
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhanced capacitive performance of cathodically reduced titania nanotubes pulsed deposited with Mn2O3 as supercapacitor electrode
Muhammad Muhammad Muzakir ab,
Zulkarnain Zainal*ac,
Hong Ngee Limac,
Abdul Halim Abdullahac and
Noor Nazihah Bahrudina
ab,
Zulkarnain Zainal*ac,
Hong Ngee Limac,
Abdul Halim Abdullahac and
Noor Nazihah Bahrudina
aDepartment of Chemistry, Faculty of Science, Universiti Putra Malaysia, Serdang, Selangor 43400, Malaysia. E-mail: zulkar@upm.edu.my; Tel: +6-03-97696810
bDepartment of Chemistry, Faculty of Science, Gombe State University, Gombe PMB 127, Nigeria
cMaterials Synthesis and Characterization Laboratory, Institute of Advanced Technology, Universiti Putra Malaysia, Serdang, Selangor 43400, Malaysia
First published on 5th August 2021
Abstract
A facile and simple pulse electrodeposition method was employed to deposit Mn2O3 nanoparticles on cathodically reduced titania nanotubes (R-TNTs) at different deposition time in the range of 3–15 min to investigate the influence of mass loading of Mn2O3 on the electrochemical performance of Mn2O3/R-TNTs nanocomposite for supercapacitor application. Mn2O3 nanoparticles were deposited on circumference of R-TNTs as well as in the nanotubes as revealed by FESEM images for all the deposited time. XPS result confirmed the presence of MnO2 (Mn4+) and MnO (Mn2+) on the Mn2O3/R-TNTs composite which provide pseudocapacitive behaviour for the electrode. Mass loading of Mn2O3 increased linearly with deposition time as confirmed by EDX analysis. The sample deposited for 12 min exhibits the highest areal capacitance of 51 mF cm−2 (which is 22 times enhancement over R-TNTs) at a current density of 0.1 mA cm−2 and specific capacitance of 325 F g−1 at 6 A g−1. The sample also show a high-rate capability by retaining 80% of its capacitance even at higher current density of 30 A g−1. Interestingly, it retained 98% of the capacitance over 5000 charge discharge cycles at 10 A g−1 after initial drop to 95% at 200th cycles suggesting an excellent long-term chemical stability. A considerably low equivalent series resistance (ESR) and charge transfer resistance (Rct) of 9.6 Ω and 0.4 Ω respectively was deduced from electrochemical impedance spectroscopy (EIS) analysis indicating good conductivity and improved charge transfer efficiency of Mn2O3/R-TNTs nanocomposite.
Introduction
There is a rapidly increasing demand for energy conversion and storage devices such as supercapacitors, batteries, and fuel cells due to environmental issues posed by fossil fuels and the appearance of serious energy crisis.1,2 Supercapacitors have attracted a wide research attention because of their unique properties, such as fast charge–discharge rates and longer cycle life than batteries, and higher energy density than conventional capacitors. Based on their charge–discharge mechanism, supercapacitors can be majorly categorised into electrochemical double layer capacitors (EDLCs) in which charge storage occurs at the electrode–electrolyte interface and pseudocapacitors where charge storage occurs via fast and reversible surface reactions.3–5 In general, carbon-based materials such as graphene, activated carbon, carbon nanotubes (CNTs), are the most employed electrode material in EDLCs whereas metal oxides such as MnO2, RuO2, NiO, Fe3O4, Co3O2, SnO2 are the frequently used materials in pseudocapacitors.6–14Among the pseudocapacitive material, MnO2 has attracted a great deal of attention because of its low cost, availability, wide electrochemical potential window, environmentally-friendly and high theoretical capacitance value (1370 F g−1).5,15,16 However, the practical capacitance value of MnO2 has deviated far from the theoretical value because of its poor electrical conductivity of 10−5 to 10−6 S cm−1 and densely packed nonporous structure.17,18 To over-come this drawback, considerable efforts were devoted to synthesised nanoscale MnO2 and incorporate it into a more electrically conductive, stable, and highly surface area nanostructured materials such as CNTs,19 carbon nanofibers,20 and titania nanotubes (TNTs).17
Highly ordered TNTs synthesized by the electrochemical anodization method, have received great attention due to their simple fabrication method, excellent controllability, high electrical conductivity (10−5 to 10−2 S cm−1), good chemical stability, and most importantly it offers an extremely large and solvated ions accessible surface area in addition to acting as a binder-free electrode as well as a substrate to support electroactive materials such as MnO2 nanoparticles.5,16,21
Deposition of nanosized MnO2 onto TNTs have numerous advantages to the electrochemical performance of the MnO2/TNTs composite. Contrary to the planar substrate, the nanotubular structure of TNTs offers unique properties conducive for rapid transfer of electrolytes ions during charge discharge processes in the electrode/electrolyte interface. It also provides an increased electronic and ionic sites that will enhance energy density of the MnO2/TNTs composite.4,5
However, it was established that TNTs suffer from a very low areal capacitance less than 1 mF cm−2 due to its n-type semiconductor nature.6 Several strategies have been employed to improve the capacitive performance of the TNTs electrode via thermal treatments to change the amorphous phase to anatase or rutile,22 hydrogenation to increase the donor densities,23 and electrochemical cathodic polarization to induced oxygen vacancies and reduced Ti4+ to Ti3+.24 Significant enhancement in the conductivity and capacitance of TNTs have been achieved through these various modifications which make it a suitable substrate or current collector that could serve as an easy path for fast transportation of ions and electrons, resulting in a reduced internal resistance and improved pseudocapacitive performance.25–27 Thus, attempts on the deposition of MnO2 onto the modified TNTs have been made by hydrothermal,8 sono-chemical,10 and chemical bath deposition,15,21 these methods involved intensive control conditions such as high temperatures or longer preparation time.
Electrodeposition is advantageous compared to chemical solution methods as a facile, simple, and single step method, it can control the composition and thickness of the thin film with good adhesion to the current collector. Moreover, the desired morphology can be easily controlled by adjusting the deposition parameters.4,28 For electrodeposition of MnO2 onto the TNTs, many studies have focused on potentiostatic (chronoamperometry), galvanostatic (chronopotentiometry) or cyclic voltammetry modes of electrodepositions.5,19,20,29,30 However, these approaches resulted in the deposition of MnO2 species only on the top of the TNTs and in some cases causing the deposits to cover the nanotubes openings. This leads to non-utilization of the entire surface of the nanotubes which result in low coverage and specific capacitance values.4,31 Pulse electrodeposition (PED) is another mode of electrodeposition that is considered as a useful technique for the synthesis of novel electroactive materials.32 Smooth, homogenous, and uniform deposits along the elongated tube walls can be obtained by this technique. The physico-chemical properties of materials (such as morphology, electrical conductivity, porosity, adhesion, and compound content) can be controlled through modifying pulse parameters.12 Zhou et al.3 reported pulse current electrodeposition of MnO2 onto cathodically polarized TNTs with the deposit along the nanotubes wall has achieved a specific capacitance of 425 F g−1. Samsudin et al.13 have successfully deposited Mn2O3 onto reduced TNTs via the reverse pulse potential electrodeposition with areal capacitance of 18.32 mF cm−2.
In this study, PED was employed to deposit Mn2O3 nanoparticles onto the R-TNTs. The influence of deposition time on the mass loading of Mn2O3 on R-TNTs which in turn affects the morphology and electrochemical performance of the Mn2O3/R-TNTs composite as electrode in supercapacitor application has been investigated. To specifically demonstrate the role of TNTs in electrochemical performance of the Mn2O3/R-TNTs composite, Mn2O3 was also deposited on the planar Ti foil without the nanotubes.
