
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Calycindaphines A–J, Daphniphyllum alkaloids from the roots of Daphniphyllum calycinum†
Ji Yanga,
Xin Liuab,
Jing Fua,
Hao-Yuan Lyua,
Li-Ping Bai a,
Zhi-Hong Jiang*a and
Guo-Yuan Zhu*a
a,
Zhi-Hong Jiang*a and
Guo-Yuan Zhu*a
aState Key Laboratory of Quality Research in Chinese Medicine, Guangdong-Hong Kong-Macao Joint Laboratory of Respiratory Infectious Disease, Macau Institute for Applied Research in Medicine and Health, Macau University of Science and Technology, Macau, People's Republic of China. E-mail: zhjiang@must.edu.mo; gyzhu@must.edu.mo
bBiology Institute, Qilu University of Technology (Shandong Academy of Sciences), Jinan 250103, China
First published on 1st March 2021
Abstract
Ten new Daphniphyllum alkaloids, calycindaphines A–J (1–10), together with seventeen known alkaloids were isolated from the roots of Daphniphyllum calycinum. Their structures were established by extensive spectroscopic methods and compared with data from literature. Compound 1 is a novel alkaloid with a new rearrangement C22 skeleton with the 5/8/7/5/5 ring system. Compound 2 represents the second example of calyciphylline G-type alkaloids. Compound 10 is the first example of secodaphniphylline-type alkaloid absent of the oxygen-bridge between C-25/C-29. The possible biogenetic pathways of 1 and 2 were also proposed. All the isolated compounds were evaluated for their bioactivities in three cell models. Compounds 22, 23, and 26 showed significant NF-κB transcriptional inhibitory activity at a concentration of 50 μM. Compounds 16 and 18 exhibited significant TGF-β inhibitory activity in HepG2 cells. Compounds 24 and 26 induced autophagic puncta and mediated the autophagic marker LC3-II conversion in HEK293 cells.
Introduction
The Daphniphyllum alkaloids were characteristically distributed in plants of the Daphniphyllum family.1–4 To date, more than 200 Daphniphyllum alkaloids with nearly 20 skeletons have been isolated from ten species. Daphniphyllum alkaloids not only provided abundant structural types but also enlighten novel strategies for the total synthesis and biosynthesis of unusual skeletons.5–10 Daphniphyllum calycinum, an evergreen tree native to southern China, provided an abundant resource of Daphniphyllum alkaloids.11,12 The leaves, stems, and roots of D. calycinum are used in Traditional Chinese Medicine to treat several symptoms including fever, asthma, inflammation, and influenza.11–13 Previous phytochemical investigations on the leaves and stems of D. calycinum led to the isolation of more than 50 Daphniphyllum alkaloids with 15 different skeletons. However, the chemical from the roots of the D. calycinum has not been reported. To seek potential bioactive alkaloids from D. calycinum, the roots of D. calycinum were first deeply studied. As a result, ten new Daphniphyllum alkaloids, named calycindaphines A–J (1–10), as well as 17 known alkaloids (11–27) were isolated from the roots of D. calycinum. In this paper, we reported the isolation, structural elucidation, and bioactivities of these isolates.Results and discussion
Compound 1, a white amorphous powder, has a molecular formula of C23H31O3N as established by its HRESIMS data (m/z 370.2375 [M + H]+, calcd for 370.2377), with 9 degrees of unsaturation. IR absorptions implied the presence of an ester carbonyl (1733 cm−1) and a lactam (1644 cm−1).14 The 1H NMR data (Table 1) of 1 exhibited proton resonances of a methoxy (δH 3.61, s), a methyl singlet (δH 1.10, s), a methyl doublet (δH 1.15, d, J = 6.7 Hz), an olefinic proton (δH 5.42, d, J = 5.2 Hz). Its 13C NMR, DEPT, and HSQC spectra showed the occurrence of 23 carbon resonances (Table 2), which consisted of three methyls (a methoxy at δC 50.9), nine methylenes (two N-methylenes at δC 46.8 and 52.5), five methines (an olefinic methine at δC 118.9), six quaternary carbons (two carbonyls at δC 182.9 and 175.2, and three olefinic quaternary carbons at δC 140.6, 139.0, and 135.5). The above-mentioned spectroscopic