DOI:
10.1039/D1RA01035B
(Review Article)
RSC Adv., 2021,
11, 22305-22316
Progress and recent trends in the direct selenocyanation of (hetero)aromatic C–H bonds
Received
7th February 2021
, Accepted 3rd June 2021
First published on 24th June 2021
Abstract
This review covers recent advances in the direct selenocyanation of (hetero)aromatic C–H bonds with an emphasis on the reaction mechanisms. This novel approach is an effective means of preparing a variety of aromatic and heteroaromatic selenocyanates, which are extremely versatile synthetic precursors of selenium-containing compounds, such as selenols, seleninic acids, selenides, diselenides, and trifluoromethyl selenides.
 Akbar Hassanpour | Akbar Hassanpour was born in Tabriz, Iran, in 1978. He received his B.S. degree in applied chemistry from Tabriz Azad University, Iran, and his M.S. degree in organic chemistry from Tabriz University, Tabriz, Iran, in 2004 under the supervision of Prof. Kazem D. Safa. He completed his PhD degree in 2008 under the supervision of Prof. Kazem D. Safa. He was a postdoctoral fellow in the lab of Prof. W. A. Stanczyk at the Centre of Molecular and Macromolecular Studies in Łódź, Poland. Now he is working at Azad University as associate professor. His research interests include organometallic, organic synthesis, and new methodologies in nanomaterial synthesis. |
 Elham Ghavidelaghdam | Elham Ghavidelaghdam was born in Shahindezh, Iran, in 1984. She received her B.S. degree in applied chemistry from University of Mohaghegh Ardebili, Ardebil, Iran and her M.S. degree in inorganic chemistry from univer of Tabriz, Tabriz, Iran, in 2009 under the supervision of Prof. A. A. Alemi. She completed Her PhD degree in 2017 under the supervision of prof. G. Shahverdizadeh in Central Tehran Azad University, Tehran, Iran. Now she is working at Ilkhchi Branch, Islamic Azad University as Associate Professor in inorganic chemistry. Her research interests include nano materials, new methodologies in inorganic synthesis and crystallography. |
 Abdol Ghaffar Ebadi | Dr Abdol Ghaffar Ebadi finished his doctoral degree in Environmental Biotechnology (Algology) from Tajik Academy of Sciences. Now he is a researcher in TAS in Tajikistan and faculty member at the Islamic Azad University of Jouybar in Mazandaran. Dr Ebadi published more than 400 scientific papers in qualified international journals and attended more than 50 international conferences. He has cooperation with many research project teams around world such as China, Malaysia, and Thailand. His interests are Environmental Biotechnology, Biochemistry, Gene pathways in the phytoremediation processes. |
 Mohammad Reza Poor Heravi | Mohammad Reza Poor Heravi was born in Seiyahkal-Lahijan, Guilan, Iran in 1965. He received the BSc degree in pure Chemistry from Tabriz University, Iran, in 1987, and the M.Sc. degree in Organic Chemistry from Tabriz University, Iran in 1990 with Professor A. Shahrisa, and the PhD degree from Isfahan University, Iran, with Professor H. Loghmanim and visiting research in 2006, The Sheffield of University, UK, with Professor Richard F. W. Jackson. He became a sciences faculty member of Payame Noor University in 1990, and associate Professor in 2012. His research field includes applications of solvent-free conditions, ionic liquids, fluorination reaction, design fluorinating reagents and ultrasound irradiation in organic synthesis, and study of methodology in organic chemistry. |
 Esmail Vessally | Esmail Vessally was born in Sharabiyan, Sarab, Iran, in 1973. He received his B.S. degree in pure chemistry from university of Tabriz, Tabriz, Iran, and his M.S. degree in organic chemistry from Tehran University, Tehran, Iran, in 1999 under the supervision of Prof. H. Pirelahi. He completed his PhD degree in 2005 under the supervision of Prof. M. Z. Kassaee. Now he is working at Payame Noor University as full Professor in organic chemistry. His research interests include theoretical organic chemistry, new methodologies in organic synthesis and spectral studies of organic compounds. |
1. Introduction
Despite its negative reputation,1 selenium has a rich history in biology2 and biotechnology.3 This chalcogen has also been included in drug design,4 ebselen (2-phenyl-1,2-benzisoselenazol-3(2H)-one) being an outstanding example, which is endowed with anti-inflammatory, anti-oxidant and cytoprotective activity, and with potential applications in the treatment of various heart and brain diseases.5
In this family of compounds, (hetero)aromatic selenocyanates have recently attracted the interest of medicinal chemists due to their remarkable biological activities especially as anti-cancer and chemopreventive agents.6 Benefiting from high versatility of selenocyanate moiety, this specific class of organoselenium compounds can be also easily converted into many other significant seleno-organic compounds such as selenols,7 selenides,8 diselenides,9 and trifluoromethyl selenides.10 In light of the above-mentioned chemistry, many efforts have been devoted toward the development of innovative and effective methods to create C–SeCN bond in the past decades.11 Traditionally, selenocyanate substituted (hetero)aromatic compounds are obtained through the reaction of selenocyanate sources with active aryl species such as aryl halides, arylboronic acids, and aryl diazonium salt.12–14 However, toxicity and mutagenicity of organic halides,15 tedious preparing and purifying arylboronic acids,16 and high reactivity and instability of diazonium salts17 may prevent the application of these methods more or less. To overpass these limitations, the direct selenocyanation of (hetero)aromatic C–H bonds has emerged as a powerful and ideal method for the fabrication of (hetero)aryl selenocyanates which because it does not require any pre-functionalized starting materials, offers a more atom economical, greener, and shorter alternative to the classical approaches. Since a number of remarkable developments in this attractive research field have taken place during the past few years, seems it is an appropriate time to summarize those advances. In continuation of our interest on the direct functionalization of C–H bonds18–21 and modern organic synthesis,22 herein, we will try to provide a comprehensive overview of the synthesis of selenocyanated arenes and heteroarenes through the direct selenocyanation of (hetero)aryl Csp2–H bonds (Fig. 1) with an aim to inspire scientists to conduct more research in the field and develop new and creative synthetic access routes to biologically active organoselenocyanates and other important organoselenium compounds. Specifically, we have structured this review based on the selenocyanating agents and the type of catalysts.
 |
| | Fig. 1 Direct selenocyanation of (hetero)aromatic C–H bonds. | |
2. Sodium/potassium selenocyanate as selenocyanating agents
In this section, we describe the available literature on the oxidative C–H selenocyanation of (hetero)aromatic compounds using relatively inexpensive sodium/potassium selenocyanate (KSeCN) as selenocyanating agent. The section is divided into four sub-sections according to the reaction types: (i) metal-catalyzed/mediated reactions; (ii) metal-free reactions; (iii) electrochemical reactions; and (iv) photocatalyzed reactions.
2.1. Metal-catalyzed/mediated reactions
In 1936, Chao and Lyons reported Cu-mediated regioselective selenocyanation of simple aniline 1 using sodium selenocyanate (NaSeCN) as the source of selenocyano functional group directly under one-pot condition.23 Inexpensive and stable Cu(OAc)2 was used as mediator and p-selenocyanated aniline 2 was obtained in 40% yield as the sole product (Scheme 1). However, the reaction was inert towards N-methylaniline, toluene, phenol, anisol, and chlorbenzene. Although only one low yield example was communicated, this paper represents the first example of direct conversion of C–H bond to the C–SeCN bond and served as an inspiration to synthetic organic chemists for the development of general, efficient and practical strategies for the oxidative C–H selenocyanation of (hetero)aromatic scaffolds.
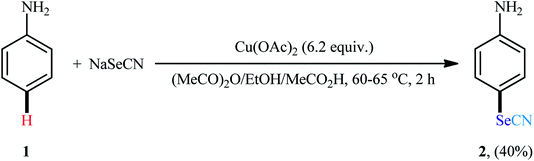 |
| | Scheme 1 Direct C4 selenocyanation of aniline 1 using NaSeCN. | |
Drawing inspiration from this work, Nair and co-workers disclosed the usefulness of KSeCN as the selenocyano source in the direct C–H selenocyanation of (hetero)arenes,24 when indoles 3 underwent regioselective selenocyanation at the C3 position with KSeCN in the presence of 2.3 equiv. of cerium(IV) ammonium nitrate ((NH4)2Ce(NO3)6) as a mediator in MeOH to form corresponding 3-(selenocyanato)indoles 4 in high yields (Scheme 2). N,N-Dimethylaniline was also efficient reactant under the optimized reaction conditions whereas 1-methylpyrrole was not suitable reactant as the corresponding selenocyanated product was isolated in only 25% yield.