Materials and methods
Preparation of reduced titania nanotubes (R-TNTs)
Titanium foil (0.25 mm thickness, 99.7% purity, Sigma Aldrich) was cut into 1 × 1 cm2 and cleaned by series of sonication in acetone, isopropanol and deionized (DI) water, for 15 min each followed by chemical etching in 3 M HNO3 (65%, MERCK) for 10 min, and finally rinsed with excess DI water and dried in air. The synthesis of TNTs by anodization was conducted in two-electrode electrochemical cell with titanium foil as the anode and high-density graphite as the cathode at room temperature. The electrolyte used was a mixture of glycerol (99.8% purity, initial water content 0.03 wt%, Fisher Scientific), 0.5 wt% NH4F (FLUKA) and 25 v/v% water. The anodization was carried out at constant voltage of 30 V for 1 h using a DC power supply (Consort Mini, Cleaver Scientific Ltd). A distance of 2 cm was maintained between the two electrodes in all the experiments. Immediately after the anodization, the samples were rinsed with DI water, dried in air, and then calcined at 500 °C under air atmosphere for 2 h at a heating rate of 2 °C min−1. Electrochemical reduction of the sample was conducted in the same electrochemical cell with the anodized sample as the cathode and high-density graphite as anode in 0.5 M Na2SO4 solution as supporting electrolyte at constant voltage of 5 V for 30 s. The sample was rinsed with DI water and dried at room temperature.Preparation of Mn2O3/R-TNTs
PED mode was used for deposition of Mn2O3 onto the R-TNTs. This was conducted in a three-electrode electrochemical cell by employing R-TNTs, Ag/AgCl (3 M KCl) and Pt wire as working, reference and counter electrode respectively using potentiostat–galvanostat (Autolab PGSTAT204/FRA32M module). 5 mM MnSO4 containing 40 mM Na2SO4 was used as deposition electrolyte. Prior to the PED, cyclic voltammetry of the electrolyte was run from −1.0 V to 1.0 V to determine the deposition potential of the Mn2O3. In the PED, one pulse consisted of applying a cathodic potential of −0.6 V for 1 s and −0.2 V for 9 s representing 10% duty cycle. The deposition was conducted for 3, 6, 9, 12 and 15 min and finally the samples were dried in an oven at 80 °C.Material characterization and electrochemical measurements
The morphology and microstructure of the prepared samples were examined by field emission scanning electron microscopy (FESEM, JSM-7600F, JOEL, Japan) equipped with an energy dispersive X-ray spectrometer (EDS). Phase identification and chemical states of the samples were investigated using X-ray diffraction (XRD, Shimadzu, D60000, Japan) with Cu Kα (λ = 1.5406 Å) radiation and X-ray photoelectron spectroscopy (XPS, PHI Quantera II) respectively. The electrochemical performance of the electrode was investigated by cyclic voltammetry (CV), galvanostatic charge–discharge (GCD) and EIS using a conventional three-electrode cell (Auto lab PGSTAT204/FRA32M) in 1 M KCl aqueous solution as electrolyte at room temperature. EIS was conducted at open circuit potential (OPC) in the frequency range of 0.01 Hz to 1 MHz with a perturbation amplitude of 5 mV. The areal capacitance (CA) and specific capacitance (SC) of the samples was calculated based on the GCD curves using eqn (1) and (2), respectively:4,33
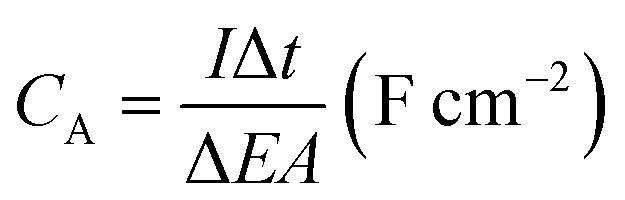 | (1) |
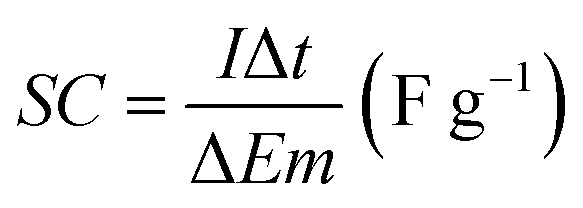 | (2) |