data suggested that 1 possesses two carbonyls and two double bonds which accounting for four out of nine indices of hydrogen deficiency, and the remaining five degrees of unsaturation were speculated for the presence of a pentacyclic system in 1. The 1H–1H COSY spectrum of 1 suggested three proton-bearing structural moieties: C-4/C-3/C-2/C-18/C-19/20, C-7/C-1/C-12/C-11, and C-13/C-14/C-15/C-16/C-17 (Fig. 2). These three fragments, quaternary carbons, and the nitrogen atom were then connected by the detailed HMBC analysis (Fig. 2). The HMBC correlations from H-13/H-14 to C-8/C-9/C-22, H-16 to C-9/C-10, H-17 to C-9/C-10/C-11, H-11 to C-10/C-17, and H-23 to C-22, led to the assignment of rings A and B. Moreover, HMBC cross-peaks from H-21 to C-4/C-5/C-6/C-8 and H-6 to C-4/C-5/C-8 indicated the presence of C-5/C-6/C-8 linkage which form a seven-membered ring (ring C). HMBC correlations from H-2/H-7/H-19 to C-1, H-3/H-4/H-6 to C-5, H-7 to C-5/C-19, and H-19 to C-7 established the connectivity of rings D and E. Thus, the planar structure of 1 was assigned as shown in Fig. 2. Compound 1 has a fused 5/8/7/5/5 ring system, which contains a 1-azabicyclo[5.2.1]decane ring (rings D/E) with a ketone at C-1 and two methyls at C-18 and C-2, a cyclohepta-1,3-diene ring (ring C) with two double bonds between C-8/C-9 and C-10/C-11, and a bicyclo[3.3.0]octane ring (rings A/B) with a methoxy carbonyl at C-14 as shown in Fig. 2. Comparing with the literature, the ring system of 1 was the same as those of daphhimalenine A.14 However, compound 1 possesses C22 carbon skeleton like most of Daphniphyllum alkaloids, while daphhimalenine A loses C-21 methyl to form a C21 carbon skeleton. The 3JH-13a/H-14 = 7.5 Hz suggest that H-15/H-14 of 1 was α-orientation and same as that of daphhimalenine A.14 Based on the same biosynthetic origin as Daphniphyllum alkaloids, the configuration of H-6 was identified as β-orientation.11,13 The relative configurations of C-20, C-21, and C-2–C-3 bond was dissented as β-orientation by the key NOESY correlations (Fig. 3) from H-21 to H-6/H-13b and H-20 to H-3 as well as the comparison with the literature.14 To determine the absolute configuration of 1, the calculated ECD were performed using the time-dependent density functional theory at the PBE0-D3(BJ)/def2-SVP level for (2R,5S,6S,14R,15R,18S)-1 and (2S,5R,6R,14S,15S,18R)-1. The ECD curve of 1 showed a negative Cotton effect at 217 (−51.89) nm, which was consistent with the calculated ECD spectrum of (2R,5S,6S,14R,15R,18S)-1 (Fig. 4). Accordingly, the absolute configuration of 1 was determined as 2R,5S,6S,14R,15R,18S. The structure of 1 was thereby established (Fig. 1) and named calycindaphine A.| No. | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 3.80 (d, 4.2) | 3.03 (s) | 2.99 (s) | 3.11 (s) | ||||||
| 2a | 2.24 (m) | 2.15 (m) | 2.65 (m) | 3.59 (m) | 1.40 (m) | 1.69 (m) | 1.03 (m) | 0.96 (m) | 1.09 (m) | |
| 2b | 0.84 (m) | 1.32 (m) | ||||||||
| 3a | 2.51 (m) | 2.00 (m) | 2.03 (m) | 2.06 (m) | 1.60 (m) | 1.70 (m) | 1.50 (m) | 1.89 (m) | 1.55 (m) | 1.50 (m) |
| 3b | 1.94 (m) | 1.66 (dd, 14.4, 4.4) | 1.74 (m) | 1.66 (m) | 2.00 (m) | 1.42 (m) | 1.40 (m) | 1.29 (m) | ||
| 4a | 2.02 (m) | 4.07 (dd, 10.5, 4.4) | 3.22 (t, 3.1) | 2.00 (m) | 1.61 (m) | 1.83 (m) | 1.81 (m) | 1.17 (m) | 1.67 (m) | 1.59 (m) |
| 4b | 1.63 (m) | 1.42 (m) | 1.76 (m) | 1.35 (m) | 1.38 (m) | 1.60 (m) | 1.15 (m) | |||
| 6 | 2.33 (m) | 2.01 (m) | 2.55 (m) | 2.79 (m) | 2.38 (m) | 1.91 (t, 5.3) | 1.99 (t, 5.3) | 1.91 (t, 5.1) | ||
| 7a | 4.32 (t, 13.1) | 2.88 (dd, 14.2, 8.9) | 3.40 (dd, 14.3, 9.8) | 4.98 (m) | 3.36 (dd, 13.6, 10.1) | 2.78 (m) | 5.97 (s) | 2.30 (d, 5.9) | 2.60 (t, 4.6) | 2.56 (d, 3.5) |
| 7b | 2.48 (m) | 3.14 (m) | 3.73 (dd, 13.6, 8.2) | |||||||
| 9 | 1.72 (m) | 1.75 (m) | 1.03 (t, 3.4) | |||||||
| 10 | 2.54 (m) | |||||||||
| 11a | 5.32 (d, 5.2) | 2.42 (m) | 2.17 (m) | 1.83 (m) | 2.24 (m) | 1.97 (m) | 2.55 (dd, 12.9, 5.8) | 1.65 (m) | 1.68 (m) | 1.67 (m) |
| 11b | 1.99 (m) | 1.29 (m) | 1.74 (m) | 2.21 (m) | 1.48 (m) | 1.55 (m) | 1.49 (m) | |||