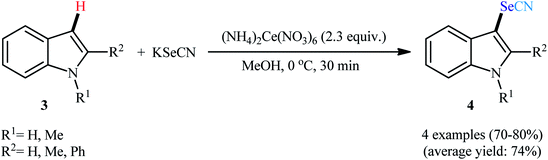 |
| | Scheme 2 Ce-mediated selenocyanation of indoles 3 with KSeCN. | |
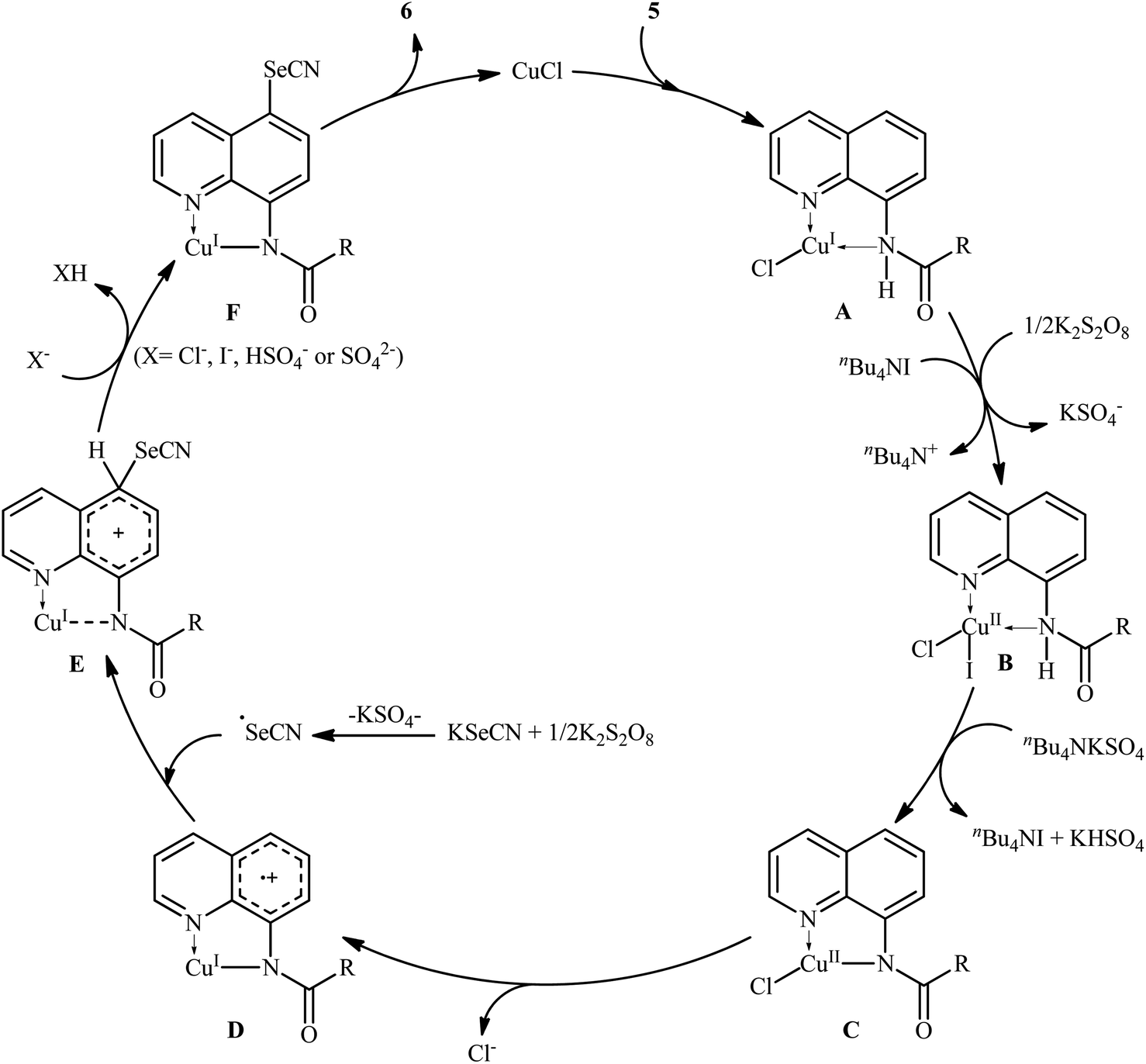
Along this line, in 2017, Wang, Xu, and co-workers demonstrated an efficient and facile method for the synthesis of C5-selenocyanated quinolines 6 by treatment of 8-aminoquinoline amides 5 with KSeCN in presence of CuCl catalyst, K2S2O8 oxidant, and tetrabutylammonium iodide (TBAI) additive in DCE solvent under refluxing condition (Scheme 3).25 Various alkyl, alkenyl, and (hetero)aryl amide-substituted quinolines were responded to the reaction under optimized conditions with moderate to high efficiency. The results indicated that quinolines having electron-donating groups afforded higher yields than that of electron-withdrawing groups. When KSeCN were replaced with KSCN, thiocyanated products were formed in satisfactory yields. It should be mentioned that the acyl protected group could be easily removed by treatment with HCl in refluxing ethanol for 1 h. The mechanistic study suggested that this transformation began with the chelation of copper catalyst by 8-aminoquinolin amide 5 to achieve Cu(I)–quinoline complex A. Next, complex A was oxidized by K2S2O8 in the presence of TBAI to from Cu(II) intermediate B. After that, deprotonation of complex B produced the amidate-Cu(II) intermediate C. Subsequently, this intermediate C experienced an intramolecular single electron transfer (SET) to form cationic radical D. The newly formed radical D underwent radical coupling reaction with the SeCN radical that was generated by the oxidation of KSeCN in the presence of K2S2O8 to generate cationic intermediate E. Thereafter, deprotonation of intermediate E produced complex F which finally led to the desired product 6 through a ligand exchange process (Scheme 4).