Results and discussion
Fig. 1a shows a CV voltammogram for MnSO4 containing Na2SO4 swept from −1.0 to 1.0 V at a scan rate of 20 mV s−1. A broad reduction peak on the forward scan with a maximum cathodic current at around −0.5 V is associated with the deposition of Mn. Meanwhile on the reverse scan, the highest anodic peak was observed at around 0.40 V which is due to the dissolution of the Mn. Based on the cathodic reduction peak from the CV, −0.6 V was selected as the deposition potential (Von) of Mn. On the other hand, the dissolution potentials (Voff) applied was −0.2 V which is the onset of Mn dissolution corresponding to zero current. Fig. 1b presents a few selected pulses from PED experiment.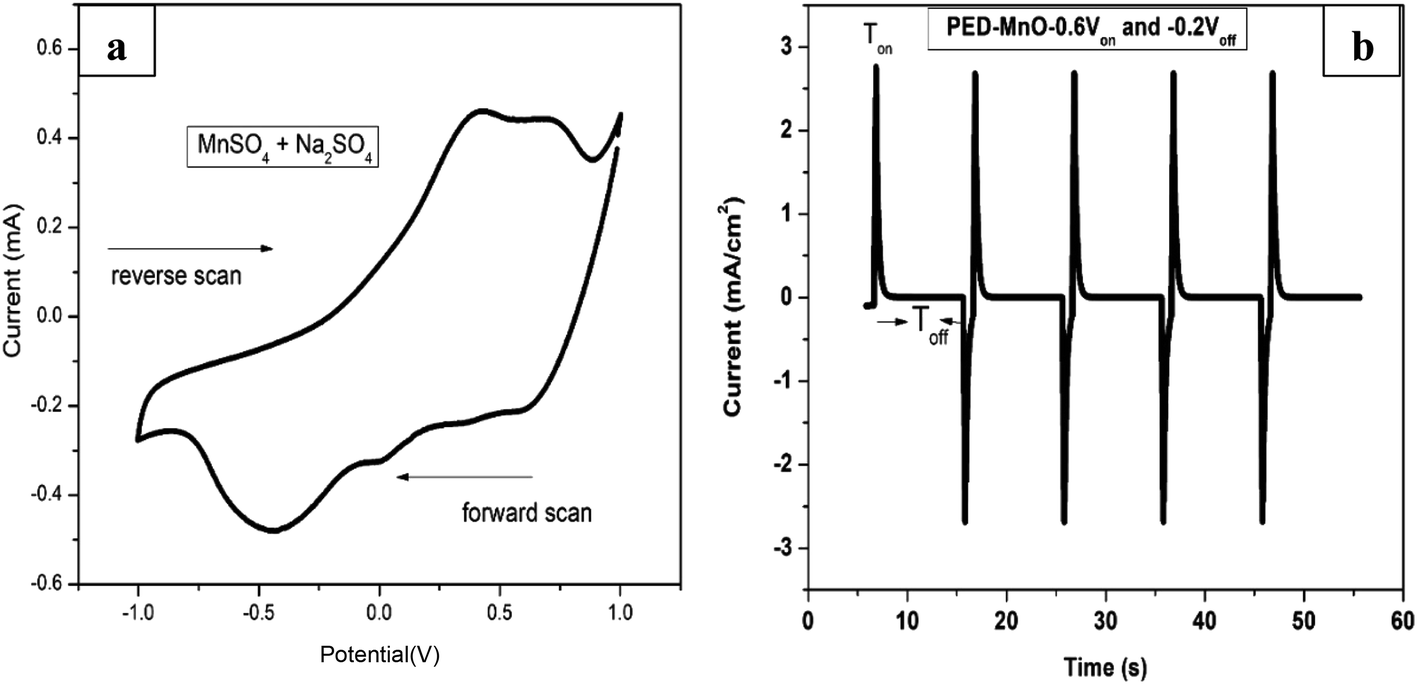 | ||
| Fig. 1 (a) Cyclic voltammogram of MnSO4 containing Na2SO4 at a scan rate of 20 mV s−1 (b) the output of pulse deposition graph for a few selected cycles. | ||
Morphology and composition analyses
Phase identification and elemental composition of the prepared samples were investigated by XRD and XPS. Fig. 2a shows the XRD patterns of TNTs, R-TNTs, and Mn2O3/R-TNTs samples. Peaks at around 35.1°, 38.4°, 40.2°, 53.2°, 63.0°, 71.0°, 76.2° and 77.4° corresponding to hexagonal Ti substrate (JCPDS: 00-044-1294) and peaks at 25.7° and 48.5° which are indexed to (101) and (200) planes of pure anatase phase (JCPDS: 01-075-1537) were found in all the samples. After cathodic reduction, the XRD pattern of the R-TNTs remained the same as the calcined TNTs with slight increment in the intensity of anatase peak. No prominent peak corresponding to the Mn2O3 was observed which may be ascribed to the dominant peaks from the substrate (R-TNTs) which retained its crystalline phase as reported elsewhere.15,21,27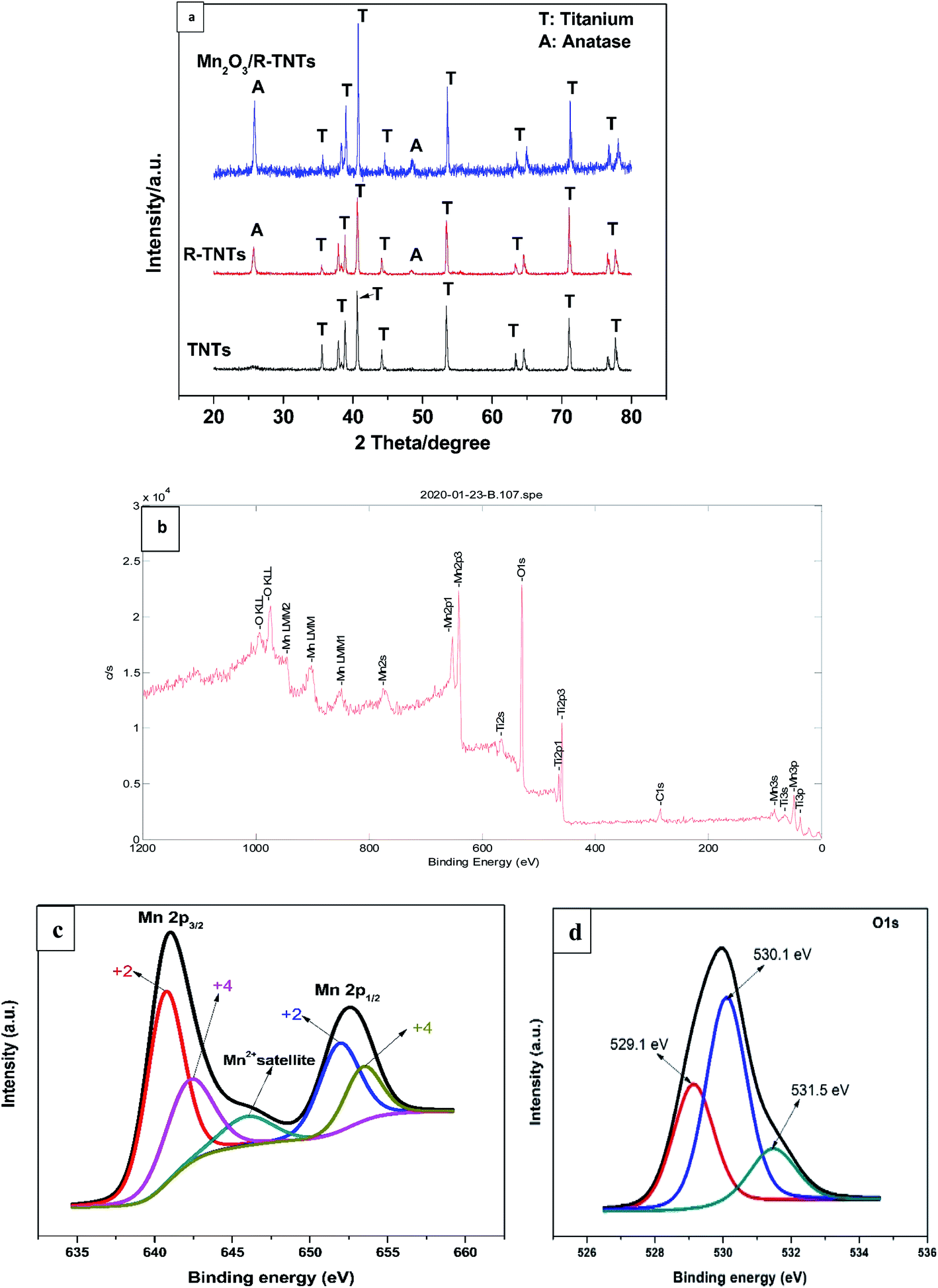 | ||
| Fig. 2 (a) XRD pattern of TNTs, R-TNTs, and Mn2O3/R-TNTs. XPS spectra of Mn2O3/R-TNTs (b) survey spectra (c) Mn 2p and (d) O 1s spectra. | ||
Mn2O3/R-TNTs sample was further characterized by XPS, the survey spectra of the sample depicted in Fig. 2b indicates the presence of four elements (Ti, O, Mn, C) in the sample. The Mn 2p spectra in Fig. 2c shows the presence of two spin–orbit doublets, where one pair of peaks at 640.8 and 651.9 eV are associated with 2p3/2 and 2p1/2 spectra of MnO, another pair of peaks centred at 642.2 and 653.4 eV are associated with 2p3/2, and 2p1/2 spectra of MnO2, respectively.15,18,30,34 This confirmed that there are two types of Mn oxidation states (Mn2+ & Mn4+) in the sample as reported elsewhere.35–38 The peak at around 646 eV could be assigned to MnO satellite feature.14 To confirm the variation of oxidation state of Mn during redox reaction, the O 1s core level spectra was analysed. The O1s spectra deconvoluted into three peaks as shown in Fig. 2d, at around 529.1, 530.1 and 531.5 eV which can be attributed to Mn–O–Mn, Mn–OH, and H–O–H bonds signals, respectively.32,39 The oxidation state of Mn can be determined from the intensities of Mn–O–Mn and Mn–OH signals by the following equation.40
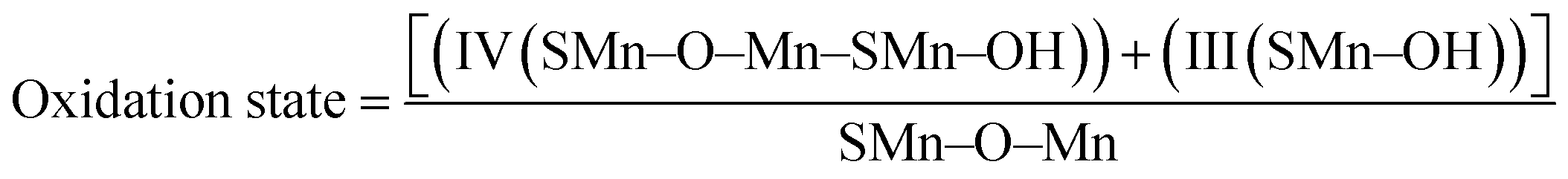 | (3) |
| Mn2+ + 2e− → Mn | (4) |
During Voff, the remaining Mn2+ in the solution is oxidized to Mn3+ and subsequently, the metastable Mn3+ is transformed into an intermediate MnOOH by hydrolysis. Consequently, MnOOH is oxidized to MnO2.