| 12a | 2.67 (m) | 2.01 (m) | 1.90 (m) | 2.07 (m) | 1.60 (m) | 2.31 (m) | 2.38 (m) | 1.59 (m) | 1.79 (m) | 1.59 (m) |
| 12b | 1.97 (m) | 1.61 (m) | 1.61 (m) | 1.87 (m) | 2.24 (m) | 1.97 (m) | 1.40 (m) | 1.61 (m) | 1.41 (m) | |
| 13a | 3.45 (m) | 2.79 (m) | 2.53 (m) | 1.84 (m) | 2.31 (m) | 2.65 (dd, 13.8, 7.9) | 2.27 (m) | 2.04 (m) | 1.65 (m) | 1.69 (m) |
| 13b | 2.89 (d, 15.5) | 2.35 (m) | 2.28 (dd, 13.5, 8.8) | 1.68 (m) | 2.61 (m) | 2.30 (m) | 2.07 (m) | 1.96 (m) | 1.54 (m) | |
| 14a | 3.18 (t, 7.6) | 2.93 (m) | 3.18 (m) | 2.74 (m) | 3.27 (dt, 11.3, 8.1) | 3.41 (dt, 12.2, 7.0) | 3.61 (m) | 2.92 (m) | 2.86 (m) | 1.30 (m) |
| 14b | 1.28 (m) | 2.65 (m) | 1.41 (m) | |||||||
| 15a | 3.63 (m) | 2.93 (m) | 3.62 (m) | 5.48 (m) | 3.38 (m) | 2.78 (m) | 2.75 (q, 8.0) | 1.78 (m) | 1.69 (m) | 1.67 (m) |
| 15b | 1.72 (m) | 1.61 (m) | 1.78 (m) | |||||||
| 16a | 1.80 (m) | 1.79 (m) | 1.83 (m) | 2.18 (m) | 1.22 (m) | 1.78 (m) | 2.29 (m) | 1.73 (m) | 1.73 (m) | 1.75 (m) |
| 16b | 1.00 (m) | 1.17 (m) | 1.17 (m) | 1.88 (dt, 12.1, 6.8) | 1.48 (m) | 2.05 (m) | 1.44 (m) | 1.46 (m) | 1.45 (m) | |
| 17a | 2.68 (m) | 2.53 (m) | 2.49 (m) | 2.09 (m) | 2.21 (m) | 2.35 (m) | 5.54 (m) | 1.66 (m) | 1.69 (m) | 1.47 (m) |
| 17b | 2.46 (m) | 2.23 (m) | 2.21 (m) | 1.42 (m) | 2.46 (m) | 1.90 (m) | 1.54 (m) | 1.56 (m) | 1.16 (m) | |
| 18 | 2.49 (m) | 2.28 (m) | 2.35 (t, 6.3) | 2.24 (m) | 2.15 (m) | 2.15 (m) | 1.49 (m) | 1.47 (m) | 1.51 (m) | |
| 19a | 3.32 (d, 9.4) | 3.22 (m) | 4.42 (s) | 2.29 (dd, 13.6, 8.6) | 4.04 (dd, 13.1, 7.9) | 4.37 (dd, 13.2, 5.6) | 0.90 (d, 6.5) | 0.91 (d, 6.5) | 0.94 (d, 6.5) | |
| 19b | 3.27 (d, 9.4) | 2.02 (m) | 4.46 (dd, 13.6, 7.7) | 3.89 (dd, 13.1, 8.9) | 2.31 (m) | |||||
| 20 | 1.15 (d, 6.7) | 1.02 (d, 7.1) | 1.13 (d, 6.7) | 1.49 (s) | 0.99 (d, 6.7) | 0.93 (d, 6.9) | 0.87 (d, 6.8) | 0.89 (d, 6.5) | 0.90 (d, 6.5) | 0.89 (m) |
| 21a | 1.10 (s) | 1.22 (s) | 0.97 (s) | 1.47 (s) | 1.16 (s) | 1.22 (s) | 1.08 (s) | 0.78 (s) | 3.72 (d, 10.6) | 0.76 (s) |
| 21b | 3.49 (d, 10.6) | |||||||||
| 22 | 3.94 (m) | 1.68 (m) | ||||||||
| 23 | 3.61 (s) | 3.61 (s) | 3.62 (s) | 3.65 (s) | 3.70 (s) | 3.68 (s) | ||||
| 24 | 3.25 (s) | 0.59 (s) | 0.78 (s) | 0.89 (s) | ||||||
| 25a | 3.58 (dd, 11.8, 1.5) | 4.25 (d, 12.1) | 3.65 (m) | |||||||
| 25b | 3.51 (d, 11.8) | 3.52 (d, 12.1) | 3.49 (d, 10.3) | |||||||
| 26 | 4.49 (d, 6.0) | 4.66 (d, 6.9) | 3.74 (dd, 11.2, 4.0) | |||||||
| 27a | 2.07 (m) | 2.07 (m) | 1.74 (m) | |||||||
| 27b | 1.96 (m) | 1.03 (m) | 1.50 (m) | |||||||
| 28a | 2.05 (m) | 2.07 (m) | 1.76 (m) | |||||||
| 28b | 1.83 (m) | 1.87 (m) | 1.47 (m) | |||||||
| 30 | 1.47 (s) | 1.43 (s) | 1.20 (s) |
| No. | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 182.9 | 73.6 | 98.7 | 63.6 | 174.4 | 175.7 | 175.6 | 48.2 | 48.6 | 47.7 |
| 2 | 46.3 | 220.8 | 45.5 | 47.8 | 72.9 | 32.3 | 32.3 | 42.7 | 42.6 | 42.6 |
| 3 | 30.2 | 33.3 | 23.5 | 22.9 | 37.1 | 26.2 | 23.0 | 20.8 | 20.0 | 20.7 |
| 4 | 41.6 | 79.1 | 82.8 | 27.3 | 42.7 | 48.3 | 40.0 | 39.2 | 32.3 | 39.1 |
| 5 | 42.9 | 51.0 | 45.9 | 84.2 | 57.1 | 41.9 | 40.7 | 36.9 | 41.7 | 36.7 |
| 6 | 46.5 | 36.0 | 39.1 | 33.9 | 40.0 | 53.5 | 130.6 | 47.8 | 45.7 | 47.6 |
| 7 | 46.8 | 44.5 | 44.7 | 47.1 | 50.9 | 176.9 | 126.9 | 60.1 | 59.7 | 59.8 |
| 8 | 140.6 | 60.6 | 34.1 | 49.0 | 41.8 | 62.9 | 65.5 | 36.9 | 45.7 | 37.2 |
| 9 | 135.5 | 143.1 | 143.2 | 143.0 | 139.1 | 96.0 | 97.2 | 53.9 | 52.8 | 54.2 |
| 10 | 139.0 | 136.4 | 135.0 | 44.7 | 133.5 | 84.7 | 145.9 | 50.5 | 50.5 | 50.6 |
| 11 | 118.9 | 26.3 | 25.9 | 25.4 | 24.5 | 32.5 | 31.3 | 39.9 | 39.4 | 40.0 |
| 12 | 33.7 | 27.0 | 22.9 | 21.2 | 25.5 | 24.1 | 27.8 | 22.9 | 23.2 | 23.0 |
| 13 | 42.1 | 33.7 | 38.2 | 23.1 | 34.8 | 31.5 | 31.1 | 25.0 | 25.8 | 36.2 |
| 14 | 46.7 | 55.1 | 42.8 | 26.4 | 42.0 | 43.2 | 43.7 | 30.1 | 34.1 | 20.7 |
| 15 | 54.2 | 43.0 | 54.5 | 124.8 | 52.9 | 54.0 | 56.8 | 30.8 | 29.7 | 30.6 |
| 16 | 28.4 | 28.6 | 28.5 | 31.0 | 28.5 | 20.2 | 31.4 | 26.8 | 26.7 | 26.9 |
| 17 | 38.9 | 42.5 | 42.6 | 34.5 | 42.1 | 34.8 | 130.9 | 36.3 | 36.2 | 36.3 |