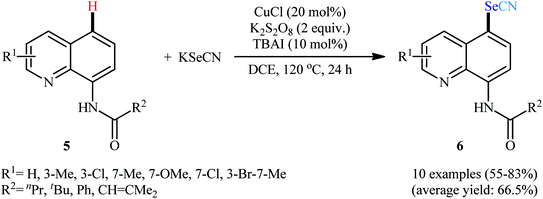 |
| | Scheme 3 Cu-catalyzed regioselective C5-selenocyanation of 8-aminoquinolin amides 5 with KSeCN. | |
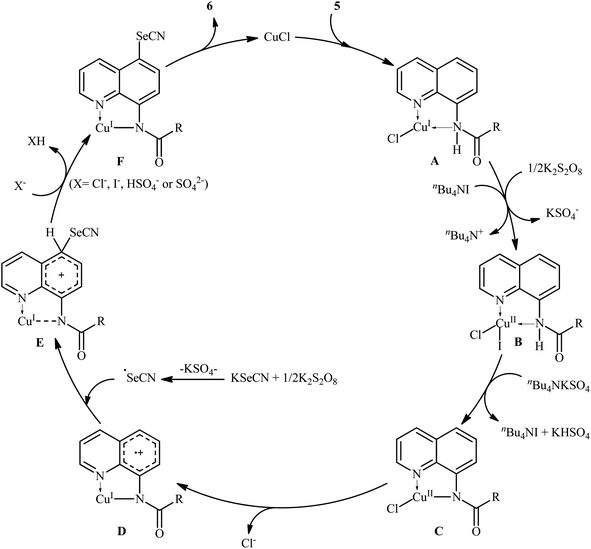 |
| | Scheme 4 Possible mechanism of the selenocyanation of 8-aminoquinolin amides 5. | |
2.2. Metal-free reactions
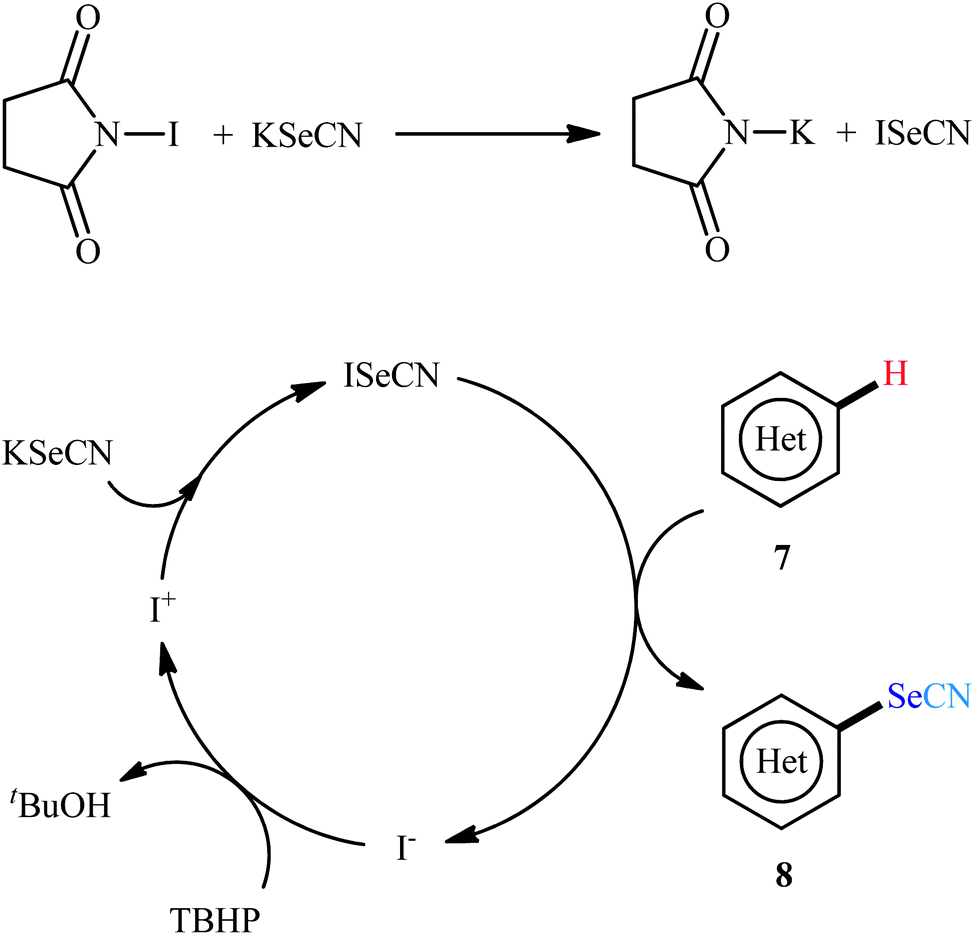
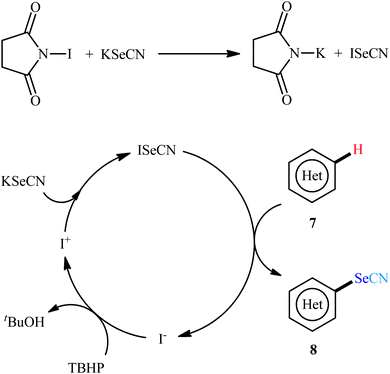
In 2016, Prabhu and co-workers informed for the first time the usefulness of halide ions as catalysts for oxidative selenocyanation of (hetero)aromatic compounds with KSeCN.26 To assess the catalytic performance of different halogen sources, 5-methoxyindole was chosen as the model substrate. Screening of various classical halogen sources such as NCS, NBS, NIS, TBAI, KI, I2 conclusively led to N-iodosuccinimide (NIS) as the superior catalyst, whereas THBP was found to be more effective oxidant compared to H2O2. In a pursuit to further improve the yield, AcOH was added as an additive to the reaction mixture. Under the optimized conditions, various (hetero)arenes including indole, phenol, aniline, and anisole derivatives 7 reacted efficiently with KSeCN to give the corresponding selenocyanated products 8 in 30–98% yields (Scheme 5). The relative reaction rates of the examined substrates followed the order: indoles > anilines ≥ phenols ≥ anisoles. When replacing KSeCN with KSCN, aryl thiocyanates were obtained. This protocol was also applicable for gram-scale synthesis of selenocyanated (hetero)arenes without further optimization. The suggested mechanism for this reaction started with the generation of iodoselenocyanate (ISeCN) through the reaction of KSeCN with NIS. The formed highly electrophilic ISeCN then trapped by nucleophilic indole on its C-3 position (para-position in the case of aniline, phenol, and anisole) to afford the expected product. In the next step, reduced iodide oxidized by TBHP to generate iodonium species which then subsequently reacted with another molecule of KSeCN to give iodothiocyanate and complete the catalytic cycle (Scheme 6).
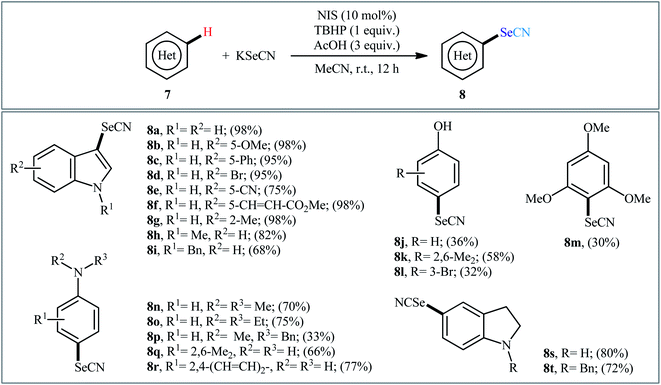 |
| | Scheme 5 Prabhu's synthesis of (hetero)aryl selenocyanates 8. | |
 |
| | Scheme 6 Presumable pathway of the formation of (hetero)aryl selenocyanates 8. | |
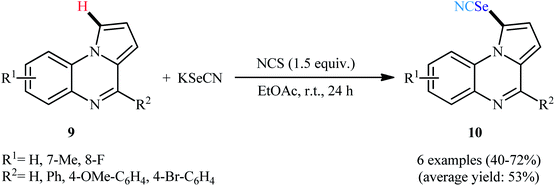
Four years later, the group of Liu-Wei disclosed the synthesis of 1-selenocyanatopyrrolo[1,2-a]quinoxalines 10 in modest to good yields and complete regioselectivity from the corresponding pyrrolo[1,2-a]quinoxalines 9 and KSeCN under the action of NCS without additional oxidants or additives in EtOAc solvent at room temperature (Scheme 7).27 This method tolerated a series of electron-donating and weak electron-withdrawing functional groups. However, the substrates bearing strong electron-withdrawing groups (e.g., NO2) were ineffective under standard condition. The authors properly solved this limitation using MeCN as solvent, which afforded decent yields of the desired products.
 |
| | Scheme 7 Direct synthesis of 1-selenocyanatopyrrolo[1,2-a]quinoxalines 10 from pyrrolo[1,2-a]quinoxalines 9 and KSeCN under the action of NCS. | |
Concurrently, Hao and Song along with their co-workers presented the molecular iodine-catalyzed direct C3-selective selenocyanation of 2-arylimidazo[1,2-a]pyridines 11 with KSeCN in the most environmentally benign solvent, water, under ambient temperature.28 The optimized condition for this C–H activation reaction is the use of 20 mol% of I2 as catalyst and 1.5 equiv. of (diacetoxyiodo)benzene (PIDA) as an oxidant. Under these conditions, the desired 3-selenocyanatoimidazo[1,2-a]pyridines 12 were obtained with up to 97% yields (Scheme 8). Importantly, imidazo[2,1-b]thiazole, aniline, and anisole derivatives were also amenable substrates in this oxidative selenocyanation and the respective mono-selenocyanated products were formed in moderate to high yields. Intriguingly, when the reaction was performed at 110 °C, only bis(aryl)selane products were obtained instead of aryl selenocyanates. The author explained this observation by the homolytical cleavage of Se–CN bond of aryl selenocyanates to generate nitrile and selenyl radicals, which the later reacts with another molecule of arene substrate to form the corresponding bis(aryl)selane.
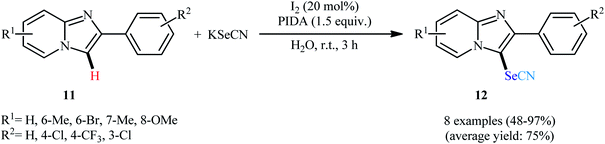 |
| | Scheme 8 I2-catalyzed C3-selective selenocyanation of 2-arylimidazo[1,2-a]pyridines 11 with KSeCN in water. | |
In a related study, Laali and colleagues reported a methodology for the synthesis of 3-selenocyanato-carbazoles 14 starting from carbazoles 13 and KSeCN in presence of 1.5 equiv. of Selectfluor under an inert atmosphere at room temperature (Scheme 9).29 The reaction afforded the highest yields when MeCN was used as a solvent. Both N-substituted and NH-free carbazoles underwent the reaction under standard conditions. In addition, indoles were also responded to the methodology with high efficiency.
 |
| | Scheme 9 Selectfluor-mediated direct selenocyanation carbazoles 13 with KSeCN. | |
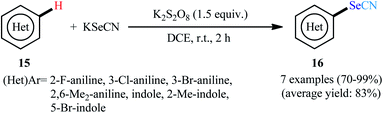
In 2020, Sorabad and Maddani reported a mild and metal-free direct selenocyanation method of NH-free aniline and indole derivatives 15 with KSeCN.30 In this case, they used K2S2O8 as the oxidizing agent and DCE as solvent. Both electron-releasing and electron-withdrawing substituent provided good yields (Scheme 10). The results indicated that indoles afforded higher yields compared to the anilines under the identical conditions. The most important point was that this reaction proceeded through a free radical pathway instead of aromatic electrophilic substitution reaction. It should be mentioned that the system was also amenable to the regioselective reaction of N-aryl enaminones, providing α-SeCN-substituted enaminone products in excellent yields.
 |
| | Scheme 10 K2S2O8-mediated oxidative selenocyanation of aniline and indole derivatives 15 with KSeCN. | |
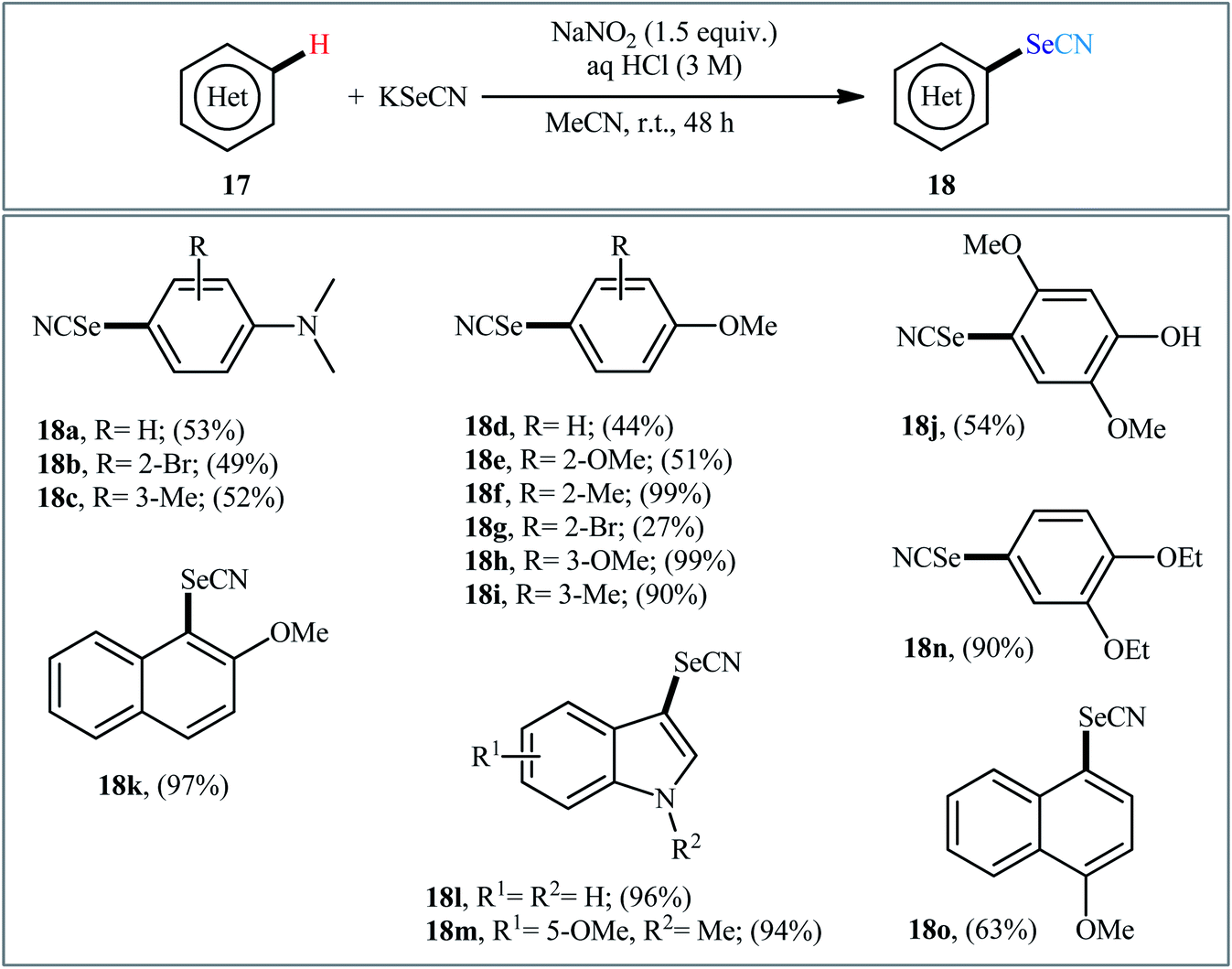
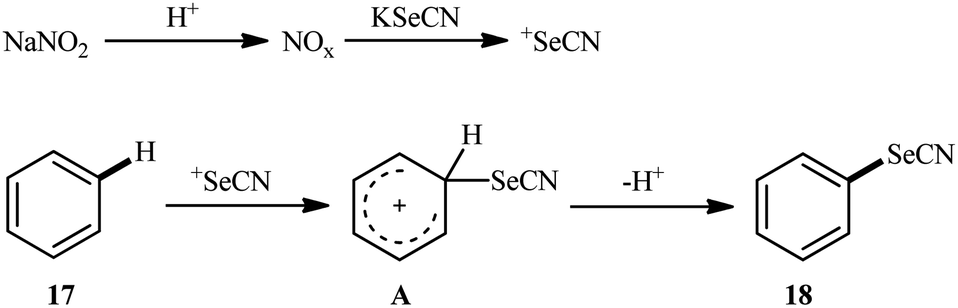
Very recently, the group of Huang synthesized a wide range of (hetero)aryl selenocyanates 18 via NaNO3-mediated oxidative selenocyanation of the corresponding (hetero)aromatic compounds 17 with KSeCN employing HCl as an additive in MeCN at room temperature.31 As shown in Scheme 11, various electron-rich aniline, anisole, and indole derivatives were compatible with this process and produced up to 99% yields of desired selenocyanated products. However, the presence of electron-withdrawing groups in the substrate considerably decreased the efficiency of this reaction. For example, while anisole having a methyl group on the ortho-position gave the corresponding C4-selenocyanated product in 99% yield, the anisole having a bromo group on this position afforded the desired product in only 27% yield. According to the authors, the mechanism of this process includes the generation of nitrogen oxide active species (NOx) through the reaction of sodium nitrite with aqueous hydrochloric acid, its reaction with KSeCN to give +SeCN ion, and the direct electrophilic selenocyanation of electron-rich arenes with +SeCN (Scheme 12).
