
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGNPS-guided discovery of xylacremolide C and D, evaluation of their putative biosynthetic origin and bioactivity studies of xylacremolide A and B†
Felix Schalk‡
a,
Janis Fricke‡ a,
Soohyun Uma,
Benjamin H. Conlonb,
Hannah Mausc,
Nils Jägerd,
Thorsten Heinzeld,
Tanja Schirmeisterc,
Michael Poulsenb and
Christine Beemelmanns*a
a,
Soohyun Uma,
Benjamin H. Conlonb,
Hannah Mausc,
Nils Jägerd,
Thorsten Heinzeld,
Tanja Schirmeisterc,
Michael Poulsenb and
Christine Beemelmanns*a
aChemical Biology of Microbe-Host Interactions, Leibniz Institute for Natural Product Research and Infection Biology, Hans Knöll Institute (HKI), Beutenbergstraße 11a, 07745 Jena, Germany. E-mail: Christine.beemelmanns@hki-jena.de
bSection for Ecology and Evolution, Department of Biology, University of Copenhagen, Universitetsparken 15, 2100 Copenhagen East, Denmark
cInstitute of Pharmaceutical and Biomedical Sciences, Johannes Gutenberg University Mainz, Staudingerweg 5, 55128 Mainz, Germany
dInstitute of Biochemistry and Biophysics at Center for Molecular Biomedicine (CMB), Department of Biochemistry, Friedrich Schiller University Jena, Hans-Knöll-Straße 2, 07745 Jena, Germany
First published on 24th May 2021
Abstract
Targeted HRMS2-GNPS-based metabolomic analysis of Pseudoxylaria sp. X187, a fungal antagonist of the fungus-growing termite symbiosis, resulted in the identification of two lipopeptidic congeners of xylacremolides, named xylacremolide C and D, which are built from D-phenylalanine, L-proline and an acetyl-CoA starter unit elongated by four malonyl-CoA derived ketide units. The putative xya gene cluster was identified from a draft genome generated by Illumina and PacBio sequencing and RNAseq studies. Biological activities of xylacremolide A and B were evaluated and revealed weak histone deacetylase inhibitory (HDACi) and antifungal activities, as well as moderate protease inhibition activity across a panel of nine human, viral and bacterial proteases.
Introduction
While most fungal species belonging to the genus Xylaria (Ascomycota: Xylariaceae) are of saprotrophic nature and globally abundant,1,2 members of the subgenus Pseudoxylaria thrive predominantely on deteriorating comb material of fungus-cultivating termites that live in an obligate symbiosis with specialized fungal cultivars in the genus Termitomyces (Basidiomycotina).3,4 Our recent studies focused on the relationship between the termite's fungal cultivar and the competitive and/or antagonistic behaviour of Pseudoxylaria to better understand their coevolutionary relationship.5,6 Based on the metabolomic analysis of fungus–fungus co-cultures, we found that isolate Pseudoxylaria sp. X802 produces several biologically active small molecules including the antibacterial tetracyclic peptides pseudoxylallemycins and epoxy-cytochalasins.7,8 Following up on this, we performed a high resolution tandem mass spectrometry (HRMS2)-based analysis of guttation droplets that appear on aging aerial hyphae of the isolate X187 (Fig. 1a).9 Processing of the MS2-data using Global Natural Products Social Molecular Networking Analysis (GNPS)10 revealed the production of four linear peptides called pseudoxylaramides A–D and two PKS-NRPS based hybrids named xylacremolides A and B.11 The identified xylacremolides share similar structural features with acremolides12 and FR235222,13 a known histone deacetylase inhibitor, isolated from Acremonium spp. and saroclides from the mangrove-derived fungus Sarocladium kiliense HDN11-112.14 Intrigued by the high abundance of xylacremolide A and B in guttation droplets of Pseudoxylaria sp. X187 (from now on named X187), and inspired by the findings that fungal PKS-based megaenzymes are often inherently promiscuous, we intensified our metabolomic and genomic analysis of X187. Here, we report for the first time on the characterization of two novel xylacremolide congeners named xylacremolide C and D, and on detailed bioactivity studies of xylacremolide A and B. In silico analysis of genomic and RNA sequencing data of X187 allowed us to propose a putative biosynthetic PKS-NRPS-based gene cluster of xylacremolides in Pseudoxylaria for the first time.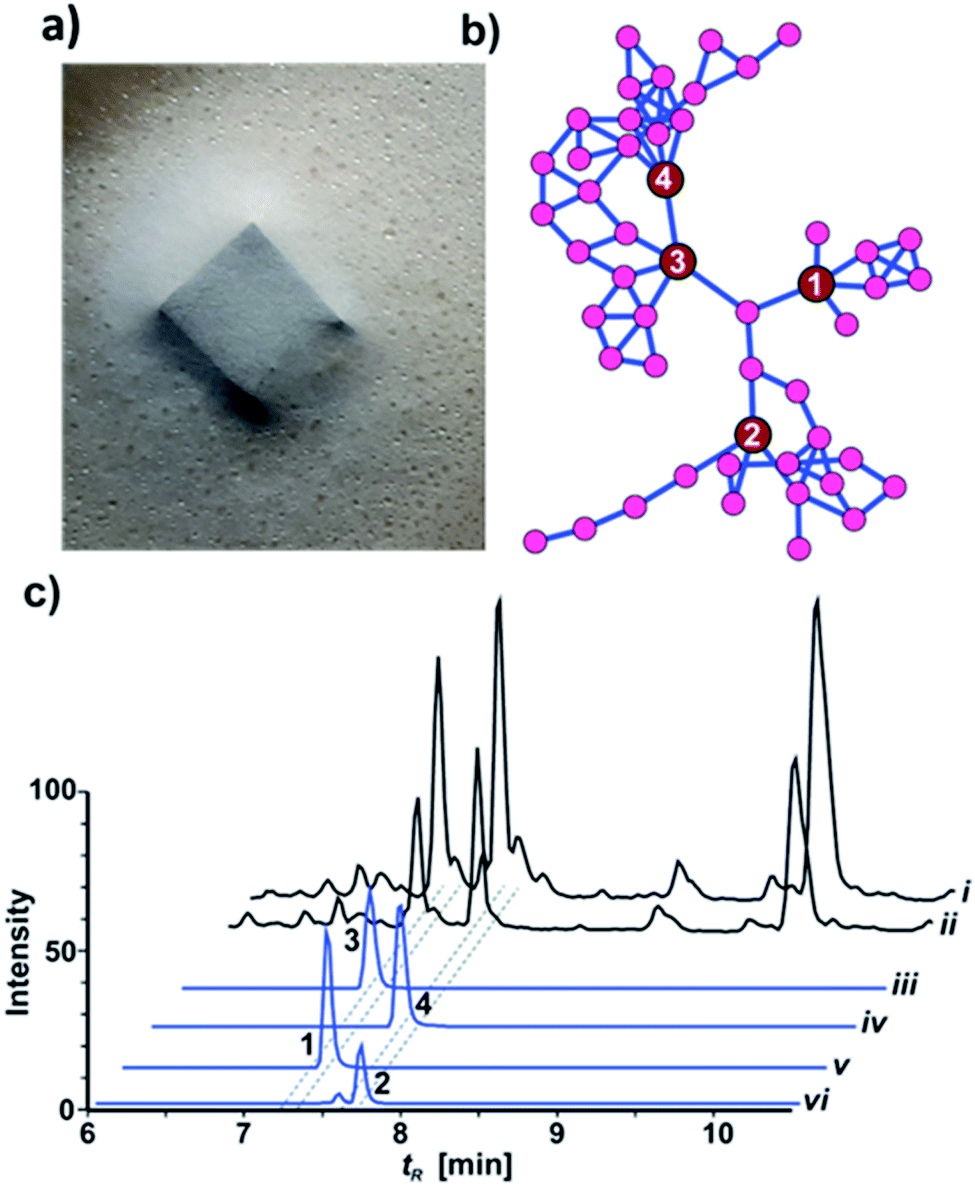 | ||
