
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelectivity for water isotopologues within metal organic nanotubes†
Maurice K. Payne,
Lindsey C. Applegate,
Priyanka Singh,
Ashini S. Jayasinghe,
George B. Crull,
Andrea B. Grafton,
Christopher M. Cheatum and
Tori Z. Forbes *
*
Department of Chemistry, University of Iowa, Iowa City, IA 52242, USA. E-mail: tori-forbes@uiowa.edu
First published on 6th May 2021
Abstract
Through a combination of many analytical approaches, we show that a metal organic nanotube (UMON) displays selectivity for H2O over all types of heavy water (D2O, HDO, HTO). Water adsorption experiments combined with vibrational and radiochemical analyses reveal significant differences in uptake and suggest that surface adsorption processes may be a key driver in water uptake for this material.
While the mass differences between isotopologues of water (H2O, H2DO, 2D2O, H3TO) are small, the overall isotopic effects can be significant and result in important differences in physical and chemical properties. The natural abundance water consists of mostly H2O with one deuterium (2D) or tritium (3T) atom for every 6.42 × 103 or 5.41 × 1016 H atoms, respectively.1 Heavy water enriched in 2D and/or 3T forms stronger hydrogen bonds, which causes smaller amplitude intermolecular motions and more ordering within the solution phase.2 This effect imparts differences in the physical properties of light and heavy water, including variations in the viscosity,3 self-diffusion coefficient,4 phase behavior,5 hydration energies,6 and exchange kinetics7 that have observable effects on protein stability,8 biological activity,7 toxicity,9 and chemical reactivity.7 Nuclear quantum effects of water isotopologues are an active area of research and the effects on kinetics and thermodynamics in complex systems are critical if difficult to untangle.10
Even with the substantial differences in the properties of light and heavy water, selectively partitioning these isotopologues remains both challenging and important. Evaporation in the environment leads to slight fractional separation in surface waters where variation of 2D concentrations ranging from 0.0130% in the Arctic ocean to 0.0162% in the Nile River, and these differences can be used for climatic reconstructions.11 In a more anthropogenic context, isotopic fractionation is also important for heavy water nuclear reactors. Although distillation is the simplest engineering procedure for separation of the isotopologues, a separation factor of 1.015 requires extensive multi-stage systems and large energy costs (10 MW h/kg D2O).11,12 Alternatively, chemical processes for heavy water production, such as the Girdler sulfide and monothermal ammonium hydrogen methodologies, offer a marginal increase in the separation factors (2.3 to 2.8) with similar energy demands.13 Clearly, a fractionation process for isotopic separation of water that involves modest energy consumption and high separation factors would offer substantial benefits. So, it is essential to identify materials capable of such large separation factors and to understand the molecular mechanisms by which these processes occur.
In previous studies we showed that a metal organic nanotube (UMON) contains 1.2 nm diameter nanopores composed of six U(VI) nodes connected through iminodiacetate linkers to create macrocyclic units that stack into nanotubular arrays by hydrogen bonding (Fig. 1). The crystalline material is stabilized by charge-balancing piperazinium cations and is durable to 200 °C.14 Within the nanotubular cavity, water molecules spontaneously arrange in hexagonal arrays resembling crystalline ice that can be removed without collapse or structural degradation of the nanotube. Dehydration of the weakly-bound, confined water within the UMON nanochannels occurs between 40–70 °C and complete removal of the H2O accounts for 5.87% weight loss in thermogravimetric analysis (TGA).14 Rehydration is spontaneous under humid conditions. Following the loss of water from the nanochannel, an additional 1.2% weight gain can be observed under high humidity conditions due to adsorption of surface water onto the material.15 The UMON surface contains ample hydrophilic groups (i.e. free carboxyl oxygen atoms and protonated amines) that are available for hydrogen bonding with water molecules to facilitate this additional surface adsorption. Removal of the more strongly bound surface water takes place between 80–150 °C. Here we show that, while UMON allows H2O to enter its the nanochannels, we observe divergent behaviour for water containing 3T or 2D. This fractionation process occurs at room-temperature with no additional energy sources or pressurization. We have employed a range of analytical methods to confirm the isotope effects observed for this material including spectroscopic and radiochemical techniques.
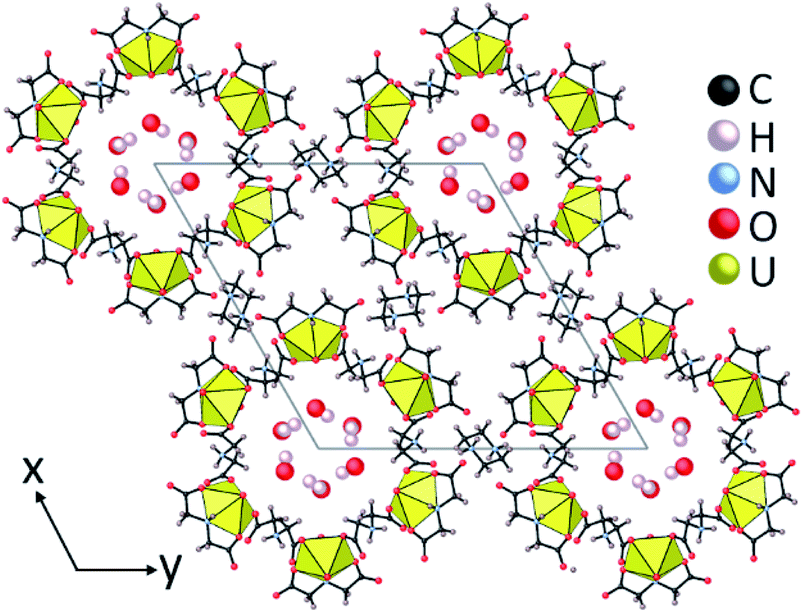 | ||
| Fig. 1 (a) UMON crystals are composed of oriented metal organic nanotube, with U(VI) (yellow polyhedra) linked by iminodiacetate molecules form the nanotubular arrays and ordered H2O molecules are located within the nanotubes. | ||
To investigate the isotopic fractionation of UMON, we synthesized the material according to the previously reported procedure.14 We initially examined the UMON behaviour in deuterated systems (D2O/HDO) using both flow-through and batch studies. In situ flow-through experiments (also see ESI† for details) using a custom-built vapor sorption instrument that was been previously described and validated for reproducibility and reliability. For the batch experiments, dehydrated UMON samples were exposed to D2O in a N2 gas filled glove bag to prevent contamination by H2O (described in detail in the ESI†). After 18 hours, the samples were analysed by a TA instruments thermogravimetric analyser (TGA) equipped with a ThermoScientific FTIR spectroscopy for the evolved gases to identify the isotopic composition of the adsorbate. A second set of dehydrated UMON were used as controls to confirm that H2O was not present in the hydration chamber or introduced as part of the developed procedure. No weight loss or evolved vapor of any kind was observed for the control samples throughout the experiments, which validated the experimental design of the batch experiments as free from protic contamination.
Flow-through experiments demonstrate difference in uptake kinetics and total uptake in the system (Fig. 2). Full uptake (5.8 ± 0.2 wt%) was observed in the H2O flow-through experiments (80% RH) whereas only 1.2 ± 0.2 wt% uptake occurs in the presence of D2O over the course of the 60 minute experiment. Additional TGA analysis of the UMON sample exposed to H2O finds a 5.1 ± 0.5% weight loss between 25 to 100 °C. A similar residual weight is observed for the material exposed to D2O, where 0.6 ± 0.5% mass loss occurs between 25 to 100 °C. Result from the flow-through experiments demonstrate differences in uptake between D2O and H2O and the related weight loss data finds that residual water (0.6–0.7 wt%) remains strongly bound to the material.
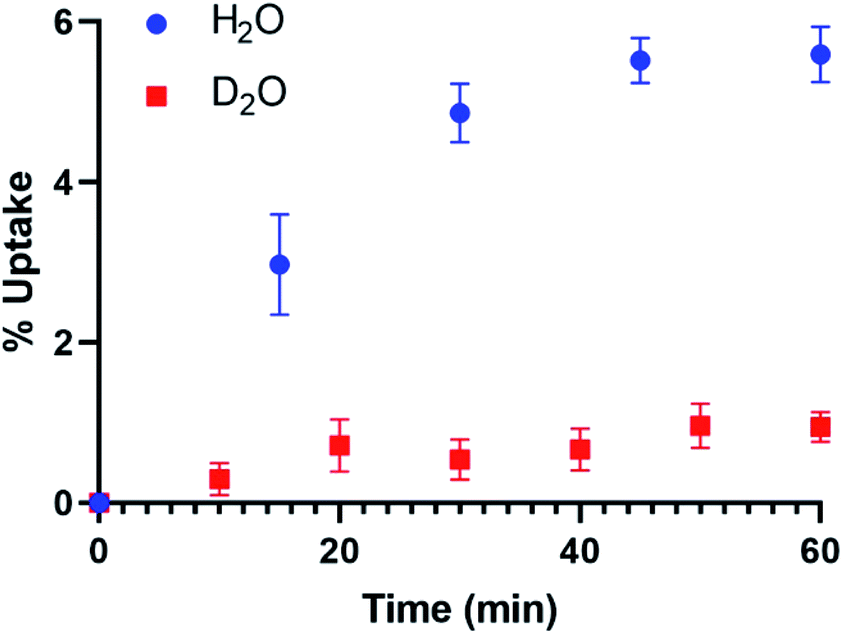 | ||
| Fig. 2 Uptake of H2O and D2O vapor into the UMON material using the flow-through apparatus. | ||
TGA of the dehydrated metal organic nanotubes exposed to D2O vapor (99.96% purity) in the batch experiments indicates a 6.1 ± 0.5% weight loss occurring between 25 and 100 °C, but the FTIR spectrum of the evolved gases only provides evidence for the presence of H2O in the channels (Fig. 3A). The observed weight loss is slightly higher than expected for water confined only within the nanotubes (5.8%); thus, we assume that the additional water must correspond to surface adsorbed species. Evolved water vapor released during the heating process is evaluated using a coupled FTIR spectrometer to determine the isotopic composition. Initially, only O–H stretching (ν1 and ν3; 3400–4000 cm−1) and bending modes (ν2; 1250–1900 cm−1) are present in the spectra for the first 5.8% of the total weight loss, indicating that the vapor removed during the initial heating process is H2O. The presence of D2O or HDO vapor would exhibit characteristic signals at 2671 or 2723 cm−1, respectively. These bands are noticeably absent in the initial heating stage, but a weak signal at 2721 cm−1 does arise during the final 5.8–6.1% portion of the total weight loss (>80 °C) (Fig. 3B). We also conducted an extended vapor sorption experiment (6 hours) and found identical uptake (6.1%), mass loss (5.8%), and no evidence of D2O in the FTIR of the evolved gas (Fig. S17†). These data suggest that H2O is present inside the nanochannel and additional heating results in the removal of surface adsorbed D2O/HDO species.
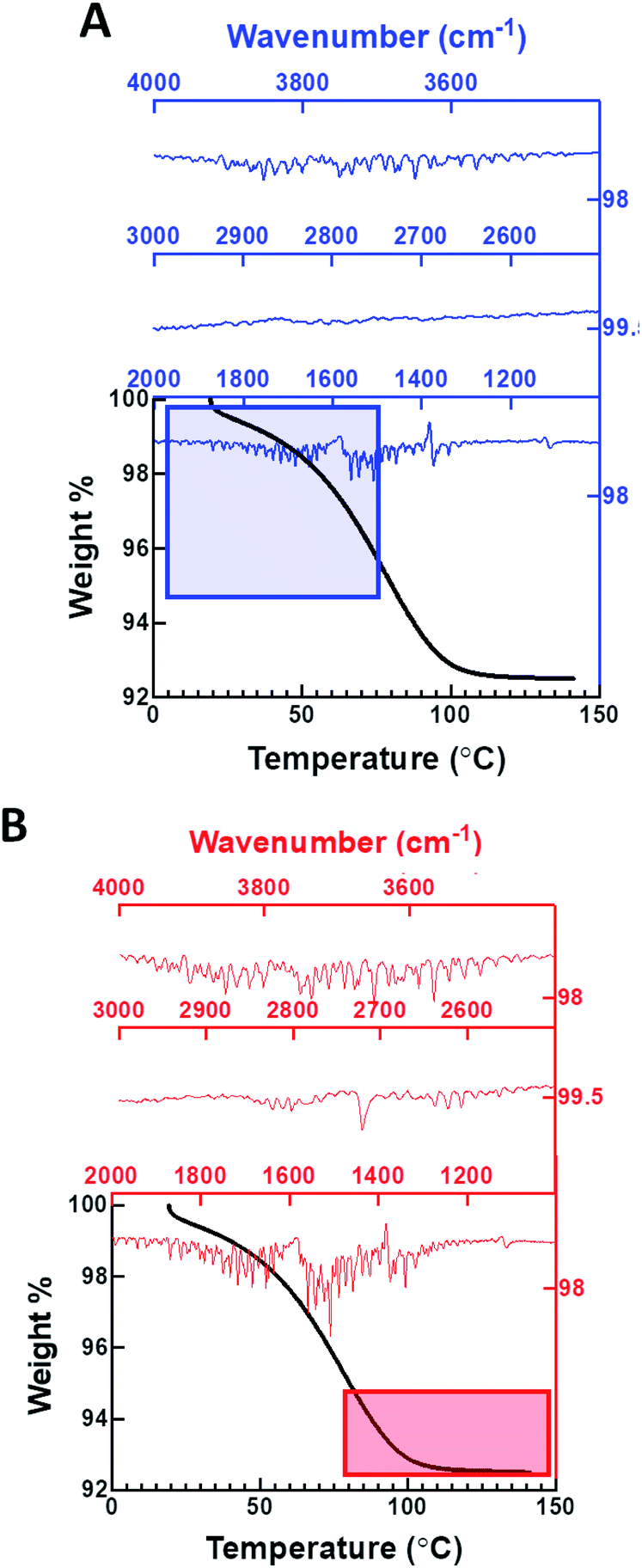 | ||
| Fig. 3 Thermogravimetric analysis combined with FTIR spectroscopy of the evolved gases determine the presence of water associated with the UMON sample exposed to pure D2O vapor. (A) Initially gases evolved during the heating process are confirmed as H2O. (B) Evolved gases observed at higher temperatures are identified as HDO by the appearance of a band at 2721 cm−1. | ||
Our previous calorimetric study supports the idea that the water in the channels desorbs before the surface waters because the interaction between the confined water and the interior walls of the nanotube is weak (−7.8 ± 0.9 kJ mol−1) whereas multiple, strong H-bonding interactions on the surface increases the binding energy, and therefore the temperature required to remove the water.17 In addition, water adsorption calorimetry experience noted two different types of water environments and related adsorption energies. Initially, we described this as two different water sites within the channel, but an alternative explanation is that the first value (−59.8 ± 1.1 kJ mol−1) corresponds to interactions with the UMON surface. After initial strong adsorption to the surface (0.25 mol H2O = 0.7 wt%), filling begins, and the differential enthalpy increases sharply to 50 kJ mol−1 and remains at this value until complete hydration.17
One puzzling aspect of the batch experiments is the full uptake of H2O in the presence of saturated D2O environment. We further characterized the dehydrated UMON samples exposed in the batch reactor (99.96% D2O vapor) by FTIR analysis and solid-state 2D NMR measurements. The infrared spectrum of dehydrated UMON exposed to D2O (Fig. S18†) shows the broad O–H stretching band (ν1 and ν3, 3400–4000 cm−1) and an additional band at 2280 cm−1 that does not correspond to any HDO or D2O spectral modes. Instead, we propose that this band is due to hydrogen/deuterium exchange with –NH2 functional groups within iminodiacetate or piperazinium cations. This spectral interpretation agrees with a study by Heacock and Marion showing that deuteration of secondary amines results in bands between 2000–2400 cm−1.18 The solid-state 2D-NMR confirms H/D exchange on the UMON surface (Fig. S21 and S22†). UMON hydrated with H2O serves as the control sample and does not produce a detectable 2D-NMR resonance, as expected. In contrast, the spectrum collected from the dehydrated UMON sample exposed to D2O vapor indicates the presence of a relatively immobile D species. The fit to the data shows that the major resonance (>99%) has a chemical shift of 3.8 ppm with respect to TMS. This shift is lower in frequency than bulk water (4.8 ppm) and is consistent with a change in the chemical environment of the D species that was detected in the UMON material. This data also provides evidence of H/D exchange, likely on the UMON surface, that produces H2O that subsequently enters the channels even in the presence of 99.96% pure D2O. We hypothesize D2O encounters the UMON surface and adsorbs through H-bonding interactions with iminodiacetate and piperazinium molecules (Fig. S23†). The selectivity of the channels prevents a majority of the surface-bound D2O from entering the channel, although it can undergo proton exchange with the –NH2 groups associated with the organic molecules on the surface. Ultimately, surface exchange with 1H atoms results in the formation of light water molecule and this molecule can enter the channel. This hypothesis explains the uptake of H2O even in the presence of 99.96% pure D2O (Fig. 3A) and the fraction of D2O/HDO molecules that thermally desorb from the surface at high temperatures in the TGA experiments (Fig. 3B). In the flow-through experiments, surface adsorption can readily occur, but the exchange reaction requires longer time periods and can only be observed when the experiment is extended to 6 hours. The mass difference between the uptake (6.1%) and release (5.8%) for the flow-through experiments suggests that in this particular experiment, exchangeable 2D is still present on the surface.
The one caveat of these results is the relatively modest limits of detection for both the FTIR of the evolved gases and condense phased characterization. To enhance the limit of detection and more rigorously assess the isotope selectivity, we expose dehydrated UMON crystallites to HTO vapor in a closed hydration chamber to assess the amount of 3T adsorption on the hydrophilic surfaces or within the channels of the UMON material. Natural abundance hydrated samples are used as controls because the protons on the surface in the hydrated samples will exhibit exchange with 3T but the water in the channels of the fully hydrated samples will not exchange because the confined water does not exchange with the bulk on this time scale. We have previously shown that there is minimal exchange of the confined water inside the channels on the timescale of these experiments, making the hydrated UMON material an ideal control.16 Liquid scintillation counting quantifies the activities of both the experimental and control samples, and the experimental samples show activities that are, within error, identical to those of the control samples (Fig. S24 and S25†). Using the measured specific activity of the tritiated water and subtracting the background (surface adsorption) allows us to calculate the average amount of HTO present at 80 ± 40 nmol HTO per mg UMON (Fig. S26†). If the tritiated water were to occupy both the nanochannels and the surface for the initially dehydrated samples, then we would expect to observe 3222 nmol HTO per mg UMON. The amount we observe represents 2.48 ± 1.24 wt%, which falls within the range observed for surface bound water (0.6–3%). Due to the radioactivity of the sample, we could not further evaluate the material using TGA analysis, but our results also suggest selectivity between isotopologues.
Although we have considerable experimental evidence to support both the conclusion that the UMON material selectively prefers light water over heavy forms and a proposed model for surface exchange and uptake of light water even in an overwhelming isotopic abundance of heavy water, the mechanism of the isotopic selectivity of the channel remains unclear. In prior studies on hydration of UMON, we showed that the water molecules in the fully hydrated UMON nanochannel adopt an ice-like structure that is well ordered based on high resolution X-ray crystallographic analysis. There are multiple mechanisms for isotope effects within materials and both kinetic and chemical affinity quantum sieving have been explored within metal organic frameworks.19–26 Kinetic quantum sieving can occur when the difference between the pore size and the molecular hard core of the guest molecule becomes comparable to the de Broglie wavelength of the guest molecule.19 Work by Heine and Hirscher and others indicate that materials with pore sizes of ∼2.5–9.1 Å can selectively bind heavier D2 isotopes over lighter H2 molecules at separation factors of 11–12 at temperatures <100 K.20–24 Nevertheless, this level of selectivity would not be enough to explain our observations and is only achievable in the previously studied systems at low temperatures. Chemical affinity quantum sieving relies on a difference in the adsorption enthalpy within the channels as a result of the mass effect on the zero-point energy differences for the bound isotopologues, but is challenging to precisely design the optimal system for this process to be used effectively and also yields modest separation factors and only at relatively low temperatures.25,26 Thus, it seems unlikely that either of these quantum sieving mechanisms could be responsible for the observed selectivity of the UMON nanopores for light water over all forms of heavy water.
A recent study of hydrophobic nanoconfinement of light and heavy water at interfaces shows significant nuclear quantum effects on the stability and ordering of the hydrogen bond network at hydrophobic interfaces that arise as a purely nanoscale phenomenon that does not significantly affect the bulk properties of the liquid–solid interface.27 Interestingly, this study shows greater stabilization of D2O relative to H2O under nanoconfinement, which is in contrast to the observations reported here. Still, the observation of distinctive nuclear quantum effects as a result of nanoscopic confinement illustrates the potential for these kinds of effects to ultimately lead to the level of selectivity that we report in our experiments. Although the precise molecular mechanisms that lead to the high level of isotope fractionation that we observe in the UMON material remain uncertain, it seems likely that nuclear quantum effects on the structure and stability of hydrogen bond networks under confinement in the UMON pores is central to the reported results. Future studies will focus on untangling the contributions to this effect by probing the structure–function relationships that determine the isotope effect observed within the UMON material.
Author contributions
T. Z. F., M. K. P., A. L. J. and P. S. designed the experiments; M. K. P., A. L. J., P. S., L. C. A., A. G., G. C. performed the experiments; T. Z. F., M. K. P., P. S., G. C., C. M. C. analysed data; T. Z. F. and M. K. P. wrote the manuscript. All authors edited and revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. K. P., L. C. A., A. S. J., and T. Z. F. acknowledge funding from the NSF CAREER Award (DMR1252831) for the batch experiments and chemical characterization and DMR2004220 for the related flow-through studies. C. M. C., P. S., and A. B. G. acknowledge funding from CHE-1707598 for the solid-state FTIR and tritiated water experiments.References
- International Association for the Properties of Water and Steam, Guideline on the Use of Fundamental Physical Constants and Basic Constants of Water IAPWS G5-01, 2016 Search PubMed
.
- A. K. Soper and C. J. Benmore, Phys. Rev. Lett., 2008, 101, 065502 CrossRef CAS PubMed
- C. H. Cho, J. Urquidi, S. Singh and G. Wilse Robinson, J. Phys. Chem. B, 1999, 103, 1991 CrossRef CAS
- R. Mills, J. Phys. Chem., 1973, 77, 685 CrossRef CAS
- V. B. Polyakov, J. Horita and D. R. Cole, Geochim. Cosmochim. Acta, 2006, 70, 1904 CrossRef CAS
- D. H. Davies and G. C. Benson, Can. J. Chem., 1965, 43, 3100 CrossRef CAS
- A. Kohen and H. H. Limbach, Isotope Effects in Chemistry and Biology, CRC Press, Boca Raton, FL, 2005 Search PubMed
- S. S. Stadmiller and G. J. Pielak, Protein Sci., 2018, 27, 1710 CrossRef CAS PubMed
- Y. Sinyak, A. Grigoriev, V. Gaydadimov, T. Gurieva, M. Levinskih and B. Pokrovskii, Acta Astronaut., 2003, 52, 575 CrossRef CAS PubMed
- M. Ceriotti, W. Fang, P. G. Kusalik and R. H. McKenzie, Chem. Rev., 2016, 116, 7529 CrossRef CAS PubMed
- A. I. Miller, CNS Bulletin, 2001, 22, 1 Search PubMed
- M. Lozada-Hidalgo, S. Zhang, S. Hu, A. Esfandiar, I. V. Grigorieva and A. K. Geim, Nat. Commun., 2017, 8, 15215 CrossRef CAS PubMed
- H. K. Rae, Separation of Hydrogen Isotopes, ed. H. K. Rae, ACS, Washington, D. C., 1978, vol. 68, pp. 1–26 Search PubMed
- D. K. Unruh, K. Gojdas, A. Libo and T. Z. Forbes, J. Am. Chem. Soc., 2013, 135, 7398 CrossRef CAS PubMed
- A. S. Jayasinghe, D. K. Unruh, A. Kral, A. Libo and T. Z. Forbes, Cryst. Growth Des., 2015, 15, 4062 CrossRef CAS
- A. S. Jayasinghe, M. K. Payne, D. K. Unruh, A. Johns, J. Leddy and T. Z. Forbes, J. Mater. Chem. A, 2018, 6, 1531–1539 RSC
- S. K. Sahu, D. K. Unruh, T. Z. Forbes and A. Navrotsky, Chem. Mater., 2014, 26, 5105–5112 CrossRef CAS
- R. A. Heacock and L. Marion, Can. J. Chem., 1956, 34, 1782 CrossRef CAS
- J. Cai, Y. Xing and X. Zhao, RSC Adv., 2012, 2, 8579 RSC
- H. Oh, I. Savchenko, A. Mavrandonakis, T. Heine and M. Hirscher, ACS Nano, 2014, 8, 761 CrossRef CAS PubMed
- J. Teufel, H. Oh, M. Hirscher, M. Wahiduzzaman, L. Zhechkov, A. Kuc, T. Heine, D. Denysenko and V. Volkmer, Adv. Mater., 2013, 25, 635 CrossRef CAS PubMed
- I. Weinrauch, I. Savchenko, D. Denysenko, S. M. Souliou, H.-H. Kim, M. Le Tacon, L. L. Daemen, Y. Cheng, A. Mavrandonakis, A. J. Ramirez-Cuesta, D. Volkmer, G. Schütz, M. Hirscher and T. Heine, Nat. Commun., 2017, 8, 14496 CrossRef CAS PubMed
- D. W. Cao, H. Huang, Y. Lan, X. Chen, Q. Yang, D. Liu, Y. Gong, C. Xiao, C. Zhong and S. Peng, J. Mater. Chem. A, 2018, 6, 19954 RSC
- H. Oh and M. Hirscher, Eur. J. Inorg. Chem., 2016, 4278 CrossRef CAS
- J. Y. Kim, R. Balderas-Xicohténcatl, L. Zhang, S. G. Kang, M. Hirscher, H. Oh and H. R. Moon, J. Am. Chem. Soc., 2017, 139, 15135 CrossRef CAS PubMed
- J. Y. Kim, L. Zhang, R. Balderas-Xicohténcatl, J. Park, M. Hirscher, R. H. Moon and H. Oh, J. Am. Chem. Soc., 2017, 139, 17743 CrossRef CAS PubMed
- B. R. Shrestha, S. Pillai, A. Santana, S. H. Donaldson Jr, T. A. Pascal and H. Mishra, J. Phys. Chem. Lett., 2019, 10, 5530 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra00602a |
| This journal is © The Royal Society of Chemistry 2021 |