| 18 | 28.1 | 43.8 | 36.3 | 77.3 | 38.4 | 33.8 | 28.8 | 28.8 | 28.7 | 28.8 |
| 19 | 52.5 | 54.2 | 96.5 | 174.4 | 52.4 | 45.1 | 51.9 | 21.2 | 21.0 | 21.0 |
| 20 | 13.3 | 11.8 | 10.8 | 20.5 | 18.1 | 26.1 | 20.4 | 21.1 | 21.0 | 21.0 |
| 21 | 33.3 | 19.1 | 20.6 | 19.7 | 25.0 | 21.4 | 22.9 | 21.3 | 66.2 | 21.1 |
| 22 | 175.2 | 176.3 | 176.5 | 170.8 | 176.1 | 174.1 | 174.4 | 75.5 | 213.0 | 52.2 |
| 23 | 50.9 | 51.0 | 51.1 | 51.3 | 51.7 | 51.5 | 39.2 | 49.8 | 43.8 | |
| 24 | 50.4 | 14.8 | 17.7 | 10.7 | ||||||
| 25 | 67.6 | 65.4 | 72.0 | |||||||
| 26 | 80.0 | 81.0 | 76.7 | |||||||
| 27 | 25.0 | 24.6 | 28.7 | |||||||
| 28 | 33.6 | 33.8 | 40.7 | |||||||
| 29 | 105.1 | 105.3 | 73.9 | |||||||
| 30 | 23.8 | 23.7 | 23.9 |
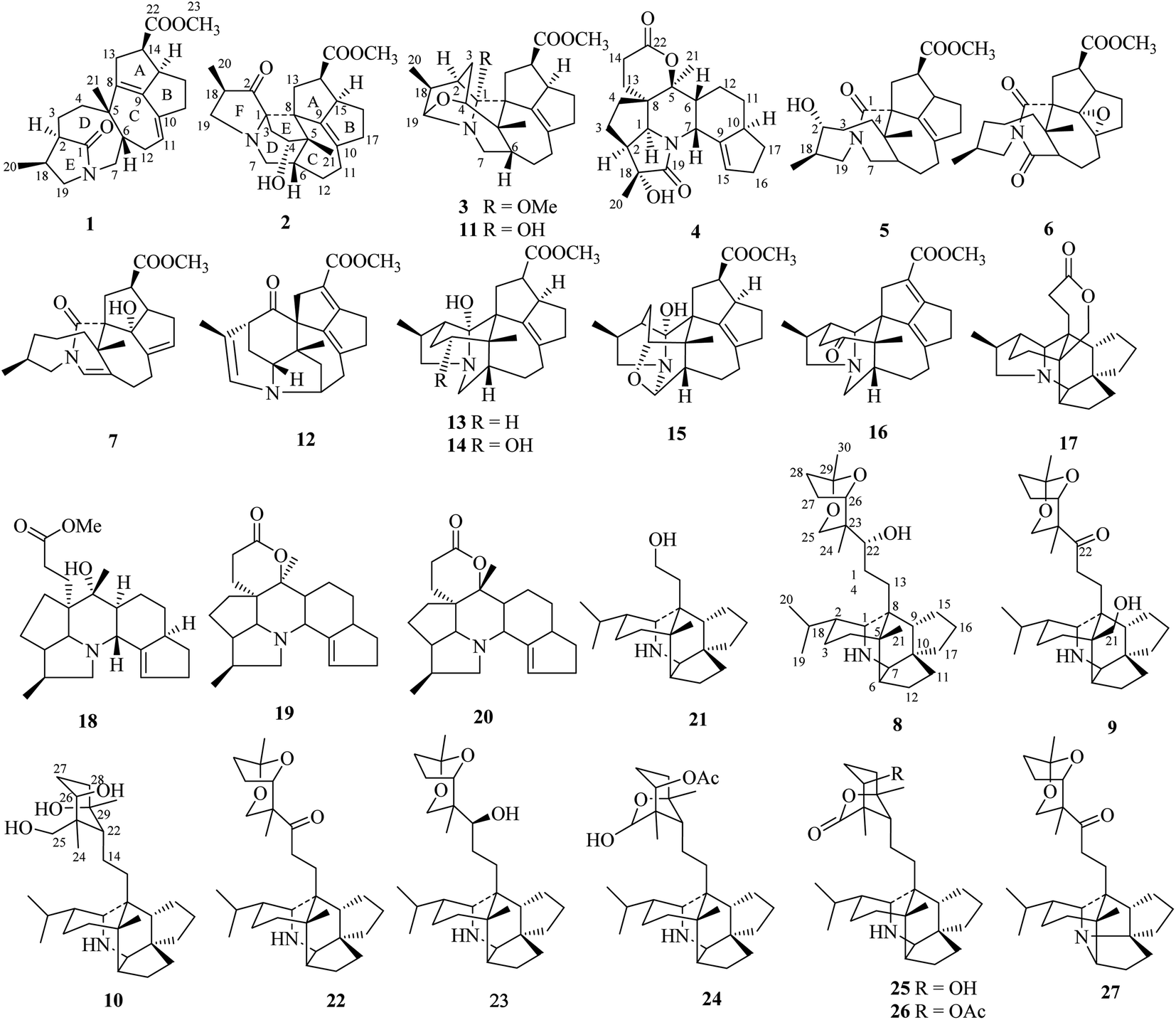 | ||
| Fig. 1 Chemical structures of compounds 1–27. | ||
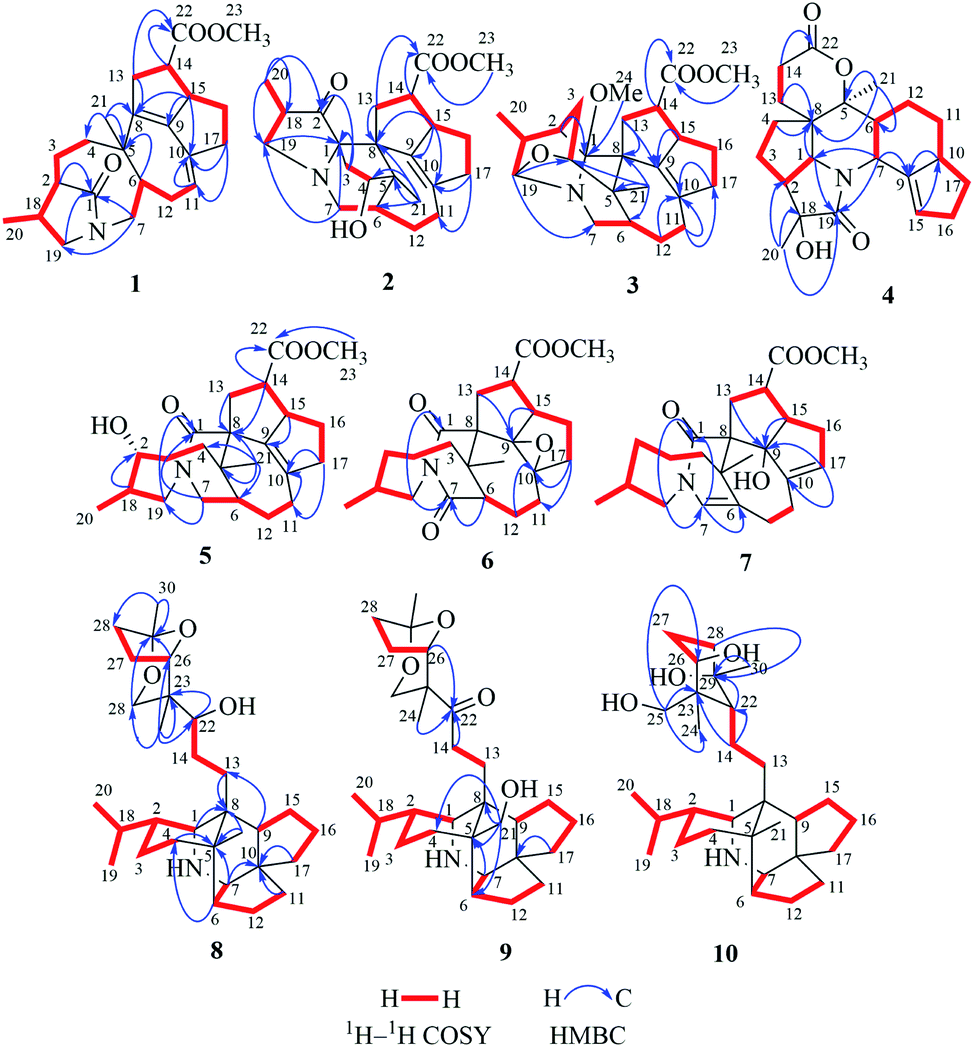 | ||
| Fig. 2 Key 1H–1H COSY and HMBC correlations of compounds 1–10. | ||
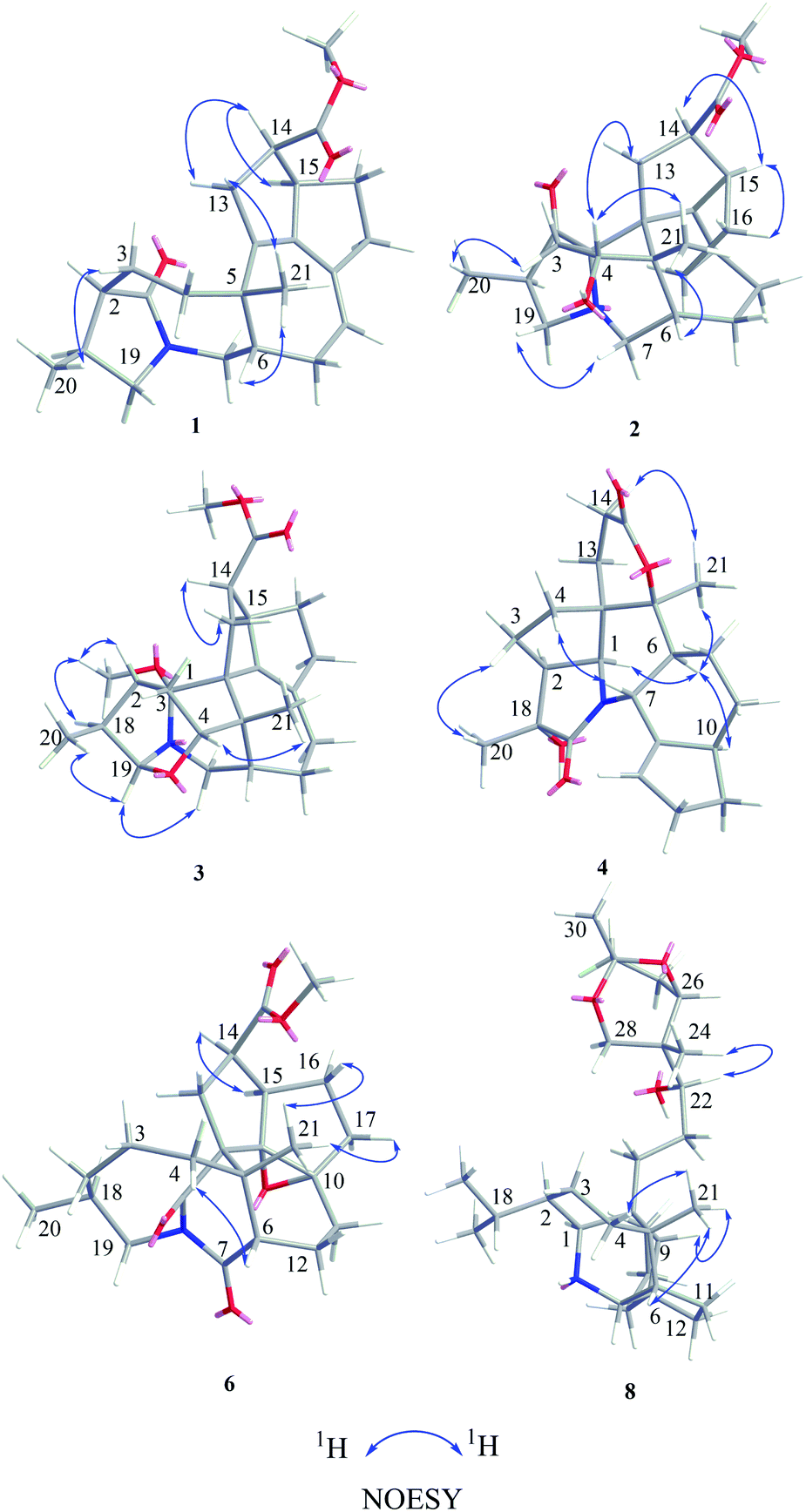 | ||
| Fig. 3 Key NOESY correlations of compounds 1–4, 6, and 8. | ||
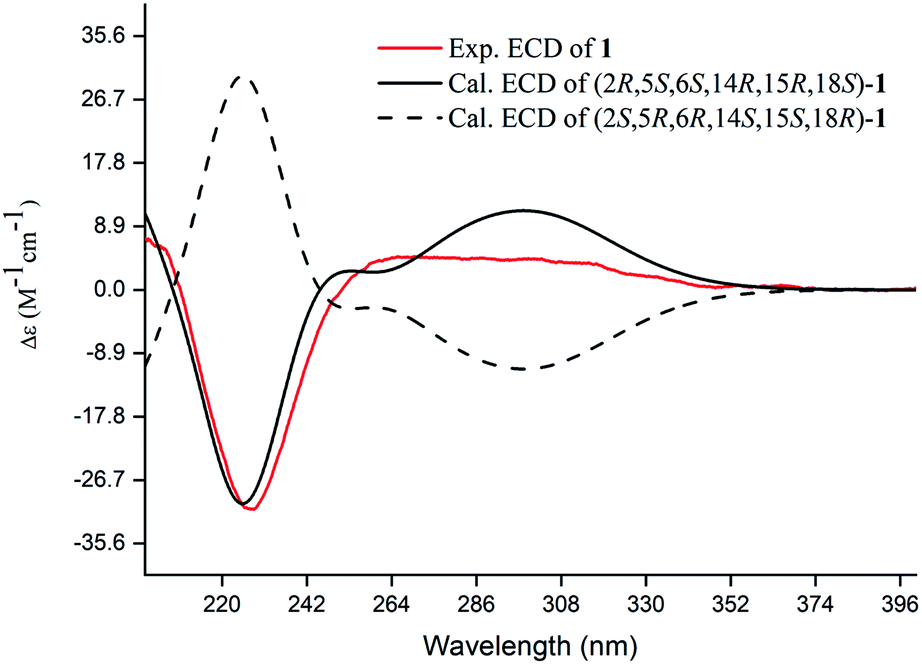 | ||
| Fig. 4 Experimental and calculated ECD spectra of compound 1. | ||
In the early report, the possible biogenetic pathway of daphhimalenine A was proposed that its precursor daphhimalenine B underwent multi-step oxidation to lose the C-21 and rearranged to form 1-azabicyclo[5.2.1]decane ring system.14 Calycindaphine A (1) has the same ring system with daphhimalenine A but reserving the key C-21 methyl. According to the structure of 1, another possible biogenetic pathway to form 1-azabicyclo[5.2.1]decane ring system was proposed, and shown in Scheme 1. The biogenetic origin of 1 and daphhimalenine A seems to be modified from a yuzurimine-type alkaloid, yunnandaphnine A (13).7 Yunnandaphnine A might undergo the oxidation of C-1 and the breakdown of C-1/C-8 bond to form 1-azabicyclo[5.2.1]decane ring. Then, the intermediate I should undergo the dehydrogenation and sigmatropic rearrangement procedures to yield 1 (C22 skeleton). The C-21 methyl of 1 should further be oxidated to give II. Then the oxidative decarboxylation and sigmatropic rearrangement of II afford daphhimalenine A (C21 skeleton).
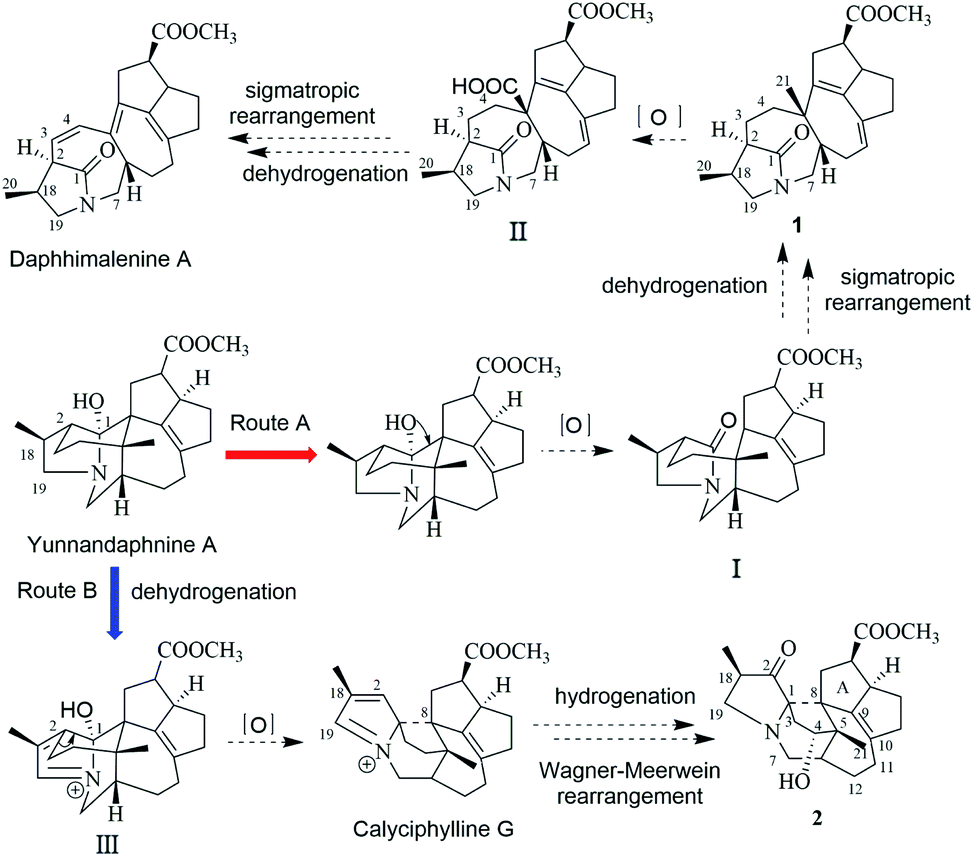 | ||
| Scheme 1 Biogenetic pathway proposed for compounds 1 and 2, daphhimalenine A, and calyciphylline G. | ||
Compound 2 was isolated as a white amorphous powder. The molecular formula of 2 was assigned as C23H31O4N based on the quasi-molecular ion peak at m/z 386.2324 [M + H]+ in its HRESIMS spectrum, requiring nine indices of hydrogen deficiency. The 1H NMR spectrum (Table 1) of 2 revealed a methoxy (δH 3.61, s), a methyl singlet (δH 1.22, s), a methine doublet (δH 1.02, d, J = 7.1 Hz), an oxidized methine (δH 4.07, dd, J = 10.5, 4.4 Hz). The 13C, DEPT, and HSQC spectra of 2 described 23 carbon resonances constituting with three methyls (a methoxy at δC 51.0), eight methylenes (two N-methylenes at δC 54.3 and 44.5), five methines (an oxidized methine at δC 79.2) and seven quaternary carbons (a carbonyl at δC 220.8, an ester carbonyl at δC 176.3, a couple of double bond carbons at δC 143.1 and 136.4, and an O/N-quaternary carbon at δC 73.6), shown in Table 2. Comparing with the known Daphniphyllum alkaloids, the spectroscopic data of 2 is similar to those of calyciphylline G15 except for the absence of Δ2,18 and Δ19,N and the presence of additional carbonyl and hydroxyl groups at C-2 and C-4 in 2, respectively. These function groups were assigned by the HMBC correlations from H-3/H-18/H-19/H-20 to C-2 and H-3/H-6/H-21 to C-4 as well as the chemical shift of C-4 (Fig. 2). The relative stereochemistry of 2 was elucidated by the NOESY experiment. The correlations of H-4/H-21/H-13b and H-21/H-6 suggested that they are on the same side and β-oriented (Fig. 3), which is the same as those of calyciphylline G.15 Accordingly, the hydroxyl group at C-4 was placed at α-orientation. Thus, the structure of 2 was determined as shown in Fig. 1 and named calycindaphine B.
