
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNanostructured RuO2–Co3O4@RuCo-EO with low Ru loading as a high-efficiency electrochemical oxygen evolution catalyst†
Lingjun Tan,
Ailian Zhang,
Ziyi Liu,
Ping'an Wei,
Panpan Yang,
Huan Guo,
Hua Fang,
Juanjuan Han,
Yuchan Zhu * and
Zhandong Ren
* and
Zhandong Ren
School of Chemical and Environmental Engineering, Wuhan Polytechnic University, Wuhan, 430023, P. R. China. E-mail: zhuyuchan@163.com
First published on 23rd March 2021
Abstract
Electrochemical water splitting technology is considered to be the most reliable method for converting renewable energy such as wind and solar energy into hydrogen. Here, a nanostructured RuO2/Co3O4–RuCo-EO electrode is designed via magnetron sputtering combined with electrochemical oxidation for the oxygen evolution reaction (OER) in an alkaline medium. The optimized RuO2/Co3O4–RuCo-EO electrode with a Ru loading of 0.064 mg cm−2 exhibits excellent electrocatalytic performance with a low overpotential of 220 mV at the current density of 10 mA cm−2 and a low Tafel slope of 59.9 mV dec−1 for the OER. Compared with RuO2 prepared by thermal decomposition, its overpotential is reduced by 82 mV. Meanwhile, compared with RuO2 prepared by magnetron sputtering, the overpotential is also reduced by 74 mV. Furthermore, compared with the RuO2/Ru with core–shell structure (η = 244 mV), the overpotential is still decreased by 24 mV. Therefore, the RuO2/Co3O4–RuCo-EO electrode has excellent OER activity. There are two reasons for the improvement of the OER activity. On the one hand, the core–shell structure is conducive to electron transport, and on the other hand, the addition of Co adjusts the electronic structure of Ru.
Introduction
Hydrogen is an ideal green energy carrier to replace fossil fuels. It has the characteristics of high energy density, storage, and no carbon emissions. Electrochemical water splitting technology is considered to be the most reliable method for converting renewable energy such as wind and solar energy into hydrogen. It is a very promising, sustainable, clean and efficient energy application strategy.1–10 The electrolysis of water contains two half reactions, the oxygen evolution reaction (OER) at the anode and the hydrogen evolution reaction (HER) at the cathode. The main challenge of this technique is to reduce the excessive overpotential of HER and OER. The high overpotential of the OER reaction is the most important factor affecting the efficiency of water electrolysis.11–20 Up to now, despite a lot of research, RuO2 is still considered to be the most active electrocatalyst for OER. However, its overpotential is still not low enough, and it has disadvantages such as poor stability and high price, which still needs to be further improved.21–25The introduction of abundant transition metal elements can improve the composition and structural of electrocatalysts, thereby improving OER activity and reducing costs.26–30 Early literature reports have proved that the doping/alloying of transition metals such as Fe, Co, Ni, and Cu can optimize the local electronic and geometric structure of the original metal through the strain/ligand effect.31–34 These changes will facilitate the chemical adsorption of intermediate adsorbed species (OH* and OOH*) on the surface of the electrocatalyst, which can increase the catalytic activity in an alkaline solution.35 At the same time, all intermediate products interact with the metal oxide surface through oxygen atoms, and the bonding interaction (M–O) in the intermediate products (M–OH, M–O, and M–OOH) is very important for the overall OER activity. At present, more researches are focused on introducing metals into metal oxides, which can generate oxygen vacancies, thus regulating the adsorption of oxygen-containing intermediates at active sites. At the same time, the integrated electrocatalyst of metal and semiconductor can benefit the charge transfer process, that is, Mott–Schottky effect.
Previous literatures have shown that by changing the electronic structure of Ru, RuCo alloy can exhibit better OER activity.36,38,40 At the same time, related literature has also proven that the rich metal–insulator interface structure is beneficial to improve the OER activity of Ru.37,39 Therefore, it is hoped that both the Mott–Schottky effect will improve the charge transfer rate and the alloying will also optimize the electronic structure of Ru. This paper pays more attention to the preparation of the RuCo alloy base and the construction of the special surface morphology in the research method. At present, for the preparation of this kind of catalyst, hydrothermal method combined with high-temperature calcination method is commonly used. But it has the disadvantages of long preparation time, high loading capacity, and the morphology of the obtained catalysts is mostly nanoparticles. Based on the above analysis, a new preparation method, that is, magnetron sputtering combined with electrochemical oxidation is investigated in this article. The method has the characteristics of short preparation time and low loading capacity. The prepared RuO2/Co3O4–RuCo-EO electrode has a nanosheet and nanorod structure with Ru loading of 0.064 mg cm−2, resulting in excellent OER activity with a low overpotential of only 220 mV at the current density of 10 mA cm−2 and Tafel slope of 59.9 mV dec−1 for OER.
Experimental
Electrode preparation
The Ti plate processed above was placed on the sample stage of the vacuum chamber. During the experiment, a Ru target with a purity of 99.99% and a Co target with a purity of 99.99% were used as sputtering targets. Evacuate the vacuum chamber to 4 × 10−4 Pa before sputtering. Then high-purity Ar gas was introduced, and the flow rate of argon gas was 30 mL min−1. Adjust the pressure of the vacuum chamber to 1.0–1.2 Pa, and then the Ru target and the Co target were connected to the DC power supply for co-sputtering at the same time. The sputtering power of the Ru target was 60 W and the sputtering power of the Co target was 80 W. The co-sputtering was performed for 10 min. After the sputtering was completed, the resulting sample was recorded as the RuCo alloy catalyst. Next, it was calcined in a muffle furnace at 400 °C for 2 h, and the resulting sample was recorded as the RuO2/Co3O4–RuCo catalyst.The above RuO2/Co3O4–RuCo catalyst was subjected to electrochemical oxidation (EO) process in a three-electrode system with the electrolyte 1.0 mol L−1 KOH solution. Before the EO process, adjust the temperature of the solution to 25 °C, and then pour argon gas with a purity of 99.999% into the solution for 15 minutes to remove dissolved oxygen from the solution. Then, using the RuO2/Co3O4–RuCo catalyst as the working electrode, a multi-segment cyclic voltammetry (CV) scan was performed. The scanning range is 0–1.4 V (vs. RHE) and the scan rate is 100 mV s−1. After the EO process, the obtained sample is recorded as RuO2/Co3O4–RuCo-EO electrode. Among them, the samples with the number of CV scan of 200, 400, 600, 800 and 1000 segments are denoted as RuO2/Co3O4–RuCo-EO-x (x = 200, 400, 600, 800 and 1000) electrodes. The preparation process of RuO2/Ru and RuO2 electrodes is described in ESI.†
Material characterization
X-ray diffraction (XRD) patterns were acquired on an XRD-7000 X-ray diffractometer. Scanning Electron Microscope (SEM) images were taken with a Zeiss ΣIGMA field-emission SEM. X-ray photoelectron spectrometry (XPS: ESCALAB 250Xi, Thermo Scientific) with monochromatized Al Kα radiation was used to analyze the electronic properties. Analysis of the composition of the electrode was carried out by X-ray fluorescence (XRF: EDX-7000, Shimadzu, Japan).Electrochemical measurements
In the electrochemical experiment, a three-electrode system was used for testing on the CHI660 instrument. Among them, the working electrode are RuO2/Co3O4–RuCo-EO, RuO2/Co3O4–RuCo RuO2/Ru and RuO2 electrodes. In this paper, the Ru loading is about 0.064 mg cm−2 for different types of catalysts. The counter electrode is a carbon paper electrode. In order to avoid electrolyte contamination, the reference electrode is HgO/Hg/KOH (1.0 mol L−1). The electrochemical characteristics of electrode materials were analyzed using cyclic voltammetry (CV) technology in 1.0 mol L−1 KOH solution. The OER activity was characterized by linear voltammetry scanning (LSV) in 1.0 mol L−1 KOH solution at a scanning speed of 5 mV s−1 in range of 1.2–1.8 V.Results and discussion
Fig. 1 shows the different surface morphologies of RuO2/Co3O4–RuCo before and after EO treatment. Fig. 1a and b are the scanning electron microscope (SEM) images of RuO2/Co3O4–RuCo without EO treatment. It can be seen that the surface of the electrode is very smooth and the Ru and Co elements of RuO2/Co3O4–RuCo electrode are evenly distributed (Fig. S1†). This is because the nanoclusters prepared by magnetron sputtering are very small, neatly arranged, and have good film-forming properties. However, after EO treatment, part of the cobalt on the electrode surface was oxidized and stripped at a high potential, resulting in a great change in the surface morphology. As shown in Fig. 1c and d, after CV scanning of 200 segments, a large number of two-dimensional nanosheets with a width of 200 nm and a thickness of 10 nm are formed on the surface of the RuO2/Co3O4–RuCo-EO-200 electrode. After CV scanning of 1000 segments, most of the two-dimensional nanosheets were broken up, leaving a large number of small one-dimensional nanorods with a length of 100 nm on the surface of the RuO2/Co3O4–RuCo-EO-1000 electrode (Fig. 1e and f). This can prove that the degree of oxidative exfoliation becomes more obvious as the number of CV cycles increases. In addition, the composition of Ru and Co of the electrodes before and after EO treatment is listed in Table S1.† Before EO treatment, the molar ratio of Ru and Co is 56![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :44. After CV scanning of 200, 400, 600, 800, and 1000 segments, the molar ratios of Ru and Co are 61:39, 66:34, 67:33, 67:33 and 65:35 respectively. This indicates that the EO treatment can reduce the Co content to a certain extent, but the composition ratio of Ru–Co under different CV cycles does not change much, but the morphology changes greatly.
:44. After CV scanning of 200, 400, 600, 800, and 1000 segments, the molar ratios of Ru and Co are 61:39, 66:34, 67:33, 67:33 and 65:35 respectively. This indicates that the EO treatment can reduce the Co content to a certain extent, but the composition ratio of Ru–Co under different CV cycles does not change much, but the morphology changes greatly.
 | ||
| Fig. 1 SEM images of RuO2/Co3O4–RuCo (a and b), RuO2/Co3O4–RuCo-EO-200 (c and d) and RuO2/Co3O4–RuCo-EO-1000 electrodes (e and f). | ||
Fig. 2 are the X-ray diffraction patterns (XRD) of RuO2/Co3O4–RuCo-EO, accompanied by the comparison of those of RuO2/Co3O4–RuCo and RuO2/Ru. For RuO2/Ru electrode, the diffraction peak at 44.0° is corresponding to the Ru(101) crystal plane (Fig. 2d), and the diffraction peak at 28.1° corresponds to the RuO2(110) crystal plane (Fig. 2b). For RuO2/Co3O4–RuCo electrode, the RuO2(110) crystal plane is still observed at 28.1°. Meanwhile, due to the addition of Co, the (111) and (311) planes of Co3O4 are observed at 18.9° and 36.7° (Fig. 2a and c), respectively. However, these diffraction peaks above cannot be observed for RuCo electrode without thermal oxidation (Fig. S2†). Therefore, it can be confirmed that the RuCo alloy is first formed on the substrate by magnetron sputtering, and then the RuO2/Co3O4–RuCo with a core–shell structure is obtained on the electrode surface with thermal oxidation of 400 °C. In addition, Ru and Co have formed bulk alloy and the characteristic crystal plane of RuCo alloy is located at 45.3° (Fig. S2†). After thermal oxidation and electrochemical oxidation, the peak of RuCo alloy is negatively shifted to 44.6° (Fig. 2d), which is still significantly positively shifted by 0.6° relative to the Ru(101) crystal plane. After EO treatment, no diffraction peak of Co3O4(111) crystal plane was found in the XRD pattern of RuO2/Co3O4–RuCo-EO electrode, and the diffraction peak intensity of Co3O4(311) crystal plane was also not obvious. This is because Co3O4 on the surface of RuO2/Co3O4–RuCo-EO electrode was oxidized and peeled off after EO treatment. At the same time, in order to verify the effect of EO treatment on the dissolution of Co on the surface, the XRD spectra of RuO2/Co3O4–RuCo-EO after 200, 400, 600, 800 and 1000 segments of CV scans were also analyzed in Fig. 3. It is found that with the increase of the number of CV scan, the diffraction peak of Co3O4(111) crystal plane gradually weakens and disappears (Fig. 3a), which indicates that the number of CV scan is related to the degree of Co dissolution. Although the EO treatment has a significant effect on the structure of Co, it has no significant effect on the stable RuO2 and RuCo alloy structure. The diffraction peaks of RuO2(110) and RuCo alloy in the RuO2/Co3O4–RuCo-EO electrode are still at 28.1° and 44.6° (Fig. 3b and d).
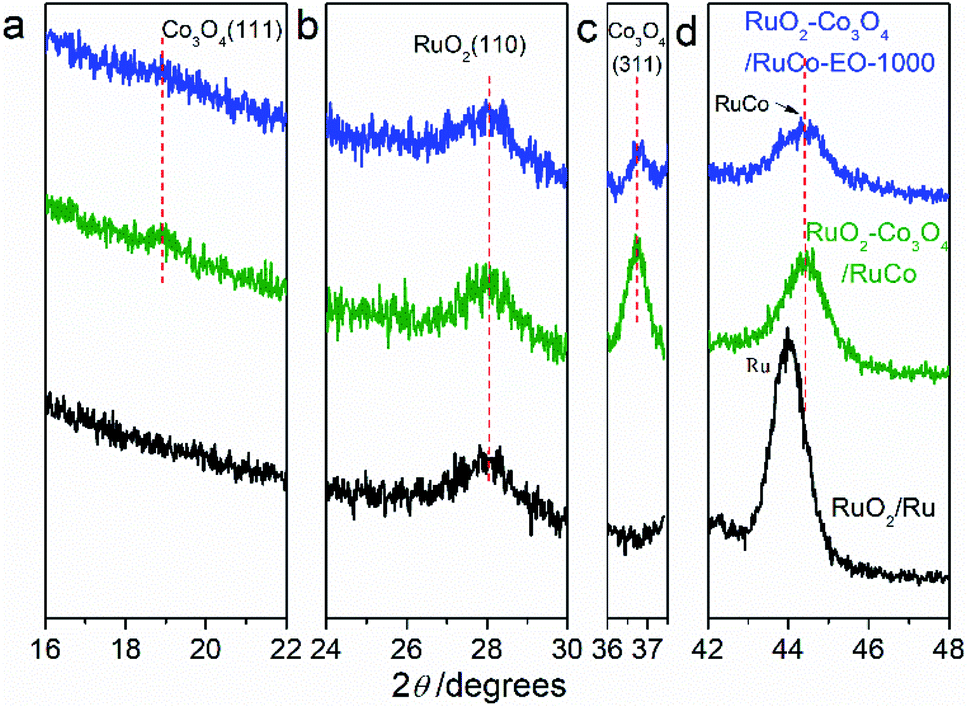 | ||
| Fig. 2 X-ray diffraction patterns of RuO2/Co3O4–RuCo-EO, RuO2/Co3O4–RuCo and RuO2/Ru electrodes. (a) Co3O4(111), (b) RuO2(110), (c) Co3O4(311), (d) RuCo or Ru(101). | ||
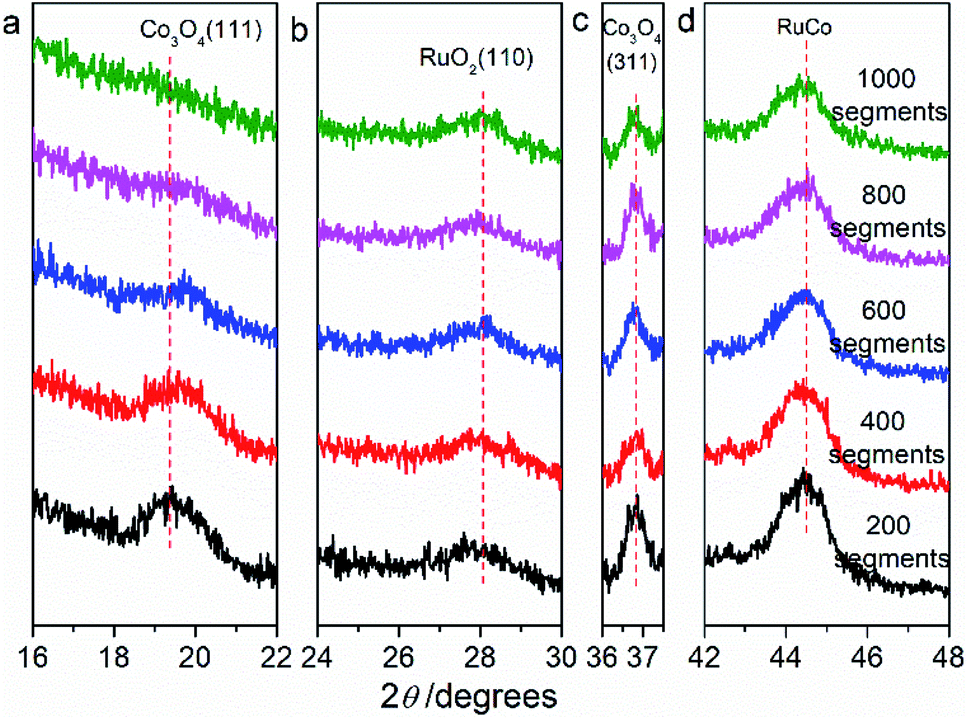 | ||
| Fig. 3 X-ray diffraction patterns of RuO2/Co3O4–RuCo-EO electrodes after CV scan of 200, 400, 600, 800 and 1000 segments. (a) Co3O4(111), (b) RuO2(110), (c) Co3O4(311), (d) RuCo. | ||
Fig. 4 are high-resolution X-ray photoelectron spectroscopy (XPS) spectra of RuO2/Ru and RuO2/Co3O4–RuCo-EO electrodes. As for RuO2/Ru in Fig. 4a, the peaks at binding energies (BEs) of 484.5 and 462.2 eV are corresponding to the spin splitting peaks of 3p1/2 and 3p3/2 orbitals of Ru0. Meanwhile, the peaks at 486.8 and 464.6 eV belong to the Ru4+ 3p1/2 and 3p3/2 orbitals, respectively. For RuO2/Co3O4–RuCo-EO, the peaks position of Ru4+ spin orbitals are the same as those of RuO2/Ru. However, the content of Ru4+ in RuO2/Co3O4–RuCo-EO electrode is significantly higher than that of RuO2/Ru electrode, indicating a higher degree of surface oxidation. In addition, the spin splitting peaks of Ru0 3p1/2 and 3p3/2 orbitals of RuO2/Co3O4–RuCo-EO are located at 484.5 and 462.2 eV, respectively, which are positively shifted by 0.3 eV from those of RuO2/Ru. In the high-resolution XPS spectrum of Co 2p of RuO2/Co3O4–RuCo-EO (Fig. 4b), two peaks at binding energies (BEs) of 779.7 (781.1) and 794.6 (796.1) eV are corresponding to the spin orbitals of Co3+ (Co2+) 2p3/2 and Co 2p1/2, accompanied by two weak satellite peaks at 789.1 and 804.7 eV. In Fig. 4c, the peak of 529.3 eV, 530.6 eV and 531.9 eV of RuO2/Ru is attributed to O 1s orbital in O2−, OH− and H2O, respectively. The peak of 529.8 eV, 531.0 eV and 532.1 eV of RuO2/Co3O4–RuCo-EO is attributed to O 1s orbital in O2−, OH− and H2O, respectively.
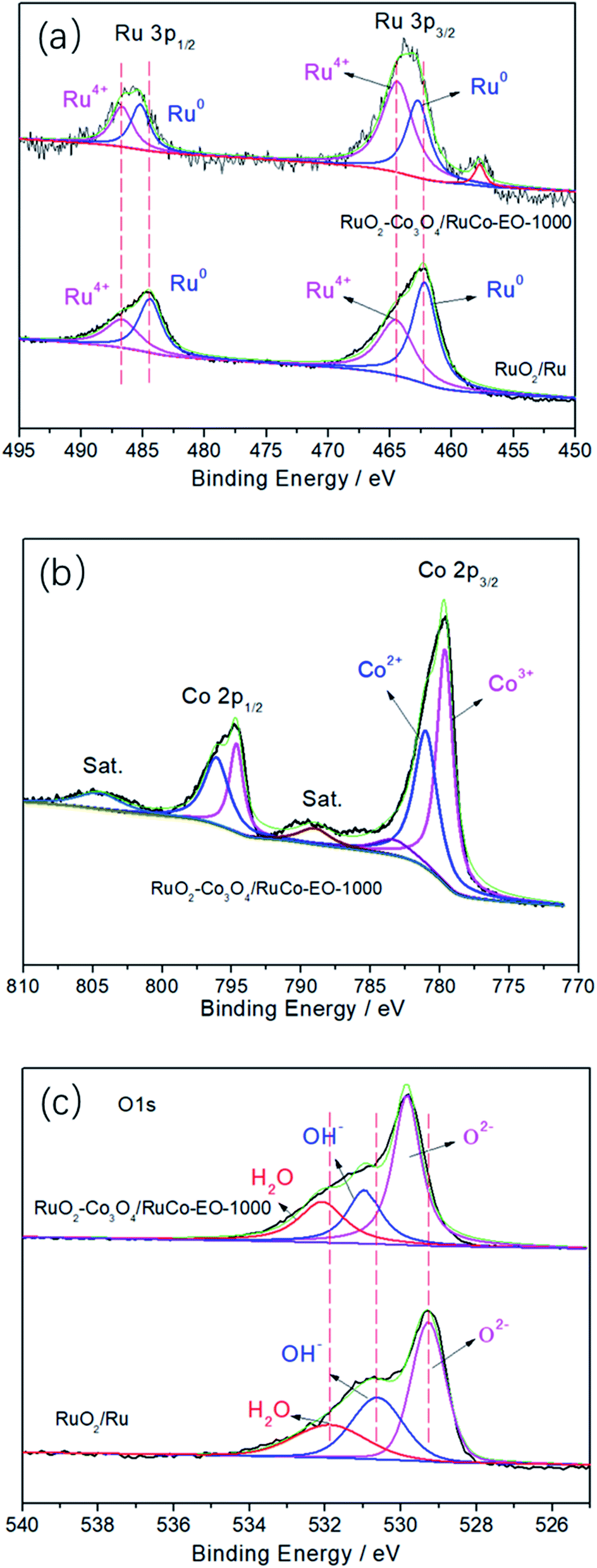 | ||
| Fig. 4 High-resolution XPS spectra of Ru 3p (a), Co 2p (b) and O 1s (c) in the RuO2/Co3O4–RuCo-EO electrode compared with those in RuO2/Ru electrode. | ||
Fig. 5 is the cyclic voltammetry (CV) curves of RuO2/Ru, RuO2/Co3O4–RuCo and RuO2/Co3O4–RuCo-EO electrodes. First of all, it can be clearly seen that the double-layer current of the RuO2/Co3O4–RuCo electrode is obviously lower than that of RuO2/Ru, indicating that the electrochemical surface area (ECSA) of the RuO2/Co3O4–RuCo electrode is small. The reason is that the surface of RuO2/Co3O4–RuCo electrode obtained by high vacuum magnetron co-sputtering technology is extremely flat and smooth, as shown by SEM characterization in Fig. 1. However, after the EO treatment, the double-layer current of the RuO2/Co3O4–RuCo-EO electrode has increased significantly, which is significantly larger than those of the other two electrodes. This indicates that the RuO2/Co3O4–RuCo-EO electrode has a larger ECSA and can provide more electrochemically active sites. In addition, it can be seen from the Fig. 5 that the electrochemical characteristics of the RuO2/Co3O4–RuCo-EO electrode are also obvious. Among them, the redox peaks at the potentials of 0.50 and 0.54 V are corresponding to the transition between Ru3+–Ru4+. And the redox process of Ru4+–Ru6+ occur at 0.82 and 0.90 V. In the potential of 1.30 and 1.31 V, the redox reaction of Ru4+–Ru8+ undergoes in this potential range. For the electrochemical characteristics of Co, the oxidation and reduction reactions of Co2+–Co3+ occur at 1.12 and 1.14 V, respectively.
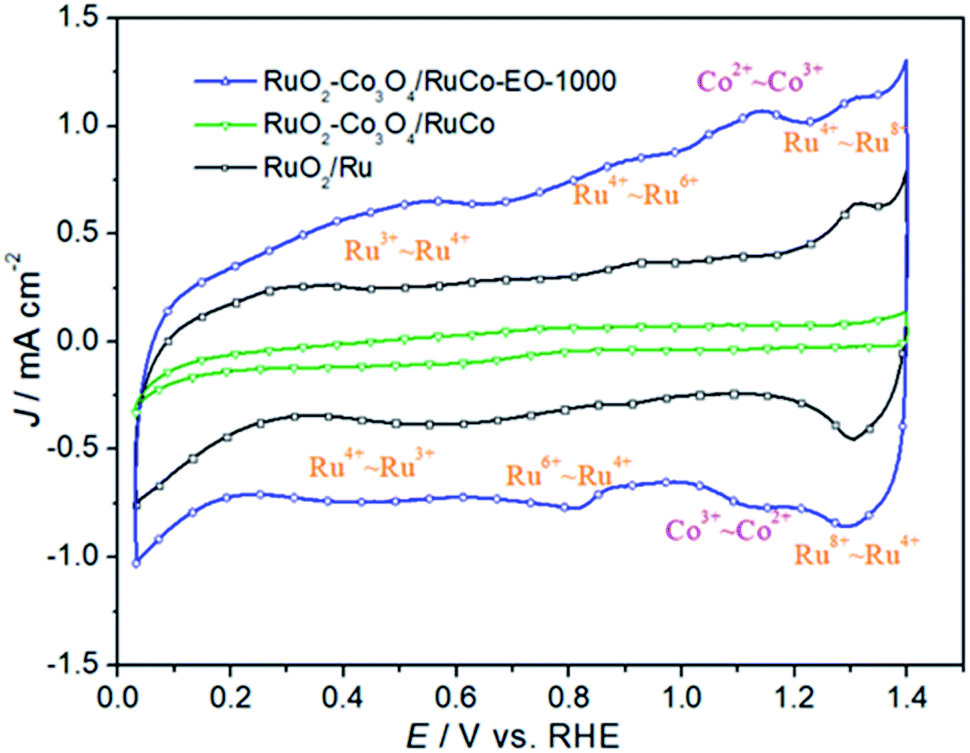 | ||
| Fig. 5 The cyclic voltammetry curves of RuO2/Co3O4–RuCo-EO, RuO2/Co3O4–RuCo and RuO2/Ru electrodes. | ||
In order to investigate the oxygen evolution reaction (OER) activity of three catalysts, a linear sweep voltammetry tests were performed, as shown in Fig. 6. It can be seen from the figure that the OER activity of RuO2/Co3O4–RuCo-EO electrode is significantly higher than that of RuO2/Co3O4–RuCo electrode. For example, the current density of RuO2/Co3O4–RuCo electrode at 1.8 V is 53.55 mA cm−2, while the current density of RuO2/Co3O4–RuCo-EO electrode is increased to 124.20 mA cm−2, which is 2.32 times that of RuO2/Co3O4–RuCo electrode. At the same time, when the current density reaches 10 mA cm−2, the overpotential of RuO2/Co3O4–RuCo and RuO2/Co3O4–RuCo-EO electrodes are 394 and 220 mV, respectively. The latter is 174 mV lower than the former, indicating a significant increase in OER activity. This indicates that electrochemical oxidation can improve the OER activity of the catalyst. Furthermore, compared with the OER activity of RuO2 prepared by thermal decomposition (Fig. S3†), the overpotential of RuO2/Co3O4–RuCo-EO electrode is reduced by 82 mV. Compared with the OER activity of RuO2 prepared by magnetron sputtering (Fig. S4†), the overpotential of RuO2/Co3O4–RuCo-EO electrode is reduced by 74 mV. Compared with the OER activity of RuO2/Ru (the overpotential of 244 mV), the overpotential of RuO2/Co3O4–RuCo-EO electrode is still decreased by 24 mV. Therefore, it is proved once again that the OER activity of RuO2/Co3O4–RuCo-EO electrode is indeed excellent.
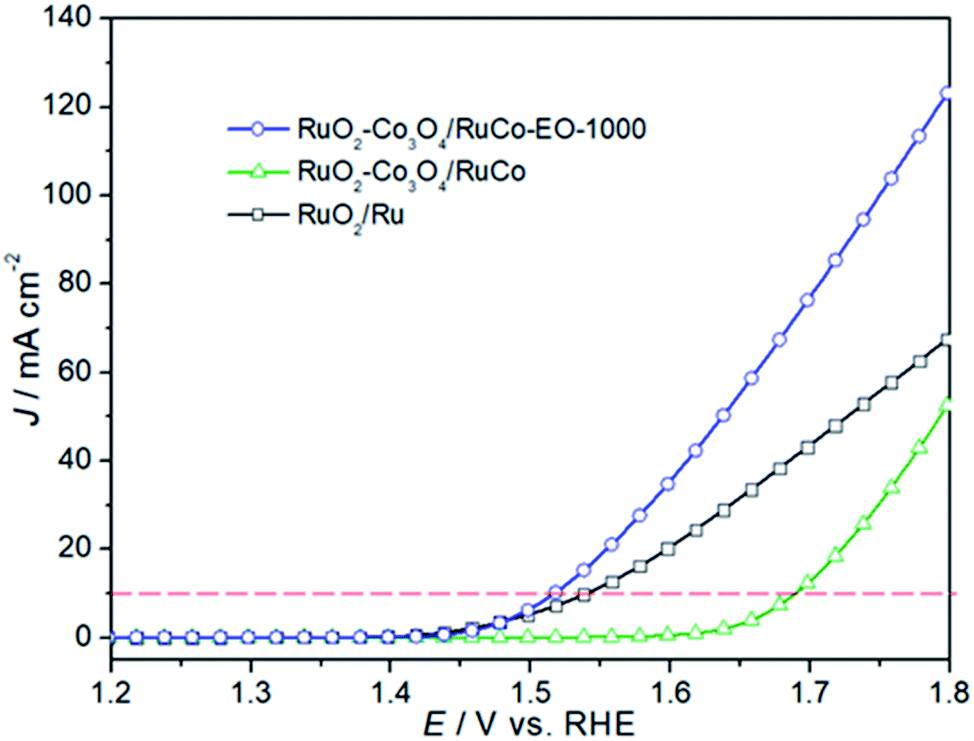 | ||
| Fig. 6 The OER activities of RuO2/Co3O4–RuCo-EO, RuO2/Co3O4–RuCo and RuO2/Ru electrodes. | ||
There are two reasons for the improvement of the electrochemical activity of RuO2/Co3O4–RuCo-EO electrode. On the one hand, the special core–shell structure of the electrode itself can facilitate the charge transfer process, and on the other hand, the addition of Co can change the electronic structure of Ru. For ordinary RuO2, whether it is prepared by magnetron sputtering or thermal decomposition, there is only an oxide phase in its structure, but no metal phase (Fig. S5 and S6†). Therefore, their activity is generally lower than that of catalysts with a metal oxide/metal core–shell structure. Generally speaking, due to the good electronic interaction and charge transfer between metal and semiconductor at the metal–semiconductor interface, that is, the Mott–Schottky effect, the combination of metal and semiconductor in the electrocatalyst can facilitate the charge transfer process. On the other hand, RuO2/Co3O4–RuCo-EO electrode has a high content of Ru oxidation state. It can be seen from the XPS spectrum (Fig. 4a) that the ratio of peak area of Ru4+/Ru0 in RuO2/Co3O4–RuCo-EO electrode is significantly greater than the ratio of peak area in RuO2/Co3O4–RuCo electrode. The spin splitting peaks of Ru0 3p1/2 and 3p3/2 of RuO2/Co3O4–RuCo-EO are positively shifted by 0.3 eV compared to those of RuO2/Ru, indicating that Ru transfers electrons to Co. Therefore, the d-band center of Ru will rise, and the adsorption of intermediate products will be enhanced, thereby increasing the OER activity. In addition, the increase in ECSA is also conducive to improving OER activity. As shown in Fig. S7,† the increase of ECSA is mainly reflected in the double-layer capacitance of RuO2/Co3O4–RuCo-EO electrode, which is obviously larger than that of RuO2/Co3O4–RuCo and RuO2/Ru electrodes.
In order to further reveal the influence of the ECSA on the OER activity of the catalyst, the OER activities of RuO2/Co3O4–RuCo-EO electrodes with different number of CV scan were studied. As shown in Fig. 7, when the CV scan is 200, 400, 600, 800, and 1000 segments, the OER current density of RuO2/Co3O4–RuCo-EO electrode is 107.1, 114.5, 125.4, 128.8 and 124.2 mA cm−2, respectively, at the potential of 1.8 V (vs. RHE). When the CV scan is between 200 and 800 segments, the current density of OER increases by about 10 mA cm−2 for every additional 200 segments. At the same time, the corresponding double-layer capacitance gradually increases, which means that the ECSA also gradually increases (Fig. S8†). This indicates that the OER activity increases continuously with the increase of the ECSA. When the CV scan is 1000 segments, the surface of RuO2/Co3O4–RuCo-EO electrode tends to be stable, then the ECSA no longer increases, and the OER activity no longer increases. In addition, it can also be observed in Fig. S9† the oxidation potential of Co gradually increases with the increase of CV scan, which means that the oxidation stripping is more thorough. In addition, the Co content also has an important influence on OER activity (Fig. S10†). When the Co content is low, it has little effect on the structure and morphology of the RuO2/Co3O4–RuCo-EO electrode, and the OER activity is low. However, when the Co content is high, the OER activity of RuO2/Co3O4–RuCo-EO electrode is also poor, because the OER activity of Co itself is poor. When the Co content is 35% after EO treatment of 1000 segments, the OER activity of the RuO2/Co3O4–RuCo-EO electrode is the best.
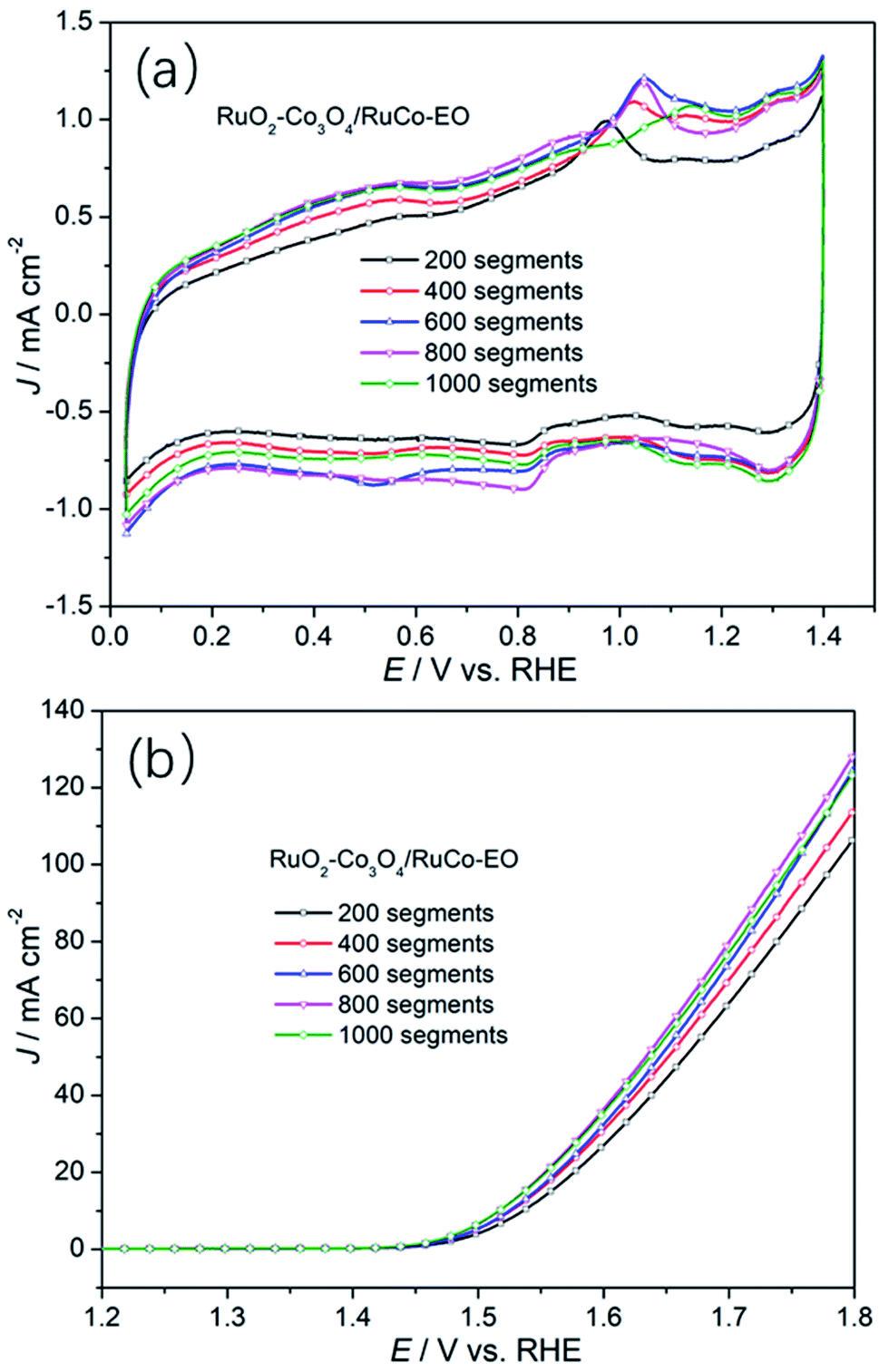 | ||
| Fig. 7 The cyclic voltammetry curves (a) and OER activities (b) of RuO2/Co3O4–RuCo-EO with different segment of CV scan. | ||
Finally, the surface morphology of the catalyst also has a significant impact on the activity. Regarding the RuO2 electrode, whether it is prepared by magnetron sputtering or thermal decomposition, the surface appears as a nanosphere with a diameter of 50–100 nm (Fig. S11 and S12†). The surface morphology of RuO2/Ru electrode prepared by magnetron sputtering also presents a smooth surface, with a particle size of 20–50 nm (Fig. S13†). However, the RuO2/Co3O4–RuCo-EO electrode prepared above has exhibited a porous nanosheet (Fig. 1c and d) or nanorod structure (Fig. 1e and f). The difference in morphology will bring about different OER activities.
In order to explore the reason for the increase in its intrinsic activity, Tafel analysis was finally performed to determine the reaction mechanism (Fig. 8). The Tafel slopes of RuO2/Co3O4–RuCo and RuO2/Ru are 79.6 and 69.1 mV dec−1, respectively. However, the Tafel slope of RuO2/Co3O4–RuCo-EO is only 59.9 mV dec−1. The lower Tafel slope indicates that the RuO2/Co3O4–RuCo-EO electrode has higher OER activity. The rate determination step of the OER process should be determined as the formation of OHads. The above discussion will be studied in detail in subsequent experiments. Finally, the stability of the RuO2/Co3O4–RuCo-EO electrode was also investigated in Fig. S14.† After 13 h of chronopotentiometry measurement at current density of 10 mA cm−2, the OER activity was basically stable ().
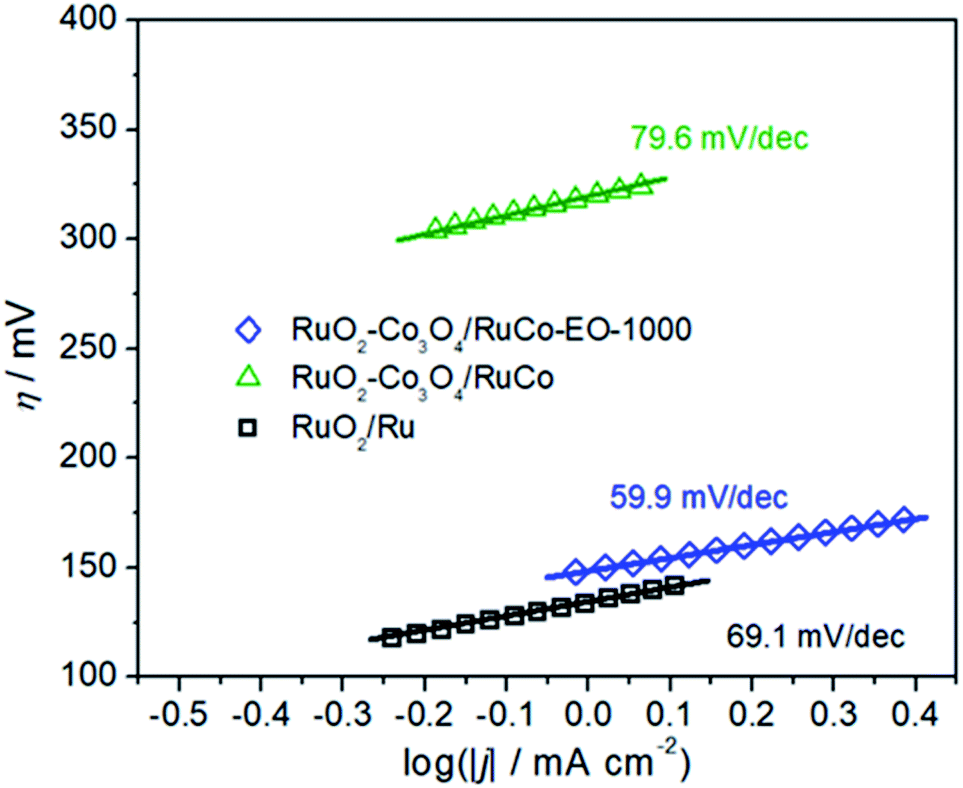 | ||
| Fig. 8 The Tafel slopes of RuO2/Co3O4–RuCo-EO, RuO2/Co3O4–RuCo and RuO2/Ru electrodes. | ||
Conclusions
In this article, a new RuO2/Co3O4–RuCo-EO electrode is designed via a magnetron sputtering combined with electrochemical oxidation for OER in an alkaline medium. The optimized RuO2/Co3O4–RuCo-EO electrode has exhibited excellent electrocatalytic performance with a low overpotential of 220 mV and Tafel slope of 59.9 mV dec−1 for OER at the current density of 10 mA cm−2. Compared with RuO2 prepared by thermal decomposition, its overpotential is reduced by 82 mV. Meanwhile, compared with RuO2 prepared by magnetron sputtering, its overpotential is also reduced by 74 mV. Compared with RuO2/Ru (the overpotential of 244 mV), the overpotential is still decreased by 24 mV. There are two reasons for the improvement of the electrochemical activity of RuO2/Co3O4–RuCo-EO electrode. On the one hand, the metal oxide–metal with core–shell structure is conducive to electron transport, and on the other hand, the addition of Co changes the electronic structure of Ru. The positive shift of the spin splitting peaks of Ru indicates that Ru transfers electrons to Co. Therefore, the d-band center of Ru will rise, and the adsorption of intermediate products will be enhanced, thereby increasing the OER activity. In addition, the increase in ECSA is also conducive to improving OER activity.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge financial support from the Natural Science Foundation of Hubei Province (2020CFB777).Notes and references
- J. Wang, W. Cui, Q. Liu, Z. Xing, A. M. Asiri and X. Sun, Adv. Mater., 2016, 28, 215–230 CrossRef CAS PubMed.
- X. Tian, P. Zhao and W. Sheng, Adv. Mater., 2019, 31, 1808066 CrossRef PubMed.
- E. Liu, J. Li, L. Jiao, H. Doan, Z. Liu, Z. Zhao, Y. Huang and K. M. Abraham, J. Am. Chem. Soc., 2019, 141, 3232–3239 CrossRef CAS PubMed.
- Z. Yao, J. Yan, V. Anthony and S. Z. Qiao, Angew. Chem., Int. Ed., 2018, 57, 7568–7579 CrossRef PubMed.
- J. Liu, Y. Liu, N. Liu, Y. Han, X. Zhang, H. Huang, Y. Lifshitz, S. Lee, J. Zhong and Z. Kang, Science, 2015, 347, 970 CrossRef CAS PubMed.
- S. T. Hunt, M. Milina, Z. Wang and Y. R. Leshkov, Energy Environ. Sci., 2016, 9, 3290–3301 RSC.
- N. Yao, P. Li, Z. Zhou, Y. Zhao, G. Cheng, S. Chen and W. Luo, Adv. Energy Mater., 2019, 9, 1902449 CrossRef CAS.
- X. Zou and Y. Zhang, Chem. Soc. Rev., 2015, 44, 5148–5180 RSC.
- J. Wang, F. Xu, H. Jin, Y. Chen and Y. Wang, Adv. Mater., 2017, 29, 1605838 CrossRef PubMed.
- M. Wang, L. Chen and L. Sun, Energy Environ. Sci., 2012, 5, 6763–6778 RSC.
- S. Zhao, J. Huang, Y. Liu, J. Shen, H. Wang, X. Yang, Y. Zhu and C. Li, J. Mater. Chem. A, 2017, 5, 4207–4214 RSC.
- D. Li, Z. Zong, Z. Tang, Z. Liu, S. Chen, Y. Tian and X. Wang, ACS Sustainable Chem. Eng., 2018, 6, 5105–5114 CrossRef CAS.
- L. Li, B. Wang, G. Zhang, G. Yang, T. Yang, S. Yang and S. Yang, Adv. Energy Mater., 2020, 10, 2001600 CrossRef CAS.
- G. Li, S. Li, J. Ge, C. Liu and W. Xing, J. Mater. Chem. A, 2017, 5, 17221–17229 RSC.
- L. Gloag, T. M. Benedetti, S. Cheong, R. F. Webster, C. E. Marjo, J. J. Gooding and R. D. Tilley, Nanoscale, 2018, 10, 15173–15177 RSC.
- Z. Wang, S. M. Xu, Y. Xu, L. Tan, X. Wang, Y. Zhao, H. Duan and Y. F. Song, Chem. Sci., 2019, 10, 378–384 RSC.
- F. M. Mota, C. H. Choi, R. Boppella, J. E. Lee and D. H. Kim, J. Mater. Chem. A, 2019, 7, 639–646 RSC.
- Z. Liu, M. Zha, Q. Wang, G. Hua and L. Feng, Chem. Commun., 2020, 56, 2352–2355 RSC.
- Z. Li, S. Wang, Y. Tian, B. Li, H. Yan, S. Zhang, Z. Liu, Q. Zhang, Y. Lin and L. Chen, Chem. Commun., 2020, 56, 1749–1752 RSC.
- L. Fagiolari, F. Zaccaria, F. Costantino, R. Vivani, C. Mavrokefalos, G. R. Patzked and A. Macchioni, Dalton Trans., 2020, 49, 2468–2476 RSC.
- S. Laha, Y. Lee, F. Podjaski, D. Weber, V. Duppel, L. M. Schoop, F. Pielnhofer, C. Scheurer, K. Müller, U. Starke, K. Reuter and B. V. Lotsch, Adv. Energy Mater., 2019, 9, 1803795 CrossRef.
- J. Yu, Q. He, G. Yang, W. Zhou, Z. Shao and M. Ni, ACS Catal., 2019, 9, 9973–10011 CrossRef CAS.
- J. Su, R. Ge, K. Jiang, Y. Dong, F. Hao, Z. Tian, G. Chen and L. Chen, Adv. Mater., 2018, 30, 1801351 CrossRef PubMed.
- K. Yang, P. Xu, Z. Lin, Y. Yang, P. Jiang, C. Wang, S. Liu, S. Gong, L. Hu and Q. Chen, Small, 2018, 14, 1803009 CrossRef PubMed.
- M. Retuerto, L. Pascual, C. Federico, P. Ferrer, D. Gianolio, A. G. Pereira, Á. García, J. Torrero, T. F. María, P. Bencok, M. A. Peña, J. L. Fierro and S. Rojas, Nat. Commun., 2019, 10, 2041 CrossRef PubMed.
- R. Farhat, J. Dhainy and L. Halaoui, ACS Catal., 2020, 10, 20–35 CrossRef CAS.
- Q. Yang, P. Jin, B. Liu, L. Zhao, J. Cai, Z. Wei, S. Zuo, J. Zhang and L. Feng, J. Mater. Chem. A, 2020, 8, 9049–9057 RSC.
- Y. Wang, P. Zheng, M. Li, Y. Li, X. Zhang, J. Chen, X. Fang, Y. Liu, X. Yuan, X. Dai and H. Wang, Nanoscale, 2020, 12, 9669–9679 RSC.
- T. Zhu, J. Huang, B. Huang, N. Zhang, S. Liu, Q. Yao, S. Haw, Y. Chang, C. W. Pao, J. M. Chen, Q. Shao, Z. Hu, Y. Ma and X. Huang, Adv. Energy Mater., 2020, 10, 2002860 CrossRef CAS.
- A. Yu, C. Lee, M. H. Kim and Y. Lee, ACS Appl. Mater. Interfaces, 2017, 9, 35057–35066 CrossRef CAS PubMed.
- C. Gao, H. Wang, S. Li, B. Liu, J. Yang, J. Gao, Z. Peng, Z. Zhang and Z. Liu, Electrochim. Acta, 2019, 327, 134958 CrossRef CAS.
- Q. Wu, M. Luo, J. Han, W. Peng, Y. Zhao, D. Chen, M. Peng, J. Liu, F. M. F. Groot and Y. Tan, ACS Energy Lett., 2020, 5, 192–199 CrossRef CAS.
- X. Sun, F. Liu, X. Chen, C. Li, J. Yu and M. Pan, Electrochim. Acta, 2019, 307, 206–213 CrossRef CAS.
- H. Wang, Y. Yang, F. DiSalvo and H. D. Abruna, ACS Catal., 2020, 10, 4608–4616 CrossRef CAS.
- R. P. Gautam, H. Pan, F. Chalyavi, M. J. Tucker and C. J. Barile, Catal. Sci. Technol., 2020, 10, 4960–4967 RSC.
- H. Liu, X. Li, L. Ge, C. Peng, L. Zhu, W. Zou, J. Chen, Q. Wu, Y. Zhang, H. Huang, J. Wang, Z. Cheng, Z. Fu and Y. Lu, Catal. Sci. Technol., 2020, 10, 8314–8324 RSC.
- X. Gao, J. Chen, X. Sun, B. Wu, B. Li, Z. Ning, Z. Li and N. Wang, ACS Appl. Nano Mater., 2020, 3, 12269–12277 CrossRef CAS.
- Y. Bao, J. Dai, J. Zhao, Y. Wu, C. Li, L. Ji, X. Zhang and F. Yang, ACS Appl. Energy Mater., 2020, 3, 1869–1874 CrossRef CAS.
- Z. Fan, J. Jiang, L. Ai, Z. Shao and S. Liu, ACS Appl. Mater. Interfaces, 2019, 11, 47894–47903 CrossRef CAS PubMed.
- T. Feng, G. Yu, S. Tao, S. Zhu, R. Ku, R. Zhang, Q. Zeng, M. Yang, Y. Chen, W. Chen, W. Chen and B. Yang, J. Mater. Chem. A, 2020, 8, 9638–9645 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra00271f |
| This journal is © The Royal Society of Chemistry 2021 |