| Mn2+ → Mn3+ + e− | (5) |
| Mn3+ + 2H2O → MnOOH + 3H+ | (6) |
| MnOOH → MnO2 + H+ + e− | (7) |
As the pulse cycle continues, MnO2 may be reduced to intermediate product MnOOH and further to MnO. But MnO is unstable in the presence of oxygen and combine with MnO2 to form Mn2O3.
| MnO2 + H+ + e− → MnOOH | (8) |
| MnOOH + H+ + e− → MnO + H2O | (9) |
| MnO + MnO2 → Mn2O3 | (10) |
Fig. 3a presents the FESEM images of R-TNTs with average inner diameter, wall thickness and tube length (inset) of 86 nm, 21 nm and 1.2 μm, respectively. Mn2O3 was successfully deposited onto the circumference as well as inside the R-TNTs for all the deposition times investigated as shown in Fig. 3b–f. The distinctive change in surface morphology before and after electrodeposition of Mn2O3 can be clearly observed. Obviously, as the deposition time increased the inner diameter of the nanotubes decreases while the wall thickness increases. The tube length become shorter after electrodeposition, for example at 12 min deposition time the average tube length is 850 nm. This may be due to the dense deposit of Mn2O3 formed on the nanotube circumference as the amount and size of Mn2O3 deposit increased with deposition time. For comparison, Mn2O3 was deposited on planar Ti for 12 min. As seen in Fig. 3g and h, the morphology and dimension of Mn2O3 is comparable to the one deposited on top of R-TNTs at 12 min deposition time even though the shape and structure of Mn2O3 deposits are more visible on the planar Ti. However, more uniform distribution of Mn2O3 deposits was obtained on R-TNTs as compared to Ti. This can be ascribed to the nanotubular structure of larger surface area that provides more nucleation sites than the Ti substrate. EDX analysis of Mn2O3/R-TNTs samples deposited at different times revealed the presence of four elements (Ti, O, C, Mn) in the Mn2O3/R-TNTs composite as confirmed by the XPS. The amount of each element in weight% is presented in Table 1.
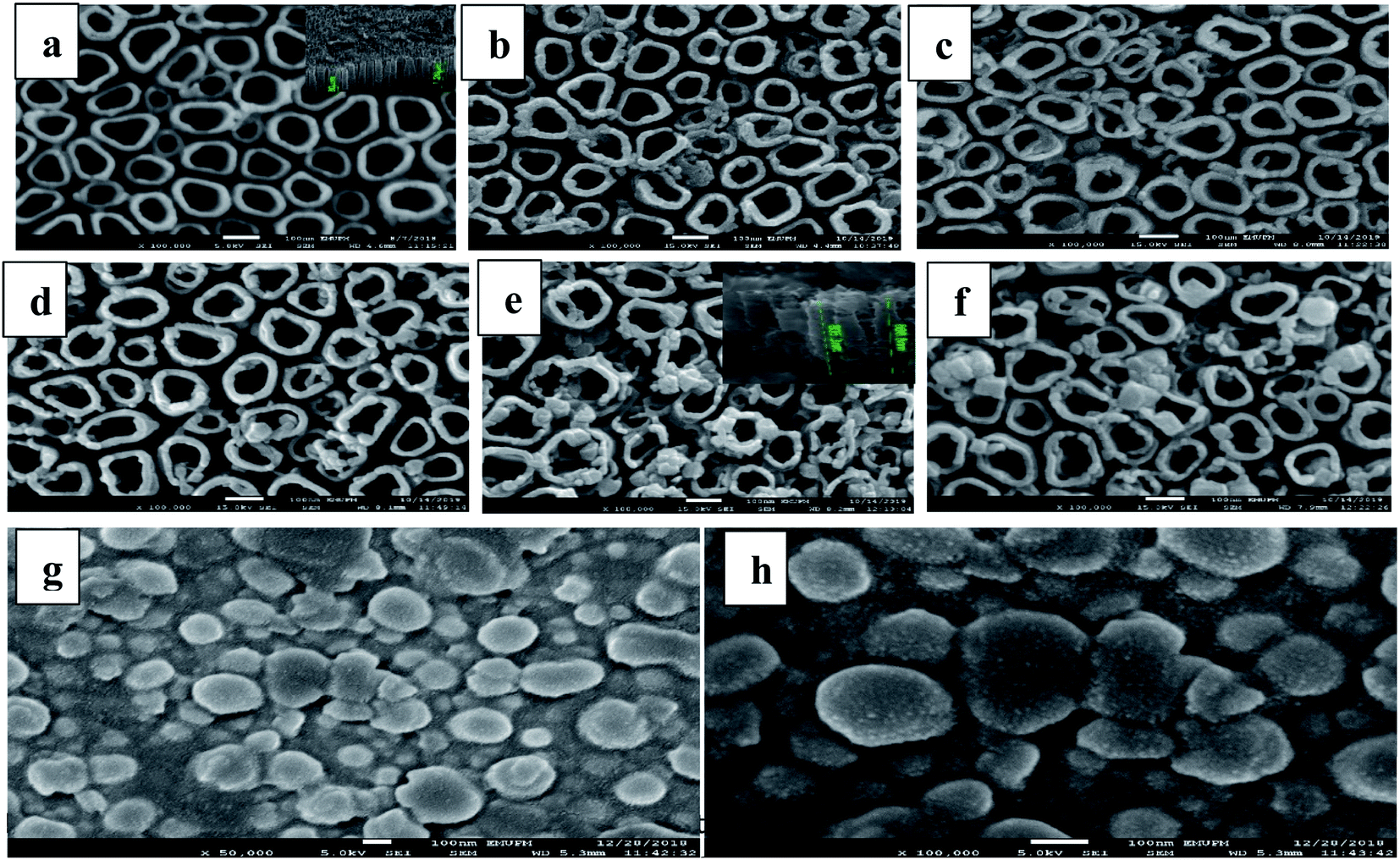 | ||
| Fig. 3 Top view FESEM images of (a) R-TNTs and cross-sectional view (inset), and Mn2O3/R-TNTs electrodeposited at (b) 3 min, (c) 6 min, (d) 9 min, (e) 12 min (inset is cross-sectional view) and (f) 15 min. Mn2O3 deposited on Ti foil for 12 min (g) lower magnification, and (h) higher magnification. | ||
| Deposition time (min) | Weight% | |||
|---|---|---|---|---|
| C | O | Ti | Mn | |
| 3 | 3.25 | 46.85 | 48.18 | 1.71 |
| 6 | 3.13 | 52.36 | 41.93 | 2.58 |
| 9 | 5.48 | 51.70 | 39.40 | 3.42 |
| 12 | 3.37 | 51.93 | 38.80 | 5.90 |
| 15 | 4.04 | 52.10 | 35.96 | 7.90 |
The carbon present in the Mn2O3/R-TNTs sample is from the residual of glycerol used in anodization. The amount of Mn increases as the deposition time is increased as shown in Table 1. This is because of more pulse deposition cycles are applied as deposition time increases which leads to the increase in the amount of Mn deposits.