The calyciphylline G was isolated as a quaternary amine alkaloid containing a 5-azatricyclo[6.2.1.01,5]undecane ring in 2007.15 However, the possible biogenetic pathway of this unprecedented fused-hexacyclic skeleton has not been described. Comparison of the structural features of calyciphylline G and 2 suggested that the calyciphylline G might be regarded as the key intermediate for 2. A plausible biogenetic pathway for this fused-hexacyclic skeleton is proposed as shown in Scheme 1. Calycindaphine B (2) and calyciphylline G might also be generated from yunnandaphnine A,7 which might be dehydrated in ring E to form intermediated III. Then, the intermediated III should be reduced and undergo the Wagner–Meerwein rearrangement to yield calyciphylline G. Following, hydrogenation of Δ2,18/Δ19,N and oxidation of C-2 and C-4 result in the formation of 2.
Compound 3 has a molecular formula of C24H33O4N with nine degrees of unsaturation. Analysis of spectroscopic data of 3 suggested that 3 have the same skeleton as that of calyciphylline E (11).16 The major difference is the presence of an additional methoxy group in 3. Based on the HMBC correlation from H-methoxy (δH 3.25, s) to C-1 (δC 98.3), the methoxy group was placed at C-1 (Fig. 2). The NOESY correlations (Fig. 3) between H-methoxy to H-2/H-18 suggest that they are on the same side and assigned as an α-orientation. Consequently, the structure of 3 was identified as shown in Fig. 1, and named calycindaphine C.
The molecular formula of compound 4 was deduced as C22H29O4N on the basis of its HRESIMS data. The NMR spectroscopic data of 4 were closely related to that of oldhamiphylline A17 except that the hydroxylated methine at δC 75.8, the methylene at δC 37.1, and the N-substituted methylene at δC 61.34 in oldhamiphylline A are replaced by a methylene at δC 25.4 (C-11), a hydroxylated quaternary carbon at δC 77.4 (C-18), and a lactam carbonyl carbon at δC 174.4 (C-19) in 4, respectively. These changes were proved by the HMBC correlations from H-10/H-12 to C-11, H-1/H-2/H-3/H-20 to C-18, and H-1/H-7/H-20 to C-19 (Fig. 2). The significant NOESY cross-peak of H-21/H-1 demonstrated the H-21 and H-1 were cofacial and placed C-21 to α-orientation (Fig. 3). Furthermore, the NOESY correlation from H-20 to H-3 indicated there are on the same side and placed C-20 to β-orientation. Accordingly, the hydroxyl group attaching to C-18 was assigned as β-orientation. Therefore, the structure of 4 was identified as shown in Fig. 1, and named calycindaphine D.
Calycindaphines E–G (5–7) possess the molecular formula C23H33O4N, C23H31O5N, and C23H31O4N, respectively. Their NMR data analysis suggested that compounds 5–7 belong to daphnezomine F-type skeleton.18 The NMR data of 5 are similar to those of daphlongeranine C18 except that the hydroxy methylene at C-21 in daphlongeranine C was replaced by a singlet methyl in 5. This change was supported by the HMBC correlations from H-21 to C-4/C-5/C-6/C-8 and H-4/H-6 to C-5/C-21. Compound 6 is structurally like 5 except that the C-9/C-10 double bond, the methylene at C-7, and the O-methine at C-2 in 5 are absented in 6, and an acylamide, two oxygenated quaternary carbons, and a methylene were presented in 6, respectively. The oxygenated C-9/C-10 in 6 were devised as an epoxy three-membered ring by the chemical shifts of C-9 and C-10 combined with the exclusive molecular formula from the exact result of HRESIMS. In addition, the HMBC correlations from H-6/H-12/H-19 to the extra acylamide (C-7), H-15/H-16/H-17 to the pair of oxygenated quaternary carbons (C-9/C-10), and H-20/H-18/H-4/H-3 to C-2 supported the above conjectures. Comparison of the chemical shifts of C-9 and C-10 with those of alkaloids containing epoxy group at C-9 and C-10 suggested that the epoxy group in 6 is α-oriented.19–21 Careful analysis of NMR data of 7 indicated that 7 is a daphnezomine F-type alkaloid with two double bonds and a hydroxylated quaternary carbon. HMBC correlations from H-7 to C-1/C-5/C-6/C-12/C-19, H-4/H-12/H-21 to C-6, H-15/H-16/H-11 to C-10/C-17, and H-14/H-15 to C-9 implied that two double bonds were placed at C-7/C-6 and C-10/C-17, and the hydroxylated quaternary carbon was fixed at C-9. Thus, the structures of 5–7 were determined as shown (Fig. 1).
The 1D NMR data of 8 suggested that compound 8 was closely related to 23.11 The major differences between 8 and 23 were that the chemical shift of H-22 was down-shielded from δH 3.33 (in 23) to δH 3.94 (in 8), which might be caused by the different configuration of C-22. Furthermore, analysis of the 1H–1H COSY and HMBC spectra implied that 8 and 23 have same planar structure. The NOESY cross-peak (Fig. 3) between H-22/H-24 in 8 illustrating that these protons are in cofacial and assigned to be β-orientation, which is opposite to that of 23. Furthermore, the optical value of 8 and 23 was measured as [α]22.5D +26.8 (c = 0.5, MeOH) and [α]22.5D −49.4 (c = 0.5, MeOH) respectively, which also provided evidence for the different configuration of C-22 in 8 and 23. Accordingly, compound 8 was elucidated as shown in Fig. 1 and named calycindaphine H.
Calycindaphine I (9) has a molecular formula C30H47O4N. Comparison of its 1D NMR spectra with those of 8 showed that the hydroxylated methine at δC 75.5 (C-22) and methyl at δC 21.3 (C-21) in 8 are replaced by a carbonyl at δC 213.0 (C-22) and a hydroxylated methylene carbon at δC 66.2 (C-21) in 9, respectively. These changes were further confirmed by the HMBC correlations from H-21 to C-4/C-5/C-6/C-8 and H-13/H-14/H-24 to C-22 (Fig. 2). Thus, the structure of 9 was determined as shown in Fig. 1.
Compound 10 showed a protonated [M + H]+ molecular ion at m/z 474.3958, corresponding to a molecular formula of C30H51O3N, with six indices of hydrogen deficiency. The 1D NMR data of 10 are similar to those of daphnioldhanine F,22 except that the characteristic hemiacetal carbon at C-25 in daphnioldhanine F was replaced by an oxidized methylene [δC 72.0, and δH 3.65 (m), 3.49 (d, J = 10.3 Hz)] in 10. The HMBC correlations (Fig. 3) from H-25 to C-22/C-23/C-24 and H-24 to C-22/C-23/C-25/C-26 confirmed the assignment. The data of HRESIMS suggested that 10 has one less degree of unsaturation and a more H2O unit than that of the daphnioldhanine F. The chemical shift of C-29 in 10 appears in upfield shift (ca. 11 ppm) than the similar alkaloids possessing the linkage C-25–O–C-29,22,23 which supported that the specific linkage of C-25–O–C-29 in secodaphniphylline-type alkaloids is broken to form a hydroxy at C-25 and C-29 in 10, respectively. The above spectroscopic evidence deduced the structure of 10 as depicted in Fig. 1, and named calycindaphine J.
