
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew protein tyrosine phosphatase inhibitors from fungus Aspergillus gorakhpurensis F07ZB1707†
Yannan Ji a,
Qiqi Zhoua,
Guosheng Liua,
Tianhui Zhua,
Yufang Wanga,
Yan Fua,
Yeying Lib,
Ruolan Lib,
Xuexia Zhangb,
Mei Dongc,
Françoise Sauriold,
Yucheng Gue,
Qingwen Shi*ac,
Xinhua Lu*b and
Zhiyu Ni*f
a,
Qiqi Zhoua,
Guosheng Liua,
Tianhui Zhua,
Yufang Wanga,
Yan Fua,
Yeying Lib,
Ruolan Lib,
Xuexia Zhangb,
Mei Dongc,
Françoise Sauriold,
Yucheng Gue,
Qingwen Shi*ac,
Xinhua Lu*b and
Zhiyu Ni*f
aSchool of Pharmaceutical Sciences, Hebei Medical University, Shijiazhuang 050017, China. E-mail: shiqingwen405@163.com
bNew Drug Research & Development Center of North China Pharmaceutical Group Corporation, National Microbial Medicine Engineering & Research Center, Hebei Industry Microbial Metabolic Engineering & Technology Research Center, Key Laboratory for New Drug Screening Technology of Shijiazhuang City, Shijiazhuang 050015, China. E-mail: luxinhua89@yeah.net
cHebei Key Laboratory of Forensic Medicine, Collaborative Innovation Center of Forensic Medical Molecular Identification, Hebei Medical University, Shijiazhuang 050017, China
dDepartment of Chemistry, Queen's University, Kingston, K7L 3N6, Canada
eSyngenta Jealott's Hill International Research Centre, Bracknell, Berkshire RG42 6EY, UK
fThe Affiliated Hospital of Hebei University, School of Basic Medical Science, Hebei University, Baoding 071000, China. E-mail: nizhiyu@hbu.edu.cn
First published on 10th March 2021
Abstract
Twelve new compounds, aspergorakhins A–L (1–12) coupled with one known xanthone leptosphaerin D (13), were isolated from the extract of soil-derived fungus Aspergillus gorakhpurensis F07ZB1707. Their structures were elucidated by spectroscopic data analysis including UV, IR, NMR, and HRESIMS. The absolute configurations of 5 and 8–11 were identified using ECD and OR calculations. All compounds were tested by enzyme inhibitory activity assay in vitro. Aspergorakhin A (1) showed selective activities against PTP1B and SHP1 over TCPTP with IC50 values 0.57, 1.19, and 22.97 μM, respectively. Compounds 1 and 2 exhibited modest cytotoxicity against tumor cell lines A549, HeLa, Bel-7402, and SMMC-7721 with IC50 values in the range of 6.75–83.4 μM.
Introduction
Protein tyrosine phosphatases (PTPs) are signaling enzymes that regulate tyrosine phosphorylation. They have shown an important role in cellular processes including proliferation, metabolism, motility, and survival. The disorder of PTPs could induce human diseases such as tumor, diabetes, autoimmune diabetes, and infectious diseases. A number of PTPs such as PTP1B and SHP1 have been explored as targets for drug discovery.1–3 However the highly conserved active site of PTPs' family members increases the difficulty of achieving selective inhibitors and drug candidates.4Natural products have play an important role in the history of drug discovery. Over 50% of all current clinically approved drugs are derived from natural products.5 Since Alexander Fleming discovered penicillin in 1928, fungi have become a considerable resource for novel compounds and new drugs.6,7 In our continuous search for active compounds from fungi, twelve new compounds were obtained from the extract of soil-derived fungus Aspergillus gorakhpurensis F07ZB1707, which was fermented by a salt-containing medium. These compounds included one 5α,8α-epidioxysterol aspergorakhin A (1), three nortriterpenoids aspergorakhins B–D (2–4) with a rare carboxyl or carbonyl groups in C-19, eight aromatic polycyclic polyketides aspergorakhins E–J, L, and leptosphaerin D (5–10, 12, 13), and one cyclohexanone derivative aspergorakhin K (11) (Fig. 1). Compounds 1, 2, and 11 showed inhibitory effect against PTPs including PTP1B, SHP1, and TCPTP in vitro. Among them, compound 1 might exhibit selective activities against PTP1B and SHP1 over TCPTP with IC50 values 0.57, 1.19, and 22.97 μM, respectively. Their inhibitory effects against cathepsin B and cytotoxicity in the human tumor cell lines were also screened. Herein, we report the isolation, structural elucidation, and biological evaluation of these compounds.
 | ||
| Fig. 1 The chemical structures of 1–13. | ||
Results and discussion
Compound 1 was obtained as a white amorphous powder. The molecular formula was determined as C27H40O3 on the basis of the HRESIMS peak at m/z 413.3047 [M + H]+ (calcd for 413.3056), indicates at the eight degrees of unsaturation. The NMR data (Table 1) of 1 showed 27 carbon resonances including five methyl carbons, seven methylene carbons, five methine carbons, five protonated olefinic carbons, and five quaternary carbons with one sp2 quaternary carbon. The IR spectrum showed a strong absorption at νmax 3386 cm−1, implying the existence of a hydroxyl group. The five sp2 olefinic and one sp2 quaternary carbons account for three degrees of unsaturation; then, the remaining five degrees of unsaturation suggest that 1 had five rings. Furthermore, the characteristic signals of four singlet methyl groups at δH 0.83 (s, H-18), 0.88 (s, H-19), 1.73 (s, H-26), and 1.75 (s, H-27) and one doublet methyl at δH 1.03 (d, J = 6.7 Hz, H-21) (Table 1), indicated the steroid skeleton of 1.8,9 Two contiguous coupled olefinic protons at δH 6.24 (d, J = 8.5 Hz, H-6) and 6.50 (d, J = 8.5 Hz, H-7), and an oxygenated methine at δH 3.97 (m, H-3) were the characteristics of a 3β-hydroxy-6-en-5α,8α-epidioxysterol skeleton,10 which was also corresponding to the 13C NMR signals at δC 66.5 (CH, C-3), 82.1 (C, C-5), 135.4 (CH, C-6), 130.7 (CH, C-7), and 79.4 (C, C-8). All these data showed great similarity with the known compound yalongsterol A11 except for different side chains at C-17. The COSY correlations H-20/H-21, 22, H-23/H-22, 24, and the HMBC correlations H-26, 27/C-24, 25 disclosed the presence of the 6-substitued 2-methylhepta-2,4-diene side chain, which was linked with C-17. The configuration of the C-22/C-23 double bond was assigned as E on the basis of the coupling constant (J = 15.0 Hz) of olefinic protons H-22 and H-23.| No. | 1 b | 2 b | 3 c | 4 c | ||||
|---|---|---|---|---|---|---|---|---|
| δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | |
| a 700 MHz for 1H, 175 MHz for 13C. Tetramethylsilane (TMS) as an internal standard.b Recorded in CDCl3.c Recorded in CD3OD.d Extracted from HMBC. | ||||||||
| 1α | 34.7 | 1.96, m | 29.0 | 2.23, m | 34.8 | 2.69, m | 34.3 | 2.49, m |
| 1β | 1.69, m | 1.76, m | 1.20, m | 1.02, m | ||||
| 2α | 30.1 | 1.86, m | 23.0 | 2.12, m | 29.8 | 1.95, m | 29.8 | 1.96, m |
| 2β | 1.55, m | 2.00, m | 1.72, m | 1.67, m | ||||
| 3 | 66.5 | 3.97, m | 84.4 | 4.13, dd (3.7, 1.6) | 78.9 | 3.21, dd (12.0, 4.6) | 79.3 | 3.19, dd (12.0, 4.6) |
| 4α | 36.9 | 2.12, m | 36.9 | 40.3 | 40.4 | |||
| 4β | 1.91, dd (13.7, 11.5) | |||||||
| 5 | 82.1 | 47.8 | 1.61, m | 50.1 | 1.29, dd (11.9, 4.3) | 52.3 | 1.25, m | |
| 6α | 135.4 | 6.24, d (8.5) | 25.2 | 2.13, m | 24.6 | 2.15, m | 18.6 | 2.42, m |
| 6β | 1.87, m | 2.71, m | 1.71, m | |||||
