |
84.5 |
81.8 |
239.0 |
26.3 |
21.8 |
13.6 |
| 4-5′′′-OH |
83.6 |
82.0 |
84.2 |
81.5 |
238.6 |
25.9 |
21.5 |
13.2 |
| Compounds |
PA |
ETE |
| Gas |
Water |
Methanol |
Acetone |
Gas |
Water |
Methanol |
Acetone |
| 1-4′-OH |
333.2 |
51.6 |
48.6 |
41.7 |
61.7 |
73.3 |
77.1 |
68.4 |
| 1-3′′-OH |
342.6 |
55.2 |
52.4 |
45.7 |
75.9 |
74.8 |
78.4 |
69.6 |
| 1-5′′-OH |
342.3 |
55.1 |
52.2 |
45.4 |
57.3 |
74.6 |
78.3 |
69.5 |
| 1-3′′′-OH |
346.9 |
56.0 |
53.2 |
46.5 |
72.1 |
91.9 |
95.7 |
87.0 |
| 1-5′′′-OH |
346.3 |
55.7 |
52.9 |
46.2 |
53.0 |
73.7 |
77.2 |
68.4 |
| 2-4′-OH |
336.1 |
51.8 |
48.9 |
42.1 |
58.6 |
73.0 |
76.7 |
68.0 |
| 2-3′′-OH |
345.2 |
55.4 |
52.6 |
46.0 |
54.8 |
74.6 |
78.1 |
69.2 |
| 2-5′′-OH |
345.0 |
55.3 |
52.4 |
45.8 |
54.5 |
74.4 |
77.9 |
69.1 |
| 3-4′-OH |
335.0 |
51.5 |
48.5 |
41.1 |
59.5 |
73.0 |
76.8 |
68.7 |
| 3-3′′′-OH |
347.6 |
55.5 |
52.7 |
46.1 |
51.9 |
74.3 |
77.8 |
69.0 |
| 3-5′′′-OH |
347.0 |
55.2 |
52.3 |
45.7 |
52.2 |
74.2 |
77.8 |
68.9 |
| 4-3′′-OH |
344.0 |
55.1 |
52.3 |
45.6 |
55.9 |
74.9 |
78.5 |
69.7 |
| 4-5′′-OH |
343.9 |
55.2 |
52.3 |
45.6 |
56.0 |
74.8 |
78.4 |
69.6 |
| 4-3′′′-OH |
346.1 |
56.0 |
53.1 |
46.5 |
53.6 |
73.8 |
77.4 |
68.5 |
| 4-5′′′-OH |
345.6 |
55.7 |
52.8 |
46.2 |
53.8 |
73.8 |
77.4 |
68.5 |
SET–PT and SPL–ET mechanisms
The first step of the SET–PT route is characterized by the IP values. From Table 1, the IP values for all studied compounds 1–4 can be arranged in the following order: gas > methanol > water > acetone. Again, this suggests that polar or weak liquids are immensely useful environments to reduce either BDE or IP parameters. However, comparing compound 1 to its products 2–4, the lack of moieties at carbons C-2, C-3, or C-5 does not appear to be a good strategy for IP reduction. PDE analysis was applied to consider the deprotonation of the cation radical A-OH+˙. The arrangement of the PDE values of the four studied compounds follows gas ≫ water > methanol > acetone. The weak polar solvent acetone dramatically decreased the PDE parameter in comparison with the non-polar gas, medium polar solvent methanol, and strong polar solvent water. The lowest PDE value of 12.0 kcal mol−1 was obtained for 3-4′-OH. In the same manner as the BDE results, 1-3′′-OH and 1-3′′′-OH are the two groups that yielded the highest PDE values in all four environments, and the deletion of functional groups at carbons C-3 and/or C-5 of compound 1 also helped to reduce the PDE values.
In the first step of the SPL–ET route, the parent molecule A-OH was deprotonated to afford A-O−, and this process can be estimated by the PA analysis. The environmental change greatly affected the PA outcomes. The lowest PA values of 41.1–42.1 kcal mol−1 were assigned to the 4′-OH of compounds 1–3, especially 3-4′-OH (41.1 kcal mol−1). The PA values in all cases show the same trend as the PDE outcomes, namely gas ≫ water > methanol > acetone (Table 1). The highest PA values were found in 1-3′′′-OH in liquids (56.0 kcal mol−1 in water, 53.2 kcal mol−1 in methanol, and 46.5 kcal mol−1 in acetone) and 3-3′′′-OH in gas (347.6 kcal mol−1). Modifications at carbons C-2, C-3, or C-5 do not appear to be a good approach to reduce the gaseous PA values. For instance, it is obvious that 2-3′′-OH and 4-3′′-OH generate 344.0–345.2 kcal mol−1 higher PA values than 1-3′′-OH (342.6 kcal mol−1). Recently, Boulebd (2020) provided a DFT/B3LYP/6-311G(d,p)-calculated approach to determine the antioxidative capacity of benzofuran moracin T, in which the OH groups of this compound possessed PA values of 352.60–356.53 kcal mol−1 in gas, 104.47–120.59 kcal mol−1 in benzene, and 44.44–45.14 kcal mol−1 in water. It was found that polar solvents, such as methanol, acetone, and water, promote the PA decrease better than weak polar solvent or gas mediums.21 The ETEs are remarkable descriptors representing the electron transfer in the second step of the SPL–ET route. Not only the charge species but also the bond breakage energy are especially sensitive to environmental change. In contrast to the PDE and PA results, the ETE values of most hydroxy groups can be arranged in order as gas < acetone < water < methanol. The lowest and highest ETE values of 51.9 and 95.7 kcal mol−1 were obtained from 3-3′′′-OH and 1-3′′′-OH in the gaseous and methanolic phases, respectively. It can again be observed that 1-3′′′-OH in each medium induces a higher ETE value than 3-′′′-OH and 4-3′′′-OH. The same phenomenon was observed when 1-3′′-OH was compared to 2-3′′-OH and 4-3′′-OH. Therefore, the total-synthetic procedure aimed to produce compounds 2–4 from bulk derivative 1 set a great mark for pharmacological purposes and molecular size reduction.
Preferable mechanisms
The preferable antioxidative HAT, SET–PT or SPL–ET routes assigned to the four compounds 1–4 are mainly based upon the minimal values among the BDE, IP, and PA parameters.9–13 The above outcomes demonstrate that the SPL–ET route is the main action in solvents because the PA values of all studied compounds 1–4 have the lowest energies in methanol and water and especially in acetone. The PA values are also much lower than those of BDE and IP. However, if the antioxidative reaction occurs in gas, the HAT route will be preferable due to BDE < IP ≪ PA.
The kinetic study
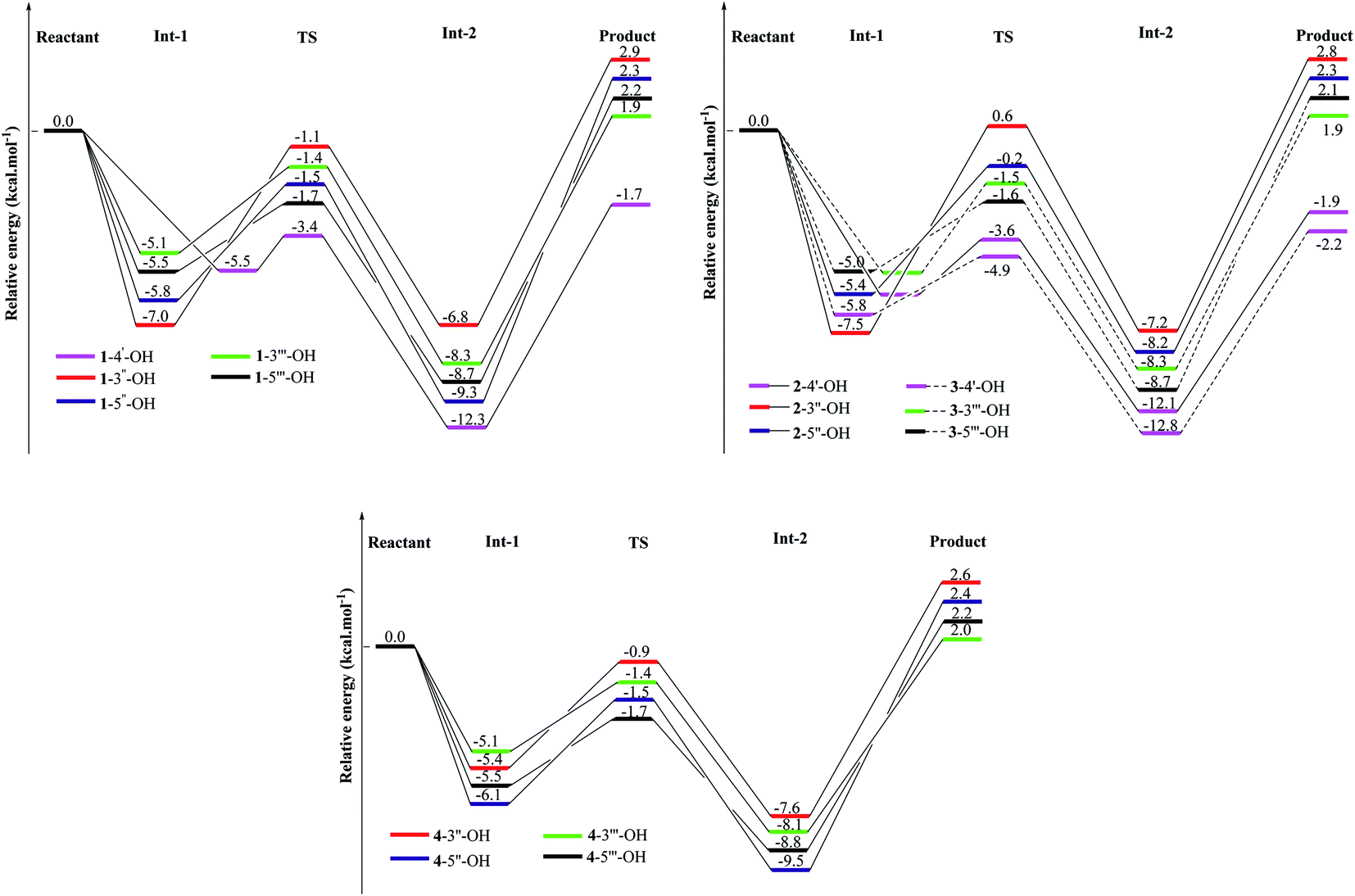
In addition to thermal analysis, understanding how HOO˙ radicals attack OH groups would help to explain the role of OH groups in kinetic actions. At the same theoretical level of B3LYP/6-311G(d,p), to eliminate the solvent effects, the kinetic reactions were considered by comprehensive analysis of ΔG# and the K in the gaseous phase. From eqn (18), the HOO˙ radical quenching reaction by compounds 1–4 becomes advantageous with a lower ΔG# value but a higher K value.9–11 The results of HOO˙ + compounds 1–4, including one transition state (TS) and two intermediates (Int-1 and Int-2), have been provided in Fig. 4 and S5† and in Table 2. In agreement with the BDE and spin density calculations, the 4′-OH groups are good sites for the free radical quenching reaction, in which 3-4′-OH (the lowest ΔG# = 4.7 kcal mol−1 and the highest K = 5.702 × 1010 L mol−1 s−1) is better than 2-4′-OH (ΔG# = 6.1 kcal mol−1 and K = 1.366 × 1010 L mol−1 s−1) and 1-4′-OH (ΔG# = 6.2 kcal mol−1 and K = 1.176 × 1010 L mol−1 s−1). This is further confirmed by the relative energies of the TS states, in which TS of 3-4′-OH + HOO˙ was associated with the lowest energy of −4.9 kcal mol−1 compared to those of 1-4′-OH + HOO˙ (−3.6 kcal mol−1) and 2-4′-OH + HOO˙ (−3.6 kcal mol−1). 1-3′′-OH induces the highest ΔG# = 10.4 kcal mol−1 and the lowest K = 1.044 × 108 L mol−1 s−1; however, these values are better in the cases of 2-3′′-OH and 4-3′′-OH (Table 2). Similar results can be observed when comparing 3′′′-OH and 5′′′-OH of compound 1 with the respective 3′′′-OH and 5′′′-OH groups of compounds 3–4. This reflects the effects of the functional groups at carbons C-2 and C-5. However, 1-5′′-OH has a K value of 6.797 × 108 L mol−1 s−1, which is better than those of 2-5′′-OH (K = 3.599 × 108 L mol−1 s−1) and 4-5′′-OH (K = 1.371 × 108 L mol−1 s−1). These findings reflect the fact that the benzofuran–stilbene hybrid derivative compound 3 appears to be better than benzofuran derivative compound 2 and benzofuran–stilbene hybrid derivative compound 4 in antioxidative treatment.
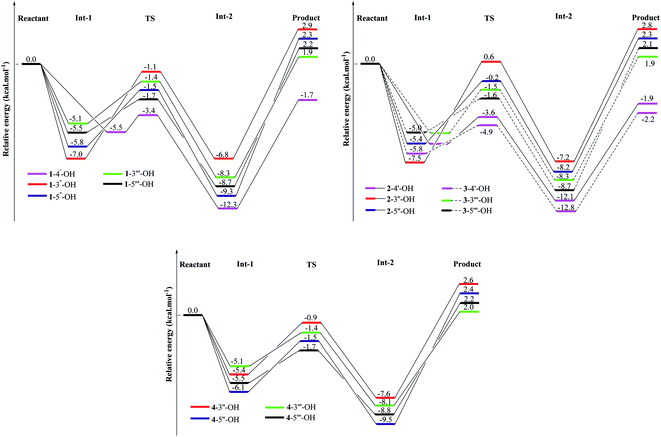 |
| | Fig. 4 Energy diagram for the reaction of HOO˙ radical attack on the studied compounds 1–4 at the B3LYP/6-311G(d,p) level of theory. | |
Table 2 The calculated ΔG# and k values in the gaseous phase and 298.15 K at the B3LYP/6-311G(d,p) level of theory for the HOO˙ radical attack
| Reaction |
Imaginary frequency (cm−1) |
κWigner |
ΔG# (kcal mol−1) |
K (L mol−1 s−1) |
Reaction |
Imaginary frequency (cm−1) |
κWigner |
ΔG# (kcal mol−1) |
K (L mol−1 s−1) |
| 1-4′-OH |
−1600 |
1.352 |
6.2 |
1.176 × 1010 |
3-4′-OH |
−1490 |
1.337 |
4.7 |
5.702 × 1010 |
| 1-3′′-OH |
−1649 |
1.350 |
10.4 |
1.044 × 108 |
3-3′′′-OH |
−1643 |
1.355 |
7.8 |
2.164 × 109 |
| 1-5′′-OH |
−1708 |
1.366 |
8.9 |
6.797 × 108 |
3-5′′′-OH |
−1609 |
1.347 |
8.1 |
1.565 × 109 |
| 1-3′′′-OH |
−1634 |
1.351 |
8.4 |
1.142 × 109 |
4-3′′-OH |
−1680 |
1.360 |
8.8 |
7.526 × 108 |
| 1-5′′′-OH |
−1642 |
1.353 |
8.3 |
1.272 × 109 |
4-5′′-OH |
−1734 |
1.372 |
8.5 |
1.371 × 108 |
| 2-4′-OH |
−1871 |
1.412 |
6.1 |
1.366 × 1010 |
4-3′′′-OH |
−1645 |
1.355 |
7.8 |
2.164 × 109 |
| 2-3′′-OH |
−1748 |
1.372 |
9.7 |
2.926 × 108 |
4-5′′′-OH |
−1631 |
1.352 |
8.0 |
1.746 × 109 |
| 2-5′′-OH |
−1715 |
1.366 |
9.5 |
3.599 × 108 |
|
|
|
|
|
Metal complex chelation
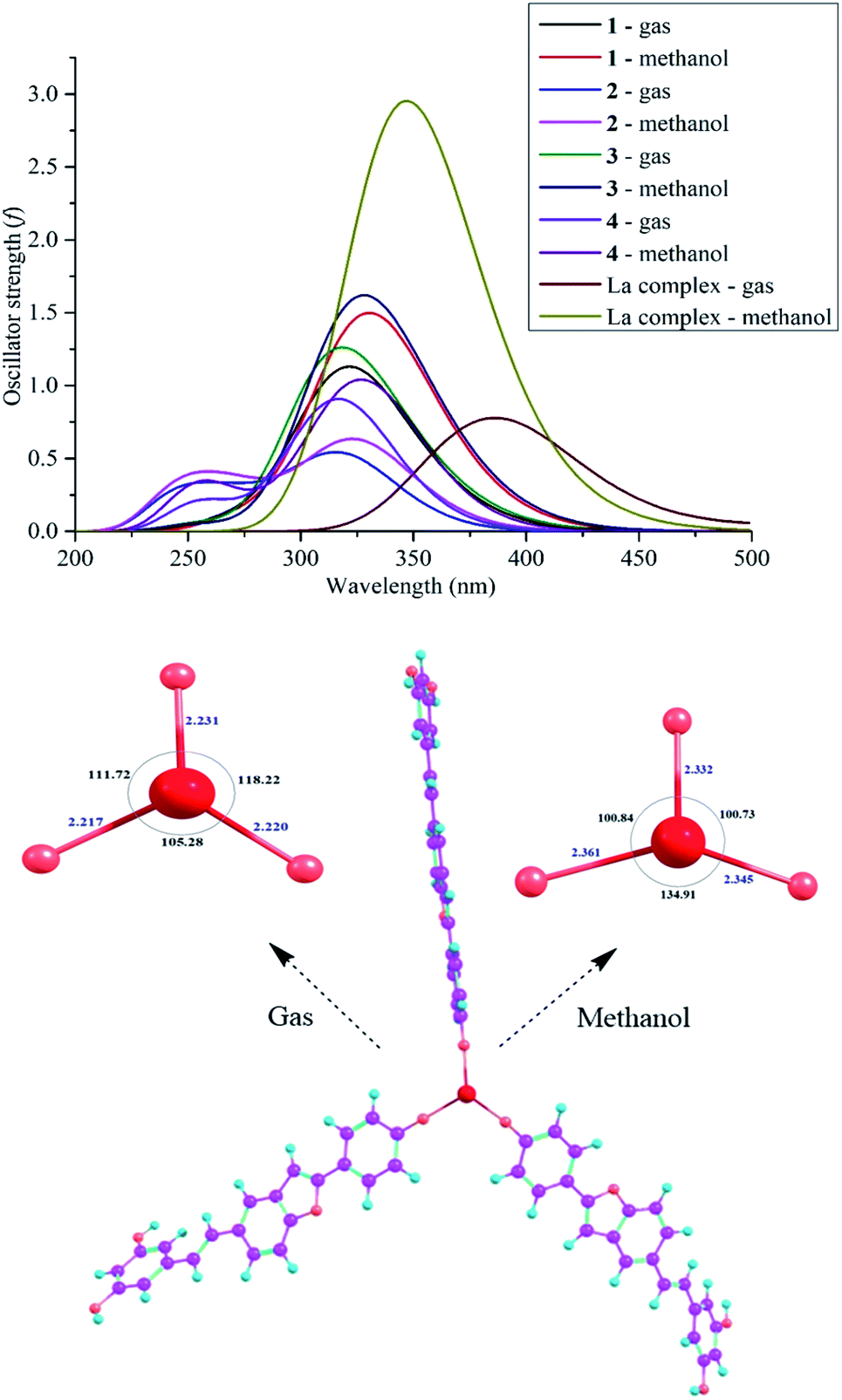
The UV-Vis spectroscopic data of the four synthetic compounds 1–4 have not been made available in the literature to date. Table 3 and Fig. 5 provide in-depth information on the UV-Vis results in gas and methanol, including λmax, vertical transition energy Evert, oscillator strength f, and transition types, where the calculations were based on the TD-DFT/B3LYP/6-311G(d,p) level.22–25 The use of gas and methanol is always recognized to be the best choice in both experimental and theoretical UV-Vis models.26–30 In both studied mediums, each compound has three main λmax peaks in the 247–353 nm region (Table 3). Comparing compounds 2–4 to compound 1, a blue-shift was observed (λmax decreased, but Evert increased) in both gas and methanol. The longest λmax in each compound is mainly due to the H → L (>73%) transition. For instance, H → L (>96%) shows the longest gaseous λmax of 319 nm (Evert = 3.893 eV and oscillator strength f = 0.5253) of compound 2.
Table 3 The UV-Vis data of compounds 1–4 at the TD-DFT/B3LYP/6-311G(d,p) level of theory and of complex [La(compound 3)3] at the B3LYP/6-311G(d,p)/LANL2DZ level of theory
| No |
Methanol |
Gas |
Transition type |
| λmax (nm) |
Evert (eV) |
Oscillator strength f |
Transition |
λmax (nm) |
Evert (eV) |
Oscillator strength f |
Transition |
| 1 |
353 |
3.514 |
0.4582 |
H → L (88%) |
348 |
3.565 |
0.3511 |
H → L (76%) |
π → π* |
| 328 |
3.777 |
1.5774 |
H−1 → L (60%) |
319 |
3.885 |
1.1639 |
H−1 → L+1 (51%) |
| 316 |
3.919 |
0.1103 |
H−1 → L+1 (79%) |
311 |
3.992 |
0.1099 |
H−2 → L (80%) |
| 2 |
325 |
3.816 |
0.6435 |
H → L (97%) |
319 |
3.893 |
0.5253 |
H → L (96%) |
| 274 |
4.530 |
0.1265 |
H−2 → L (75%) |
274 |
4.532 |
0.1230 |
H−2 → L (68%) |
| 247 |
5.021 |
0.1653 |
H → L+3 (63%) |
246 |
5.042 |
0.1197 |
H → L+3 (62%) |
| 3 |
353 |
3.508 |
0.6079 |
H → L (86%) |
348 |
3.566 |
0.4822 |
H → L (73%) |
| 325 |
3.821 |
1.6907 |
H → L+1 (69%) |
316 |
3.919 |
1.3786 |
H → L+1 (56%) |
| 303 |
4.092 |
0.0964 |
H−1 → L+1 (67%) |
299 |
4.144 |
0.1330 |
H → L+2 (53%) |
| 4 |
329 |
3.772 |
1.1821 |
H → L (98%) |
319 |
3.889 |
0.9279 |
H → L (91%) |
| 313 |
3.966 |
0.0799 |
H−2 → L (93%) |
312 |
3.978 |
0.1576 |
H−1 → L (55%) |
| 253 |
4.901 |
0.1512 |
H−5 → L (53%) |
253 |
4.909 |
0.0924 |
H−5 → L (62%) |
| Complex |
347 |
3.573 |
3.309 |
H−2 → L+5 (25%) |
385 |
3.220 |
0.796 |
H−3 → L+1 (47%) |
| H−1 → L+4 (32%) |
H−2 → L+1 (40%) |
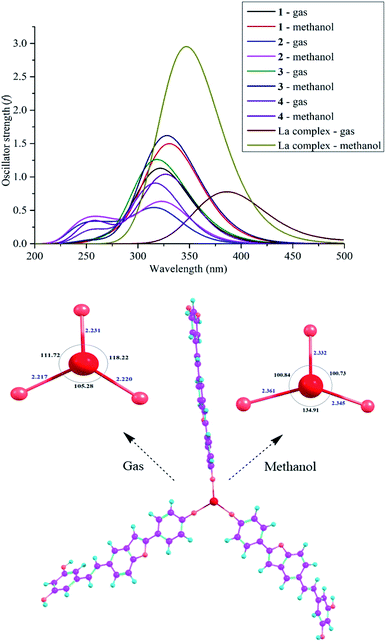 |
| | Fig. 5 . The UV-Vis spectra of compounds 1–4 at the TD-DFT/B3LYP/6-311G(d,p) level of theory and the optimized structures and UV-Vis spectrum of complex [La(compound 3)3] at the B3LYP/6-311G(d,p)/LANL2DZ level of theory. | |
Searching for bioactive compounds to remove toxic metals from living bodies is the first aim of chelation therapies.31–33 Selvaraj et al. (2013) suggested the antioxidant activity of the flavonoid chrysin because of its OH chelation with La.33 Based on this conclusion, we suggest that the antioxidative capacity of the promising compound 3 is also caused by its metal chelation; thereby, we proposed a complex model [La(compound 3)3] because the most active site 3-4′-OH covalently binds to the metal La (Fig. 5). UV-Vis spectroscopic analysis is a promptly helpful approach to view successful complex formation. The theoretical level B3LYP/6-311G(d,p)/LANL2DZ was used for optimization, whereas the TD-DFT method aided by the B3LYP functional using the 6-311G (d,p) basis set (for the light atoms oxygen, hydrogen, and carbon) and the LANL2DZ basis set (for the metal La center) is highly recommended for UV-Vis prediction.34 As shown in Fig. 5, the transformation of compound 3 into a complex gave rise to a loss of planarity between ring E and the benzofuran nucleus. Significantly, the lengths of the La–O bonds (2.217–2.231 Å in gas and 2.332–2.361 Å in methanol) drastically increased compared with those the of 3-4′-OH bonds. The environmental application places much of the influence on the bond angles OLaO (105.28°–118.22° in gas, and 100.73°–134.91° in methanol). The complex formation can be also defined from the UV-Vis data. The complex contains one peak in both gas and methanol, in which the H−2 → L+5 (25%) and H−1 → L+4 (32%) transitions are reasonable for the methanolic λmax of 347 nm (Evert = 3.573 eV and f = 3.309); however, the gaseous λmax of 385 nm (Evert = 3.220 eV and f = 0.796) arises from H−3 → L+1 (47%) and H−2 → L+1 (40%) transitions. Resembling the four compounds 1–4, the transition type π → π* is characteristic of a complex, and the complex formation is associated with the redshift in gas.
Conclusion
In the current study, we first set out to investigate the thermodynamics and kinetics of radical quenching for synthetic compounds 1–4 based on the DFT method. The antioxidative routes assigned to these compounds are more dependent on their environmental uses. The SPL-ET route should be suitable for compounds 1–4 in three solvents of water, methanol, and acetone, whereas the HAT route becomes more favorable in the gaseous phase. The structural modifications at carbons C-2, C-3, and C-5 of bulk compound 1 greatly affected the antioxidative results. Physicochemical and structural analyses indicated that compound 3 with planarity and benzofuran–stilbene hybrid properties is better than the non-planar compounds 1, 2, and 4. The 4′-OH group of compound 3 successfully possesses the lowest gaseous BDE values of 76.6–78.6 kcal mol−1 and the lowest liquid PA value of 41.1–51.5 kcal mol−1. 3-4′-OH group + HOO˙ also exerts the lowest ΔG# = 4.7 kcal mol−1 and the highest K = 5.702 × 1010 L mol−1 s−1. At the theoretical level of B3LYP/6-311G(d,p)/LANL2DZ, in the UV-Vis spectra, complex [La(compound 3)3] causes a redshift in gas. This communication is a useful guideline for experimental assays.
Conflicts of interest
The authors declare that we do not have any conflict of interest.
Acknowledgements
This work was supported by a grant from Vietnam Academy of Science and Technology-2021. This work was supported by a grant from Vietnam Academy of Science and Technology-2021 and Ministry of Education and Training for support under the project B2021-DHV-07.
References
- N. T. Son, D. T. Mai Thanh and N. V. Trang, J. Mol. Struct., 2019, 1193, 76–88 CrossRef CAS.
- N. T. Dung, D. M. Thanh, N. T. Huong, P. T. Thuy, N. T. Hoan, D. T. Mai Thanh, N. V. Trang and N. T. Son, Struct. Chem., 2020, 31, 2435–2450 CrossRef CAS.
- K. C. Nicolaou, D. Vourloumis, N. Winssinger and P. S. Baran, Angew. Chem., Int. Ed., 2000, 39, 44–122 CrossRef CAS PubMed.
- K. Chanda, R. A. Hiremathad, M. Singh, M. A. Santos and R. S. Keri, Pharmacol. Rep., 2017, 69, 281–295 CrossRef PubMed.
- S. N. The, Evid-Based Complement, Altern. Med., 2017, 2017, 7142370 Search PubMed.
- B. H. Teng, Q. B. Zhu, Y. Y. Fan and C. S. Yao, J. Asian Nat. Prod. Res., 2020, 22, 947–955 CrossRef CAS PubMed.
- G. Catinella, L. M. Mattio, L. Musso, S. Arioli, D. Mora, G. L. Beretta, N. Zaffaroni, A. Pinto and S. Dallavalle, Int. J. Mol. Sci., 2020, 21, 2168 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision C.01, Inc., Wallingford CT, 2010 Search PubMed.
- P. T. Thuy and N. T. Son, J. Chem., 2020, 2020, 8869023 Search PubMed.
- N. T. Son, P. T. Thuy and N. V. Trang, J. Mol. Struct., 2021, 11224, 12902 Search PubMed.
- P. T. Thuy, N. V. Trang and N. T. Son, RSC Adv., 2020, 10, 6315–6332 RSC.
- A. I. Elshamy, M. Hama, N. V. Trang, N. T. Son, Y. Okamoto, M. Noji, S. Ban and A. Umeyama, J. Mol. Struct., 2020, 1200, 127061 CrossRef CAS.
- N. T. Son, D. M. Thanh and N. V. Trang, J. Chem., 2019, 2019, 4360175 Search PubMed.
- P. T. Thuy and N. T. Son, J. Mol. Model., 2021, 27, 6 CrossRef CAS PubMed.
- L. T. T. Anh, N. T. Son, N. V. Tuyen, P. T. Thuy, P. M. Quan, N. T. T. Ha and N. T. Tra, Mol. Diversity, 2021 DOI:10.1007/s11030-021-10206-6.
- W. Cai, Y. Chen, L. Xie, H. Zhang and C. Hou, Eur. Food Res. Technol., 2014, 238, 121–128 CrossRef CAS.
- M. Saqib, S. Iqbal, A. Mahmood and R. Akram, Int. J. Food Prop., 2016, 19, 745–751 CrossRef CAS.
- L. Lu, M. Qiang, F. Li, H. Zhang and S. Zhang, Dyes Pigm., 2014, 103, 175–182 CrossRef CAS.
- K. Sadasivam and R. Kumaresan, Mol. Phys., 2011, 109, 839–852 CrossRef.
- K. Sadasivam and R. Kumaresan, Comput. Theor. Chem., 2011, 963, 227–235 CrossRef CAS.
- H. Boulebd, Free Radical Res., 2020, 54, 221–230 CrossRef CAS PubMed.
- V. Deepha, R. Praveena and K. Sadasivam, J. Mol. Struct., 2015, 1082, 131–142 CrossRef CAS.
- S. Chithiraikumar, S. Gandhimathi and M. A. Neelakantan, J. Mol. Struct., 2017, 1137, 569–580 CrossRef CAS.
- S. Sebastian and N. Sundaraganesan, Spectrochim. Acta, Part A, 2010, 75, 941–952 CrossRef CAS PubMed.
- T. U. Rahman, G. Uddin, R. U. Nisa, R. Ludwig, W. Liaqat, T. Mahmood, G. Mohammad, M. I. Choudhary and K. Ayub, Spectrochim. Acta, Part A, 2015, 148, 375–381 CrossRef CAS PubMed.
- N. T. Son, T. Yamamoto and Y. Fukuyama, Biochem. Syst. Ecol., 2018, 78, 98–101 CrossRef CAS.
- N. T. Son, Y. Fukuyama and N. M. Cuong, Chem. Nat. Compd., 2019, 55, 735–736 CrossRef.
- A. Dang, Y. Liu and F. Turecek, J. Phys. Chem. A, 2019, 123, 3272–3284 CrossRef CAS PubMed.
- N. T. Son, M. Kamiji, T. T. Huong, M. Kubo, N. M. Cuong and Y. Fukuyama, Med. Chem. Res., 2019, 28, 1441–1447 CrossRef CAS.
- E. Viglino, C. J. Shaffer and F. Turecek, Angew. Chem., Int. Ed., 2016, 55, 7469–7473 CrossRef CAS PubMed.
- M. L. Hegde, P. Bharthi, S. Anitha, V. Chitra, J. Ramya, P. Pankaj, S. Pulabhatla, S. Kumar, R. K. Jagannatha, S. Janez, M. Luigi and Z. Paolo, J. Alzheimer’s Dis., 2009, 17, 457–468 CAS.
- J. J. Kim, Y. S. Kim and V. Kumar, J. Trace Elem. Med. Biol., 2019, 54, 226–231 CrossRef CAS PubMed.
- S. Selvaraj, S. Krishnaswamy, V. Devashya, S. Sethuraman and U. Maheswari Krishnan, Med. Res. Rev., 2014, 34, 677–702 CrossRef CAS PubMed.
- A. D. S. Leonardo, M. S. Malucia and B. D. A. Wagner, Int. J. Quantum Chem., 2018, 118, e25773 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S5 and Tables S1–S4. See DOI: 10.1039/d1ra01076j |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *d
*d