By NMR data analysis and comparison of the reported spectroscopic data, 17 known compounds (11–27) were identified as calyciphylline E (11),16 calyciphylline Q (12),24 yunnandaphnine A (13),7 macrodaphniphyllamine (14),7 yunnandaphnine E (15),7 caldaphnidine A (16),11 (−)-bukittinggine (17),25 longistylumphylline C (18),26 deoxyisocalyciphylline B (19),27 deoxycalyciphylline B (20),25,27 caldaphnidine D (21),11 secodaphniphylline (22),28 caldaphnidine E (23),11 daphnioldhanin G (24),22 daphnioldhanin D (25),29 daphnioldhanin E (26),22 and calyciphylline D (27).30
Previous phytochemical studies have shown that Daphniphyllum alkaloids from D. calycinum mainly focused on the anti-cancer effect.15,31 But, in the traditional folk medicine, the plants of D. calycinum are extensively used to treat different diseases which are closely related to inflammation.11–13 To provide more evidences for the pharmacological action of alkaloids from D. calycinum, all isolated alkaloids were evaluated for their effects on TNFα-induced NF-κB activation, TGF-β pathway, and cell autophagy. The bioassay results showed that compounds 22, 23, and 26 inhibited TNFα-induced NF-κB activation in a dose dependent manner (Fig. 5a). Compounds 16 and 18 exhibited significant inhibitory activity on TGF-β/SMAD pathway at a concentration of 50 μM in HepG2 cells (Fig. 5b). Two compounds (24 and 26) revealed their autophagy modulating activities by inducing autophagic puncta and upregulating the autophagy marker LC3-II levels in HEK293 cells (Fig. 5c/d).
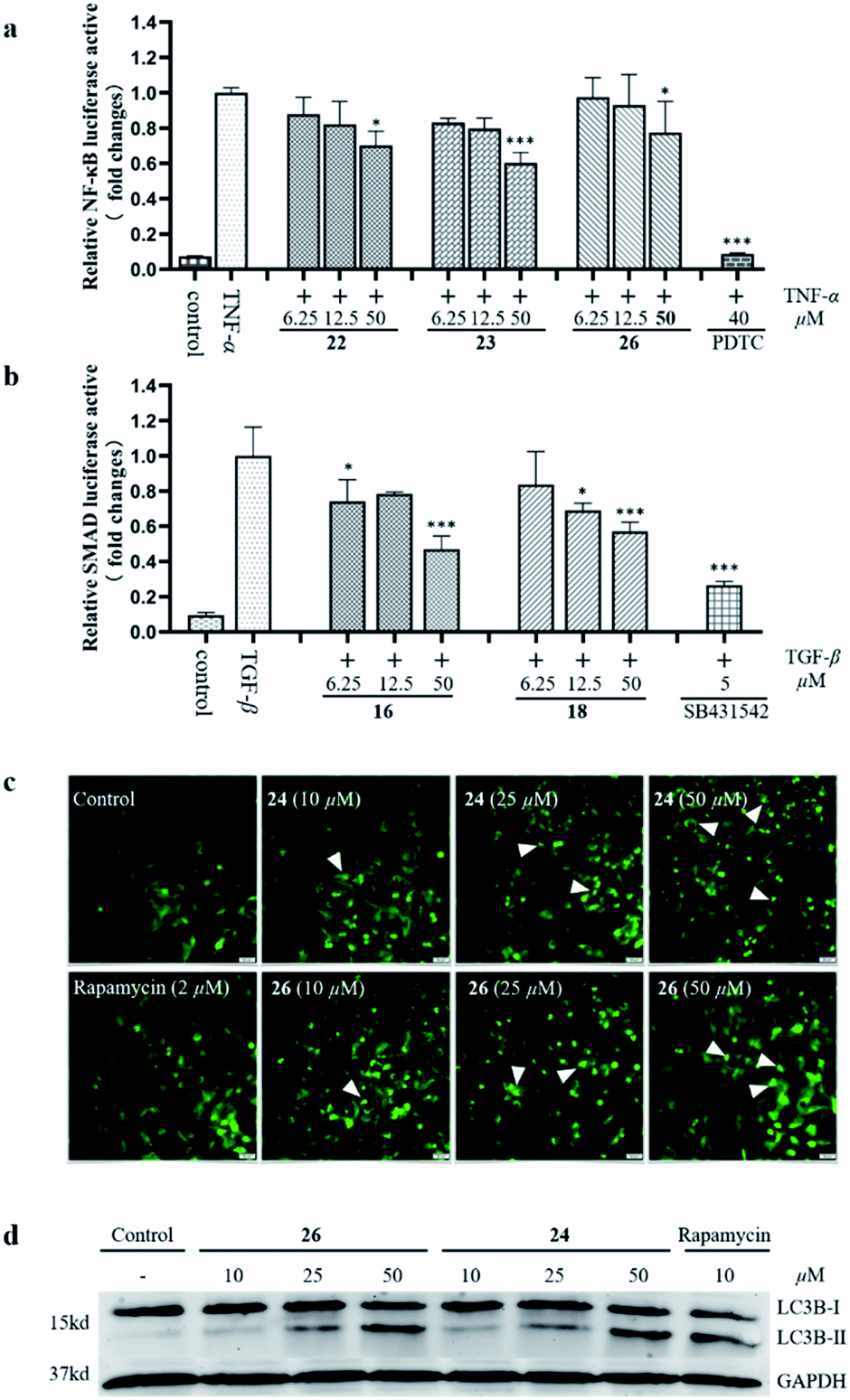 | ||
| Fig. 5 The effects of isolated alkaloids on TNFα-induced NF-κB activation (a), TGF-β/SMAD pathway (b), and cell autophagy (c and d). | ||
Conclusions
In conclusion, ten new Daphniphyllum alkaloids, calycindaphines A–J (1–10), together with 17 known alkaloids were isolated from the roots of D. calycinum. Compound 1 is a novel alkaloid with a new C22 skeleton with a rare 5/8/7/5/5 ring system containing a unique 1-azabicyclo[5.2.1]decane. Compound 2 is the second example for the unique skeleton with 5/6/5/8/5/5 ring system. Compound 10 is the first example of secodaphniphylline-type alkaloid absent of the oxygen-bridge between C-25/C-29.Compounds 16, 18, 22–24, and 26 exhibited their potential bioactivities on NF-κB or TGF-β inhibition and/or cell autophagic induction. Our findings not only revealed the chemicals from the roots of D. calycinum for the first time, but also give a new insight into the complex polycyclic skeletons and structural diversity of Daphniphyllum alkaloids.
Experimental section
General experimental procedures
Optical rotations were measured on a Rudolph Research Analytical Autopol I automatic polarimeter. Ultraviolet (UV) and CD spectra were recorded on a Jasco J-1500 Circular Dichroism Spectrometer. IR spectra were carried on an Agilent Cary 660 series FT-IR spectrometer (KBr). HRESIMS spectra were obtained on an Agilent 6230 HRESIMS spectrometer. 1D and 2D NMR spectra were performed on a Bruker Ascend 600 NMR spectrometer. The chemical shifts were expressed in δ (ppm) with TMS as an internal reference. Column chromatography was performed on Silica gel (40–60 mesh, Grace, USA) column. Thin layer chromatography was carried on precoated silica gel 60 F254 plates (200 μm thick, Merck KGaA, Germany). MPLC was performed using a Buchi Sepacore flash system with a RP-18 column (SilicBond C18, 36 × 460 mm ID, 40–63 μm particle size). Semi-preparative HPLC was conducted on an Agilent 1100/1200 liquid chromatography instrument with a Waters Xbridge Prep C18 column (10 × 250 mm, 5 μm) or Xbridge Prep C8 column (10 × 250 mm, 5 μm). UHPLC analyses were conducted on an Agilent 1290 system using a ZORBAX RRHD Eclipse Plus C18 column (1.8 μm, 2.1 × 50 mm, Agilent).Plant material
The roots of D. calycinum were collected from Guigang city of Guangxi Province, People's Republic of China, in October 2018, and identified by one of the authors, Dr Zhu G.-Y. A voucher specimen (DC-201810) was deposited at State Key Laboratory of Quality Research in Chinese Medicine, Macau University of Science and Technology.Extraction and isolation
The air-dried, powdered roots (30 kg) of D. calycinum were extracted three times with 80% EtOH. The combined filtrates were concentrated under vacuum to afford a dark extract, which was adjusted to pH 2 with HCl. The acidic mixture was centrifuged to remove dark brown precipitates. The aqueous layer was basified to pH 10 with NaHCO3 and then exhaustively extracted with EtOAc to afford the crude alkaloids (170.0 g). The crude alkaloids were subjected to a Silica gel column (CHCl3/MeOH, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0–0:1) to obtain 10 fractions (Fr.A–Fr.F).
:0–0:1) to obtain 10 fractions (Fr.A–Fr.F).