Electrochemical measurements
The electrochemical measurements of all the synthesised samples were conducted in 1 M KCl within the potential window of 0.0 to 0.8 V vs. Ag/AgCl. The CV profiles for R-TNTs recorded at different scan rates are displayed in Fig. 4a. It shows a symmetrical rectangular curves at all the scan rate measured and maintained the same shape even at high scan rate of 600 mV s−1. This is a typical characteristics of EDLC that exhibits excellent reversibility. The areal capacitance of R-TNTs evaluated at 100 mV s−1 is 3.82 mF cm−2. The CV curves of Mn2O3/R-TNTs synthesised at different deposition time, measured at a scan rate of 5 mV s−1 are presented in Fig. 4b. All the cyclic voltammograms display near symmetrical rectangular shape, which is a typical of a mixture of EDLC and pseudocapacitive behaviour of the electrodes. The redox waves are obvious at 0.35, 0.40 and 0.52 V were due to the transition between Mn4+/Mn2+ on the surface of the electrode.26,37 The presence of both ions was confirmed by XPS. The Mn2O3/R-TNTs sample synthesised for 12 min shows higher integrated area and current response than the other samples which implies higher capacitive performance, therefore, was selected for further evaluation. CV at varying scan rates from 20 mV s−1 to 100 mV s−1 was performed as shown in Fig. 4c. The CV curves show an increased current density with the increase in scan rate, indicating the quasi-reversibility of the redox reactions and thus excellent capacitive behaviour. Even at high scan rate of 100 mV s−1 it still retains its shape and symmetry, indicating good ions diffusion into the electrode material from the electrolyte.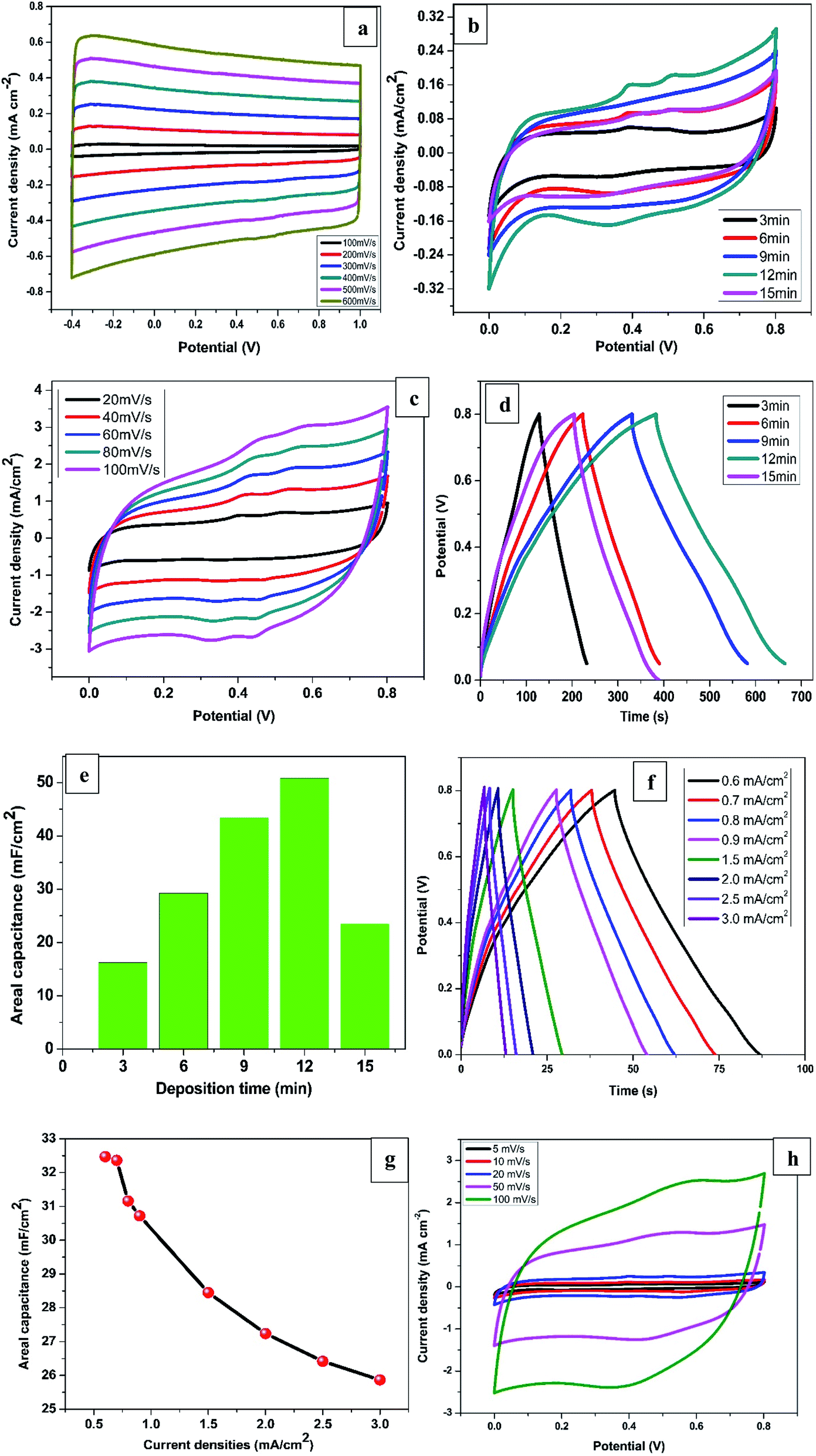 | ||
| Fig. 4 CV curves of (a) R-TNTs at different scan rate, Mn2O3/R-TNTs (b) at different deposition time (c) as a function of scan rate for Mn2O3/R-TNTs synthesised for 12 min. (d) GCD curves of Mn2O3/R-TNTs at different deposition time. (e) Areal capacitance as a function of deposition time. (f) GCD curves of Mn2O3/R-TNTs synthesised for 12 min at different current densities, (g) areal capacitance as a function of current densities and (h) CV curves of Mn2O3/Ti at different scan rate. | ||
To further confirmed the capacitive performance of the samples, GCD measurement was performed at current density of 0.1 mA cm−2 as presented in Fig. 4d. All the GCD curves are linear and triangular suggesting good capacitive behaviour and coulombic efficiency. The sample synthesised for 12 min shows a longer discharging time indicating a higher capacitive performance which agrees with the result from the CV measurement. The areal capacitance as a function of deposition time is shown in Fig. 4e. The value increases from 16.2 mF cm−2 to 50.8 mF cm−2 as deposition time was increased from 3 min to 12 min due to the increase in mass loading of the Mn2O3 as confirmed by EDX. However, the areal capacitance for a sample synthesised for 15 min decreased by more than half (23.5 mF cm−2) despite an increase in Mn2O3 mass loading. This can be ascribed to the increase in size of Mn deposits at 15 min deposition time which leads to partial coverage of nanotubes openings thereby reducing the electrolyte ions diffusion into the film.13
Fig. 4f displays the GCD curves of Mn2O3/R-TNTs synthesised for 12 min at different current densities. The GCD curves retained its shape even at high current density of 3.0 mA cm−2 with small IR drop (0.08 V) indicating excellent capacitive behaviour and good reversibility.41
Interestingly, the sample retained about 80% of the initial capacitance even at high current density of 3.0 mA cm−2 proving high-rate capability.21,28 The areal capacitance as a function of current densities are depicted in Fig. 4g, the decrease in capacitance at higher current densities is as a result of inability of the electrolyte ions to fully accessed the electrode material.15
Fig. 4h displayed the CV curves at different scan rates for Mn2O3 deposited on planar Ti substrate for 12 min to further study the role of the nanotubes in electrochemical performance of the Mn2O3/R-TNTs composite. The areal capacitance of the sample measured at a scan rate of 5 mV s−1 is 35.51 mF cm−2 which is about 30% decrease in capacitance compared to the same sample deposited on R-TNTs. This indicates a superior capacitive performance due to the nanotubular channels that enhanced diffusion of electrolytes ions to the current collector. Furthermore, the CV curve at higher scan rate of 100 mV s−1 for Mn2O3/Ti deviated slightly from rectangular compared to Mn2O3/R-TNTs sample measured at the same scan rate. This can be attributed to good reversibility of the sample due to the rapid electrons transfer across electrode/electrolytes interface as reported elsewhere.16
On the other hand, specific capacitance based on the mass of Mn2O3 obtained by average weight difference using analytical balance (0.1 mg accuracy) before and after electrodeposition for 12 min was evaluated at different current densities. The specific capacitance decreased from 324.7 F g−1 at 6 A g−1 to 258.7 F g−1 at 30 A g−1 as shown in Fig. 5a. The values are greater than most of the previously reported capacitances for MnxOx for different composite materials as shown in Table 2.
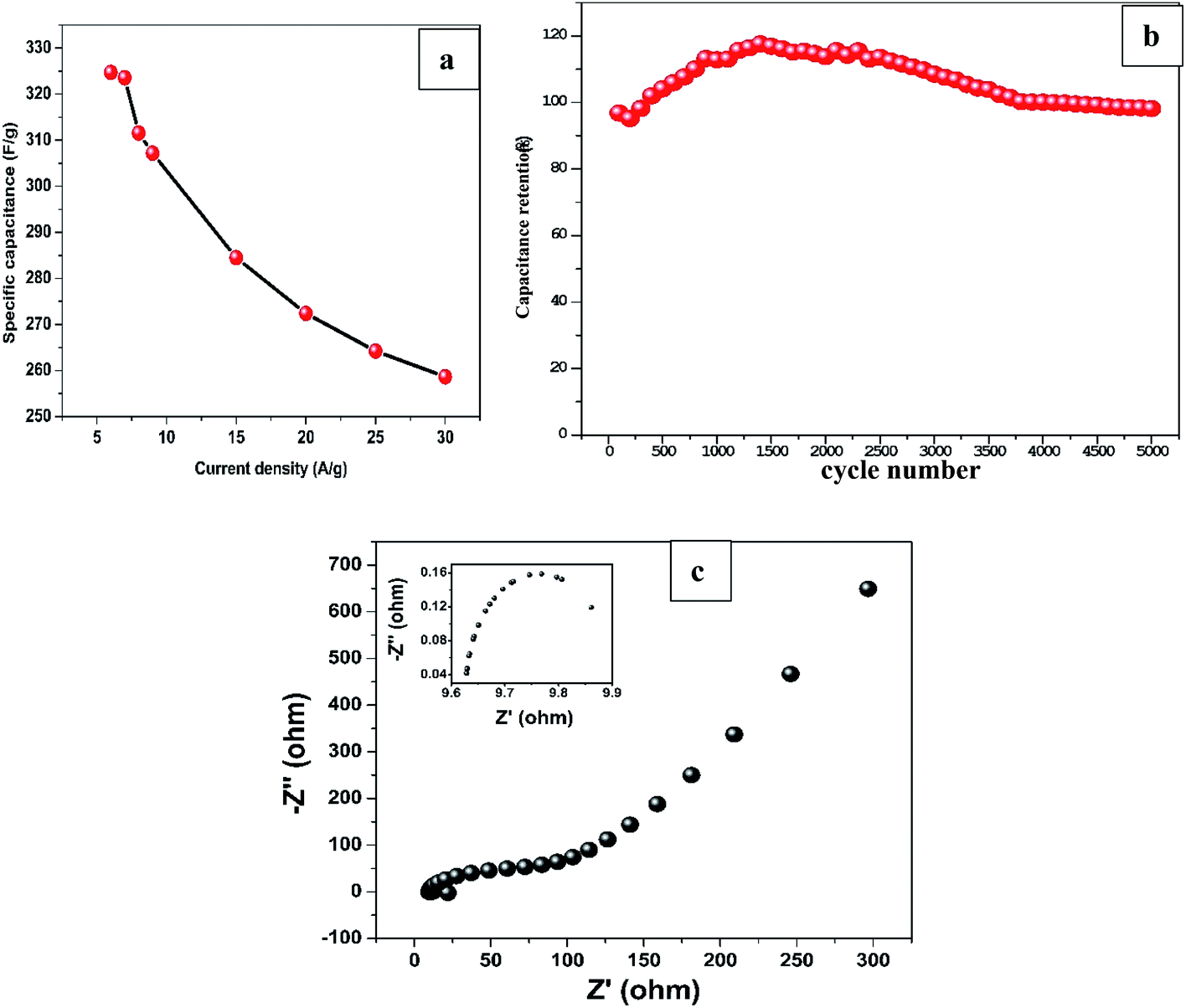 | ||
| Fig. 5 (a) Specific capacitance of Mn3O4/R-TNTs-12 as a function of current density. (b) Cyclic stability of Mn3O4/R-TNTs-12 over 5000 cycles at 1 mA cm−2. (c) Nyquist plot of Mn3O4/R-TNTs-12 with inset showing magnified high frequency region. | ||
| Material | Preparation method | Areal/specific capacitance | Current density/scan rate | Electrolyte | Capacitance retention | Ref. |
|---|---|---|---|---|---|---|
| Graphene/CNT/MnOx | Dispersion & hydrothermal | 210 F g−1 | Not mentioned | 1 M Na2SO4 | 91% after 1000 cycles | 2 |
| MnO2 nanoparticles | Precipitation method | 250 F g−1 | 1 mA cm−2 | 1 M Ca(NO3)2 | 67% after 1000 cycles | 17 |
| Mn3O4 nanoparticles | Cathodic electrodeposition | 416 F g−1 | 1 A g−1 | 1 M NaOH | 47.1% after 1000 cycles | 15 |
| MnO2/PEDOT | Anodic electrodeposition | 159 F g−1 | 5 mV s−1 | 0.5 M Na2SO4 | 91% after 500 cycles | 45 |
| TiO2@MnO2 | Hydrothermal | 320 mF cm−2 | 2 mA cm−2 | 1 M Na2SO4 | 17.7% after 1000 cycles | 18 |
| MnO2–TNTAs | Galvanostatic electrodeposition | 40.4 mF cm−2 | 0.032 mA cm−2 | 0.1 M KOH | 85.6% after 100 cycles | 19 |
| MnO2–TiO2 | SCBD | 175 mF cm−2 | 10 mV s−1 | 0.5 M Na2SO4 | 82.5% after 1000 cycles | 16 |
| MnO2–TNTAs | Pulse current electrodeposition | 425 F g−1 | 0.5 A g−1 | 0.5 M Na2SO4 | 71.4% after 3000 cycles | 3 |
| PPy/MnO2 | Cyclic voltammetry | 596.3 F g−1 | 0.5 A g−1 | 0.5 M Na2SO4 | 87.6% after 1000 cycles | 20 |
| TiO2@MnO2 | Hydrothermal | 22.19 mF cm−2 | 5 mV s−1 | 1 M Na2SO4 | 85% after 4000 cycles | 21 |
| MnO2/GA | Potentiostatic electrodeposition | 410 F g−1 | 2 mV s−1 | 1 M Na2SO4 | 95% after 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles 000 cycles |
22 |
| MnO2 nanoparticles | Pulse current electrodeposition | 290 F g−1 | 5 mV s−1 | 0.5 M Na2SO4 | 72% after 250 cycles | 11 |
| Mn2O3 nanoparticles | Pulsed laser deposition | 210 F g−1 | 1 mV s−1 | 0.1 M Na2SO4 | Not mentioned | 46 |
| MnO2–TiO2 NTs | SILAR | 436.2 mF cm−2 | 0.1 mA cm−2 | 0.5 M Na2SO4 | 85.7% after 3000 cycles | 5 |
| MnO2 microspheres | Solvothermal | 190 F g−1 | 0.5 A g−1 | 1 M Na2SO4 | ∼100% after 1000 cycles | 47 |
| MnOx/TiO2/CFP | Galvanostatic electrodeposition | 327 mF cm−2 | 0.25 mA cm−2 | 1 M Na2SO4 | 96% after 5000 cycles | 23 |
| MnO2–TNT | Sono-chemical | 65 mF cm−2 | 1 mV s−1 | 1 M KCl | 95% after 2500 cycles | 10 |
| MnO2/C | Dispersion | 205 F g−1 | 50 mV s−1 | 2 M NaCl & 0.5 M TBAP/ACN | 98% after 300 cycles | 24 |
| MnOx–CDGs | Sonication & chemical reduction | 280 F g−1 | 1 A g−1 | 1 M Na2SO4 | 94.7% after 10000 cycles |
48 |
| MnO2/AC | Grafting oxidation | 332.6 F g−1 | 2 mV s−1 | 0.5 M Na2SO4 | 87% after 2000 cycles | 25 |
| CQDs/MnO2 | Sono-chemical | 210 F g−1 | 20 A g−1 | 1 M Na2SO4 | 90.3% after 10000 cycles |
49 |
| MnO2 microspheres | Ultrasonic spray pyrolysis | 320 F g−1 | 2.5 mA cm−2 | 1 M LiClO | 98% after 1000 cycles | 50 |
| H–TiO2/C/MnO2 | Hydrothermal | 299.8 F g−1 | 0.5 A g−1 | 1 M Na2SO4 | 87% after 2000 cycles | 8 |
| Mn2O3/R-TNTs | Pulse reverse electrodeposition | 18.32 mF cm−2 | 0.1 mA cm−2 | 1 M KCl | Not mentioned | 13 |
| MnOx | Pulse current electrodeposition | 252 F g−1 | 10 mV s−1 | 3 M KCl | Not mentioned | 12 |
| Mn2O3/R-TNTs | Pulse electrodeposition | 324.72 F g−1 | 6 A g−1 | 1 M KCl | 98% after 5000 cycles | Present work |
To evaluate long-term cycling stability of the sample, GCD test was performed at 1 mA cm−2 which is ∼10 A g−1 current density for 5000 cycles as shown in Fig. 5b. The GCD curves maintained its triangular shape even at 5000th cycle demonstrating excellent capacitive performance and superior reversibility. The sample retained 98% of the capacitance at the end of 5000 cycles after initial decrease to 95% at ∼200th cycle which can be ascribed to the surface activation of the nanostructured material in the electrode induced by electrolyte ions penetration.42,43