| Fig. 1 (a) Pseudoxylaria sp. X187 with guttation droplets on PDA; (b) GNPS network showing three distinct clades of substructures containing molecular ion peaks related to xylacremolides A (3), B (4), C (1) and D (2); (c) top to bottom: LC-MS chromatogram comparing TICs of crude culture extracts of X187 grown on PDA (i) or ISP2 medium (ii); XICs of compounds 1–4 (iii–vi), ±5 ppm range. Intensity (100%) was set to 4 × 109 for (i)–(iv) and 4 × 108 for (v) and (vi). | ||
Results and discussion
Metabolomic analysis and isolation
HRMS2-based metabolomic analysis of guttation droplet of X187 and subsequent GNPS-based visualization of the acquired MS2-data was performed according to an established procedure (Fig. 1b and S1†).11 GNPS-networks were screened for molecular ion peaks belonging to xylacremolide A and B and putative structural congeners that varied in chain length and oxidation of the PKS moiety. The identified m/z subnetwork related to xylacremolide A and B (m/z 403.223 (3), m/z 417.238 (4)) contained a total of 50 connected nodes with molecular ion peaks that were partially assigned to congeners carrying additional methylene units (Δm/z 14.016, Δm/z 28.032) and derivatives with varying C2H4OH moieties (Δm/z 45.057). The subcluster containing molecular ion peaks of 3 and 4 was connected by a single node (m/z 443.252) to two additional subclusters containing the molecular ion peaks m/z 461.263 and m/z 459.248, respectively. Subsequent dereplication of HRMS values using Antibase and Scifinder indicated that X187 might produce yet unreported xylacremolide congeners.11We then evaluated the relative abundance of identified molecular ion peaks in culture extracts obtained from different growth conditions. Methanolic extracts were obtained from cultures grown on potato-dextrose-agar (PDA; samples taken after 18, 28 and 42 days) and ISP2 medium (samples taken after 18 and 42 days). Subsequent comparative HRMS-analysis of culture extracts revealed a very stable xylacremolide composition within all extracts with xylacremolide B as the main product, followed by non-methylated derivative xylacremolide A (Fig. 1c, S2 and Table S1†). Although the abundance of other molecular ion peaks belonging to putative new congeners was found to be about one order of magnitude lower compared to xylacremolide B, we pursued a MS-guided isolation of those molecular ions peaks (Fig. 1).
For this purpose X187 was grown on 30 plates (PDA, 26 °C) and mycelium-covered agar plates were extracted with methanol after 28 days. Culture extracts were subjected to MS-guided semi-preparative liquid chromatography yielding two new xylacremolide congeners, xylacremolide named C (1) and D (2) in analytical purity (Fig. 2 and S3†). The first isolated molecule (HR-ESI-MS m/z [M + H]+ calcd for C25H37O6N2 461.2646, found 461.2644, Δppm = −0.434) was structurally analyzed by comparative 1D (1H and 13C) and 2D (HSQC, COSY, and HMBC) NMR spectroscopy (Tables 1, S2 and Fig. S6–S11†). On the basis of the molecular formula, nine degrees of unsaturation were conjectured. The 1H NMR spectrum of 1 showed features of a peptidic compound bearing two α-proton signals [δH 4.85 and 4.25] and one NH signal [δH 8.46]. Also, three carbonyl carbon signals [δC 174.9, 171.9, and 169.8] and two α-carbon signals [δC 59.7 and 51.3] based on the HSQC and HMBC spectra supported that compound 1 is a peptide-derived compound (Table S1†). Comparative analysis of 1H and 2D NMR data of 1 and revealed two amino acid residues including a proline (Pro) and a phenylalanine (Phe) and 3,5,7-trihydroxy-2-methyldecanoic acid (TMDA). The α-proton [H-2, δH 4.85] of Pro showed homonuclear correlations to H-3 [δH 2.02 and 1.79]. Additional 1H–1H correlations of H-3/H-4 [δH 1.80 and 1.45], H-4/H-5 [δH 3.79 and 3.37] and respective HMBC correlations from H-2 to C-3 [δC 29.5], H-3 to C-2 and C-4 [δC 59.7 and 24.9], and H-5 [δH 3.79, 3.72] to C-3 and C-4 supported the assumption of a ring structure (Fig. 2b).