Fr.A (22.5 g) was further chromatographed on a silica gel (40–60 mesh) column (CHCl3/MeOH, 1:0–1:20) to give 10 subfractions (Fr.A1–Fr.A10). Compounds 16 (560.0 mg) and 17 (11.0 mg) were isolated and purified by semipreparative reversed-phase (RP) HPLC with C-18 column eluted with 75% MeCN/0.1% DEA from the fraction Fr.A1. The fraction Fr.A2 was separated into two subfractions (Fr.A2a and Fr.A2b) by MPLC on C-18 column eluting with a gradient of MeCN/0.1% DEA–H2O (20:80 to 100:0, v/v). Fr.A2a was subjected to RP-HPLC with C-18 column (75% MeCN/0.1% DEA) to obtain 4 (6.2 mg). Fr.A2b was purified by semi-preparative HPLC with 72% MeCN/0.1% DEA to give 5 (29.0 mg) and 6 (5.2 mg). Fr.A3 was submitted to CC on silica gel to yield the subfraction (Fr.A3a). Fr.A3a was further purified by RP-HPLC with C-8 column eluted with 80% MeCN/0.1% DEA to afford 18 (95.0 mg), 24 (11.1 mg), 25 (11.0 mg), and 26 (35.0 mg). Fr.A4 was purified by semi-preparative HPLC with a C-18 column to give 22 (88.0 mg). Compound 27 (24.0 mg) was acquired by means of recrystallization from Fr.A10.
Fr.B (20.1 g) was subjected to MPLC on C-18 column and eluted with a gradient of MeCN/0.1% DEA–H2O (20:80 to 100:0, v/v) to give four subfractions (Fr.B1–Fr.B4). Fr.B1 was isolated by semi-preparative HPLC with C-18 column (60% MeCN/0.1% DEA) to obtain three alkaloids, 2 (3.6 mg), 19 (76.8 mg), and 20 (16.3 mg). Fr.B2 was separated by RP-HPLC with C-18 column (75% MeCN/0.1% DEA) to yield Fr.B2a and Fr.B2b. Fr.B2a was purified by RP-HPLC with C-18 column (54% MeCN/0.1% DEA) to give 7 (3.1 mg). Fr.B2b was purified by RP-HPLC (62% MeCN/0.1% DEA) to give 1 (7.0 mg). Fr.B3 was isolated by RP-HPLC with C-8 column (65% MeCN/0.1% DEA) to yield 13 (15.7 mg). Fr.C (19.3 g) was isolated by the MPLC on C-18 to give Fr.C8 and then was purified by RP-HPLC with C-18 column (55% MeCN/0.1% DEA) to afford 12 (2.6 mg). Fr.D (9.2 g) was separated by MPLC with a C-18 column and eluted with a gradient of MeCN/0.1% DEA–H2O (10:80 to 100:0, v/v) to give the main subfraction of Fr.D1. And then, the subfraction was separated by the RP-HPLC with C-18 column (58% MeCN/0.1% DEA) to afford 3 (2.5 mg), 9 (4.0 mg), 11 (16.0 mg), 14 (3.6 mg), 15 (45.0 mg), 21 (6.2 mg), and 23 (22.0 mg). Alkaloids of Fr.F (6.0 g) were enriched by the MPLC to give the main subfraction Fr.F7 which was purified by RP-HPLC with C-18 column to obtained 8 (2.7 mg) and 10 (5.2 mg).
ε): 200.4 (7.45), 304.8 (0.620) nm; IR (KBr) νmax: 3730, 3700, 3625, 3595, 3444, 2956, 1768, 1708, 1448, 1388, 1266, 1200, 754 cm−1; ECD (MeOH) λmax (logε): 217.2 (−51.89), 270.6 (32.30), 352.2 (−36.27) nm; 1H NMR and 13C NMR see Tables 1 and 2; HRESIMS m/z 370.2340 [M + H]+ (calcd for C23H31NO4 370.2375).ε): 201.1 (6.66), 346.1 (1.24) nm; IR (KBr) νmax: 3730, 3595, 3442, 2929, 1733, 1645, 1562, 1442, 1387, 1268, 1169, 754 cm−1; ECD (MeOH) λmax (logε): 200.4 (−52.76), 226.8 (166.29) nm; 1H NMR and 13C NMR see Tables 1 and 2; HRESIMS m/z 386.2324 [M + H]+ (calcd for C23H32NO4 386.2326).ε): 201.3 (1.26), 304.8 (0.66) nm; IR (KBr) νmax: 3730, 3701, 3626, 3596, 3449, 2933, 2865, 1735, 1649, 1545, 1393, 1358, 756 cm−1; ECD (MeOH) λmax (logε): 214.2 (−33.48), 247.8 (5.28), 283.2 (−13.25) nm; 1H NMR and 13C NMR see Tables 1 and 2; HRESIMS m/z 400.3485 [M + H]+ (calcd for C24H34NO4 400.2482).ε): 199.5 (12.9) nm; IR (KBr) νmax: 3730, 3701, 3625, 3596, 3380, 2949, 2858, 1770, 1699, 1546, 1425, 1388, 1350, 1268, 1128, 1093, 1043, 754 cm−1; ECD (MeOH) λmax (logε): 209.5 (533.12), 230.2 (−181.22) nm; 1H NMR and 13C NMR see Tables 1 and 2; HRESIMS m/z 372.2166 [M + H]+ (calcd for C22H30NO4 372.2169).ε): 204.1 (8.95) nm; IR (KBr) νmax: 3730, 3701, 3625, 3595, 3426, 2929, 1730, 1650, 1484, 1434, 1355, 1316, 1280, 1170, 1036, 741 m−1; ECD (MeOH) λmax (logε): 205.2 (271.64), 230.4 (−310.91) nm; 1H NMR and 13C NMR see Tables 1 and 2; HRESIMS m/z 388.2480 [M + H]+ (calcd for C23H34NO4 388.2482).ε): 200.7 (4.12), 222.8 (6.62) nm; IR (KBr) νmax: 3730, 3701, 3625, 3595, 3446, 2927, 2875, 1725, 1680, 1447, 1380, 1270, 1173, 755 cm−1; ECD (MeOH) λmax (logε): 229.1 (−413.27), 256.8 (72.60) nm; 1H NMR and 13C NMR see Tables 1 and 2; HRESIMS m/z 402.2293 [M + H]+ (calcd for C23H32NO5 402.2275).ε): 202.4 (4.54), 264.8 (1.03) nm; IR (KBr) νmax: 3730, 3701, 3626, 3596, 3446, 3927, 1731, 1644, 1457, 1391, 1271, 755 cm−1; ECD (MeOH) λmax (logε): 198.0 (−217.81), 234.6 (68.96), 264.0 (−150.08) nm; 1H NMR and 13C NMR see Tables 1 and 2; HRESIMS m/z 386.2332 [M + H]+ (calcd for C23H32NO4 386.2326).ε): 223.2 (10.89), 265.8 (−14.92), 294.6 (−4.72), 313.2 (10.71) nm; 1H NMR and 13C NMR see Tables 1 and 2; HRESIMS m/z 472.3786 [M + H]+ (calcd C30H50NO3 for 472.3785).ε): 202.4 (3.28), 300.5 (1.76) nm; IR (KBr) νmax: 3730, 3701, 3625, 3595, 3446, 2934, 2868, 2316, 1703, 1647, 1545, 1458, 1392, 1268, 755 cm−1; ECD (MeOH) λmax (logε): 210.6 (−29.16), 232.2 (−16.40), 288.6 (−16.97) nm; 1H NMR and 13C NMR see Tables 1 and 2; HRESIMS m/z 486.3562 [M + H]+ (calcd for C30H48NO4 486.3578).ECD calculations of compound 1
Conformational analyses were carried out via random searching in the Sybyl-X 2.0 using the MMFF94S force field with an energy cutoff of 2.5 kcal mol−1. The results showed the nine lowest energy conformers for both compounds. Subsequently, the conformers were re-optimized using DFT at the PBE0-D3(BJ)/def2-SVP level in MeOH using the polarizable conductor calculation model (SMD) by the ORCA4.2.1 program. The energies, oscillator strengths, and rotational strengths (velocity) of the first 60 electronic excitations were calculated using the TDDFT methodology at the PBE0/def2-TZVP level in MeOH. The ECD spectra were simulated by the overlapping Gaussian function (half the bandwidth at 1/e peak height, sigma = 0.30 for all). To get the final spectra, the simulated spectra of the conformers were averaged according to the Boltzmann distribution theory and their relative Gibbs free energy (ΔG).Cell lines and cell cultures
The HepG2-NF-κB-Luc cell line is stably transfected with the NF-κB-luciferase gene, which was generously provided by Dr C. H. Leung (University of Macau). Cells were cultivated with DMEM medium supplemented with 10% fetal bovine serum (FBS), 100 U mL−1 penicillin, and 100 μg mL−1 streptomycin at 37 °C with 5% CO2 and 95% air incubator.SMAD 2/3 responsive luciferase reporter HepG2 stable cell line was purchased from Signosis. Cells were cultivated with DMEM medium supplemented with 5% FBS, 100 U mL−1 penicillin, 100 μg mL−1 streptomycin, and 100 μg mL−1 hygromycin B at 37 °C with 5% CO2 and 95% air incubator.
HEK293 cell line stable transfected with GFP-LC3 was kindly provided by Dr X. M. Zhu (Macau University of Science and Technology). The cells were cultured in an α-MEM medium supplemented with 10% FBS under a humidified atmosphere containing 5% CO2 at 37 °C.
NF-κB luciferase assay
The NF-κB activity was determined by NF-κB luciferase assay as described in our previous publication with a slight modification.32 Briefly, HepG2-NF-κB-Luc cells were seeded on a 96-well microplate with 1 × 104 cells per well and cultured at 37 °C with 5% CO2 incubator for 18 h. Then, cells were pretreated with compounds (6, 12.5, and 50 μM) for 12 h and induced with TNF-α (10 ng mL−1) for 4 h. Ammonium pyrrolidinedithiocarbamate (PDTC) was used as the positive control. The firefly luciferase signal was measured with the Bright-Glo Luciferase Reporter Assay System (Promega, Madison, WI) according to the manufacturer's instruction using a multimode reader (SpectraMax iD5, Danaher).TGF-β induced SMAD luciferase assay
The effects of compounds on the TGF-β/SMAD were determined by TGF-β/SMAD luciferase assay. Briefly, HepG2/SMAD-Luc cells were seeded on a 96-well microplate with 1 × 104 cells per well and cultured at 37 °C with 5% CO2 incubator for 18 h. After adhesion, the cells were pretreated with compounds at different concentration for 6 h and induced with TGF-β (10 ng mL−1) for 18 h. Meanwhile, SB-431542, a specific inhibitor of TGF-β Receptor Kinase, was applied as the positive control. The firefly luciferase signal was measured with the Bright-Glo Luciferase Reporter Assay System (Promega, Madison, WI) according to the manufacturer's instruction using a multimode reader (SpectraMax iD5, Danaher).LC3 puncta counting
The HEK293-GFP-LC3 cells were applied for visualizing autophagosome formation after treatment with various compounds for 24 hours. During autophagosome formation, GFP-LC3 is processed and recruited to the autophagosome membrane, where it can be imaged as cytophasmic puncta by IncuCyte ZOOM live cell imaging (Olympus, coupled with Hamama Tsu ORCA-Flash 40 LT Plus Scientific CMOS Digital Camera). The percentage of GFP-LC3 positive cells can be determined and is indicative of autophagosome formation.Western blot analysis
Primary antibody against LC3II was purchased from Cell Signaling Technology. Primary antibody against glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was purchased from Abcam Inc. (Cambridge, MA). Secondary antibodies, and goat anti-mouse/anti-rabbit IgG H&L (IRDye 800CW) were purchased from Abcam Inc. (Cambridge, MA).The cells were seeded onto 6-well plates with α-MEM medium and cultured for 24 hours. After treating with various concentrations of compounds 24 and 26, cells were collected and lysed in lysis buffer on ice for 30 minutes. Protein samples were electrophoresed using 15% SDS-PAGE gel and transferred onto a nitrocellulose membrane (NC membrane). The membranes were blocked by a 5% BSA and incubated with the primary antibody overnight at 4 °C, followed by the secondary antibody for 1 hour at room temperature. Protein bands were detected by the LI-COR Odyssey imaging system (Lincoln, NE).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Macao Science and Technology Development Funds (0008/2019/A, 0075/2019/AGJ, 0023/2019/AKP, and 125/2017/A3).Notes and references
- S. Yamamura, H. Irikawa, Y. Okumura and Y. Hirata, Bull. Chem. Soc. Jpn., 1975, 48, 2120–2123 CrossRef CAS.
- J. i. Kobayashi and T. Kubota, Nat. Prod. Rep., 2009, 26, 936–962 RSC.
- M. Cao, Y. Zhang, H. He, S. Li, S. Huang, D. Chen, G. Tang, S. Li, Y. Di and X. Hao, J. Nat. Prod., 2012, 75, 1076–1082 CrossRef CAS.
- M.-M. Cao, L. Wang, Y. Zhang, H.-P. He, Y.-C. Gu, Q. Zhang, Y. Li, C.-M. Yuan, S.-L. Li and Y.-T. Di, Fitoterapia, 2013, 89, 205–209 CrossRef CAS.
- H. Wu, X. Zhang, L. Ding, S. Chen, J. Yang and X. Xu, Planta Med., 2013, 79, 1589–1598 CrossRef CAS.
- T. Nakano, M. Hasegawa and Y. Saeki, J. Org. Chem., 1973, 38, 2404–2405 CrossRef CAS.
- Y.-T. Di, H.-P. He, C.-S. Li, J.-M. Tian, S.-Z. Mu, S.-L. Li, S. Gao and X.-J. Hao, J. Nat. Prod., 2006, 69, 1745–1748 CrossRef CAS.
- H. Morita and J. i. Kobayashi, Org. Lett., 2003, 5, 2895–2898 CrossRef CAS.
- G. A. Wallace and C. H. Heathcock, J. Org. Chem., 2001, 66, 450–454 CrossRef CAS.
- T. He, Y. Zhou, Y. H. Wang, S. Z. Mu and X. J. Hao, Helv. Chim. Acta, 2011, 94, 1019–1023 CrossRef CAS.
- Z.-J. Zhan, C.-R. Zhang and J.-M. Yue, Tetrahedron, 2005, 61, 11038–11045 CrossRef CAS.
- C.-R. Zhang, S.-P. Yang and J.-M. Yue, J. Nat. Prod., 2008, 71, 1663–1668 CrossRef CAS.
- S.-M. Shen, H. Li, J.-R. Wang, Y.-B. Zeng and Y.-W. Guo, Tetrahedron, 2020, 131616 CrossRef CAS.
- Y. Zhang, Y.-T. Di, Q. Zhang, S.-Z. Mu, C.-J. Tan, X. Fang, H.-P. He, S.-L. Li and X.-J. Hao, Org. Lett., 2009, 11, 5414–5417 CrossRef CAS.
- S. Saito, T. Kubota and J. i. Kobayashi, ChemInform, 2007, 48, 5693–5695 CAS.
- S. Saito, T. Kubota and J. i. Kobayashi, Tetrahedron Lett., 2007, 48, 3809–3812 CrossRef CAS.
- X. Chen, Z. J. Zhan and J. M. Yue, Chem. Biodiversity, 2004, 1, 1513–1518 CrossRef CAS.
- T. Q. Yang, Y. T. Di, H. P. He, Q. Zhang, Y. Zhang and X. J. Hao, Helv. Chim. Acta, 2011, 94, 397–403 CrossRef CAS.
- C. R. Zhang, H. B. Liu, S. H. Dong, J. Y. Zhu, Y. Wu and J. M. Yue, Org. Lett., 2009, 11, 4692–4695 CrossRef CAS.
- C. S. Li, Y. T. Di, S. Z. Mu, H. P. He and X. J. Hao, J. Nat. Prod., 2008, 71, 1202–1206 CrossRef CAS.
- Z. Li, S. Peng, L. Fang, Y. Yang and Y. Guo, Chem. Biodiversity, 2010, 6, 105–110 CrossRef.
- S. Z. Mu, X. W. Yang, Y. T. Di, H. P. He, Y. Wang, Y. H. Wang, L. Li and X. J. Hao, Chem. Biodiversity, 2007, 4, 129–138 CrossRef CAS.
- S.-Z. Mu, J.-S. Wang, X.-S. Yang, H.-P. He, C.-S. Li, Y.-T. Di, Y. Wang, Y. Zhang, X. Fang and L.-J. Huang, J. Nat. Prod., 2008, 71, 564–569 CrossRef CAS.
- H. Yahata, T. Kubota and J. i. Kobayashi, J. Nat. Prod., 2009, 72, 148–151 CrossRef CAS.
- D. Arbain, L. Byrne, J. Cannon, V. Patrick and A. White, Aust. J. Chem., 1990, 43, 185–190 CrossRef CAS.
- X. Chen, Z. J. Zhan and J. M. Yue, Helv. Chim. Acta, 2005, 88, 854–860 CrossRef CAS.
- X. Zhang, J. Zhang, Y. Tan, Q. Liu and M. Liu, Molecules, 2012, 17, 9641–9651 CrossRef CAS.
- M. Toda, Y. Hirata and S. Yamamura, Tetrahedron, 1972, 28, 1477–1484 CrossRef CAS.
- Z. J. Zhan, G. W. Rao, X. R. Hou, C. P. Li and W. G. Shan, Helv. Chim. Acta, 2009, 92, 1562–1567 CrossRef CAS.
- S. Saito, T. Kubota, E. Fukushi, J. Kawabata, H. Zhang and J. i. Kobayashi, Org. Lett., 2007, 9, 1207–1209 CrossRef CAS.
- S. Saito, H. Yahata, T. Kubota, Y. Obara and J. I. Kobayashi, Tetrahedron, 2008, 64, 1901–1908 CrossRef CAS.
- G.-Y. Zhu, L.-P. Bai, L. Liu and Z.-H. Jiang, Phytochemistry, 2014, 107, 175–181 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra00107h |
| This journal is © The Royal Society of Chemistry 2021 |