EIS was conducted to explore the kinetic properties of Mn2O3/R-TNTs electrode as represented by a Nyquist plot in Fig. 5c. The intercept at real axis at high frequency region represents equivalent series resistance (ESR) which corresponds to the combination of bulk electrolyte resistance, contact resistance at the interface between current collector and active material and internal resistance of the active material. At mid frequency region, the diameter of the semicircle (shown as inset in Fig. 5c) represents the charge transfer resistance (Rct) at the electroactive material/electrolyte interface, while the linear part at low frequency region corresponds to the combination of ion diffusion from electrolyte to the active material and accumulation of charges at the electrode surface.20,44 The sample exhibits ESR and Rct of 9.6 Ω and 0.4 Ω respectively, these considerably low resistance values indicate improved conductivity and charge storage capability of Mn2O3/R-TNTs sample.
Conclusion
A facile and cost-effective PED method was successfully used to deposit Mn2O3 nanoparticles onto R-TNTs at different deposition time to investigate the mass loading of Mn2O3 and its effect on the electrochemical performance of Mn2O3/R-TNTs. Mn2O3 nanoparticles have been uniformly deposited at the nanotubes circumference as shown by FESEM images which allowed for maximum utilization of the nanotubes surface area as pathway for ion diffusion across the current collector–electrolyte interface. The sample exhibited superior capacitive performance over the Mn2O3 deposited on planar Ti at the same deposition time. The mass loading and size of Mn2O3 deposits for Mn2O3/R-TNTs increased with increase in deposition time as revealed by EDX and FESEM analysis, respectively. However, the sample deposited for 12 min recorded the highest capacitance. CV and GCD measurement at different scan rates and current densities revealed pseudocapacitive characteristics, excellent rate capability and good reversibility of Mn2O3/R-TNTs and interestingly, a superior stability of 98% retention over 5000 long-term charging discharging cycles with considerably low ESR and Rct values as deduced from EIS analysis. Therefore, these good electrochemical properties suggest that Mn2O3/R-TNTs composite can serve as a potential electrode for supercapacitor application.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by Putra Grant (UPM/700-2/1/GPB/2017/9555700). M.M.M acknowledges the sponsorship from Tertiary Education Trust Fund (TETFUND) and Gombe State University, Nigeria.References
- M. He, J. Li, W. Xu, Z. Dong, Y. Wu and L. Lv, Nano, 2019, 14, 1–8 CrossRef.
- M. A. A. Mohd Abdah, N. H. N. Azman, S. Kulandaivalu, N. Abdul Rahman, A. H. Abdullah and Y. Sulaiman, J. Power Sources, 2019, 444, 227324 CrossRef CAS.
- H. Zhou, X. Zou and Y. Zhang, Electrochim. Acta, 2016, 192, 259–267 CrossRef CAS.
- J. Li, X. Wang, X. Yu, C. Ma and J. Zhao, Int. J. Hydrogen Energy, 2016, 41, 22162–22170 CrossRef CAS.
- Z. Li, X. Wang, X. Wang, T. Xiao, L. Zhang, P. Lv and J. Zhao, Int. J. Hydrogen Energy, 2018, 43, 8859–8867 CrossRef CAS.
- M. Salari, K. Konstantinov and H. K. Liu, J. Mater. Chem., 2011, 21, 5128–5133 RSC.
- M. Salari, S. H. Aboutalebi, A. T. Chidembo, I. P. Nevirkovets, K. Konstantinov and H. K. Liu, Phys. Chem. Chem. Phys., 2012, 14, 4770–4779 RSC.
- J. Di, X. Fu, H. Zheng and Y. Jia, J. Nanopart. Res., 2015, 17, 255, DOI:10.1007/s11051-015-3060-z.