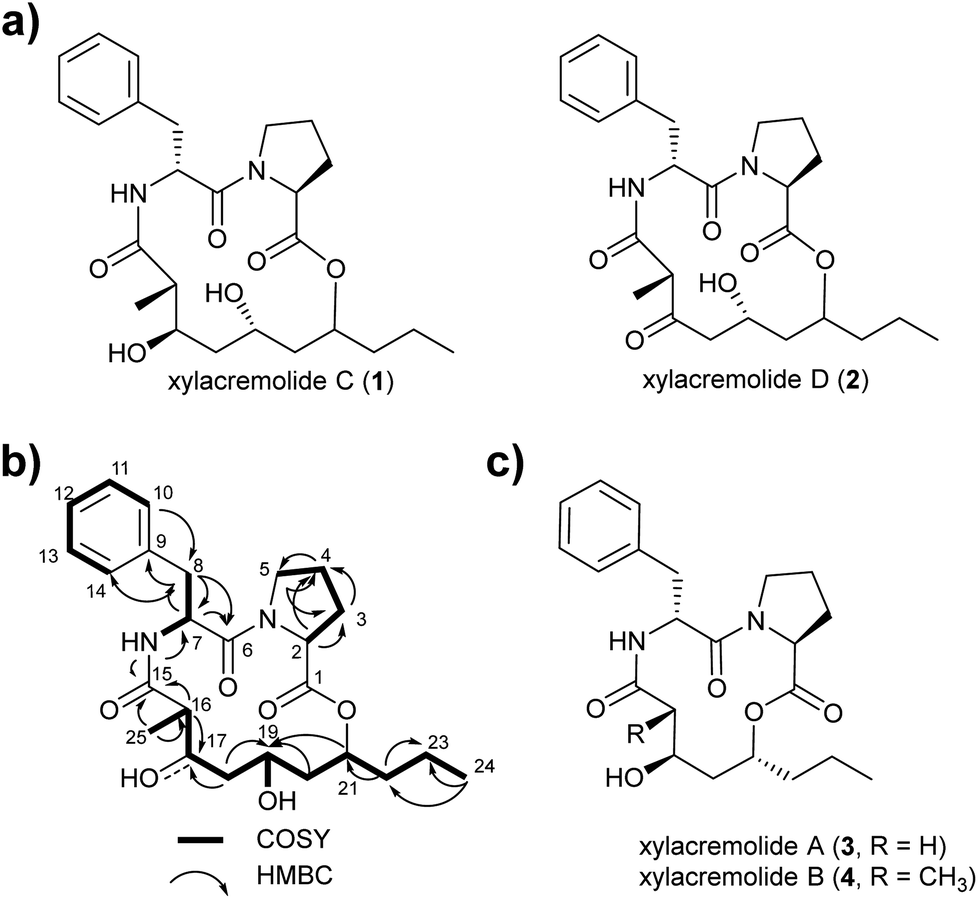 | ||
| Fig. 2 (a) Structures of isolated xylacremolides C (1) and D (2); (b) representative COSY and HMBC correlation of xylacremolide C (1); (c) structures of previously isolated xylacremolides A (3) and B (4) from X187. | ||
| Nr | 1 | 2 | ||
|---|---|---|---|---|
| 13C | 1H (J in Hz) | 13C | 1H (J in Hz) | |
| 1 | 171.9, s | — | 174.6, s | — |
| 2 | 59.7, d | 4.26, dd (8.5, 2.5) | 59.4, d | 4.32, m |
| 3 | 29.5, t | 2.02, m | 26.8, t | 2.02, m |
| 1.79, m | 1.79, m | |||
| 4 | 24.9, t | 1.80, m | 24.3, t | 1.80, m |
| 1.45, m | 1.54, m | |||
| 5 | 47.1, t | 3.79, m | 47.0, t | 3.78, m |
| 3.37, m | 3.43, m | |||
| 6 | 169.8, s | 167.1, s | ||
| 7 | 51.3, d | 4.85, ddd (9.0, 7.5, 6.0) | 51.4, d | 4.94, ddd (9.0, 7.5, 6.0) |
| 8 | 36.7, t | 2.92, dd (13.5, 6.0) | 36.0, t | 2.92, dd (13.5, 6.0) |
| 2.86, dd (13.5, 9.0) | 2.86, dd (13.5, 9.0) | |||
| 9 | 138.2, s | — | 138.1, s | — |
| 10, 14 | 129.7, d | 7.25, m | 129.7, d | 7.26, m |
| 11, 13 | 128.4, d | 7.25, m | 128.4, d | 7.26, m |
| 12 | 126.7, d | 7.17, m | 126.7, d | 7.18, m |
| NH | 8.46, d (7.5) | 8.96, d (7.5) | ||
| 15 | 174.7, s | 170.8, s | ||
| 16 | 42.1, d | 2.61, m | 51.5, d | 3.58, q (6.5) |
| H3-25 | 13.5, q | 0.91, d (7.0) | 13.6, q | 0.91, d (6.5) |
| 17 | 71.3, d | 3.74, m | 205.5, s | — |
| 18 | 43.4, t | 1.46, m | 50.6, t | 2.70, m |
| 1.37, m | 2.18, m | |||
| 19 | 64.4, d | 3.77, m | 63.2, d | 4.28, m |
| 20 | 41.7, t | 1.83, m | 33.9, t | 1.85, m |
| 1.53, m | 1.53, m | |||
| 21 | 74.3, d | 4.64, m | 74.3, d | 4.68, m |
| 22 | 33.8, t | 1.60, m | 39.2, t | 1.63, m |
| 23 | 19.0, t | 1.30, m | 39.2, t | 1.31, m |
| 1.23, m | 1.28, m | |||
| 24 | 13.5, q | 0.89, dd (7.5, 7.5) | 13.4, q | 0.87, dd (7.5, 7.5) |
Similarly, the α-proton [H-7, δH 4.85] of Phe revealed 1H–1H-COSY correlations to H-8 [δH 2.92 and 2.86] and the amide proton [δH 8.46]. The connectivities were supported by HMBC correlations from the α-proton H-7 to C-8 [δC 36.7], from the H-8 to C-7 [δC 51.3], and from the NH proton to C-7; and the heteronuclear multi-bond correlations from H-8 to C-9 [δC 138.2] and C-10/C-14 [δC 129.7] of Phe. The benzene ring in the phenylalanine was determined by COSY and HMBC correlations between H-10/H-14 [δH 7.25] and C-12 [δC 126.7] and C-9, as well as from H-12 [δH 7.17] to C-10/C-14 [δC 129.7].
The trihydroxy-methyldecanoic acid (TMDA) was deduced by 1H–1H correlations from the methyl protons [H3-25, δH 0.89] to the methine proton [H-16, δH 2.61] bearing three hydroxyl carbons [C-17, C-19, and C-21; δC 71.3, 64.4, and 74.3] and one methyl group [C-25, δC 13.5; δH 0.91] at C-16 [δC 42.1] supported by HMBC correlations. The connectivity of the two amino acid residues was confirmed with the NOESY correlation between H-5 of Pro and H-7 of Phe. The amide proton [δH 8.46] of Phe also showed HMBC correlation to the carbonyl carbon C-15 of TMDA. The nine degrees of unsaturation were in line with the weak HMBC correlation from the methine proton [C-21, δH 4.64] signal to the carbonyl carbon [C-1, δC 171.9] of Pro closing the 14-membered ring.
Due to its structural similarity to xylacremolide B (4), compound 1 was named xylacremolide C (1). Subsequently xylacremolide D (2) was also purified as an amorphous white powder and its molecular formula was deduced as C25H33O6N2 (HR-ESI-MS m/z [M + H]+ calcd for C25H34O6N2 459.2490, found 459.2485, Δppm = −1.088). Comparative 1H and 2D NMR analysis of 2 with those of 1 revealed a similar core structure with a carbonyl moiety at position at C-17 [δC 205.5], which was supported by HMBC correlations of H-16, 16-CH3 and H-18 (Table S2 and Fig. S12–S15†).
Based on the assumption that xylacremolides share the same biosynthetic origin and the similar NMR chemical shift pattern, we deduced the identical stereochemical assignment for compound 1 and 2 as shown for xylacremolide B (4) by X-ray crystallography,11 while the configuration at position C-21 remained unsolved.
Biosynthesis
To gain better insights into the enzymatic machinery responsible for the production of the xylacremolides, we thus sequenced X187 using Ilumina MiSeq and PacBio sequencing technology that yielded 499 contigs in a 39 Mbp draft genome assembly. We then set out to identify the putative biosynthetic pathway that was deduced to be of NRPS and PKS-based origin.15–17 Mining of the obtained draft genome sequence using FungiSmash18 revealed only one biosynthetic gene cluster (BGC) region, designated as the xya cluster (Fig. 3; GenBank accession number MW579544), that encodes for a highly-reducing PKS (HRPKS) xyaA (KS-AT-DH-CMet-ER-KR), a bimodular nonribosomal peptide synthase (NRPS) xyaB (C1-A1-T1-E1-C2-A2-T2-CT), a major-facilitator superfamily (MFS) transporter xyaC and two short-chain dehydrogenases/reductases (SDR) xyaD/xyaE. Subsequent RNA-Seq analysis showed that all xya genes were transcribed under producing conditions. In silico analysis of the putative substrate preference of the A domains of XyaB using NRPSPredictor2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 19 revealed a preference for D-Phe for A1 and a hydrophobic amino acid with L-Pro being suggested by the closest neighbour prediction for A2, which matches the structure of the xylacremolides. Additionally, the encoded proteins XyaA and XyaB showed high similarity (70% and 65% similar amino acid sequence) to the characterized ValA and ValB from Aspergillus terreus ATCC 20542, responsible for the biosynthesis of the structurally related lipodepsipeptides valactamide A–H20 (Fig. 3a). Furthermore, homologues BGCs have been identified in silico in other ascomycetes (Fig. 3b and Table S2†). Among them Tolypocladium capitatum, Colletotrichum gloeosporioides and Acremonium chrysogenum, produce tolyprolinol,21 colletotrichamides22 and acremolides,12 which are all lipopeptides comprised of two amino acids and an aliphatic part pointing towards a similar biosynthetic origin (Fig. 3a). Although these three predicted BGCs have not yet been biochemically characterized, our findings support the assignment of the xya BGC and could set the stage for further biomolecular and biotechnological investigations.
19 revealed a preference for D-Phe for A1 and a hydrophobic amino acid with L-Pro being suggested by the closest neighbour prediction for A2, which matches the structure of the xylacremolides. Additionally, the encoded proteins XyaA and XyaB showed high similarity (70% and 65% similar amino acid sequence) to the characterized ValA and ValB from Aspergillus terreus ATCC 20542, responsible for the biosynthesis of the structurally related lipodepsipeptides valactamide A–H20 (Fig. 3a). Furthermore, homologues BGCs have been identified in silico in other ascomycetes (Fig. 3b and Table S2†). Among them Tolypocladium capitatum, Colletotrichum gloeosporioides and Acremonium chrysogenum, produce tolyprolinol,21 colletotrichamides22 and acremolides,12 which are all lipopeptides comprised of two amino acids and an aliphatic part pointing towards a similar biosynthetic origin (Fig. 3a). Although these three predicted BGCs have not yet been biochemically characterized, our findings support the assignment of the xya BGC and could set the stage for further biomolecular and biotechnological investigations.
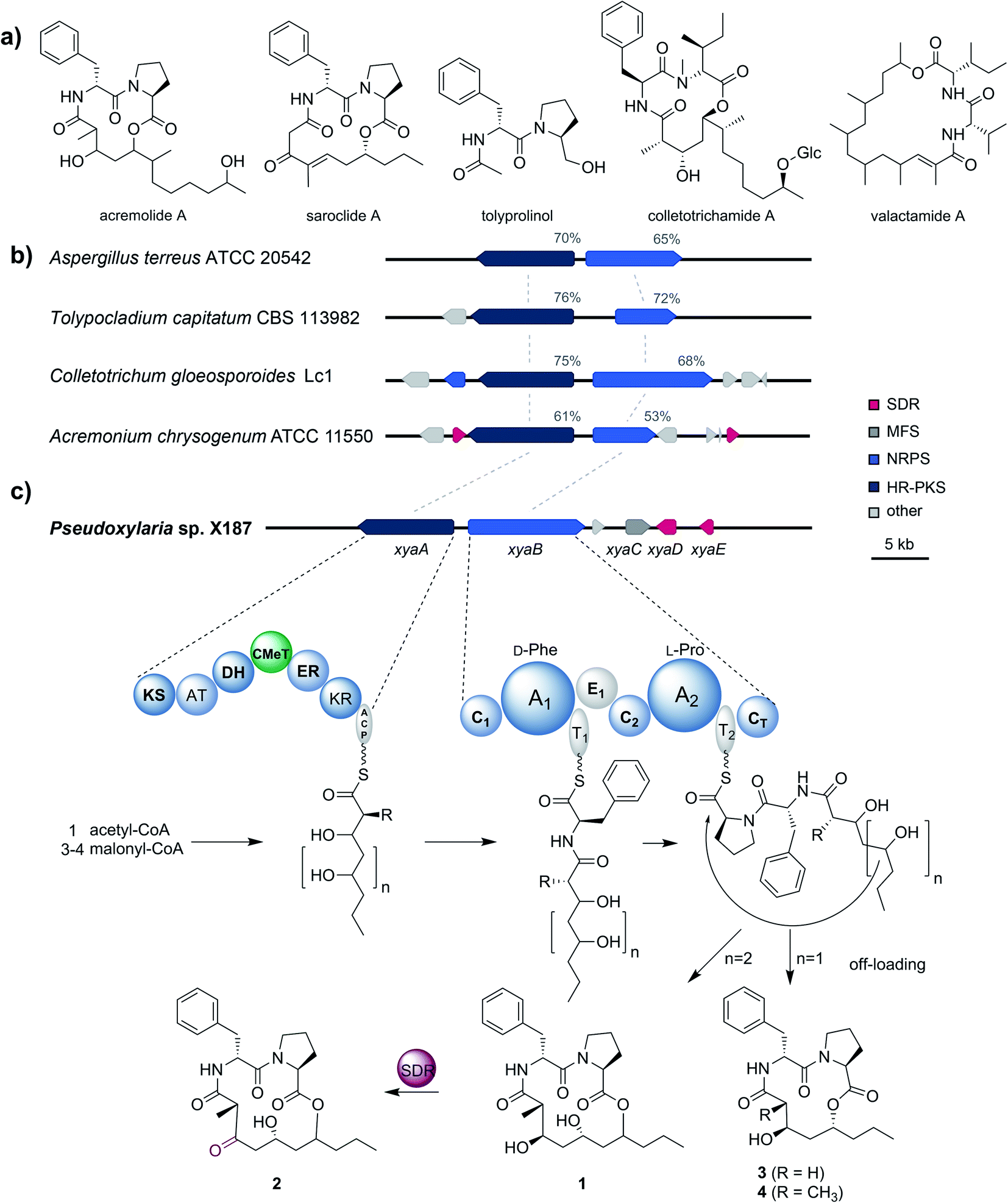 | ||
| Fig. 3 (a) Structures of acremolide (Acremonium sp.), saroclide (Sarocladium kiliense), tolyprolinol (Tolypocladium sp. FKI-7981), colletotrichamide (Colletotrichum gloeosporioides JS419), and valactamide (Aspergillus terreus). (b) Schematic representation of the xya BGC in Pseudoxylaria sp. X187 and comparison to homologues BGCs from Aspergillus terreus ATCC 20542, Tolypocladium capitatum CBS 113982, Colletotrichum gloeosporioides Lc1 and Acremonium chrysogenum ATCC 11550, which were identified using BLAST. Shown are genes coding for highly-reducing polyketide synthases (dark blue; HR-PKS), non-ribosomal peptide synthases (light blue; NRPS), short-chain dehydrogenases/reductases (red; SDR) and major facilitator superfamily transporters (dark grey; MFS). Non related genes are shown in light grey. Sequence similarities of the corresponding proteins of the homologues BGCs regarding XyaA/B are shown in percentages. (c) Proposed biosynthesis of the xylacremolides. | ||
The proposed biosynthetic assembly line for xylacremolides (Fig. 3c) is consistent with both the domain architectures of XyaA and XyaB and their molecular structure. Here, we propose that the KS domain of the iterative HRPKS XyaA incorporates an acetyl starter unit, followed by subsequent prolongation with either three or four malonyl-CoA derived ketide units. The first ketide is fully reduced to an alkane by the KR, DH and ER domains, whereas the following units are only partially reduced to the corresponding alcohols. Notably, the CMeT domain catalyzes the α-methylation of five membered polyketide chains, but can bypass this reaction in the four-membered products, which leads to the production of 3. The C1 domain of XyaB then catalyzes the amide bond formation of the tetra- and pentaketide chains with D-Phe, which was isomerized by the included E domain. Chain elongation is then catalyzed by the C2 domain incorporating L-Pro, followed by macrolactonization by the CT domain releasing the main products, which is typical for fungal NRPSes.16–18 Subsequent tailoring oxidation reaction of the secondary alcohol (1) to the keto functionality of 2 could be catalyzed by either XyaD or XyaE; however incomplete reduction by XyaA cannot be excluded at this stage. Here, it is again noteworthy that congeners 1 and 2 are produced in a 2:1 ratio almost independently from the tested cultivation conditions (Table S1†).