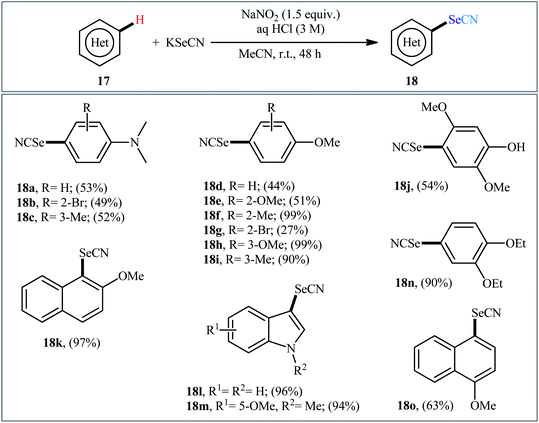 |
| | Scheme 11 NaNO2-mediated selenocyanation of (hetero)aromatic compounds 17 with KSeCN. | |
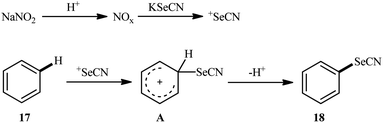 |
| | Scheme 12 Mechanistic proposal for the reaction in Scheme 11. | |
2.3. Electrochemical reactions
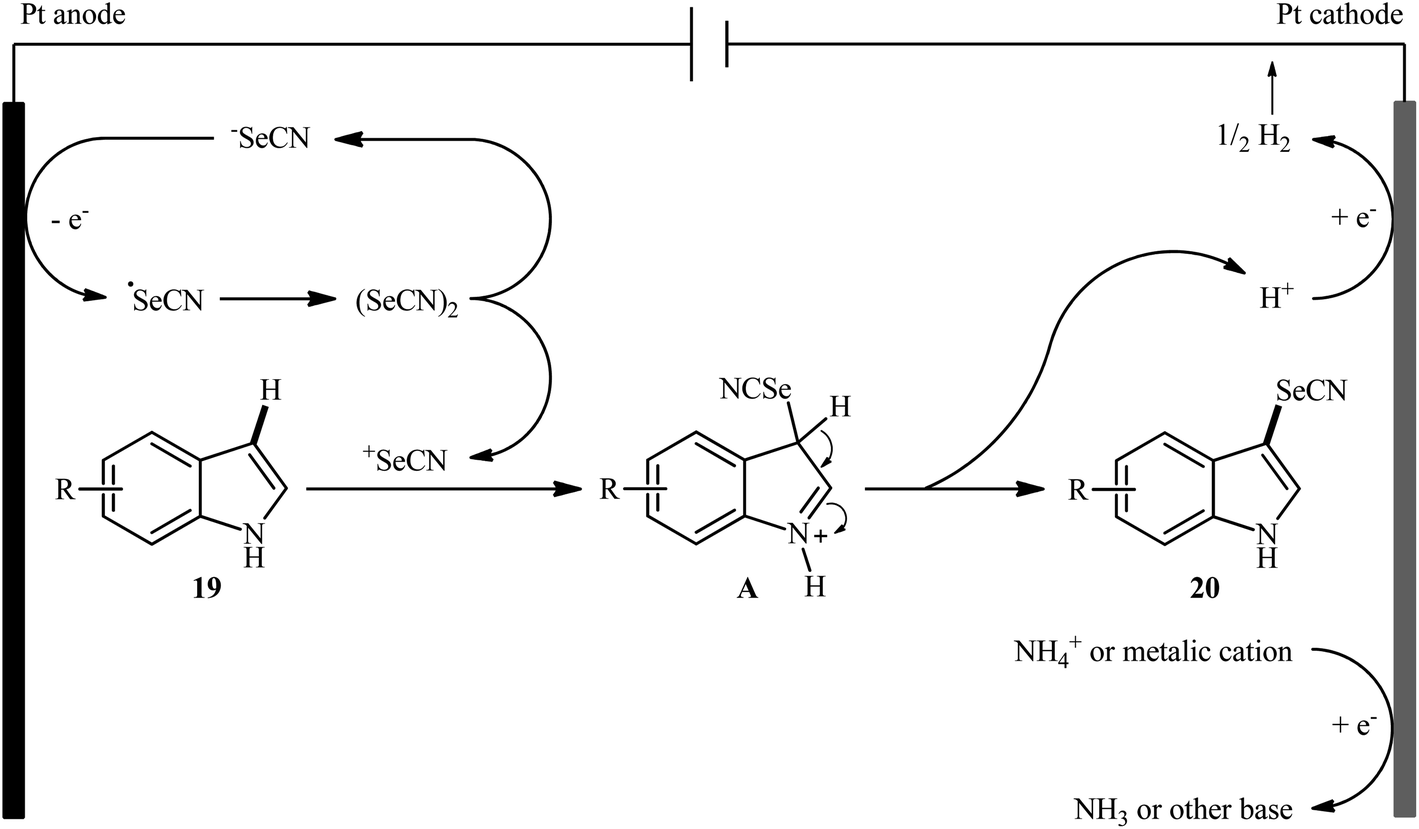
After pioneering works by Melnikov32 and Cauquis33 research groups of the electrochemical selenocyanation of small libraries of electron-rich arenes using KSeCN as the selenocyanting agent in the years of 1947 and 1971, respectively, the first general report of the synthesis of (hetero)aryl selenocyanates through the direct and site-selective selenocyanation of hetero(aromatic) C–H bonds under electrochemical conditions was published in 2018 by Sun et al.34 In this investigation, nine 3-selenocyanato-indole derivatives 20 were efficiently synthesized through the treatment of respective C3-unsabstituted indoles 19 with KSeCN in an undivided cell with platinum electrodes under constant current of 18 mA at room temperature. The reactions were carried out in MeCN without supporting electrolyte under an inert atmosphere, tolerated multiple groups of substituents (e.g., F, Cl, NO2, OMe), and provided the target products in moderate to excellent yields within 3 h (Scheme 13). Notably, the protocol was applicable to gram-scale synthesis without any difficulty. The methodology was also shown to be compatible with aniline derivatives. However, similar reaction conditions could not be generalized to pyrroles and anisoles. The suggested mechanism for this reaction is represented in Scheme 14. Initially, selenocyanate anion (−SeCN) gets oxidized by a single-electron-transfer process at the anode to form ˙SeCN radical, which then undergoes homocoupling to generate selenocyanogen ((SeCN)2). A subsequent heterolytic Se–Se bond cleavage of (SeCN)2 provides the electrophile +SeCN and regenerates the nucleophilic −SeCN. In the next step, +SeCN reacts with indole 19 to afford the hydroindole cation intermediate A. Finally, deprotonation of this cation D gives the expected product 20.
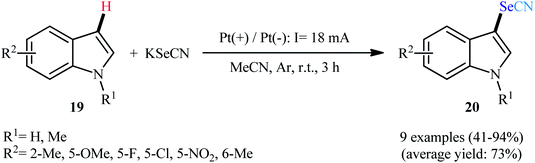 |
| | Scheme 13 Electrosynthesis of 3-selenocyanato-indoles 20 reported by Sun. | |
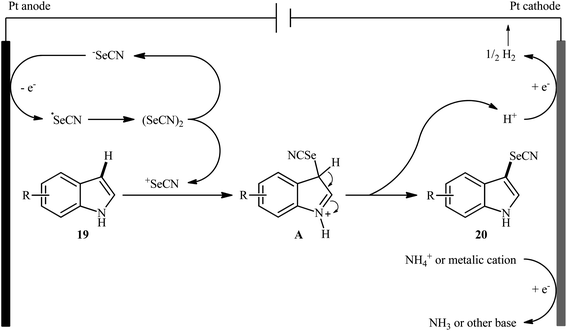 |
| | Scheme 14 Plausible mechanism for electrosynthesis of 3-selenocyanato-indoles 20. | |
In 2020, Liu and Sun along with their co-workers proposed the electrosynthesis of 3-selenocyanated imidazopyridines 22 via regioselective oxidative C–H selenocyanation of imidazopyridines 21 with KSeCN using an undivided electrolytic cell under transition-metal- and oxidant-free conditions.35 With platinum plate electrodes and nBu4NPF6 supporting electrolyte, the reaction was conducted at a constant current density of 5 mA cm−2 in MeCN at room temperature for 6 h and yields of up to 65% were obtained (Scheme 15). The authors proposed mechanism for this transformation is analogous to the one depicted for indoles.
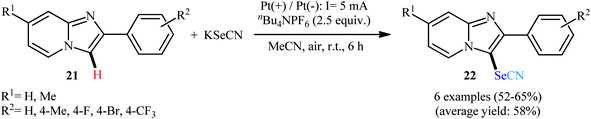 |
| | Scheme 15 Electrolytic C–H selenocyanation of imidazopyridines 21 with KSeCN. | |
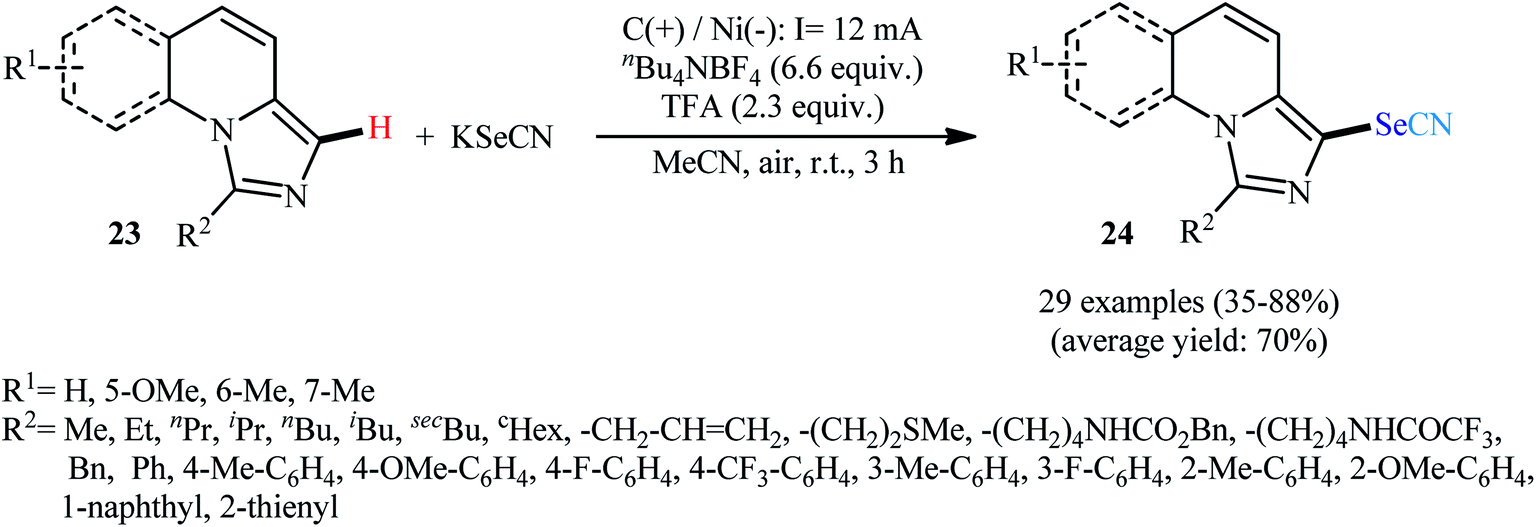
Along this line, very recently, Xu and colleagues devised a method for electrolytic C–H selenocyanation of imidazo[1,5-a]quinolines 23 with KSeCN in an undivided cell with Ni and graphite plates as the electrodes using nBu4NBF4 as a supporting electrolyte and TFA as an additive in MeCN solvent at room temperature to afford C3-selenocyanated imidazo[1,5-a]quinolines 24 (Scheme 16).36 The reaction scope was extensively investigated and 29 examples were obtained in modest to high yields, ranging from 35% to 88%. Notably, a wide panel of important functional groups such as fluoro, trifluoromethyl, ether, amide and urethane functionalities were well tolerated by this reaction which might provide potential opportunities for further manipulation of the end products.
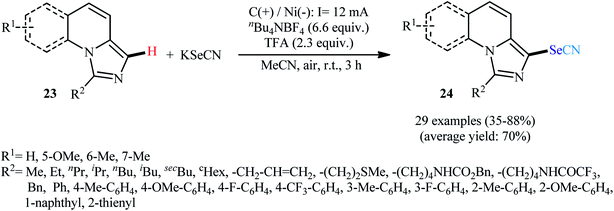 |
| | Scheme 16 Electrochemical synthesis of C3-selenocyanated imidazo[1,5-a]quinolines 24. | |
2.4. Photocatalyzed reactions
The possibility of synthesizing (hetero)aryl selenocyanates through the photocatalyzed oxidative selenocyanation of Caryl–H bonds using KSeCN was first realized by Hajra and co-workers, who showed that the treatment of 2-phenylimidazo[1,2-a]pyridine 25 with 3 equiv. of KSeCN in the presence of a catalytic amount of eosin Y under blue LED irradiation at room temperature afforded 2-phenyl-3-selenocyanatoimidazo[1,2-a]pyridine 26 in 51% yield after 3 h (Scheme 17a).37 Later, Bettanin, Lenardão, and co-workers tried to extend this chemistry to indoles.38 By employing unsubstituted indole 27 as the model reactant, the authors carefully studied the reaction variables such as photocatalyst, light source and solvent. It was found that performing the process in the presence of a catalytic amount of rose bengal (5 mol%) in MeCN at room temperature under irradiation of blue LEDs was the optimum reaction condition, giving the expected C3-selenocyanated indole 28 in a yield of 91% (Scheme 17b). Nevertheless, no comment was made by the authors regarding the substrate scope or possible mechanistic pathway of this synthetic strategy.
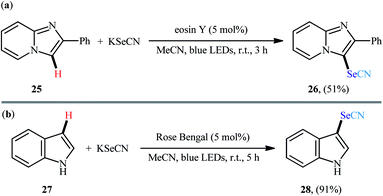 |
| | Scheme 17 (a) Hajra' synthesis of 2-phenyl-3-selenocyanatoimidazo[1,2-a]pyridine 26; (b) photocatalyzed regioselective selenocyanation of indole 27 with KSeCN. | |
3. Triselenodicyanide as selenocyanating agent
In 2004, Kachanov and co-workers disclosed for the first time the usefulness of triselenodicyanide, Se3(CN)2, as an efficient selenocyanating agent for the direct selenocyanation of (hetero)aromatic C–H bonds without the requirement for any catalyst or additive.39 They showed that aniline derivatives 29 in the treatment with in situ generated Se3(CN)2 (from the reaction of selenium dioxide with malononitrile) in DMSO at room temperature, underwent C4–H selective selenocyanation to afford p-selenocyano anilines 30 in good to quantitative yields within 15–45 min (Scheme 18). Notably, presence of either electron-donating or electron-withdrawing groups in the phenyl ring periphery did not have much impact in the yield of the reactions. In addition, site-selective C3–H selenocyanation of indole derivatives have also been demonstrated in this report. Unfortunately, the authors did not propose a plausible mechanism for this C–Se bond formation reactions. A decade later, Palop's research group applied this protocol in the synthesis of a library of bioactive hybrid selenosulfonamides.40
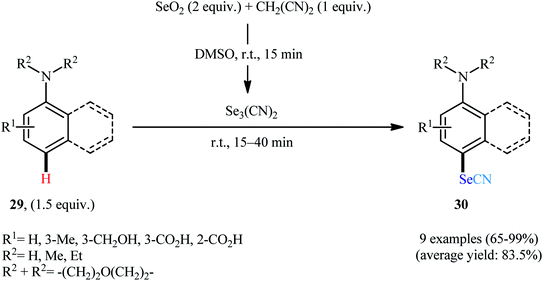 |
| | Scheme 18 Regioselective selenocyanation of aniline derivatives 29 using Se3(CN)2. | |
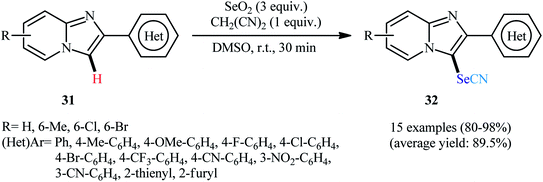
Recently, Redon and Vanelle along with their co-workers developed a similar protocol for regioselective selenocyanation of imidazo[1,2-a]pyridines 31 at the C3 position using the merge of SeO2 with CH2(CN)2 as the selenocyano source.41 The reaction was undertaken under an air atmosphere at ambient temperature, tolerated by various important functional groups (e.g., OMe, F, Cl, Br, NO2, CN), and generally afforded the desired C3-selenocyanated products 32 in high to excellent yields (Scheme 19). The protocol was also equally applicable to other imidazoheterocyclic compounds such as imidazopyrimidine, imidazothiazole and imidazobenzothiazole. This transformation was also compatible for the scale up reaction as exemplified by the formation of 2-phenyl-3-selenocyanatoimidazo[1,2-a]pyridine on a 1.5 g scale (84%). Furthermore, the authors were able to demonstrate that the SeCN functional group in 2-phenyl-3-selenocyanatoimidazo[1,2-a]pyridine could be easily transformed into SeO2H, SeCF3, Se–Se, SePh, and SeMe groups. However, they were silent on the possible mechanistic pathway for this transformation.
 |
| | Scheme 19 C3–H selenocyanation to imidazo[1,2-a]pyridine scaffolds 31 developed by Redon and Vanelle. | |
4. Combination of elemental selenium and TMSCN as selenocyanating agent
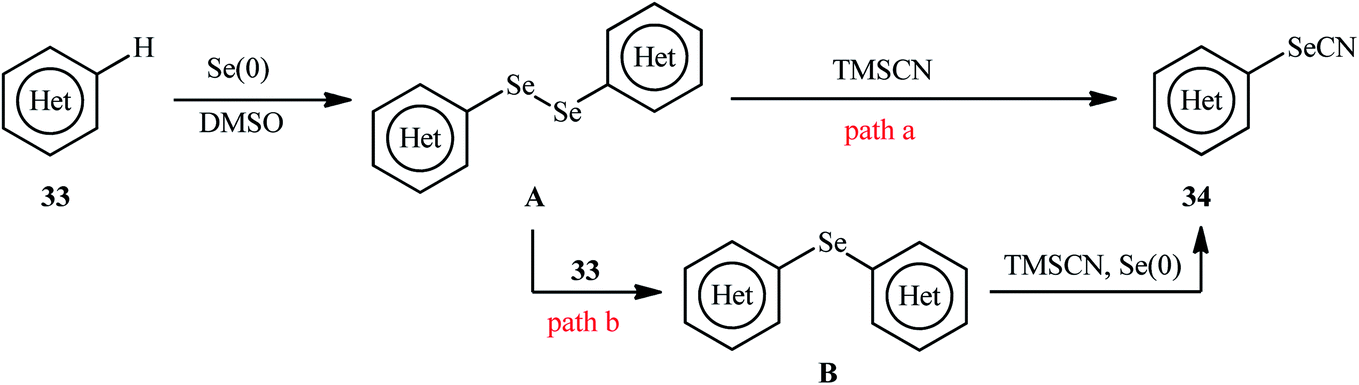
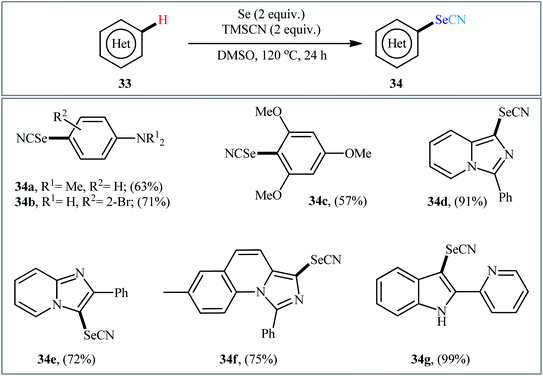
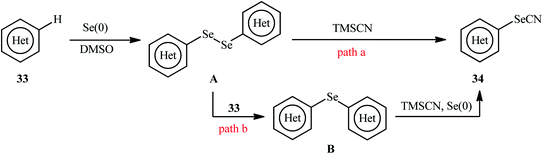
In 2018, Yan and colleagues reported an innovative methodology for regioselective oxidative C–H selenocyanation of (hetero)arenes 33 by using elemental selenium and trimethylsilyl cyanide (TMSCN) as a novel combined selenocyanation source in DMSO under catalyst and additive-free conditions.42 Gratifyingly, various electron-rich arenes and N-heteroarenes were compatible with this reaction and produced corresponding mono-selenocyanated products 34 in moderate to excellent yields (Scheme 20). In this reaction DMSO served as dual role as the solvent and the mild oxidant. Interestingly, when elemental selenium was replaced by elemental sulfur, the reaction furnished the thiocyanated products without difficulty. According to the author proposed mechanism this reaction may proceeds through the formation of di(hetero)aryl diselenide A via C–Se cross-coupling reaction of (hetero)arene 33 with elemental selenium in the presence of DMSO followed by direct cyanation with TMSCN via the C–Se bond cleavage (Scheme 21, path a). In another possibility, nucleophilic attack of another molecule of (hetero)arene 33 on diselenide intermediate A affords symmetrical selenoether B, which in the presence of Se(0) and TMSCN converts to the final product 34 (Scheme 21, path b).
 |
| | Scheme 20 Catalyst-free oxidative C–H selenocyanation of (hetero)arenes 33 with elemental selenium and TMSCN. | |
 |
| | Scheme 21 Plausible mechanism for the reaction in Scheme 20. | |
5. Conclusion
The (hetero)aryl selenocyanates have been identified as a potentially bioactive class of compounds that exhibit a broad range of biological activities such as antimicrobial, antileishmanial, anticancer and chemopreventive activities. Hence, it is of interest to seek convenient and efficient synthetic methodologies to selectively access this class of organoselenium compounds. Recently, direct selenocyanation of (hetero)aromatic C–H bonds has been emerging as one of the extremely robust methodologies for the construction of the titled compounds within a single click. Since this page of (hetero)aryl selenocyanates synthesis don't need to the pre-functionalized substrates, it is greatly greener, shorter, and atom economical compared to the traditional procedures which rely on the use of active aryl species such as aryl metal reagents, aryl halides, aryl diazonium salt, and arylboronic acids. However, the majority of reported examples of this chemistry are limited to the use of odorous and air-sensitive KSeCN as selenocyanating agent. In addition, the scope of (heter)arenes that underwent reaction is limited to nitrogen-containing heteroarene and electron-rich arene substrates. Consequently, further research should be focused on development of powerful, stable, and odorless sources of selenocyanating agents and highly active catalytic systems with much broader substrate scope. We hope that this review will be beneficial to stimulate scientists to further research and thinking on this topic.
Conflicts of interest
There are no conflicts to declare.
References
-
(a) C. G. Wilber, Clin. Toxicol., 1980, 17, 171–230 CrossRef CAS PubMed
 ;
(b) M. Mézes and K. Balogh, Acta Biol., 2009, 53, 15–18 Search PubMed .
;
(b) M. Mézes and K. Balogh, Acta Biol., 2009, 53, 15–18 Search PubMed . - M. P. Rayman, Lancet, 2012, 379, 1256–1268 CrossRef CAS .
- T. M. Sakr, M. Korany and K. V. Katti, J. Drug Delivery Sci. Technol., 2018, 46, 223–233 CrossRef CAS .
- M. Vinceti, T. Filippini, C. Del Giovane, G. Dennert, M. Zwahlen, M. Brinkman, M. P. Zeegers, M. Horneber, R. D'Amico and C. M. Crespi, Cochrane Database Syst. Rev., 2018, 1, Cd005195 Search PubMed .
- M. J. Parnham and H. Sies, Biochem. Pharmacol., 2013, 86, 1248–1253 CrossRef CAS PubMed .
- W. Ali, M. Álvarez-Pérez, M. A. Marć, N. Salardón-Jiménez, J. Handzlik and E. Domínguez-Álvarez, Curr. Pharmacol. Rep., 2018, 4, 468–481 CrossRef CAS .
-
(a) J. Müller and A. Terfort, Inorg. Chim. Acta, 2006, 359, 4821–4827 CrossRef ;
(b) Y. Ie, T. Hirose, A. Yao, T. Yamada, N. Takagi, M. Kawai and Y. Aso, Phys. Chem. Chem. Phys., 2009, 11, 4949–4951 RSC .
- N. Mukherjee, D. Kundu and B. C. Ranu, Adv. Synth. Catal., 2017, 359, 329–338 CrossRef CAS .
- A. Krief, W. Dumont and C. Delmotte, Angew. Chem., Int. Ed., 2000, 39, 1669–1672 CrossRef CAS PubMed .
-
(a) T. Billard, S. Large and B. R. Langlois, Tetrahedron Lett., 1997, 38, 65–68 CrossRef CAS ;
(b) P. Nikolaienko and M. Rueping, Chem.–Eur. J., 2016, 22, 2620–2623 CrossRef CAS PubMed .
- J.-C. Guillemin, Curr. Org. Chem., 2011, 15, 1670–1687 CrossRef CAS .
-
(a) C. Paulmier and F. Outurquin, J. Heterocycl. Chem., 1983, 20, 113–119 CrossRef CAS ;
(b) A. Frolov, E. Smirnov, O. Kul'bitskaya and A. El'tsov, Zh. Org. Khim., 1980, 16, 2302–2309 CAS ;
(c) V. Nefedov, L. Tarygina, L. Kryuchkova and Y. S. Ryabokobylko, Zh. Org. Khim., 1981, 17, 570–584 CAS .
-
(a) S. Redon, A. R. O. Kosso, J. Broggi and P. Vanelle, Synthesis, 2019, 51, 3758–3764 CrossRef CAS ;
(b) D. He, J. Yao, B. Ma, J. Wei, G. Hao, X. Tuo, S. Guo, Z. Fu and H. Cai, Green Chem., 2020, 22, 1559–1564 RSC .
-
(a) H. Rheinboldt and E. Giesbrecht, Chem. Ber., 1956, 89, 631–636 CrossRef CAS ;
(b) J. Gosselck and E. Wolters, Chem. Ber., 1962, 95, 1237–1244 CrossRef CAS ;
(c) S. Yavuz, A. Dişli, Y. Yıldırır and L. Türker, Molecules, 2005, 10, 1000–1004 CrossRef CAS PubMed .
- M. Abdoli and H. Saeidian, J. Sulfur Chem., 2015, 36, 556–582 CrossRef CAS .
- A. Monfared, R. Mohammadi, S. Ahmadi, M. Nikpassand and A. Hosseinian, RSC Adv., 2019, 9, 3185–3202 RSC .
-
(a) J. Hofmann and M. R. Heinrich, Tetrahedron Lett., 2016, 57, 4334–4340 CrossRef CAS ;
(b) A. Roglans, A. Pla-Quintana and M. Moreno-Manas, Chem. Rev., 2006, 106, 4622–4643 CrossRef CAS PubMed .
- Selected reviews: Y. Liu, A. G. Ebadi, L. Youseftabar-Miri, A. Hassanpour and E. Vessally, RSC Adv., 2019, 9, 25199–25215 RSC .
- J. Wang, P. Su, S. Abdolmohammadi and E. Vessally, RSC Adv., 2019, 9, 41684–41702 RSC .
- Y. Yang, D. Zhang and E. Vessally, Top. Curr. Chem., 2020, 378, 37 CrossRef CAS PubMed .
- Z. Liu, A. Ebadi, M. Toughani, N. Mert and E. Vessally, RSC Adv., 2020, 10, 37299–37313 RSC .
- Selected reviews:
(a) S. Ebrahimiasl, F. Behmagham, S. Abdolmohammadi, R. N. Kojabad and E. Vessally, Curr. Org. Chem., 2019, 23, 2489–2503 CrossRef CAS ;
(b) L. Feng, X. Li, B. Liu and E. Vessally, J. CO2 Util., 2020, 40, 101220 CrossRef CAS ;
(c) I. Söğütlü, E. A. Mahmood, S. A. Shendy, S. Ebrahimiasl and E. Vessally, RSC Adv., 2021, 11, 2112–2125 RSC ;
(d) S. Sarhandi, M. Daghagheleh, M. Vali, R. Moghadami and E. Vessally, Chem. Rev. Lett., 2018, 1, 9–15 Search PubMed ;
(e) M. R. J. Sarvestani, N. Mert, P. Charehjou and E. Vessally, J. Chem. Lett., 2020, 1, 93–102 Search PubMed ;
(f) M. Daghagheleh, M. Vali, Z. Rahmani, S. Sarhandi and E. Vessally, Chem. Rev. Lett., 2018, 1, 23–30 Search PubMed ;
(g) L. Sreerama, E. Vessally and F. Behmagham, J. Chem. Lett., 2020, 1, 9–18 Search PubMed ;
(h) S. Shahidi, P. Farajzadeh, P. Ojaghloo, A. Karbakhshzadeh and A. Hosseinian, Chem. Rev. Lett., 2018, 1, 37–44 Search PubMed ;
(i) S. Mohammadi, M. Musavi, F. Abdollahzadeh, S. Babadoust and A. Hosseinian, Chem. Rev. Lett., 2018, 1, 49–55 Search PubMed ;
(j) S. Majedi, L. Sreerama, E. Vessally and F. Behmagham, J. Chem. Lett., 2020, 1, 25–31 Search PubMed ;
(k) S. Farshbaf, L. Sreerama, T. Khodayari and E. Vessally, Chem. Rev. Lett., 2018, 1, 56–67 Search PubMed ;
(l) S. Farshbaf, L. Sreerama, T. Khodayari and E. Vessally, Chem. Rev. Lett., 2018, 1, 56–67 Search PubMed ;
(m) E. A. Mahmood, B. Azizi and S. Majedi, Chem. Rev. Lett., 2020, 3, 2–8 Search PubMed ;
(n) N. Salehi and B. Azizi, et. al, J. Chem. Lett., 2021, 2, 2–8 Search PubMed ;
(o) D. Tayde and M. Lande, Chem. Rev. Lett., 2021, 4, 30–36 Search PubMed ;
(p) A. Bakhtiary, M. R. P. Heravi, A. Hassanpour, I. Amini and E. Vessally, RSC Adv., 2020, 11, 470–483 RSC .
- T. Chao and R. Lyons, Proc. Indiana Acad. Sci., 1937, 46, 105–106 CAS .
- V. Nair, A. Augustine and T. G. George, Eur. J. Org. Chem., 2002, 2363–2366 CrossRef CAS .
- J. Chen, T. Wang, T. Wang, A. Lin, H. Yao and J. Xu, Org. Chem. Front., 2017, 4, 130–134 RSC .
- N. Muniraj, J. Dhineshkumar and K. R. Prabhu, ChemistrySelect, 2016, 1, 1033–1038 CrossRef CAS .
- Z. Yang, J. He, Y. Wei, W. Li, P. Liu, J. Zhao and Y. Wei, Org. Biomol. Chem., 2020, 18, 9088–9094 RSC .
- Y. S. Zhu, Y. Xue, W. Liu, X. Zhu, X. Q. Hao and M. P. Song, J. Org. Chem., 2020, 85, 9106–9116 CrossRef CAS PubMed .
- R. Abonia, L. F. Gutierrez, A. T. Zwarycz, S. Correa Smits and K. K. Laali, Heteroat. Chem., 2019, 2019, 1459681 CrossRef .
- G. S. Sorabad and M. R. Maddani, New J. Chem., 2020, 44, 2222–2227 RSC .
- J. Cui, M. Wei, L. Pang, J. Xiao, C. Gan, J. Guo, C. Xie, Q. Zhu and Y. Huang, Tetrahedron, 2020, 76, 130978 CrossRef CAS .
- N. N. Mel'nikov and E. M. Cherkasova, Zh. Obshch. Khim., 1946, 16, 1025–1028 Search PubMed .
- G. Cauquis and G. Pierre, C. R. Acad. Sci., 1971, 272, 609 CAS .
- X. Zhang, C. Wang, H. Jiang and L. Sun, RSC Adv., 2018, 8, 22042–22045 RSC .
- T. Cui, X. Zhang, J. Lin, Z. Zhu, P. Liu and P. Sun, Synlett, 2021, 32(03), 267–272 CrossRef CAS .
- J. Zhang, H. Wang, Y. Chen, H. Xie, C. Ding, J. Tan and K. Xu, Chin. Chem. Lett., 2020, 31, 1576–1579 CrossRef CAS .
- S. Mitra, M. Ghosh, S. Mishra and A. Hajra, J. Org. Chem., 2015, 80, 8275–8281 CrossRef CAS PubMed .
- L. Bettanin, F. Penteado, L. H. Dapper and E. J. Lenardão, Chem. Proc., 2020, 2(1), 29 Search PubMed .
- A. V. Kachanov, O. Y. Slabko, O. V. Baranova, E. V. Shilova and V. A. Kaminskii, Tetrahedron Lett., 2004, 45, 4461–4463 CrossRef CAS .
- Y. Baquedano, E. Moreno, S. Espuelas, P. Nguewa, M. Font, K. J. Gutierrez, A. Jimenez-Ruiz, J. A. Palop and C. Sanmartin, Eur. J. Med. Chem., 2014, 74, 116–123 CrossRef CAS PubMed .
- S. Redon, A. R. O. Kosso, J. Broggi and P. Vanelle, Tetrahedron Lett., 2017, 58, 2771–2773 CrossRef CAS .
- C. Feng, Y. Peng, G. Ding, X. Li, C. Cui and Y. Yan, ChemComm, 2018, 54, 13367–13370 RSC .
|
| This journal is © The Royal Society of Chemistry 2021 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence c,
Mohammad Reza Poor Heravi
c,
Mohammad Reza Poor Heravi