- N. A. Samsudin, Z. Zainal, H. N. Lim, Y. Sulaiman, S. K. Chang, Y. C. Lim and W. N. Mohd Amin, J. Nanomater., 2018, 9509126, DOI:10.1155/2018/9509126.
- H. R. Barai, A. N. Banerjee, F. Bai and S. W. Joo, J. Ind. Eng. Chem., 2018, 62, 409–417 CrossRef CAS.
- H. Adelkhani and M. Ghaemi, J. Alloys Compd., 2010, 493, 175–178 CrossRef CAS.
- F. Xiao and Y. Xu, Int. J. Electrochem. Sci., 2012, 7, 7440–7450 CAS.
- N. A. Samsudin, Z. Zainal, H. N. Lim, Y. Sulaiman, S. K. Chang, Y. C. Lim, A. K. Ayal and W. N. Mohd Amin, RSC Adv., 2018, 8, 23040–23047 RSC.
- H. A. El-Fattah, I. El-Mahallawi, M. Shazly and W. Khalifa, in Energy Technology, the Minerals, Metals & Materials Series, ed. T. Wang, et al., Springer, 2019, part III, pp. 253–263, DOI: DOI:10.1007/978-3-030-06209-5_2.
- T. Nguyen, M. João Carmezim, M. Boudard and M. Fátima Montemor, Int. J. Hydrogen Energy, 2015, 40, 16355–16364 CrossRef CAS.
- H. Zhou and Y. Zhang, J. Power Sources, 2014, 272, 866–879 CrossRef CAS.
- R. Aswathy, Y. Munaiah and P. Ragupathy, J. Electrochem. Soc., 2016, 163, A1460–A1468 CrossRef.
- X. Y. Liu, M. Zhou, C. Chen and Y. X. Zhang, Ceram. Int., 2017, 43, 10595–10600 CrossRef CAS.
- Y. Huang, X. Zhang, Xi. Chen, H. Wang, J. Chen, X. Zhong and Q. Li, Int. J. Hydrogen Energy, 2015, 40, 14331–14337 CrossRef CAS.
- M. He, Y. Zheng and Q. Du, Nano, 2013, 8, 1–7 Search PubMed.
- A. Ramadoss and S. J. Kim, Int. J. Hydrogen Energy, 2014, 39, 12201–12212 CrossRef CAS.
- C. C. Wang, H. C. Chen and S. Y. Lu, Chem.–Eur. J., 2014, 20, 517–523 CrossRef CAS PubMed.
- L. Li, X. Zhang, G. Wu, X. Peng, K. Huo and P. K. Chu, Adv. Mater. Interfaces, 2015, 2, 1–10 CAS.
- V. M. Tran, A. T. Ha and M. L. P. Le, Adv. Nat. Sci.: Nanosci. Nanotechnol., 2014, 5, 025005, DOI:10.1088/2043-6262/5/2/025005.
- J. W. Wang, Y. Chen and B. Z. Chen, J. Electrochem. Soc., 2015, 162, A1654–A1661 CrossRef CAS.
- M. Krajewski, P. Y. Liao, M. Michalska, M. Tokarczyk and J. Y. Lin, J. Energy Storage, 2019, 26, 101020 CrossRef.
- A. Y. Lo, L. Saravanan, C. M. Tseng, F. K. Wang and J. T. Huang, ACS Omega, 2020, 5, 578–587 CrossRef CAS.
- I. I. Gurten Inal and Z. Aktas, Appl. Surf. Sci., 2020, 514, 145895 CrossRef CAS.
- M. Hakamada, A. Moriguchi and M. Mabuchi, J. Power Sources, 2014, 245, 324–330 CrossRef CAS.
- M. Elrouby, A. M. Abdel-Mawgoud and R. A. El-Rahman, J. Mol. Struct., 2017, 1147, 84–95 CrossRef CAS.
- M. H. Pham, A. Khazaeli, G. Godbille-Cardona, F. Truica-Marasescu, B. Peppley and D. P. J. Barz, J. Energy Storage, 2020, 28, 101210 CrossRef.
- W. Cong, R. Miao, F. Miao and B. Tao, Mater. Res. Express, 2017, 40, 245–257 Search PubMed.
- A. Moses Ezhil Raj, S. G. Victoria, V. B. Jothy, C. Ravidhas, J. Wollschläger, M. Suendorf, M. Neumann, M. Jayachandran and C. Sanjeeviraja, Appl. Surf. Sci., 2010, 256, 2920–2926 CrossRef CAS.
- D. Wang, Y. Li, Q. Wang and T. Wang, Eur. J. Inorg. Chem., 2012, 628–635 CrossRef CAS.
- G. An, P. Yu, M. Xiao, Z. Liu, Z. Miao, K. Ding and L. Mao, Nanotechnology, 2008, 19, 275709, DOI:10.1088/0957-4484/19/27/275709.
- J. Zhang, Y. Wang, Y. Qin, C. Yu, L. Cui, X. Shu, J. Cui, H. Zheng, Y. Zhang and Y. Wu, J. Solid State Chem., 2017, 246, 269–277 CrossRef CAS.
- Y. Xu, L. Wang, Y. Zhou, J. Guo, S. Zhang and Y. Lu, J. Electroanal. Chem., 2019, 852, 113507 CrossRef CAS.
- J. Xu, K. Hou, Z. Ju, C. Ma, W. Wang, C. Wang, J. Cao and Z. Chen, J. Electrochem. Soc., 2017, 164, A430–A437 CrossRef CAS.
- R. Tholkappiyan, A. N. Naveen, K. Vishista and F. Hamed, J. Taibah Univ. Sci., 2018, 12, 669–677 CrossRef.
- S. Yang, K. Cheng, J. Huang, K. Ye, Y. Xu, D. Cao and X. Z. G. Wang, Electrochim. Acta, 2014, 120, 416–422 CrossRef.
- N. Ain, M. Amirul, A. Mohd, N. Hawa, N. Azman and Y. Sulaiman, J. Electroanal. Chem., 2020, 867, 114188 CrossRef.
- A. E. Elkholy, F. E. Heakal and N. K. Allam, RSC Adv., 2017, 7, 51888–51895 RSC.
- W. Teng, X. Wang, T. Wang, L. Su, T. Tesfamichael and F. Yu, J. Alloys Compd., 2019, 803, 950–957 CrossRef.
- M. P. Clark, W. Qu and D. G. Ivey, ECS Trans., 2015, 64, 57–67 CrossRef CAS.
- T. A. Ha, V. M. Tran and M. L. P. Le, Adv. Nat. Sci.: Nanosci. Nanotechnol., 2013, 4, 035004, DOI:10.1088/2043-6262/4/3/035004.
- D. Yang, J. Power Sources, 2013, 228, 89–96 CrossRef CAS.
- W. Y. Ko, L. J. Chen, Y. H. Chen and K. J. Lin, J. Mater. Res., 2014, 29, 107–114 CrossRef CAS.
- B. Unnikrishnan, C. W. Wu, I. W. P. Chen, H. T. Chang, C. H. Lin and C. C. Huang, ACS Sustainable Chem. Eng., 2016, 4, 3008–3016 CrossRef CAS.
- R. Thangappan, M. Arivanandhan, R. Dhinesh Kumar and R. Jayavel, J. Phys. Chem. Solids, 2018, 121, 339–349 CrossRef CAS.
- Y. Zhang, L. A. Huff, A. A. Gewirth and K. S. Suslick, Part. Part. Syst. Charact., 2015, 32, 899–906 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |