
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal free C-3 chalcogenation (sulfenylation and selenylation) of 4H-pyrido[1,2-a]pyrimidin-4-ones†
Prasanjit Ghosh ,
Gautam Chhetri and
Sajal Das*
,
Gautam Chhetri and
Sajal Das*
Department of Chemistry, University of North Bengal, Darjeeling-734013, India. E-mail: sajal.das@hotmail.com; Fax: +91-0353-2699-001; Tel: +91-0353-2776-381
First published on 9th March 2021
Abstract
An expeditious metal free C-3 chalcogenation of 4H-pyrido[1,2-a]pyrimidin-4-one has been devised to synthesize diversely orchestrated 3-ArS/ArSe derivatives in high yields (up to 95%). This operationally simple reaction proceeds under mild reaction conditions, can be executed in gram scale, and also highlights broad functional group tolerance. Preliminary experimental investigation suggests a radical mechanistic pathway for these transformations.
Organosulfur species and derivatives thereof, have garnered a prominent position in contemporary organic synthesis.1 They also have widespread applications in pharmaceuticals, bioactive compounds and polymer materials.2 In addition, carbon-selenium-carbon skeletons are high value core structures for their extensive use as therapeutically active agents, such as antioxidant, antihypertensive, antimicrobial, antibacterial, antiviral, anticancer agents etc.3 Moreover, organoselenium species have been identified as non-toxic compounds.4 Some of the biologically active C–S/C–Se linkage containing scaffolds is outlined in Fig. 1. Consequently, numerous efforts have been devoted to develop facile and reliable methods for the installation of a sulfenyl/selenyl group into organic frameworks.5 Apart from the cross-coupling approach, an alternative protocol for the transition-metal-catalyzed C–S bond formation via C–H bond functionalization has been established, which is known as a sulfenylation reaction.6 In this reaction, aryl sulfonyl hydrazides,7 arylsulfonyl chlorides,8 sulfinic acids,9 and sodium sulfinates10, thiols11 are mostly used as the sulfenylating agents. Although, these methods are advantageous, certain limitations comprising the use of metal catalyst and toxic reagents still persist. Complete removal of trace amounts of transition-metal residues from the anticipated bioactive products is quite challenging task, and this contamination of a transition metal also inhibits the sustainable development.12 Despite these accomplishments, development of an efficient and practical method for transition-metal-free C–S/Se bond forming reactions using thiols/diselenide as the sulfenylation/selenation reagent is an attractive and synthetically desirable.
 | ||
| Fig. 1 Representative examples of some biologically active 4H-pyrido[1,2-a]pyrimidin-4-one and diarylsulfide/diselenide scaffold. | ||
On the other hand, N-fused bicyclic heterocycles13 has received enormous interest from synthetic chemists as well as medicinal researchers due to their profound impact in agrochemicals, pharmaceuticals and material sciences.14 In this family, 4H-pyrido[1,2-a]pyrimidin-4-one (Fig. 1) exhibits versatile biological activities,15 such as CXCR3 antagonism,16 HLE inhibition,17 MexAB-OprM specific efflux pump inhibition,18 potent 5-HT6 antagonists,19 and acetylcholinesterase inhibition.20 Meanwhile, Pd catalyzed direct arylation and alkenylation of 4H-pyrido[1,2-a]pyrimidin-4-one through C–H bond functionalization has already been reported in the literature.21 Rather, only a single report for the insertion of –SAr group in 4H-pyrido[1,2-a]pyrimidin-4-one molecule using sulfonyl hydrazides as thiol surrogates is documented by Wang et al.22 Nevertheless, this protocol is effective at elevated temperature. Based on our research interests on the structural diversification of heterocyclic scaffolds, we recently reported different methodologies for the metal free direct C–H bond functionalization.23 Herein, we envisaged to disclose a straightforward and efficient protocol of sulfenylation/selenylation for 4H-pyrido[1,2-a]pyrimidin-4-one in the presence of iodine under mild conditions. Pleasingly, several thiols/organodiselenides are smoothly coupled with 4H-pyrido[1,2-a]pyrimidin-4-one and furnished the desired anticipated products in good to excellent yields (Scheme 1).
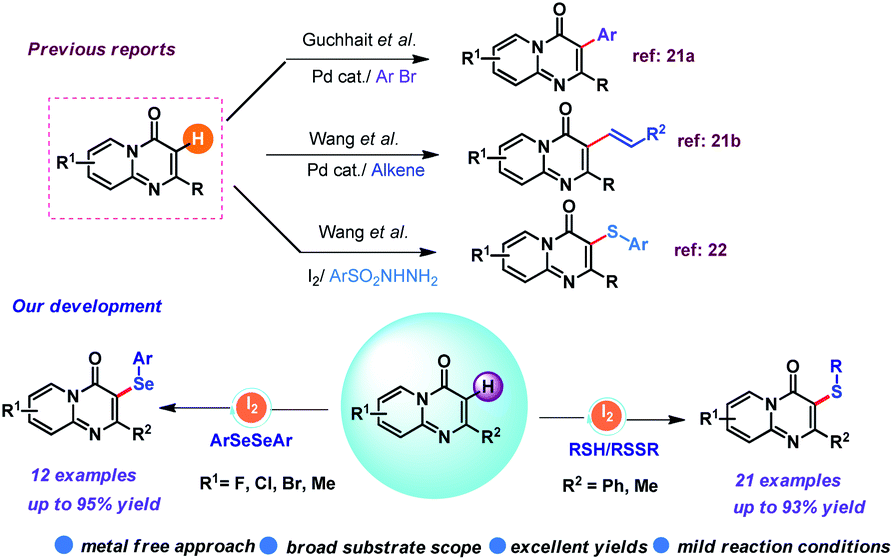 | ||
| Scheme 1 Previous approaches and the present route of C–H bond functionalization of 4H-pyrido[1,2-a]pyrimidin-4-ones. | ||
We commenced our studies with the optimization of the sulfenylation reaction where 2-phenyl-4H-pyrido[1,2-a]pyrimidin-4-one (1a) and thiophenol were used as a model coupling partner (Table 1). Initial attempts to couple them, using NaI and TBHP (as oxidant) in DMSO as a solvent at 100 °C remained unfruitful (Table 1, entry 1). No improvement was observed even after switching the solvent from DMSO to acetonitrile (Table 1; entry 2). However, yield of the expected product 2a was raised to 50% upon using TBAI/K2S2O8 in water (Table 1; entry 3). The reaction efficiency was further enhanced using one equiv. of TBAI in MeCN at 70 °C (Table 1; entry 4). Subsequently, several others inexpensive and readily available iodine/iodide additives were screened under aerobic condition (Table 1; entries 5–9). Delightedly, one equiv. of iodine provided the desired thiolated product 2a in excellent (91%) yield at 70 °C (entry 5), after 12 h, while other forms of iodine additives were unable to promote this transformation effectively (Table 1; entries 6–9). It is noteworthy that except TBHP all others oxidant appears to be redundant in this reaction (Table 1; entries 10–13). Lowering the reaction temperature (50 °C and 30 °C) had a detrimental result on the reaction outcome (Table 1; entries 14–15). Notably, in absence of potassium persulfate, no desired product was detected in TLC, indicating that the oxidant has a decisive role for this sulfenylation reaction (Table 1; entry 16). Interestingly, consistent with our previous observation (Table 1; entry 5), using of 50 mol% of iodine also afforded the 92% yield of thiolated derivative 2a (Table 1; entry 17). Other solvents were inefficient to provide decent yields (Table 1; entries 18–22). So it is evident from the optimization table that a combination of 50 mol% of iodine and K2S2O8 (2 equiv.) as an oxidant in MeCN at 70 °C was found to be optimal for the sulfenylation reaction of 2-phenyl-4H-pyrido[1,2-a]pyrimidin-4-one, which resulted in the formation of the corresponding 3-ArS derivative 2a in excellent yield (92%) after 12 h (Table 1, entry 17).
|
||||||
|---|---|---|---|---|---|---|
| Entry | Reagent (equiv.) | Oxidant (equiv.) | Temperature (°C) | Solvent (ml) | Time (h) | Yieldb (%) |
| a Reaction condition: 2-phenyl substituted 4H-pyrido[1,2-a]pyrimidin-4-ones (0.125 mmol, 1 equiv.), benzene thiol (0.1875 mmol, 1.5 equiv.), inducer (equiv./mol%), solvent (2 ml), oxidant (3 equiv.).b Isolated yields based on the reactants 1a, the reaction was run for 12–24 h. | ||||||
| 1 | NaI (3) | TBHP (3) | 100 | DMSO | 24 | NR |
| 2 | NaI (3) | TBHP (3) | 100 | CH3CN | 24 | NR |
| 3 | TBAI (2) | K2S2O8 (2) | 70 | H2O | 24 | 50 |
| 4 | TBAI (1) | K2S2O8 (2) | 70 | CH3CN | 12 | 67 |
| 5 | I2 (1) | K2S2O8 (2) | 70 | CH3CN | 12 | 91 |
| 6 | KI (1) | K2S2O8 (2) | 70 | CH3CN | 12 | NR |
| 7 | NaI (1) | K2S2O8 (2) | 70 | CH3CN | 12 | NR |
| 8 | NH4I (1) | K2S2O8 (2) | 70 | CH3CN | 12 | 60 |
| 9 | NIS (1) | K2S2O8 (2) | 70 | CH3CN | 12 | 63 |
| 10 | I2 (1) | TBHP (2) | 70 | CH3CN | 12 | 85 |
| 11 | I2 (1) | DTBP (2) | 70 | CH3CN | 12 | NR |
| 12 | I2 (1) | TBPB (2) | 70 | CH3CN | 12 | NR |
| 13 | I2 (1) | H2O2 (2) | 70 | CH3CN | 12 | 71 |
| 14 | I2 (1) | K2S2O8 (2) | 50 | CH3CN | 12 | 53 |
| 15 | I2 (1) | K2S2O8 (2) | 30 | CH3CN | 12 | NR |
| 16 | I2 (1) | — | 70 | CH3CN | 12 | NR |
| 17 | I2 (50 mol%) | K2S2O8 (2) | 70 | CH3CN | 12 | 92 |
| 18 | I2 (50 mol%) | K2S2O8 (2) | 70 | Toluene | 12 | 35 |
| 19 | I2 (50 mol%) | K2S2O8 (2) | 70 | Dioxane | 12 | 41 |
| 20 | I2 (50 mol%) | K2S2O8 (2) | 70 | DCE | 12 | 66 |
| 21 | I2 (50 mol%) | K2S2O8 (2) | 70 | EtOH | 12 | 81 |
| 22 | I2 (50 mol%) | K2S2O8 (2) | 70 | DMF | 12 | NR |

Having assimilated the robust reaction conditions for the C–S coupling of 4H-pyrido[1,2-a]pyrimidin-4-one, we sought to explore the scope and general applicability of this protocol (Table 2). A variety of 2-substituted-4H-pyrido[1,2-a]pyrimidin-4-one was treated with a broad range of thiols, and the corresponding results are represented in Table 2. Satisfyingly, both electron-rich (–OMe, –Me) and electron-deficient (–F, –Cl, –Br) groups bearing benzene thiols reacted smoothly with 2-phenyl-4H-pyrido[1,2-a]pyrimidin-4-one, affording the desired 3-sulfenylated derivatives in good to excellent yields (Scheme 1; entries 2b–2h). Noticeably, a crucial effect in the product yield was surveyed with the substituents present at the benzene thiol. 4-Methoxybenzene thiol furnished much lower yield of the corresponding coupled product (2b), compared to electron-withdrawing group, probably due to the generation of a more stable dimer [disulphide]. However, the yield of the anticipated product [2b] could further be enhanced upon/on using stoichiometric amount of catalyst (I2). To our delight, maximum productivity of the product was obtained in the case of F-substituted benzenethiol compare to the other halogens. Notably, ortho bromo-substituted benzene thiol delivered in a higher yield of the corresponding product (2h) than the corresponding chloro derivative (2c). 2,5-Dimethylbenzene thiol was endured under the current reaction conditions to provide 88% yield of 2e. Importantly, the bulkier naphthalene thiol also effectively participated in this transformation to give 82% yield of the C–S coupled product (2g). For adorning the synthetic potentiality further, we investigated the reactivity of various thiols with diverse 4H-pyrido[1,2-a]pyrimidin-4-one. Employment of both electron-neutral (–Me) and electron-deficient (–Cl) functional group substituted parent scaffold provided synthetically useful yields of the desired sulfenylated products with a wide spectrum of benzene thiols (entries 2i–2p). In this context, a suitable choice of benzene thiols is also important, since electronic bias plays a pivotal role in this transformation (entries 2i–2p). Comparatively, a higher yield of the desired ArS derivatives was always obtained in the presence of an electron-withdrawing group (–F, –Cl) at the para position of benzene thiol (entries 2k, 2m, 2n and 2p). Exposure of 2-alkyl substituted 4H-pyrido[1,2-a]pyrimidin-4-one with thiophenol and 4-chlorothiophenol was also fruitful to give intended products in acceptable yields (entries 2q and 2r). Unfortunately, benzyl thiol, 1-pentane thiol and heterocyclic congener of thiol (2-mercapto benzimidazole) did not respond under the optimal reaction conditions (entries 2s, 2t and 2u). Especially, upscale synthesis of 2a was also achieved, illuminating potential capabilities to assemble specialized 3-ArS substituted 4H-pyrido-[1,2-a]pyrimidin-4-ones. It was remarkable that PhSSPh was also amenable instead of PhSH with this catalytic system.
| a Reaction condition: substituted 4H-pyrido[1,2-a]pyrimidin-4-ones (0.125 mmol, 1 equiv.), thiol (0.1875 mmol, 1.5 equiv.), I2 (50 mol%), MeCN (2 ml), K2S2O8 (2 equiv.).b Isolated yields based on the reactants 1, the reaction was run for 12 h.c Yield at 1 g scale.d PhSSPh was used instead of PhSH.e 1 equiv. of I2 was used. |
|---|
 |
With the successful establishment of a straightforward and practical protocol for the C–S coupling reaction, we next investigated the broadness of the selenylation reaction between 2-substituted-4H-pyrido[1,2-a]pyrimidin-4-one and organodiselenides for the selective installation of the –SeAr group at the C-3 position of parent precursor (Table 3). Generally, diphenyl diselenide was smoothly coupled with diverse functionalized 2-substituted 4H-pyrido[1,2-a]pyrimidin-4-one, resulting in excellent yields of the final C–Se coupled products 3. Various organo diselenides bearing electron-donating (–OMe) and electron-withdrawing groups (–CF3 and –F) were found compatible, howbeit electron-deficient substrates showed better reactivity to render higher yields (entries 3b–3c, 3e). The methodology was successfully applied to bulkier naphthyl diselenide, delivering the desired product (3d) in 67% yield. Notably, halogen substituent (–F, –Br, –Cl) at the C-6 position of 4H-pyrido[1,2-a]pyrimidin-4-one were tolerable, which are useful synthetic handle for late stage functionalization (entries 3h–3j). Substrates having methyl functionality in the 4H-pyrido[1,2-a]pyrimidin-4-one was also amenable (3f–3g) and smoothly produced the anticipated products in high yields (81−93%). Furthermore, reaction efficiency of C-2 alkyl substituted parent molecule was also evaluated, providing an excellent yield (80%) of the targeted 3-SeAr product 3k. Besides to a broad range of aromatic diselenides, dibutyl diselenide also proved to be an efficient selenylating agent, offering the desired 3l in 71% isolated yield.

To comprehend the plausible reaction mechanism, we executed the sulfenylation reaction under inert atmosphere (N2) and isolated 76% yield of the desired product 2a (Scheme 2; eqn (1)). This observation revealed that aerial oxygen was not only the sole oxidant for this transformation. Additionally, the reaction of 2-phenyl-4H-pyrido[1,2-a]pyrimidin-4-one and iodine in presence potassium persulfate did not afford the corresponding iodo derivative (Scheme 2; eqn (2)). The result unambiguously confirmed that iodinated derivative of 4H-pyrido[1,2-a]pyrimidin-4-one was not involved in the catalytic cycle. Furthermore, the presence of stoichiometric amount of radical scavengers (TEMPO, BHT and 1,1-diphenyl ethylene) inhibited the reactivity, refuting the involvement of non-radical pathway in the reaction mechanism (Scheme 2; eqn (3) and (4)). In addition, we have trapped the in situ generated radical intermediate (PhSe˙) and isolated the compound 4 in reasonable yield (Scheme 2; eqn (4)).
 | ||
| Scheme 2 Mechanistic studies. | ||
On the basis of these findings and previous literature reports,24 a plausible mechanistic pathway is elaborated in Scheme 3. Presumably, this sulfenylation/selenylation strategy involve an initial generation of the thiyl radical or selenyl radical species A (˙SY/˙SeY, Y = R) in presence of persulfate (S2O82−) or sulfate radical anion (SO4˙−). Subsequently, the reactive sulphur/selenyl radical intermediates A coupled with 2-phenyl-4H-pyrido[1,2-a]pyrimidin-4-one substrate leading to a formation of next intermediate 1ab. Then, it underwent further oxidation by sulfate radical anion via a SET mechanism to generate a cationic intermediate 1ac which could be stabilized by resonance to 1ac′. Lastly, the final coupled product (2a/3a) was formed with the liberation of H2 species.
 | ||
| Scheme 3 Plausible mechanism (radical pathway). | ||
In summary, we have developed an efficient and straightforward transformative tool for regioselective chalcogenation of 4H-pyrido[1,2-a]pyrimidin-4-one under mild conditions. The protocol tolerated diverse common organic functional groups and resulted in good to excellent yields of the desired sulfenylated/selenylated products. Our methodology is operationally simple, regioselective, scalable and avoid the use any expensive metal catalyst. This present protocol opens a new avenue for the direct and convenient chalcogenation of 4H-pyrido[1,2-a]pyrimidin-4-one. Further, C–H bond functionalization reactions on 4H-pyrido[1,2-a]pyrimidin-4-one are currently underway in our laboratory and these observations will be forthcoming.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support (EMR/2016/001250) SERB, New Delhi is gratefully acknowledged. We also acknowledge DST-FIST for providing infrastructure facilities.Notes and references
- For selected examples: (a) C. B. Mishra, S. Kumari and M. Tiwari, Eur. J. Med. Chem., 2015, 92, 1 CrossRef CAS PubMed; (b) A. Ayati, S. Emami, A. Asadipour, A. Shafiee and A. Foroumadi, Eur. J. Med. Chem., 2015, 97, 699 CrossRef CAS PubMed; (c) P. Sun, D. Yang, W. Wei, L. Jiang, Y. Wang, T. Dai and H. Wang, Org. Chem. Front., 2017, 4, 1367 RSC; (d) M. Klečka, R. Pohl, J. Čejka and M. Hocek, Org. Biomol. Chem., 2013, 11, 5189 RSC.
- For selected reviews and examples: (a) M. E. Peach, in The Chemistry of the Thiol Group, ed. S. Patai, John Wiley & Sons, London, 1979, pp. 721–756 Search PubMed; (b) L. A. Damani, Sulfur-Containing Drugs and Related Organic Compounds: Chemistry, Biochemistry, and Toxicology, Part B: Metabolism of Sulfur Functional Groups, Ellis Horwood Ltd, Chichester, UK, 1989, vol. 1 Search PubMed; (c) R. J. Cremlyn, in An Introduction to Organosulfur Chemistry, Wiley & Sons, New York, 1996 Search PubMed; (d) M. D. McReynolds, J. M. Doughtery and P. R. Hanson, Chem. Rev., 2004, 104, 2239 CrossRef CAS PubMed; (e) J. Zhao and X. Jiang, Chin. Chem. Lett., 2018, 29, 1079 CrossRef CAS; (f) P. Chauhan, S. Mahajan and D. Enders, Chem. Rev., 2014, 114, 8807 CrossRef CAS PubMed.
- (a) D. A. Bellnier, D. N. Young, M. R. Detty, S. Camacho and A. R. Oseroff, Photochem. Photobiol., 1999, 70, 630 CrossRef CAS PubMed; (b) K. A. Leonard, J. P. Hall, M. I. Nelen, S. R. Davies, S. O. Gollnick, S. Camacho, A. R. Oseroff, S. L. Gibson, R. Hilf and M. R. Detty, J. Med. Chem., 2000, 43, 4488 CrossRef CAS PubMed.
- (a) D. Kundu, S. Ahammed and B. C. Ranu, Org. Lett., 2014, 16, 1814 CrossRef CAS PubMed; (b) S. Bhadra, A. Saha and B. C. Ranu, J. Org. Chem., 2010, 75, 4864 CrossRef CAS PubMed; (c) R. J. Cohen, D. L. Fox and R. N. Salvatore, J. Org. Chem., 2004, 69, 4265 CrossRef CAS PubMed.
- (a) G. Li, Z. Gan, K. Kong, X. Dou and D. Yang, Adv. Synth. Catal., 2019, 361, 1808 CrossRef CAS; (b) W. Wei, L. Wang, H. Yue, Y.-Y. Jiang and D. Yang, Org. Biomol. Chem., 2018, 16, 8379 RSC; (c) X. Wang, M. Yang, W. Xie, X. Fan and J. Wu, Chem. Commun., 2019, 55, 6010 RSC.
- (a) K. Inamoto, C. Hasegawa and K. Hiroya, Org. Lett., 2008, 10, 5147 CrossRef CAS PubMed; (b) L. L. Joyce and R. A. Batey, Org. Lett., 2009, 11, 2792 CrossRef CAS PubMed; (c) C. Shen, H. Xia, H. Yan, X. Chen, S. Ranjit, X. Xie, D. Tan, R. Lee, Y. Yang, B. Xing, K. Huang, P. Zhang and X. Liu, Chem. Sci., 2012, 3, 2388 RSC; (d) R. Xu, J. P. Wan, H. Mao and Y. Pan, J. Am. Chem. Soc., 2010, 132, 15531 CrossRef CAS PubMed; (e) J. Zhu, Z. Chen, H. Xie, S. Li and Y. Wu, Org. Lett., 2010, 12, 2434 CrossRef CAS PubMed; (f) C. Xu and Q. Shen, Org. Lett., 2014, 16, 2046 CrossRef CAS PubMed.
- (a) A. K. Bagdi, S. Mitra, M. Ghosh and A. Hajra, Org. Biomol. Chem., 2015, 13, 3314 RSC; (b) X. Zhao, T. Li, L. Zhang and K. Lu, Org. Biomol. Chem., 2016, 14, 1131 RSC.
- Q. Wu, D. Zhao, X. Qin, J. Lan and J. You, Chem. Commun., 2011, 47, 9188 RSC.
- C.-R. Liu and L.-H. Ding, Org. Biomol. Chem., 2015, 13, 2251 RSC.
- F. Xiao, S. Chen, J. Tian, H. Huang, Y. Liu and G.-J. Deng, Green Chem., 2016, 18, 1538 RSC.
- Y. Liao, P. Jiang, S. Chen, H. Qi and G.-J. Deng, Green Chem., 2013, 15, 3302 RSC.
- (a) D. Nair, J. Scarpello, L. White, L. M. Freitas dos Santos, I. Vankelecom and A. Livingston, Tetrahedron Lett., 2001, 42, 8219 CrossRef CAS; (b) The European Agency for the Evaluation of Medicinal Products; Committee for Proprietary Medicinal Products; London, 2002 Search PubMed; (c) J. Rivera-Utrilla, I. Bautista-Toledo, M. Ferro-Garcia and C. Moreno Castilla, Carbon, 2003, 41, 323 CrossRef CAS; (d) C. Garrett and K. Prasad, Adv. Synth. Catal., 2004, 346, 889 CrossRef CAS , For reviews on green chemistry, see: ; (e) P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, New York, 1998 Search PubMed; (f) C.-J. Li and B. M. Trost, Green chemistry for chemical synthesis, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 13197 CrossRef CAS PubMed; (g) P. J. Dunn, Chem. Soc. Rev., 2012, 41, 1452 RSC.
- (a) A. K Bagdi, S. Santra, K. Monira and A. Hajra, Chem. Commun., 2015, 51, 1555 RSC; (b) A. T. Baviskar, C. Madaan, R. Preet, P. Mohapatra, V. Jain, A. Agarwal, S. K. Guchhait, C. N. Kundu, U. C. Banerjee and P. V. Bharatam, J. Med. Chem., 2011, 54, 5013 CrossRef CAS PubMed; (c) I. V. Seregin, A. W. Schammel and V. Gevorgyan, Org. Lett., 2007, 9, 3433 CrossRef CAS PubMed; (d) D. Chernyak, S. B. Gadamsetty and V. Gevorgyan, Org. Lett., 2008, 10, 2307 CrossRef CAS PubMed; (e) O. R. Thiel, M. M. Achmatowicz, A. Reichelt and R. D. Larsen, Angew. Chem., Int. Ed., 2010, 49, 8395 CrossRef CAS PubMed.
- (a) M. E. Welsch, S. A. Snyder and B. R. Stockwell, Curr. Opin. Chem. Biol., 2010, 14, 347 CrossRef CAS PubMed; (b) L. D. Quin and J. A. Tyrell, The Importance of Heterocycles in Medicine: Fundamentals of Heterocyclic Chemistry: Importance in Nature and in the Synthesis of Pharmaceuticals, John Wiley & Sons, Inc, Hoboken, NJ, 2010, p. 196 Search PubMed.
- (a) K. Nakayama, H. Kawato, J. Watanabe, M. Ohtsuka, K. Yoshida, Y. Yokomizo, A. Sakamoto, N. Kuru, T. Ohta, K. Hoshino, K. Yoshida, H. Ishida, A. Cho, M. H. Palme, J. Z. Zhang, V. J. Lee and W. J. Watkins, Bioorg. Med. Chem. Lett., 2004, 14, 475 CrossRef CAS PubMed; (b) K. Nakayama, N. Kuru, M. Ohtsuka, Y. Yokomizo, A. Sakamoto, H. Kawato, K. Yoshida, T. Ohta, K. Hoshino, K. Akimoto, J. Itoh, H. Ishida, A. Cho, M. H. Palme, J. Z. Zhang, V. J. Lee and W. J. Watkins, Bioorg. Med. Chem. Lett., 2004, 14, 2493 CrossRef CAS PubMed; (c) G. Roma, N. Cinone, M. Di Braccio, G. Grossi, G. Leoncini, M. G. Signorello and A. Carotti, Bioorg. Med. Chem., 2000, 8, 751 CrossRef CAS PubMed.
- A.-R. Li, M. G. Johnson, J. Liu, X. Chen, X. Du, J. T. Mihalic, J. Deignan, D. J. Gustin, J. Duquette, Z. Fu, L. Zhu, A. P. Marcus, P. Bergeron, L. R. McGee, J. Danao, B. Lemon, T. Carabeo, T. Sullivan, J. Ma, L. Tang, G. Tonn, T. L. Collins and J. C. Medina, Bioorg. Med. Chem. Lett., 2008, 18, 688 CrossRef CAS PubMed.
- M. Varga, Z. Kapui, S. Bátori, L. T. Nagy, L. Vasvári-Debreczy, E. Mikus, K. Urbán-Szabó and P. Arányi, Eur. J. Med. Chem., 2003, 38, 421 CrossRef CAS PubMed.
- K. Nakayama, Y. Ishida, M. Ohtsuka, H. Kawato, K. Yoshida, Y. Yokomiza, T. Ohta, K. Hoshino, T. Otani, Y. Kurosaka, K. Yoshida, H. Ishida, V. J. Lee, T. E. Renau and W. J. Watkins, Bioorg. Med. Chem. Lett., 2003, 13, 4205 CrossRef CAS PubMed.
- S. Hu, Y. Huang, Y.-J. Wu, H. He, K. A. Grant-Young, R. L. Bertekap, V. Whiterock, P. Brassil, K. Lentz, P. Sivaprakasam, D. R. Langley, R. S. Westphal and P. M. Scola, Bioorg. Med. Chem., 2014, 22, 1782 CrossRef CAS PubMed.
- C. T. Sadashiva, J. N. N. S. Chandra, K. C. Ponnappa, T. V. Gowda and K. S. Rangappa, Bioorg. Med. Chem. Lett., 2006, 16, 3932 CrossRef CAS PubMed.
- (a) S. K. Guchhait and G. Priyadarshani, J. Org. Chem., 2015, 80, 8482 CrossRef CAS PubMed; (b) W. Liu, S. Wang, Q. Zhang, J. Yu, J. Li, Z. Xie and H. Cao, Chem.–Asian J., 2014, 9, 2436 CrossRef CAS PubMed.
- W. Liu, S. Wang, Z. Cai, Z. Li, J. Li and A. Wang, Synlett, 2018, 29, 116 CrossRef CAS.
- (a) P. Ghosh, A. K. Nandi, G. Chhetri and S. Das, J. Org. Chem., 2018, 83, 12411 CrossRef CAS PubMed; (b) P. Ghosh, G. Chhetri, A. K. Nandi, S. Sarkar, T. Saha and S. Das, New J. Chem., 2019, 43, 10959 RSC; (c) P. Ghosh, G. Chhetri, E. Perl and S. Das, Adv. Synth. Catal., 2021 DOI:10.1002/adsc.202001426.
- (a) K. Phakdeeyothin and S. Yotphan, Org. Biomol. Chem., 2019, 17, 6432 RSC; (b) B. Hu, P. Zhou, Q. Zhang, Y. Wang, S. Zhao, L. Lu, S. Yan and F. Yu, J. Org. Chem., 2018, 83, 14978 CrossRef CAS PubMed; (c) B. Hu, Q. Zhang, S. Zhao, Y. Wang, L. Xu, S. Yan and F. Yu, Adv. Synth. Catal., 2019, 361, 49 CrossRef CAS; (d) Y. Liu, Y. Hu, Z. Cao, X. Zhan, W. Luo, Q. Liu and C. Guo, Adv. Synth. Catal., 2019, 361, 1084 CrossRef CAS; (e) R. Semwal, C. Ravi, R. Kumar, R. Meena and S. Adimurthy, J. Org. Chem., 2019, 84, 792 CrossRef CAS PubMed; (f) Y. Zheng, Y. He, G. Rong, X. Zhang, Y. Weng, K. Dong, X. Xu and J. Mao, Org. Lett., 2015, 17, 5444 CrossRef CAS PubMed; (g) D. Yang, K. Yan, W. Wei, G. Li, S. Lu, C. Zhao, L. Tian and H. Wang, J. Org. Chem., 2015, 80, 11073 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra00834j |
| This journal is © The Royal Society of Chemistry 2021 |