| 7α | 130.7 | 6.50, d (8.5) | 119.0 | 5.48, d (7.2) | 123.7 | 5.64, d (6.6) | 27.3 | 2.19, m |
| 7β | 2.15, m | |||||||
| 8 | 79.4 | 143.3 | 142.3 | 140.9d | ||||
| 9 | 51.1 | 1.50, m | 131.5 | 139.2 | 128.2d | |||
| 10 | 37.0 | 46.9 | 48.0 | 49.5 | ||||
| 11α | 20.6 | 1.60, m | 124.8 | 5.58, t (4.5) | 122.7 | 5.60, d (6.2) | 23.3 | 2.18, m |
| 11β | 1.90, m | |||||||
| 12α | 39.3 | 1.95, m | 38.0 | 2.28, d (4.5) | 39.2 | 2.26, d (17.9) | 32.3 | 1.79, m |
| 12β | 1.24, m | 2.12, dd (17.9, 6.2) | 1.71, m | |||||
| 13 | 44.7 | 44.3 | 45.4 | 46.2 | ||||
| 14 | 51.6 | 1.56, m | 49.7 | 51.2 | 51.3 | |||
| 15α | 23.4 | 1.53, m | 31.3 | 1.73, m | 32.6 | 1.74, m | 32.0 | 1.74, m |
| 15β | 1.23, m | 1.35, m | 1.40, m | 1.27, m | ||||
| 16α | 28.6 | 1.78, m | 27.3 | 1.98, m | 28.3 | 2.01, m | 28.7 | 1.96, m |
| 16β | 1.36, m | 1.42, m | 1.40, m | 1.40, m | ||||
| 17 | 56.2 | 1.25, m | 47.1 | 1.70, m | 48.5 | 1.69, m | 48.2 | 1.58, m |
| 18 | 12.9 | 0.83, s | 15.7 | 0.77, s | 16.3 | 0.59, s | 16.2 | 0.74, s |
| 19 | 18.2 | 0.88, s | 175.7 | 179.1 | 179.9d | |||
| 20 | 39.7 | 2.13, m | 39.2 | 1.59, m | 40.5 | 1.51, m | 40.8 | 1.52, m |
| 21 | 20.5 | 1.03, d (6.7) | 16.5 | 1.03, d (6.5) | 17.2 | 1.01, d (6.5) | 17.4 | 1.00, d (6.5) |
| 22α | 137.7 | 5.38, dd (15.0, 8.7) | 68.1 | 3.67, dd (10.6, 2.9) | 68.0 | 3.58, dd (10.7, 3.0) | 68.1 | 3.57, dd (10.7, 3.0) |
| 22β | 3.40, dd (10.6, 7.0) | 3.27, dd (10.7, 7.1) | 3.25, dd (10.7, 7.1) | |||||
| 23 | 124.5 | 6.14, dd (15.0, 10.8) | 24.2 | 0.89, s | 25.4 | 0.91, s | 25.0 | 0.94, s |
| 24 | 125.1 | 5.75, br d (10.8) | 28.8 | 1.12, s | 27.2 | 1.00, s | 27.2 | 0.98, s |
| 25 | 133.1 | 23.2 | 1.07, s | 14.8 | 0.79, s | 14.7 | 0.77, s | |
| 26 | 18.2 | 1.73, s | ||||||
| 27 | 25.9 | 1.75, s | ||||||
The relative configuration of 1 was determined according to the NOESY correlations. The NOESY correlations of H-19/H-4β and H-2β imply that this methyl was axial and H-3 (3.97, m) was also axial from the J values. The strong NOESY correlation of H-18/H-7 indicated that the second ring was in the boat geometry with a double bond and CH3-18 was axial and in cis-configuration to CH3-19. Then, the peroxide bridge was in trans-orientation with respect to CH3-19. The NOESY correlation of H-17/H-14 together with no correlation between H-18 and H-14 indicated that H-18 was in trans-configuration with respect to H-14 and H-17. Given the NOESY correlation of H-18/H-20 and no NOESY correlation between H-18 and H-21, it suggested that H-18 and H-20 were β-oriented. A comparison of the 13C NMR data of C-20 (δC 39.7) with the literature value11 also supported the β-orientation of H-20 in 1. Furthermore, the β-orientation of H-20 was consistent with several 5α,8α-epidioxysterols in many studies from different origins, whose structures confirmed were by X-ray diffraction analysis. Thus, the structure of compound 1 was consequently identified to be (22E)-5α,8α-epidioxy-6,22,24-trien-3β-ol, named aspergorakhin A.
Compound 2 was obtained as a white amorphous powder. The molecular formula was established as C25H36O3 through the pseudo molecular ion [M + Na]+ peak at m/z 407.2556 (calcd for 407.2562), accounting for seven degrees of unsaturation. The presence of the hydroxy group was indicated by the obvious absorption at νmax 3390 cm−1 in the IR spectrum. The 1H NMR data exhibited five methyls (δH 0.77, 0.89, 1.03, 1.07, and 1.12), two olefinic protons (δH 5.48 and 5.58), one oxygenated methine (δH 4.13), and one oxygenated methylene (δH 3.67 and 3.40). The 13C NMR data revealed 25 carbons containing four olefinic carbons (δC 143.3, 131.5, 124.8, and 119.0) and two oxygenated carbons (δC 84.4 and 68.1). The NMR data of 2 was analogous to those of 23,24,25,26,27-pentanorlanost-7,9(11)-dien-3β,22-diol closely,12 except for the absence of one methyl group (δC 22.7, CH3-19) and the presence of one carbonyl group (δC 175.7), which was oxidized to carbonyl group and then formed an ester group (δC 175.7) with the 3β-hydroxyl group in 2. The key COSY correlations, as shown in Fig. 2, demonstrated clearly that 2 was a triterpenoid nucleus. The key HMBC correlations of H-23/C-8, H-11/C-9, C-10, and C-13, H-12/C-9 and C-11 implied that two double bonds were at C-7(8) and C-9(11). There were five methyls in the structure, of which two singlet methyls were germinal; they were correlated in the HMBC with C-3 (δC 84.4), a quaternary carbon (δC 36.9), and C-5 (δC 47.8). Therefore, the singlet methyls (CH3-24, CH3-25) were located at C-4 (δC 36.9). There were another two singlet protons of methyl at δH 0.89 and 0.77, which were both coupled to the same quaternary carbons (δC 44.3/49.7) (C-13/C-14). The protons of methyl at δH 0.89 were further coupled to a CH2 (C-15) and to an olefin carbon C-8 (δC 143.3); thus, the methyl should be attributed to CH3-23. The other protons of methyl at δH 0.77 was correlated to a CH2 (C-12) with the COSY correlated to the olefinic proton (H-11) at δH 5.58, while the CH carbon (C-17) was correlated in HMBC to a doublet protons of methyl at δH 0.77. Therefore, the protons of methyl (δH 0.77) were attributed to CH3-18. The remaining doublet protons of methyl (δH 1.03) was correlated to C-17, C-20, and C-22 in the HMBC spectrum, which should account for CH3-21.
 | ||
Fig. 2 Key COSY ( ), HMBC ( ), HMBC ( ), and NOESY( ), and NOESY( ) correlations of compounds 1–12. ) correlations of compounds 1–12. | ||
The relative configuration of 2 was determined by the key NOESY correlations of H-18/H-15β; H-23/H-15α, H-17, implying that CH3-18 was axial and in trans-configuration with respect to CH3-23 and H-17. Obviously, H-3 was equatorial and 3-OH was β-oriented, forming an up bridge with C-19 (δC 175.7). The key NOESY correlations of H-3/H-25 and H-5/H-25 indicated that H-3 and H-5 were cis-orientated with respect to each other. Finally, CH3-21 was assigned the β-orientation compared with the NMR data of cladosporide A, whose absolute configuration was confirmed by X-ray diffraction analysis.13 On the basis of the evidences mentioned above, 2 was consequently identified as aspergorakhin B.
Compound 3 was isolated as a white amorphous powder, whose molecular formula was C25H38O4, as determined by the HRESIMS peak at m/z 403.2850 [M + H]+ (calcd for 403.2848). The NMR data of 3 exhibited great similarity with compound 23,24,25,26,27-pentanorlanost-7,9(11)-dien-3β,22-diol except for the absence of a methyl group (δC 22.7, CH3-19), the presence of a carboxyl group (δC 179.1, C-19), the upfield shift of an olefinic carbon (δC 139.2 in 3, C-9), and the downfield shift of a quaternary carbon (δC 48.0 in 3, C-10),12 which indicated that the methyl group was oxidized to the carbonyl group. The relative configuration of 3 was assigned by the NOESY correlations of H-1α/H-3, 5, H-24/H-3, 5, H-18/H-15β, H-23/H-15α, 17, together with the NMR data comparing with those of the known compounds,12 implying that 3 had the same absolute configuration as that of 23,24,25,26,27-pentanorlanost-7,9(11)-dien-3β,22-diol. On the basis of the evidences mentioned above, the structure of 3 was consequently identified as aspergorakhin C.
Compound 4 was isolated as a white amorphous powder. Its molecular formula was confirmed to be C25H40O4 by the HRESIMS peak at m/z 405.3014 [M + H]+ (calcd for 405.3005). The NMR data of 4 was closely similar to that of 3, except for the absence of two olefinic signals (δH 5.64, δC 123.7) and (δH 5.60, δC 122.7) in 3, which indicated that there was only one couple of double bonds in 4. The key HMBC correlation of H-23/C-8 verified that the double bond was located between C-8 and C-9. The key NOESY correlations of H-3/H-5, 24, H-18/H-15β, H-23/H-15α, H-17 (Fig. 2) together with the same J values of H-3 (dd, J = 12.0, 4.6 Hz) and almost the same chemical shifts for C-20 (δC 40.5 in 3, δC 40.8 in 4) indicated that the absolute configuration of 4 was the same as that of 3. Consequently, 4 was named as aspergorakhin D.
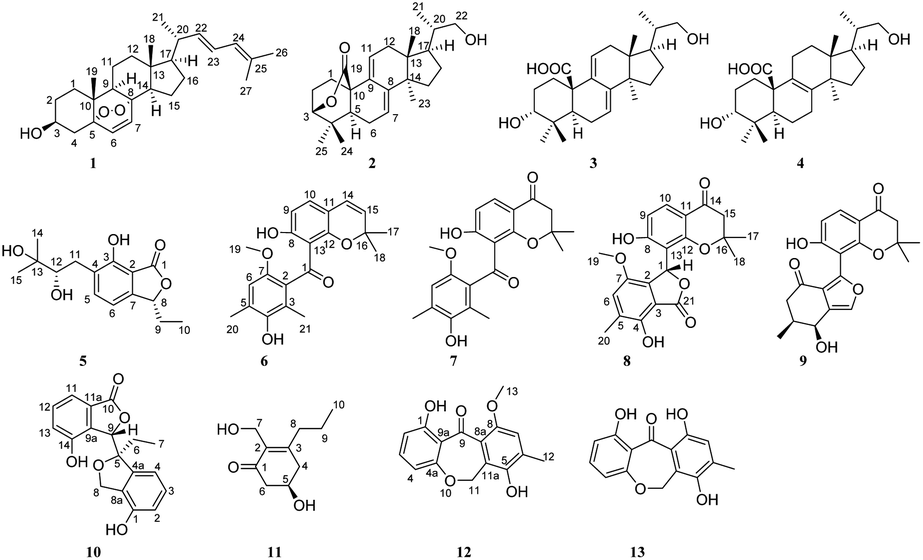
Compound 5 was isolated as a pale brown powder. The molecular formula was resolved as C15H20O5 from the [M + Na]+ peak at m/z 303.1207 (calcd for 303.1208). The strong absorption bands at 3408 and 1731 cm−1 in the IR spectrum revealed the presence of a hydroxyl group and a lactone carbonyl group. Comprehensive analysis of the NMR data (Table 2) suggested that 5 was a phthalide derivative, similar to isoochracein and (S)-3-ethyl-6-hydroxyphthalide.14 The NMR data of 5 showed two adjacent aromatic protons at δH 7.52 (d, J = 7.4 Hz, H-5) and δH 6.92 (d, J = 7.4 Hz, H-6), indicating a typical 1,2,3,4-tetra-substituted benzene moiety with a hydroxyl substituted at C-3, which was determined by the HMBC correlations of H-5/C-3 and H-11/C-3 (Fig. 2). The side chain and the lactone ring were assigned by the key HMBC data (Fig. 2). The absolute configuration of 5 was determined by the calculated ECD together with OR using the time-dependent density functional theory (TDDFT) (ESI S114† for details). Notably, the calculated ECD spectrum for (8R,12R)-5 and (8R,12S)-5 were both in accordance with the experimental curve of 5 (Fig. 3) and the quantum calculation of optical rotations (OR) for (8R,12R)-5 and (8R,12S)-5 were +22.4 and +118.6, respectively. The experimental OR of 5 was +107.7 (c 0.13, MeOH). Thus, the absolute configuration of 5 was identified to be (8R,12S).
| No. | 5 | 6 | 7 | 8 | 9 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | |
| a 600 MHz for 1H, 150 MHz for 13C, CD3OD. TMS as an internal standard. | ||||||||||
| 1 | 172.9 | 204.1 | 203.7 | 75.1 | 6.94, s | 150.6 | ||||
| 2 | 112.8 | 133.4 | 132.7 | 136.2 | 129.7 | |||||
| 3 | 156.3 | 150.4 | 150.2 | 148.0 | 120.9 | |||||
| 4 | 129.1 | 112.0 | 6.63, s | 112.0 | 6.66, s | 122.2 | 7.03, s | 65.3 | 4.88, d (3.2) | |
| 5 | 139.7 | 7.52, d (7.4) | 127.0 | 127.6 | 127.6 | 38.0 | 2.37, ddd (10.7, 3.9, 3.2) | |||
| 6 | 113.6 | 6.92, d (7.4) | 148.0 | 148.2 | 148.9 | 44.2 | 2.37, dd (16.9, 3.9) | |||
| 2.60, dd (16.9, 10.7) | ||||||||||
| 7 | 150.7 | 123.2 | 123.3 | 115.0 | 196.2 | |||||
| 8 | 83.9 | 5.45, dd (6.9, 4.2) | 164.7 | 171.4 | 165.0 | 165.6 | ||||
| 9 | 28.6 | 2.10, m | 109.9 | 6.42, d (8.4) | 112.3 | 6.62, d (8.9) | 110.5 | 6.55, d (8.8) | 110.5 | 6.55, d (8.8) |
| 1.78, m | ||||||||||
| 10 | 8.8 | 0.95, t (7.3) | 135.4 | 7.11, d (8.4) | 135.3 | 7.94, d (8.9) | 129.6 | 7.66, d (8.8) | 130.5 | 7.78, d (8.8) |
| 11 | 33.2 | 3.07, dd (14.2, 1.9) | 114.4 | 113.8 | 114.6 | 114.1 | ||||
| 2.64, dd (14.2, 10.5) | ||||||||||
| 12 | 79.8 | 3.60, dd (10.5, 1.9) | 156.9 | 164.2 | 162.6 | 162.3 | ||||
| 13 | 73.8 | 113.3 | 113.1 | 111.0 | 107.9 | |||||
| 14 | 25.0 | 1.25, s | 122.5 | 6.20, d (9.8) | 192.3 | 193.1 | 193.4 | |||
| 15 | 25.7 | 1.25, s | 128.0 | 5.38, d (9.8) | 48.4 | 2.55, s | 48.8 | 2.58, d (16.7) | 48.9 | 2.66, s |
| 2.38, d (16.7) | ||||||||||
| 16 | 78.6 | 81.9 | 80.9 | 81.1 | ||||||
| 17 | 27.5 | 0.93, s | 26.0 | 0.99, s | 24.9 | 0.77, s | 26.7 | 1.32, s | ||
| 18 | 27.5 | 0.93, s | 26.0 | 0.99, s | 27.2 | 1.14, s | 26.8 | 1.32, s | ||
| 19 | 56.5 | 3.64, s | 56.6 | 3.63, s | 57.0 | 3.60, s | 140.7 | 7.66, s | ||
| 20 | 17.1 | 2.28, s | 17.2 | 2.29, s | 15.4 | 2.30, s | 16.1 | 1.13, d (6.9) | ||
| 21 | 13.3 | 2.00, s | 13.3 | 2.03, s | 174.2 | |||||
 | ||
| Fig. 3 The chemical structures of the isomers and the comparison of the experimental and calculated ECD spectra of 5. | ||
Compound 6 was isolated as a brown oil with the molecular formula C21H22O5 and eleven degrees of unsaturation, as determined by the [M + Na]+ peak at m/z 377.1374 (calcd for 377.1365). The proton spectrum (Table 2) of 6 showed signals of five olefinic or aromatic protons (δH 6.20, 6.63, 6.42, and 7.11), an oxygenated proton of methyl group (δH 3.64), and four protons of methyl groups (δH 0.93, 0.93, 2.00, and 2.28). The 13C NMR data exhibited 21 carbons in 6, including one ketone carbonyl carbon (δC 204.1), one oxygenated non-protonated carbon (δC 78.6), and fourteen olefinic or aromatic carbons. These fragments accounted for eight degrees of unsaturation, requiring three additional rings in 6. The detailed analysis of its NMR data (Table 2) indicated that 6 contained fragments or signals of pentasubstituted and 1,2,3,4-tetrasubstituted benzene rings, a 1,2-disubstituted olefin (δC 122.5, 128.0), two aryl methyl groups (δC 17.1, 13.3), coincident or identical methyl groups (δC 27.5, 27.5) on an oxygenated quaternary carbon (δC 78.6), a methoxy group (δC 56.5), and a ketone carbon (δC 204.1), suggesting that 6 was very similar to massarinins A,15 except for the absence of an formaldehyde moiety and the difference in the substitution of the aromatic ring. Comprehensive analysis of the HSQC and HMBC data of 6 (Fig. 2) confirmed a 2-methoxyl-4,6-dimethyl-5-hydroxyl benzol unit by key HMBC correlations of H-4/C-2, C-3, and C-6, H-19/C-3, H-20/C-4, C-5, and C-6, H-21/C-2, C-6, and C-7. The ketone carbon (C-1) was as a bridge carbon connecting to C-2 and C-13 on the basis of a weak four-bond HMBC correlation of H-4/C-1.
Compound 7, a pale yellow powder, was assigned a molecular formula of C21H22O6 with eleven degrees of unsaturation by the [M + H]+ peak at m/z 371.1492 (calcd for 371.1495) in the HRESIMS spectrum. A detailed comparison of the NMR data with that of 6 suggested that 7 was similar to 6 except for a carbonyl group (δC 192.3, C-14) and a methylene (δC 48.4, C-15) in 7, replacing the 1,2-disubstituted olefin (δC 122.5, 128.0) in 6. Therefore, the substituent groups on the pentasubstituted aromatic ring in 7 were revealed. Homogeneously, C-1 was linked to C-2 and C-13 on the basis of weak 4J HMBC correlations of H-4/C-1.
Compound 8 was obtained as a pale yellow powder. The HRESIMS spectrum yielded an [M + Na]+ ion peak at m/z 407.1102 (calcd for 407.1107), which suggested its molecular formula to be C21H20O7 with twelve degrees of unsaturation. The NMR data (Table 2) indicated that 8 and 7 had the same 2,2-dimethyl-benzopyrone unit, except for the presence of a lactone ring and the difference of the pentasubstituted aromatic ring in 8. The 13C NMR data of δC 75.1 (C-1), 136.2 (C-2), 148.0 (C-3), 122.2 (C-4), 127.6 (C-5), 148.9 (C-6), 115.0 (H-7), and 174.2 (C-21) exposed the presence of an isobenzofuranone group. In addition, the key HMBC correlations of H-1/C-13, C-2, and C-21 confirmed that C-1 (δC 75.1) was connected to C-13 (δC 112.0), accomplishing the assignment of 8.
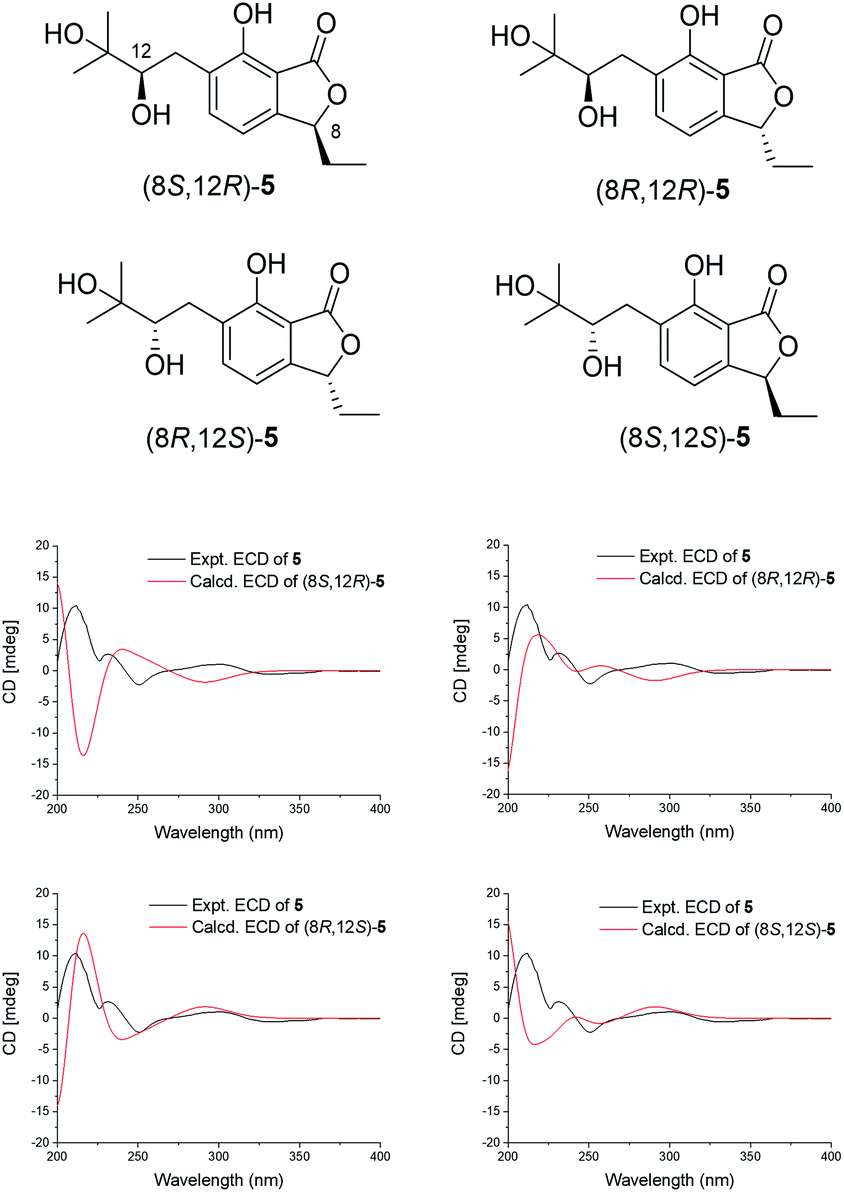
The absolute configuration of 8 was elucidated by the ECD calculation. The calculated ECD spectrum of enantiomer (1R)-8 was consistent with that of the experimental ECD spectrum of 8, which exhibited one strong negative Cotton effect at 213 nm together with two weak positive Cotton effects at 246 and 274 nm (Fig. 4). Thus, the absolute configuration of 8 was identified as (1R).
 | ||
| Fig. 4 The chemical structures of isomers of 8 and the comparison of the experimental and calculated ECD spectra of 8. | ||
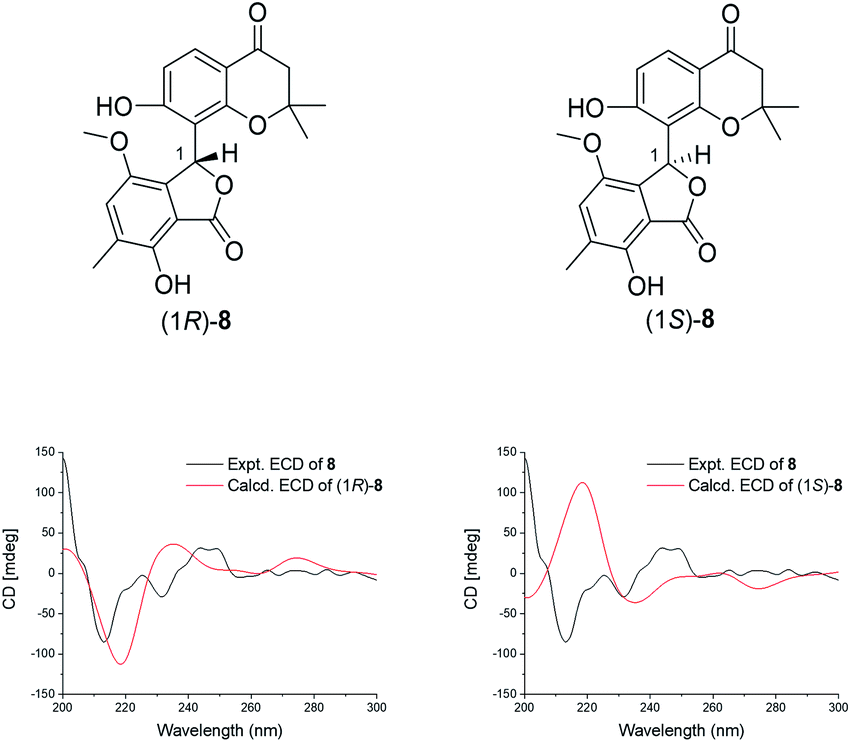
Compound 9 was a pale yellow solid and its molecular formula C20H20O6 was elucidated by the [M + H]+ peak at m/z 357.1333 (calcd for 357.1338), indicating the presence of eleven degrees of unsaturation. The 1H and 13C NMR data of 9 (Table 2) revealed a hydroxyl-substituted 2,2-dimethylbenzopyrone fragment, which was consistent with 7 and 8. Coincidentally, the comparison of the 2D NMR spectra between 9 and massarinins B indicated that the planar structure of 9 contained the upper part of 7 and the lower part of massarinins B.15 The description mentioned above was proved by the key 2D NMR correlations shown in Fig. 2. In addition, C-1 (δC 150.6) was connected to C-13 (δC 107.9) by a weak four-bond HMBC correlation of H-9 at δH 6.55 (d, J = 8.8 Hz) with C-1. The J-value for H-5 with H-6β was 10.7 Hz, which revealed that they were both axial orientations. H-4 was equatorially oriented and cis- to H-5, according to the J4,5-value (3.2 Hz) and the half chair form of the cyclohexanone group. The absolute configuration of 9 was further determined to be (4S,5S) by the comparison of the calculated ECD of (4S,5S)-9 and (4R,5R)-9 with the experimental CD curve (Fig. 5).
 | ||
| Fig. 5 The structures of the isomers and the comparison of the experimental and calculated ECD spectra of 9. | ||
Compound 10 was isolated as a light yellow solid. Its molecular formula C18H16O5 was confirmed by the [M + Na]+ peak at m/z 335.0890 (calcd for 335.0895) with eleven degrees of unsaturation. The 1D NMR (Table 3) and HSQC spectra revealed signals for twelve olefinic carbons, one methine, two methylenes, one methyl, and one carbonyl group. Moreover, the COSY correlations shown in Fig. 2, together with the HMBC correlations of H-11/C-10, H-9/C-10, H-9/C-9a, H-9/C-4a, H-8/C-4a, H-8/C-1, and H-8/C-5, implied the presence of an isobenzofuranone group and an isobenzofuran group in 10. The HMBC correlations of H-9 to C-6, H-7 to C-5, and H-9 to C-5, indicated that the isobenzofuranone group and the isobenzofuran group were linked by C5–C9 and an extra ethyl side chain was attached to C-5. The absolute configuration of 10 was determined by ECD and OR calculations. All four isomers were considered in the ECD calculations. The calculated curves of the (5S,9S)-10 and (5R,9S)-10 isomers both showed a good consistency with the experimental spectrum (Fig. 6). Furthermore, the calculated ORs for (5S,9S)-10 and (5R,9S)-10 were +31.1 and −14.7, respectively. The experimental OR of 10 was −10.4 (c 0.10, MeOH). Thus, compound 10 was identified as (5R,9S)-10, aspergorakhin J.
| No. | 10 b | 11 c | 12 d | 13 d | ||||
|---|---|---|---|---|---|---|---|---|
| δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | |
| a TMS as an internal standard.b 600 MHz for 1H, 150 MHz for 13C, CD3OD.c 700 MHz for 1H, 175 MHz for 13C, CDCl3.d 600 MHz for 1H, 150 MHz for 13C, (CD3)2CO.e Overlapped. | ||||||||
| 1 | 152.9 | 199.1 | 163.5 | 166.6 | ||||
| 2 | 115.9 | 6.75, d (7.8) | 134.4 | 110.4 | 6.56, d (8.2, 1.2) | 112.4 | 6.64, dd (8.2, 1.2) | |
| 3 | 130.3 | 7.13, t (7.8) | 158.7 | 137.2 | 7.38, t (8.2) | 138.7 | 7.48, t (8.2) | |
| 4 | 115.2 | 6.63, d (7.8) | 39.6 | 2.71, dd (17.9, 4.3) | 110.0 | 6.47, d (8.2, 1.2) | 110.5 | 6.54, dd (8.2, 1.2) |
| 2.49e | ||||||||
| 4a | 141.8 | 162.6 | 163.5 | |||||
| 5 | 94.9 | 66.1 | 4.26, tt (8.6, 4.3) | 145.5 | 144.9 | |||
| 6 | 27.6 | 1.58, dq (14.5, 7.4) | 46.7 | 2.76, dd (16.4, 4.3) | 132.3 | 137.9 | ||
| 1.85, dq (14.5, 7.4) | 2.51e | |||||||
| 7 | 7.4 | 0.65, t (7.4) | 56.9 | 4.37, d (12.4) | 117.0 | 6.99, s | 120.7 | 6.85, s |
| 4.35, d (12.4) | ||||||||
| 8 | 73.6 | 5.30, d (11.8) | 36.5 | 2.35, m | 152.5 | 156.0 | ||
| 5.20, d (11.8) | ||||||||
| 8a | 126.7 | 128.7 | 121.8 | |||||
| 9 | 85.5 | 5.63, s | 21.5 | 1.55e | 196.7 | 198.6 | ||
| 9a | 133.0 | 113.8 | 113.8 | |||||
| 10 | 172.2 | 14.1 | 0.97, t (7.4) | |||||
| 11 | 117.5 | 7.35, d (7.7) | 66.6 | 5.32, s | 67.5 | 5.35, s | ||
| 11a | 129.7 | 125.1 | 124.8 | |||||
| 12 | 132.5 | 7.46, t (7.7) | 17.3 | 2.36, s | 17.5 | 2.36, s | ||
| 13 | 122.6 | 7.18, d (7.7) | 57.1 | 3.76, s | ||||
| 14 | 154.0 | |||||||
| 1-OH | 12.55, s | 13.25, s | ||||||
| 5-OH | 7.75, s | 7.71, s | ||||||
| 8-OH | 10.49, s | |||||||
 | ||
| Fig. 6 The structures of the isomers and the comparison of the experimental and calculated ECD spectra of 10. | ||
Compound 11 was isolated as a white amorphous powder with the molecular formula of C10H16O3, as determined from the [M + Na]+ peak at m/z 207.0989 (calcd for 207.0997). Its 1D-NMR revealed a total of 10 carbons including one carbonyl, two olefinic quaternary carbons, one oxygenated methine, one methyl, and five methylene carbons, including an oxygenated methylene (δC 56.9). The COSY spectrum revealed two sets of contiguous protons of H-6/H-5/H-4 and H-8/H-9/H-10. Then, the key HMBC correlations of H-6/C-1, H-4/C-2, H-4/C-8, H-10/C-8, H-7/C-1, H-7/C-2 (Fig. 2) indicated that 11 was an α,β-unsaturated cyclohexanone derivative. Combining the molecular formula and the degree of unsaturation, we confirmed two oxygenated alkyl groups C-5 (δC 66.1) and C-7 (δC 56.9) connected to the hydroxyl groups. The absolute configuration of 11 was established by the ECD calculations. The calculated ECD spectrum of (5R)-11 was consistent with the experimental ECD, which showed one strong positive Cotton effect at 207 nm and one strong negative Cotton effect at 239 nm with one weak positive Cotton effect at 283 nm (Fig. 7). Thus, 11 was affirmed to be (5R)-11, aspergorakhin K.
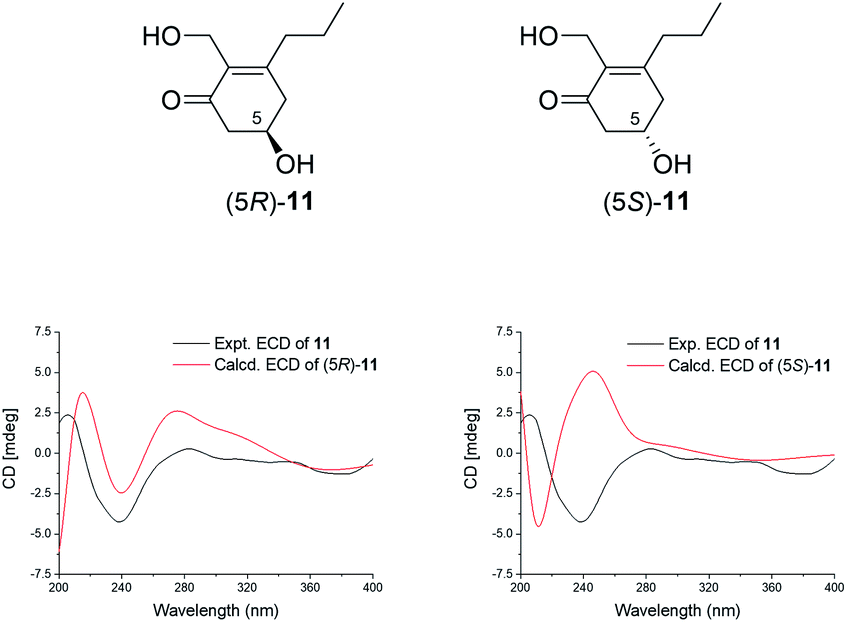 | ||
| Fig. 7 The structures of the isomers and the comparison of the experimental and calculated ECD spectra of 11. | ||
Compound 12 was obtained as a light-yellow amorphous powder and its molecular formula C16H14O5 was identified by the [M + H]+ peak at m/z 287.0924 (calcd for 287.0919). The characteristics of 12 in NMR were extremely similar to those of leptosphaerin D (13),16 except for an extra methoxyl group at C-8, which was confirmed by the HMBC correlations from H-13 to C-8.
The potential inhibitory activity of 1–13 was screened against PTP1B, SHP1, TCPTP, and cathepsin B in vitro. Cathepsin B, as an important tumor-promoting factor, acts as a target of new chemotherapeutics.17–19 Compound 1 showed significant inhibition of four enzymes with IC50 values of 0.57, 1.19, 22.97, and 8.18 μM, respectively (Table 4). Aspergorakhin B (2) inhibited SHP1 and TCPTP with IC50 values of 6.18 and 7.50 μM. Also, the IC50 value of aspergorakhin K (11) against PTP1B was 12.06 μM. However, the other compounds did not exhibit inhibitory activity with the concentration of 13.33 μg mL−1 (34.69 μM for 2 and 72.41 μM for 11). In addition, 1–4 were assayed for their cytotoxicity toward human cancer cells A549, HeLa, Bel-7402, and SMMC-7721. The results (Table 5) included that 1 and 2 exhibited modest cytotoxicity against the four cancer cells with IC50 values in the range of 6.75–83.40 μM.
Experimental section
General information
Optical rotations were recorded on a SGW-533 digital polarimeter (INESA Co., Shanghai, China). The NMR spectra were recorded on Bruker Avance III 600 or 700 spectrometers (Bruker, USA) with tetramethylsilane (TMS) or deuterated solvent as the internal standard. The ECD data were collected using a MOS-450/SFM300 spectropolarimeter (Bio-Logic, Seyssinet-Pariset, France). The ESIMS and HR-ESIMS data were recorded with an Agilent 6120 (Agilent, Santa Clara, CA, USA) or AB SCIEX TripleTOF 5600+ (AB SCIEX, Boston, MA, USA; Shimadzu LC-20A, Tokyo, Japan) LC/MS/MS system. Preparative HPLC was performed with BRIX 1860 (Orienda Ltd., Beijing, China) equipped with an UV dual wavelength detector. Preparative HPLC was carried out using GRACE Allsphere ODS-2 column (22 mm × 250 mm, 5 μm; Alltech, Chicago, IL, USA).Strain materials
The fungus Aspergillus gorakhpurensis F07ZB1707 was isolated from the mountainous region of Shennongjia Forestry District, Hubei Province of China in 2007. It was identified according to the morphological characteristics and ITS gene sequences (GenBank accession No. MT994887, ESI S117†). The fungus was preserved in New Drug Research & Development Center of North China, Pharmaceutical Group Corporation (NCPC).Fermentation, extraction, and isolation of the compounds
The strain was cultivated on PDA slant media at 26 °C for 7 days. The fungal colony scraped from the slant media was transferred to 250 mL flasks, each containing 60 mL seed media (2.0% starch, 0.2% soybean cake starch, 1.0% glucose, 0.3% yeast extract, 0.6% malt extract, 0.2% NaCl, 0.2% CaCO3, and 0.1% MgSO4, pH 7.0). Flasks with the media were cultured at 26 °C for 72 h on a rotary shaker at 200 rpm. A total of 225 Erlenmeyer flasks (1000 mL), each of which contained 200 mL liquid media (20.0 g maltose, 10.0 g monosodium glutamate, 20.0 g sorbitol, 33.0 g sea salt, 3.0 g yeast extract, 0.5 g KH2PO4, 0.3 g MgSO4, 0.5 g L-tryptophanper liter of purified water, pH 6.5) were individually inoculated with 10 mL seed broth and incubated at 26 °C on a rotary shaker at 200 rpm for 7 days. All the media were sterilized at 121 °C for 30 min before use.The culture of large-scale liquid fermentation (45.0 L) of the fungus was filtered under reduced pressure to obtain the filtrate and a mycelia cake. The filtrate and mycelia cake were extracted with EtOAc three times. The EtOAc solution was concentrated under reduced pressure at 40 °C to produce the crude extract (35.0 g). The extract was separated by silica gel column chromatography (CHCl2/MeOH, 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0–0:100) and yielded twelve subfractions (Fr. 1−Fr. 12). Fr. 5 (2.1 g) was separated by CC over Sephadex LH-20 (CH2Cl2:MeOH = 1:1) to obtain 10 subfractions (Fr. 5.1−Fr. 5.10). Fr. 5.3 (0.9 g) was subjected to silica gel column chromatography (PE/EtOAc, 40:1–1:1) to yield 8 fractions (Fr. 5.3.1–5.3.8). Fr. 5.3.7 (80.0 mg) was purified by ODS HPLC (GRACE Allsphere ODS-2, 22 mm × 250 mm, 5 μm) and eluted with CH3CN/H2O/FA (45:55:0.1 to 100:0:0.1, 20 mL min−1) to afford 1 (1.5 mg) at tR 34.0 min and 2 (4.8 mg) at tR 19.0 min. Fr. 5.5 (1.15 g) was subjected to Sephadex LH-20 CC (MeOH) again to obtain subfractions (Fr. 5.5.1−Fr. 5.5.8). Fr. 5.5.6 (127.0 mg) was purified by preparative HPLC with an ODS column and eluted with CH3CN/H2O/FA (45:55:0.1 to 60:40:0.1, 20 mL min−1) to afford 6 (5.9 mg) at tR 21.0 min, 7 (8.5 mg) at tR 13.5 min, 8 (2.3 mg) at tR 8.2 min, and 10 (5.6 mg) at tR 11.1 min. Fr. 9 (0.4 g) was directly purified by ODS HPLC and eluted with CH3CN/H2O/FA (10:90:0.1 to 65:35:0.1, 20 mL min−1) to afford 3 (15.9 mg) at tR 17.8 min, 4 (3.3 mg) at tR 19.0 min, and 11 (3.8 mg) at tR 6.9 min. Fr. 7 (2.3 g) was separated by CC over Sephadex LH-20 (MeOH) to obtain 13 subfractions (Fr. 7.1−Fr. 7.13). Fr. 7.4 (467.0 mg) was purified by ODS HPLC and eluted with CH3CN/H2O/FA (15:85:0.1 to 40:60:0.1, 20 mL min−1) to afford 5 (9.9 mg) at tR 18.2 min. Fr. 7.5 (311.0 mg) subjected to ODS MPLC (CH3CN/H2O/FA, 15:85:0.1–100:0:0.1) to obtain Fr. 7.5.B3, and then Fr. 7.5.B3 (95.0 mg) was purified by ODS HPLC and eluted with CH3CN/H2O/FA (15:85:0.1 to 45:55:0.1, 20 mL min−1) to afford 9 (3.9 mg) at tR 19.0 min. Fr. 12 (2.6 g) was separated by CC over Sephadex LH-20 (MeOH) to obtain 10 subfractions (Fr. 12.1−Fr. 12.10). Fr. 12.9 (100.0 mg) was purified by ODS HPLC abd eluted with CH3CN/H2O/FA (20:80:0.1 to 45:55:0.1, 20 mL min−1) to afford 12 (50.3 mg) at tR 14.8 min and 13 (30.5 mg) at tR 15.2 min.
:0–0:100) and yielded twelve subfractions (Fr. 1−Fr. 12). Fr. 5 (2.1 g) was separated by CC over Sephadex LH-20 (CH2Cl2:MeOH = 1:1) to obtain 10 subfractions (Fr. 5.1−Fr. 5.10). Fr. 5.3 (0.9 g) was subjected to silica gel column chromatography (PE/EtOAc, 40:1–1:1) to yield 8 fractions (Fr. 5.3.1–5.3.8). Fr. 5.3.7 (80.0 mg) was purified by ODS HPLC (GRACE Allsphere ODS-2, 22 mm × 250 mm, 5 μm) and eluted with CH3CN/H2O/FA (45:55:0.1 to 100:0:0.1, 20 mL min−1) to afford 1 (1.5 mg) at tR 34.0 min and 2 (4.8 mg) at tR 19.0 min. Fr. 5.5 (1.15 g) was subjected to Sephadex LH-20 CC (MeOH) again to obtain subfractions (Fr. 5.5.1−Fr. 5.5.8). Fr. 5.5.6 (127.0 mg) was purified by preparative HPLC with an ODS column and eluted with CH3CN/H2O/FA (45:55:0.1 to 60:40:0.1, 20 mL min−1) to afford 6 (5.9 mg) at tR 21.0 min, 7 (8.5 mg) at tR 13.5 min, 8 (2.3 mg) at tR 8.2 min, and 10 (5.6 mg) at tR 11.1 min. Fr. 9 (0.4 g) was directly purified by ODS HPLC and eluted with CH3CN/H2O/FA (10:90:0.1 to 65:35:0.1, 20 mL min−1) to afford 3 (15.9 mg) at tR 17.8 min, 4 (3.3 mg) at tR 19.0 min, and 11 (3.8 mg) at tR 6.9 min. Fr. 7 (2.3 g) was separated by CC over Sephadex LH-20 (MeOH) to obtain 13 subfractions (Fr. 7.1−Fr. 7.13). Fr. 7.4 (467.0 mg) was purified by ODS HPLC and eluted with CH3CN/H2O/FA (15:85:0.1 to 40:60:0.1, 20 mL min−1) to afford 5 (9.9 mg) at tR 18.2 min. Fr. 7.5 (311.0 mg) subjected to ODS MPLC (CH3CN/H2O/FA, 15:85:0.1–100:0:0.1) to obtain Fr. 7.5.B3, and then Fr. 7.5.B3 (95.0 mg) was purified by ODS HPLC and eluted with CH3CN/H2O/FA (15:85:0.1 to 45:55:0.1, 20 mL min−1) to afford 9 (3.9 mg) at tR 19.0 min. Fr. 12 (2.6 g) was separated by CC over Sephadex LH-20 (MeOH) to obtain 10 subfractions (Fr. 12.1−Fr. 12.10). Fr. 12.9 (100.0 mg) was purified by ODS HPLC abd eluted with CH3CN/H2O/FA (20:80:0.1 to 45:55:0.1, 20 mL min−1) to afford 12 (50.3 mg) at tR 14.8 min and 13 (30.5 mg) at tR 15.2 min.
Spectral data
ε) 203 (4.37), 230 (4.10), and 273 (3.34); IR νmax 3386, 2924, 2871, 2855, 1732, 1651, 1557, 1456, 1377, 1260, 1061, 1026, 798 cm−1; 1H and 13C NMR data, Table 1; HR-ESIMS: m/z 413.3047 ([M + H]+, calcd for C27H41O3, 413.3056).ε) 204 (4.45) and 242 (4.55); IR νmax 3390, 2924, 1739, 1673, 1607, 1464, 1355, 1257, 1131, 1060, 969 cm−1; 1H and 13C NMR data, Table 1; HR-ESIMS: m/z 407.2556 ([M + Na]+, calcd for C25H36O3Na, 407.2562).ε) 203 (4.18) and 244 (4.00); IR νmax 3353, 2924, 2873, 1687, 1557, 1373, 1238, 1032, 983, 802, 578 cm−1; 1H and 13C NMR data, Table 1; HR-ESIMS: m/z 403.2850 ([M + H]+, calcd for C25H39O4, 403.2848).ε) 203 (3.74); IR νmax 3390, 2920, 2874, 1683, 1583, 1450, 1374, 1237, 1030, 593 cm−1; 1H and 13C NMR data, Table 1; HR-ESIMS: m/z 405.3014 ([M + H]+, calcd for C25H41O4, 405.3005).ε) 206 (5.34) and 303 (4.39); IR νmax 3408, 2971, 2932, 1731, 1631, 1598, 1443, 1277, 1106, 1077, 967, 798 cm−1; 1H and 13C NMR data, Table 2; HR-ESIMS: m/z 303.1207 ([M + Na]+, calcd for C15H20O5Na, 303.1208).ε) 204 (4.55) and 278 (4.07); IR νmax 3412, 2921, 2850, 1682, 1609, 1470, 1429, 1344, 1204, 1123, 804, 724 cm−1; 1H and 13C NMR data, Table 2; HR-ESIMS: m/z 377.1374 ([M + Na]+, calcd for C21H22O5Na, 377.1365).ε) 206 (4.51), 247 (4.48), 276 (4.44), and 343 (3.88); IR νmax 3400, 2916, 2849, 1668, 1585, 1424, 1294, 1236, 1092, 1054, 829 cm−1; 1H and 13C NMR data, Table 2; HR-ESIMS: m/z 371.1492 ([M + H]+, calcd for C21H23O6, 371.1495).ε) 218 (4.24), 275 (3.68), and 320 (3.75); IR νmax 3300, 2916, 2849, 1730, 1581, 1539, 1466, 1417, 1047, 944, 873, 814, 720 cm−1; 1H and 13C NMR data, Table 2; HR-ESIMS: m/z 407.1102 ([M + Na]+, calcd for C21H20O7Na, 407.1107).ε) 204 (4.00) and 278 (3.30); IR νmax 3359, 2919, 2851, 1644, 1465, 1377, 1097, 1028 cm−1; 1H and 13C NMR data, Table 2; HR-ESIMS: m/z 357.1333 ([M + H]+, calcd for C20H21O6, 357.1338).ε) 214 (3.96) and 301 (3.11); IR νmax 3234, 2957, 2921, 2850, 1759, 1741, 1599, 1472, 1290, 1023, 789, 736 cm−1; 1H and 13C NMR data, Table 3; HRESIMS m/z 335.0890 [M + Na]+ (calcd for C18H16O5Na, 335.0895).ε) 204 (4.16) and 276 (3.18); IR νmax 3366, 2921, 2850, 1650, 1465, 1378, 1273, 1016, 600 cm−1; 1H and 13C NMR data, Table 3; HR-ESIMS: m/z 207.0989 ([M + H]+, calcd for C10H16O3Na, 207.0997).ε) 205 (4.11), 280 (3.63), and 365 (3.54); IR νmax 3359, 2919, 2851, 1644, 1465, 1377, 1097, 1028 cm−1; 1H and 13C NMR data, Table 3; HR-ESIMS: m/z 287.0924 ([M + H]+, calcd for C16H15O5, 287.0919).Electronic circular dichroism (ECD) and optical rotation (OR) calculation
Conformational searches were performed by means of the BARISTA software (CONFLEX Corporation) using the MMFF94S force field. The conformers with energy from 0–5 kcal mol−1 were used for optimization at the B3LYP/6-31G(d) level in the gas phase using the Gaussian 09 package. These conformations with 0–2.5 kcal mol−1 were further optimized at the B3LYP/6-311+G(d) level in the gas phase. The calculation of ECD and OR was performed at the B3LYP/6-311++G(2d,p) level. Compounds 5 and 8–11 (ESI S114†) were selected for the calculations.20–25 The Boltzmann sums were performed to simulate the ECD curves and the OR values for the selected chiral compounds.Inhibitory activity of the enzymes assay
Human phosphatase PTP1B, SHP1, and TCPTP were used for inhibitory activity assays. Na3VO4 was used as the positive control. The detailed procedure is placed in ESI S116,† the same as that described in the literature.26 The cathepsin B assays were carried out in triplicate according to a published method27 with modification in ESI S116.† Leupeptin was used as the positive control.Cytotoxicity assays
Human cancer cells A549, Bel-7402, SMMC-7721, and HeLa were selected for cytotoxicity assays.28 A549, Bel-7402, SMMC-7721, and HeLa cells were cultured and then dealt with various concentrations of 1–4 for 48 h and detected by the MTT assay. Taxol was used as the positive control. All the human cell lines were provided by the Hebei Key Laboratory of Forensic Medicine, College of Forensic Medicine, Hebei Medical University.Conclusions
In summary, 12 new metabolites aspergorakhin A–L (1–12) and leptosphaerin D (13) were isolated from the cultivation of the fungus Aspergillus gorakhpurensis F07ZB1707 cultured in a salt-containing medium. The structures of these compounds included: sterol (1), nortriterpenes (2–4), aromatic polycyclic polyketides (5–10, 12–13), and cyclohexanone derivative (11). All the compounds were obtained from Aspergillus gorakhpurensis without precedent and this study was helpful to fill the research gap in the field of novel compounds' discovery of this strain. Aspergorakhin A (1) showed selective activities against PTP1B and SHP1 over TCPTP with IC50 values of 0.57, 1.19, and 22.97 μM, respectively.Author contributions
Yannan Ji: conceptualization, data curation, formal analysis, investigation, validation, writing – original draft. Qiqi Zhou: data curation. Guosheng Liu: data curation. Tianhui Zhu: data curation, formal analysis. Yufang Wang: data curation, formal analysis. Yan Fu: data curation. Yeying Li: data curation, methodology. Ruolan Li: data curation, methodology, formal analysis. Xuexia Zhang: data curation, methodology, formal analysis. Mei Dong: resources, methodology, formal analysis. Françoise Sauriol: data curation, methodology, formal analysis. Yucheng Gu: funding acquisition, writing – review & editing. Qingwen Shi: conceptualization, supervision, project administration, writing – review & editing. Xinhua Lu: resources, methodology, writing – review & editing. Zhiyu Ni: conceptualization, methodology, writing – review & editing.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
We gratefully acknowledge financial support from the Natural Science Foundation of Hebei Province (H2020206599). We also wish to extend our sincere thanks for the financial support from Syngenta Ltd. (12112020-Hebei Medical University-Syngenta). In this work we thank Hebei University and Hebei Medical University Core Facilities and Centers for CD measurements.Notes and references
- A. Alonso, J. Sasin, N. Bottini, I. Friedberg, I. Friedberg, A. Osterman, A. Godzik, T. Hunter, J. Dixon and T. Mustelin, Cell, 2004, 117, 699–711 CrossRef CAS PubMed.
- N. K. Tonks, Nat. Rev. Mol. Cell Biol., 2006, 7, 833–846 CrossRef CAS PubMed.
- H. A. Watson, S. Wehenkel, J. Matthews and A. Ager, Biochem. Soc. Trans., 2016, 44, 356–362 CrossRef CAS PubMed.
- Z. Zhang, Acc. Chem. Res., 2017, 50, 122–129 CrossRef CAS PubMed.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2016, 79, 629–661 CrossRef CAS PubMed.
- M. Liu, W. Sun, J. Wang, Y. He, J. Zhang, F. Li, C. Qi, H. Zhu, Y. Xue, Z. Hu and Y. Zhang, Bioorg. Chem., 2018, 80, 525–530 CrossRef CAS PubMed.
- J. W. Blunt, A. R. Carroll, B. R. Copp, R. A. Davis, R. A. Keyzers and M. R. Prinsep, Nat. Prod. Rep., 2018, 35, 8–53 RSC.
- E. Ioannou, A. F. Abdel-Razik, M. Zervou, D. Christofidis, X. Alexi, C. Vagias, M. N. Alexis and V. Roussis, Steroids, 2009, 74, 73–80 CrossRef CAS PubMed.
- H. Sun, F. Liu, M. R. Feng, Q. Peng, X. J. Liao, T. T. Liu, J. Zhang and S. H. Xu, Nat. Prod. Res., 2016, 30, 2819–2824 CrossRef CAS PubMed.
- S. Yu, Z. Deng, L. van Ofwegen, P. Proksch and W. Lin, Steroids, 2006, 71, 955–959 CrossRef CAS PubMed.
- M. Yang, L. F. Liang, H. Li, W. Tang and Y. W. Guo, Nat. Prod. Res., 2020, 34, 1814–1819 CrossRef CAS PubMed.
- X. Y. Liu, X. L. Wang, T. Shen, D. M. Ren, H. X. Lou and X. N. Wang, Nat. Prod. Res., 2020, 34, 2179–2185 CrossRef CAS PubMed.
- T. Hosoe, H. Okada, T. Itabashi, K. Nozawna, K. Okada, G. M. Takaki, K. Fukushima, M. Miyaji and K. Kawai, Chem. Pharm. Bull., 2000, 48, 1422–1426 CrossRef CAS PubMed.
- W. Fang, J. Wang, J. Wang, L. Shi, K. Li, X. Lin, Y. Min, B. Yang, L. Tang, Y. Liu and X. Zhou, J. Nat. Prod., 2018, 81, 1405–1410 CrossRef CAS PubMed.
- H. Oh, D. C. Swenson, J. B. Gloer and C. A. Shearer, J. Nat. Prod., 2003, 66, 73–79 CrossRef CAS PubMed.
- J. Lin, S. Liu, B. Sun, S. Niu, E. Li, X. Liu and Y. Che, J. Nat. Prod., 2010, 73, 905–910 CrossRef CAS PubMed.
- A. Mitrovic, J. Kljun, I. Sosic, M. Ursic, A. Meden, S. Gobec, J. Kos and I. Turel, Inorg. Chem., 2019, 58, 12334–12347 CrossRef CAS PubMed.
- J. Kos, A. Mitrovic and B. Mirkovic, Future Med. Chem., 2014, 6, 1355–1371 CrossRef CAS PubMed.
- A. Gopinathan, G. M. Denicola, K. K. Frese, N. Cook, F. A. Karreth, J. Mayerle, M. M. Lerch, T. Reinheckel and D. A. Tuveson, Gut, 2012, 61, 877–884 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- F. Cao, Z. H. Meng, P. Wang, D. Q. Luo and H. J. Zhu, J. Nat. Prod., 2020, 83, 1283–1287 CrossRef CAS PubMed.
- J. Ren, D. Zhao, S. Wu, J. Wang, Y. Jia, W. Li, H. Zhu, F. Cao, W. Li, C. U. Pittman and X. He, Tetrahedron, 2019, 75, 1194–1202 CrossRef CAS.
- D. Zhao, Z. Q. Li, F. Cao, M. M. Liang, C. J. Pittman, H. J. Zhu, L. Li and S. S. Yu, Chirality, 2016, 28, 612–617 CrossRef CAS PubMed.
- S. Ding, C. Zhang, W. Shi, M. Liang, Q. Yang, H. Zhu and Y. Li, Tetrahedron Lett., 2016, 57, 75–79 CrossRef CAS.
- D. Zhao, J. Ren, Y. Xiong, M. Dong, H. Zhu and C. U. Pittman, Chem. Res. Chin. Univ., 2019, 35, 109–119 CrossRef CAS.
- C. Huo, Z. Zheng, Y. Xu, Y. Ding, H. Zheng, Y. Mu, Y. Niu, J. Gao and X. Lu, J. Nat. Prod., 2020, 83, 1394–1399 CrossRef CAS PubMed.
- W. Halangk, M. M. Lerch, B. Brandt-Nedelev, W. Roth, M. Ruthenbuerger, T. Reinheckel, W. Domschke, H. Lippert, C. Peters and J. Deussing, J. Clin. Invest., 2000, 106, 773–781 CrossRef CAS PubMed.
- T. Y. Huang, C. Y. Huang, C. H. Chao, C. C. Lin, C. F. Dai, J. H. Su, P. J. Sung, S. H. Wu and J. H. Sheu, Mar. Drugs, 2020, 18, 452 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: HRESIMS, 1D, 2D NMR spectra for 1–12, ECD and OR computational details of 5, 8–11. See DOI: 10.1039/d1ra00788b |
| This journal is © The Royal Society of Chemistry 2021 |