Bioactivities of xylacremolide A and B
Similar to their closest structural counterparts (acremolides), xylacremolides revealed so far no cytotoxic or antibacterial activities.11 We then questioned which other cellular and/or ecological role xylacremolides might impose. A literature survey revealed that structurally related fungal metabolites trapoxin and FR235222 were previously investigated and found to be potent inhibitors of histone deacetylase (HDACi),23,24 and are thus being investigated in cancer therapy, as well as in parasitic and inflammatory diseases. To test for HDACi activity, we selected the two most abundant xylacremolide derivatives, xylacremolides A and B, and found weak inhibitory activity of both compared to the positive controls valproic acid and trichostatin A (tested concentration range: 0.25–1.0 mM; Fig. S4†). We then tested both compounds for antifungal activity against Aspergillus nidulans RMS011 using a spore germination assay with carboxine and amphotericin B as positive controls (Fig. 4 and S5†).25 Here, a moderate inhibitory effect of both compounds on spore germination was observed when spores were incubated for less than 20 h, while longer incubation times caused a significant decline in the inhibitory activity. Thus, it can be concluded that xylacremolides delays spore germination to moderate extent, but once A. nidulans is growing, it either tolerates or even detoxifies xylacremolides.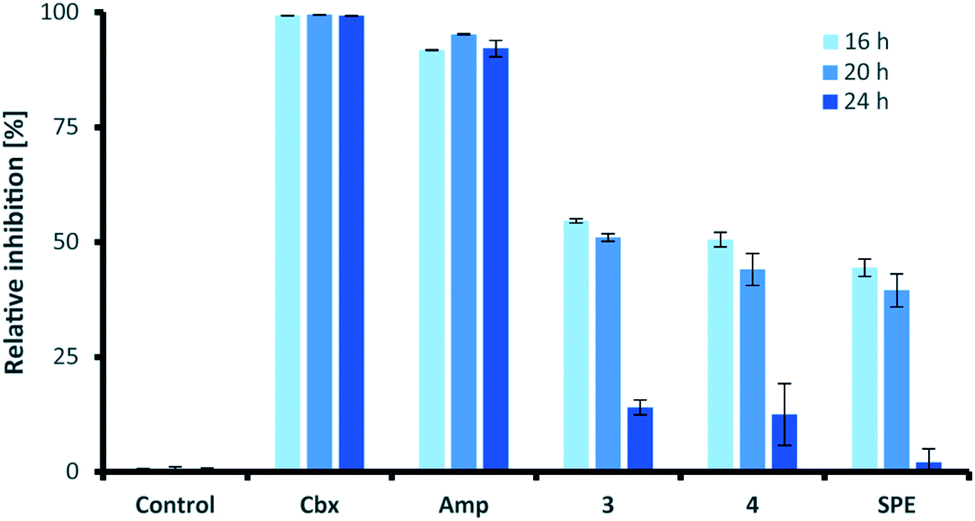 | ||
| Fig. 4 Antifungal activity assay of compound 3 and 4 and enriched SPE fraction of Pseudoxylaria sp. X187 crude extract (SPE) against Aspergillus nidulans RMS011. Carboxine (Cbx) and amphotericin B (Amp) served as positive controls. All substrates were tested at 1 μg mL−1. The percentage of 0% inhibition is considered normal growth; while 100% are considered complete growth inhibition. | ||
In light of the ecological origin of Pseudoxylaria sp. X187 (co-evolved antagonistic fungus of Termitomyces), it could be speculated at this point that the inhibitory activity of xylacremolides, while moderate, allows in combination with other metabolites an overall growth advantages and supports the successful outcompetion of the fungal cultivar when colony homeostasis is disrupted.
In line with our long-term mission to identify pharmacologically relevant protease inhibitors, we tested the inhibitory activity of the two most abundant derivatives, xylacremolides A and B, on various pharmacologically important proteases using an established fluorescence-based assay (Table 2).26–29 Both rhodesain (Rd), and cruzain, cysteine proteases belonging to the papain-family, play central roles in the life cycle of the parasitic protists Trypanosoma brucei (Human African Trypanosomiasis (HAT))30 and Trypanosoma cruzi (Chagas-disease),31 respectively. Three proteases were selected for their important functions in cancer cells (Cath L, Cath B, urokinase (uPA)).32 Sortase A (SrtA) was selected as promising antibacterial target due to its protease and transpeptidase activity related to the formation of the peptidoglycan layer of Staphylococcus aureus.33 For antiviral activity testing, we selected four proteases (ZIKV NS2B-NS3 protease,34 DENV2-NS2B-NS3,35 Mpro (also known as 3C like protease and which is the main protease of the novel corona virus SARS-CoV2),36 and PLpro).37 Initial inhibitor screening was performed at compound concentrations of 200 μM in DMSO, which revealed mild inhibition of cruzain, cathepsin L, DENV2 NS2B-NS3, SARS-CoV(2) Mpro and PLpro, SrtA and moderate inhibition of rhodesain and ZIKV NS2B-NS3. Despite overall moderate activity, it was interesting to observe that methylated xylacremolide B showed in most cases higher inhibitory activities compared to non-methylated xylacremolide A.
| Protease | Compound 3a/[%] | Compound 4a/[%] |
|---|---|---|
| a Inhibition at a compound concentration of 200 μM in DMSO as a percentage mean value ± standard deviation with n = 3. Inhibition rates below 0.5% are given as n.i. (no inhibition). | ||
| Cathepsin L | 7.3 ± 3.3 | 11 ± 5.0 |
| Cathepsin B | 4.9 ± 1.4 | n.i. |
| uPa | n.i. | n.i. |
| SrtA | 3.8 ± 6.4 | 2.5 ± 4.2 |
| Rhodesain | 5.7 ± 5.2 | 15 ± 4.6 |
| Cruzain | 3.6 ± 2.3 | 3.8 ± 5.4 |
| Mpro (SARS-CoV2) | n.i. | 11 ± 9 |
| PLpro (SARS-CoV) | n.i. | 11 ± 14 |
| DENV2 | n.i. | 4.8 ± 4.8 |
| ZIKV | 0.7 ± 1.2 | 17 ± 3.4 |
Experimental
Compound identification based on GNPS analysis
Pseudoxylaria sp. X187-2 was grown on 30 potato-dextrose (PD) agar plaes (0.8 L of PDA, 2% agar, standard plates 16 mm × 92 mm, 20 mL per plate) for four weeks at room temperature. Mycelium-covered agar plates were cut into small pieces (0.5 cm × 0.5 cm), extracted twice with 300 mL MeOH, and methanolic extracts dried under vacuum. Crude extracts were subjected to UPLC-ESI-HRMS measurements (Dionex Ultimate 3000 system combined with a Q-Exactive Plus mass spectrometer equipped with an electrospray ion (ESI) source (Thermo Scientific)). The resulting HRMS2 was subjected to MS2 fragmentation based network analysis carried out on the global natural products social molecular networking platform (GNPS). The data was filtered by removing all MS/MS peaks within ±17 Da of the precursor m/z. MS/MS spectra were window filtered by choosing only the top 6 peaks in the ±50 Da window throughout the spectrum. The data was then clustered with MS-Cluster with a parent mass tolerance of 0.02 Da and a MS/MS fragment ion tolerance of 0.02 Da to create consensus spectra. Further, consensus spectra that contained less than 2 spectra were discarded. A network was then created where edges were filtered to have a cosine score above 0.7 and more than 6 matched peaks. Further, edges between two nodes were kept in the network if each of the nodes appeared in each other's respective top 5 most similar nodes. Finally, the maximum size of a molecular family was set to 50, and the lowest scoring edges were removed from molecular families until the molecular family size was below this threshold.Analysis of production levels
Pseudoxylaria sp. X187-2 was cultivated on 30 PDA and 30 ISP2 agar plates (0.6 L, 2% agar, 16 × 92 mm plates, 20 mL per plate). Fungal plates were grown for up to six weeks at room temperature. Cultures were extracted with methanol and extracts of four biological replicates were combined and concentrated. The crude extract was re-dissolved in 20% MeOH (unless stated otherwise: ddH2O was used as second solvent) and loaded on an activated and equilibrated SPE C18ec column (1 g, Macherey-Nagel). The SPE cartridge was washed with two column volumes 20% MeOH and one column volume 50% MeOH. Metabolites were eluted in fractions by stepwise elution from 60% to 100% MeOH (two column volumes each fraction). The resulting 60% and 80% MeOH SPE fractions were combined dried under reduced pressure and re-dissolved in MeOH to yield 50 μg mL−1. Particles were removed by centrifugation for 15 min at 13.000 rpm and submitted for LC-MS based quantification. Compounds 1–4 were quantified based on integration of corresponding XICs (AUC, area under the curve, Δ5 ppm range) using the automatic peak detection feature in Xcalibur™. The AUC of xylacremolide A (3) was set to 100% and the abundance of the other derivatives was normalized based on their AUC relative to 3 in the same sample.Large-scale cultivation
Pseudoxylaria sp. X187-2 was cultivated on 30 PDA 16 × 92 mm, 20 mL per plate, 2% agar) and plates incubated for four weeks at room temperature. Mycelium-covered agar plates were cut into small pieces and extracted twice with 600 mL MeOH. Extracts were filtered through filter paper, concentrated under reduced pressure, and enriched using C18 SPE cartridges as described before. Xylacremolides A–D eluted predominantly as a mixture within the 60% MeOH fraction with some residual elution in the 80% MeOH fraction from the SPE column. Fractions were concentrated under reduced pressure to yield a white-brown solid material which was subjected to further purification by semi-preparative HPLC until analytical compounds were obtained.Structure elucidation
Buffers and substrates. Rhodesain (50 mM Na-acetate pH 5.5, 5 mM EDTA, 200 mM NaCl, 5 mM DTT, 10 μM Z-Phe-Arg-AMC); cruzain (50 mM Na-acetate pH 5.5, 5 mM EDTA, 200 mM NaCl, 5 mM DTT, 5 μM Z-Phe-Arg-AMC); cathepsin L (50 mM Tris pH 6.5, 5 mM EDTA, 200 mM NaCl, 2 mM DTT, 6.25 μM Z-Phe-Arg-AMC); cathepsin B (50 mM Tris pH 6.5, 5 mM EDTA, 200 mM NaCl, 2 mM DTT, 100 μM Z-Phe-Arg-AMC); SARS-CoV2 Mpro (20 mM Tris pH 7.5, 0.1 mM EDTA, 200 mM NaCl, 1 mM DTT, 50 μM Dabcyl-KTSAVLQS GFRKME-Edans); SARS-CoV PLpro (20 mM Tris pH 7.5, 0.1 mM EDTA, 200 mM NaCl, 1 mM DTT, 50 μM Z-Arg-Leu-Arg-Gly-Gly-AMC); DENV2 and ZIKV NS2B-NS3 (50 mM Tris pH 9.0, 20% glycerol, 1 mM Chaps, 100 μM Boc-Gly-Arg-Arg-AMC); urokinase (50 mM Tris pH 7.4, 50 mM NaCl, 0.5 mM EDTA, 240 μM Z-Gly-Gly-Arg-AMC), sortase A (5 mM Tris pH 7.5, 15 mM NaCl, 0.5 mM CaCl2, 0.5 mM Gly4, 25 μM Abz-LPETG-Dap(dnp)-OH).
Conclusions
Reinvestigation and intensified HRMS2-GNPS-based metabolomic analysis of Pseudoxylaria sp. X187 resulted in the identification of two additional lipopeptidic congeners of xylacremolides, which we named xylacremolide C and D. While xylacremolide A and B are built from three malonyl-CoA derived ketide units, the newly identified congeners are built from four malonyl-CoA derived ketide units, which indicates towards a promiscuous PKS. Evaluation of possible biological activities revealed moderate to weak antifungal and HDACi activity, as well as moderate protease inhibition activities across a panel of nine human, viral and bacterial proteases. Although their single bioactivities alone are likely not responsible for the antagonistic behaviour of Pseudoxylaria sp. X187, the combined actions might allow for a considerable growth advantage.Author contributions
FS: conceptualization, data curation, formal analysis, methodology, writing original draft, visualization. JF: formal analysis, data curation, methodology, writing original draft, visualization. SU: formal analysis, methodology. BHC: formal analysis, methodology. NJ: formal analysis, methodology. HM: formal analysis, methodology. TH: supervision, project administration, funding acquisition. TS: writing – review & editing, supervision, project administration, funding acquisition. MP: data curation, writing – review & editing, funding acquisition. CB: conceptualization, writing original draft, writing – review & editing, supervision, project administration, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy – EXC 2051 with Project-ID 390713860 and CRC 1127 with Project-ID 239748522 to CB and TH. Funded by the European Research Council (ERC-CoG – 771349) and The Danish Council for Independent Research (DFF – 7014-00178) to MP. We thank the Humboldt Foundation for a postdoctoral fellowship to SU, and Mrs Heike Heinecke (Hans Knöll Institute) for recording NMR spectra.Notes and references
- F. Song, S.-H. Wu, Y.-Z. Zhai, Q.-C. Xuan and T. Wang, Chem. Biodiversity, 2014, 11, 673–694 CrossRef CAS PubMed.
- S. E. Helaly, B. Thongbai and M. Stadler, Nat. Prod. Rep., 2018, 35, 992–1014 RSC.
- M. Poulsen, H. Hu, C. Li, Z. Chen, L. Xu, S. Otani, S. Nygaard, T. Nobre, S. Klaubauf, P. M. Schindler, F. Hauser, H. Pan, Z. Yang, A. S. M. Sonnenberg, Z. Wilhelm De Beer, Y. Zhang, M. J. Wingfield, C. J. P. Grimmelikhuijzen, R. P. De Vries, J. Korb, D. K. Aanen, J. Wang, J. J. Boomsma and G. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 14500–14505 CrossRef CAS PubMed.
- S. Otani, V. L. Challinor, N. B. Kreuzenbeck, S. Kildgaard, S. Krath Christensen, L. L. M. Larsen, D. K. Aanen, S. A. Rasmussen, C. Beemelmanns and M. Poulsen, Sci. Rep., 2019, 9, 1–10 CAS.
- A. A. Visser, V. I. D. Ros, Z. W. De Beer, A. J. M. Debets, E. Hartog, T. W. Kuyper, T. Læssøe, B. Slippers and D. K. Aanen, Mol. Ecol., 2009, 18, 553–567 CrossRef CAS PubMed.
- A. A. Visser, P. W. Kooij, A. J. M. Debets, T. W. Kuyper and D. K. Aanen, Fungal Ecol., 2011, 4, 322–332 CrossRef.
- H. Guo, N. B. Kreuzenbeck, S. Otani, M. Garcia-Altares, H. M. Dahse, C. Weigel, D. K. Aanen, C. Hertweck, M. Poulsen and C. Beemelmanns, Org. Lett., 2016, 18, 3338–3341 CrossRef CAS PubMed.
- H. Guo, A. Schmidt, P. Stephan, L. Raguž, D. Braga, M. Kaiser, H. M. Dahse, C. Weigel, G. Lackner and C. Beemelmanns, ChemBioChem, 2018, 19, 2307–2311 CrossRef CAS PubMed.
- M. Gareis and C. Gottschalk, Mycotoxin Res., 2014, 30, 151–159 CrossRef CAS PubMed.
- M. Wang, J. J. Carver, V. V. Phelan, L. M. Sanchez, N. Garg, Y. Peng, D. D. Nguyen, J. Watrous, C. A. Kapono, T. Luzzatto-Knaan, C. Porto, A. Bouslimani, A. V. Melnik, M. J. Meehan, W. T. Liu, M. Crüsemann, P. D. Boudreau, E. Esquenazi, M. Sandoval-Calderón, R. D. Kersten, L. A. Pace, R. A. Quinn, K. R. Duncan, C. C. Hsu, D. J. Floros, R. G. Gavilan, K. Kleigrewe, T. Northen, R. J. Dutton, D. Parrot, E. E. Carlson, B. Aigle, C. F. Michelsen, L. Jelsbak, C. Sohlenkamp, P. Pevzner, A. Edlund, J. McLean, J. Piel, B. T. Murphy, L. Gerwick, C. C. Liaw, Y. L. Yang, H. U. Humpf, M. Maansson, R. A. Keyzers, A. C. Sims, A. R. Johnson, A. M. Sidebottom, B. E. Sedio, A. Klitgaard, C. B. Larson, C. A. P. Boya, D. Torres-Mendoza, D. J. Gonzalez, D. B. Silva, L. M. Marques, D. P. Demarque, E. Pociute, E. C. O'Neill, E. Briand, E. J. N. Helfrich, E. A. Granatosky, E. Glukhov, F. Ryffel, H. Houson, H. Mohimani, J. J. Kharbush, Y. Zeng, J. A. Vorholt, K. L. Kurita, P. Charusanti, K. L. McPhail, K. F. Nielsen, L. Vuong, M. Elfeki, M. F. Traxler, N. Engene, N. Koyama, O. B. Vining, R. Baric, R. R. Silva, S. J. Mascuch, S. Tomasi, S. Jenkins, V. Macherla, T. Hoffman, V. Agarwal, P. G. Williams, J. Dai, R. Neupane, J. Gurr, A. M. C. Rodríguez, A. Lamsa, C. Zhang, K. Dorrestein, B. M. Duggan, J. Almaliti, P. M. Allard, P. Phapale, L. F. Nothias, T. Alexandrov, M. Litaudon, J. L. Wolfender, J. E. Kyle, T. O. Metz, T. Peryea, D. T. Nguyen, D. VanLeer, P. Shinn, A. Jadhav, R. Müller, K. M. Waters, W. Shi, X. Liu, L. Zhang, R. Knight, P. R. Jensen, B. Palsson, K. Pogliano, R. G. Linington, M. Gutiérrez, N. P. Lopes, W. H. Gerwick, B. S. Moore, P. C. Dorrestein and N. Bandeira, Nat. Biotechnol., 2016, 34, 828–837 CrossRef CAS PubMed.
- F. Schalk, S. Um, H. Guo, N. B. Kreuzenbeck, H. Görls, Z. W. de Beer and C. Beemelmanns, ChemBioChem, 2020, 21, 2991–2996 CrossRef CAS PubMed.
- R. Ratnayake, L. J. Fremlin, E. Lacey, J. H. Gill and R. J. Capon, J. Nat. Prod., 2008, 71, 403–408 CrossRef CAS PubMed.
- H. Mori, F. Abe, S. Furukawa, S. Furukawa, F. Sakai, M. Hino and T. Fujii, J. Antibiot., 2003, 56, 80–86 CrossRef CAS.
- W. Guo, S. Wang, N. Li, F. Li, T. Zhu, Q. Gu, P. Guo and D. Li, J. Nat. Prod., 2018, 81, 1050–1054 CrossRef CAS PubMed.
- A. Vassaux, L. Meunier, M. Vandenbol, D. Baurain, P. Fickers, P. Jacques and V. Leclère, Biotechnol. Adv., 2019, 37, 107449 CrossRef CAS PubMed.
- A. S. Brown, M. J. Calcott, J. G. Owen and D. F. Ackerley, Nat. Prod. Rep., 2018, 35, 1210–1228 RSC.
- D. Yu, F. Xu, S. Zhang and J. Zhan, Nat. Commun., 2017, 8, 1–11 CrossRef PubMed.
- K. Blin, S. Shaw, K. Steinke, R. Villebro, N. Ziemert, S. Y. Lee, M. H. Medema and T. Weber, Nucleic Acids Res., 2019, 47, W81–W87 CrossRef CAS PubMed.
- M. Röttig, M. H. Medema, K. Blin, T. Weber, C. Rausch and O. Kohlbacher, Nucleic Acids Res., 2011, 39, 362–367 CrossRef PubMed.
- K. D. Clevenger, J. W. Bok, R. Ye, G. P. Miley, M. H. Verdan, T. Velk, C. Chen, K. Yang, M. T. Robey, P. Gao, M. Lamprecht, P. M. Thomas, M. N. Islam, J. M. Palmer, C. C. Wu, N. P. Keller and N. L. Kelleher, Nat. Chem. Biol., 2017, 13, 895–901 CrossRef CAS PubMed.
- W. Fukasawa, N. Mori, M. Iwatsuki, R. Hokari, A. Ishiyama, M. Nakajima, T. Ouchi, K. Nonaka, H. Kojima, H. Matsuo, Ō. Satoshi and K. Shiomi, J. Antibiot., 2018, 2–4 Search PubMed.
- S. Bang, C. Lee, S. Kim, J. H. Song, K. S. Kang, S. T. Deyrup, S. J. Nam, X. Xia and S. H. Shim, J. Org. Chem., 2019, 84, 10999–11006 CrossRef CAS PubMed.
- R. Randino, I. Moronese, E. Cini, V. Bizzarro, M. Persico, M. Grimaldi, M. Scrima, A. M. D'Ursi, E. Novellino, E. Sobarzo-Sanchez, L. Rastrelli, C. Fattorusso, A. Petrella, M. Rodriquez and M. Taddei, Curr. Top. Med. Chem., 2017, 17, 441–459 CAS.
- N. J. Porter and D. W. Christianson, ACS Chem. Biol., 2017, 12, 2281–2286 CrossRef CAS PubMed.
- M. C. Monteiro, M. De La Cruz, J. Cantizani, C. Moreno, J. R. Tormo, E. Mellado, J. R. De Lucas, F. Asensio, V. Valiante, A. A. Brakhage, J. P. Latgé, O. Genilloud and F. Vicente, J. Biomol. Screening, 2012, 17, 542–549 CrossRef CAS PubMed.
- P. Klein, F. Barthels, P. Johe, A. Wagner, S. Tenzer, U. Distler, T. A. Le, P. Schmid, V. Engel, B. Engels, U. A. Hellmich, T. Opatz and T. Schirmeister, Molecules, 2020, 25, 2064 CrossRef CAS PubMed.
- F. Barthels, G. Marincola, T. Marciniak, M. Konhäuser, S. Hammerschmidt, J. Bierlmeier, U. Distler, P. R. Wich, S. Tenzer, D. Schwarzer, W. Ziebuhr and T. Schirmeister, ChemMedChem, 2020, 15, 839–850 CrossRef CAS PubMed.
- A. Welker, C. Kersten, C. Müller, R. Madhugiri, C. Zimmer, P. Müller, R. Zimmermann, S. Hammerschmidt, H. Maus, J. Ziebuhr, C. Sotriffer and T. Schirmeister, ChemMedChem, 2021, 16, 340–354 CrossRef CAS PubMed.
- B. Millies, F. von Hammerstein, A. Gellert, S. Hammerschmidt, F. Barthels, U. Göppel, M. Immerheiser, F. Elgner, N. Jung, M. Basic, C. Kersten, W. Kiefer, J. Bodem, E. Hildt, M. Windbergs, U. A. Hellmich and T. Schirmeister, J. Med. Chem., 2019, 62(24), 11359–11382 CrossRef CAS PubMed.
- D. Cullen and M. Mocerino, Curr. Med. Chem., 2017, 24, 701–717 CrossRef CAS PubMed.
- P. S. Doyle, Y. M. Zhou, I. Hsieh, D. C. Greenbaum, J. H. McKerrow and J. C. Engel, PLoS Pathog., 2011, 7, 1–11 Search PubMed.
- M. J. Duffy, P. M. McGowan, N. Harbeck, C. Thomssen and M. Schmitt, Breast Cancer Res., 2014, 16, 1–10 CrossRef PubMed.
- S. K. Mazmanian, G. Liu, E. R. Jensen, E. Lenoy and O. Schneewind, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 5510–5515 CrossRef CAS PubMed.
- W. W. Phoo, Y. Li, Z. Zhang, M. Y. Lee, Y. R. Loh, Y. B. Tan, E. Y. Ng, J. Lescar, C. Kang and D. Luo, Nat. Commun., 2016, 7, 1–8 Search PubMed.
- X. Yao, S. Guo, W. Wu, J. Wang, S. Wu, S. He, Y. Wan, K. S. Nandakumar, X. Chen, N. Sun, Q. Zhu and S. Liu, J. Pharmacol. Sci., 2018, 138, 247–256 CrossRef CAS PubMed.
- Z. Jin, X. Du, Y. Xu, Y. Deng, M. Liu, Y. Zhao, B. Zhang, X. Li, L. Zhang, C. Peng, Y. Duan, J. Yu, L. Wang, K. Yang, F. Liu, R. Jiang, X. Yang, T. You, X. Liu, X. Yang, F. Bai, H. Liu, X. Liu, L. W. Guddat, W. Xu, G. Xiao, C. Qin, Z. Shi, H. Jiang, Z. Rao and H. Yang, Nature, 2020, 582, 289–293 CrossRef CAS PubMed.
- M. Kandeel, Y. Kitade, M. Fayez, K. N. Venugopala and A. Ibrahim, J. Med. Virol., 2021, 93, 1581–1588 CrossRef CAS PubMed.
- S. W. Wingett and S. Andrews, F1000Research, 2018, 7, 1–13 Search PubMed.
- P. Ewels, M. Magnusson, S. Lundin and M. Käller, Bioinformatics, 2016, 32, 3047–3048 CrossRef CAS PubMed.
- G. Marçais and C. Kingsford, Bioinformatics, 2011, 27, 764–770 CrossRef PubMed.
- G. W. Vurture, F. J. Sedlazeck, M. Nattestad, C. J. Underwood, H. Fang, J. Gurtowski and M. C. Schatz, Bioinformatics, 2017, 33, 2202–2204 CrossRef CAS PubMed.
- A. Bankevich, S. Nurk, D. Antipov, A. A. Gurevich, M. Dvorkin, A. S. Kulikov, V. M. Lesin, S. I. Nikolenko, S. Pham, A. D. Prjibelski, A. V. Pyshkin, A. V. Sirotkin, N. Vyahhi, G. Tesler, M. A. Alekseyev and P. A. Pevzner, J. Comput. Biol., 2012, 19, 455–477 CrossRef CAS PubMed.
- M. Stanke, R. Steinkamp, S. Waack and B. Morgenstern, Nucleic Acids Res., 2004, 32, 309–312 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra00997d |
| ‡ Contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |