
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTransition-metal-free decarboxylative thiolation of stable aliphatic carboxylates†
Wei-Long Xing,
De-Guang Liu and
Ming-Chen Fu *
*
Department of Chemistry, University of Science and Technology of China, Hefei 230026, China. E-mail: fumingchen@ustc.edu.cn
First published on 22nd January 2021
Abstract
A transition-metal-free decarboxylative thiolation protocol is reported in which primary, secondary, tertiary (hetero)aryl acetates and α-CN substituted acetates undergo the decarboxylative thiolation smoothly, to deliver a variety of functionalized aryl alkyl sulfides in moderate to excellent yields. Aryl diselenides are also amenable substrates for construction of C–Se bonds under the simple and mild reaction conditions. Moreover, the protocol is successfully applied to the late-stage modification of pharmaceutical carboxylates with satisfactory chemoselectivity and functional-group compatibility.
Introduction
Organosulfur chemistry has attracted increasing attention due to the importance of sulfur-containing compounds in bioactive natural products, functional materials, and pharmaceuticals, such as butoconazole, nelfinavir and axitinib (Fig. 1).1 Therefore, the development of convenient and efficient protocols for constructing C–S bonds is of great significance in organic synthesis.2 The conventional approach to the direct construction of C(sp2)–S bonds usually involves the use of transition-metal catalysts to realize thiolation of aryl halides with thiols or disulfides.3 Significant achievements have been made by the groups of Hartwig,4 Fu and Peters et al.5 For construction of C(sp3)–S bonds, nucleophilic substitution reaction of alkyl halide with mercaptan is the most straightforward, but this is limited to primary alkyl halides. Research on the use of diaryl disulfides as sulfur sources to construct secondary and tertiary C(sp3)–S bonds remains scarce. Alkyl carboxylic acids, which are environmentally benign and widely available building blocks, have been widely used as organic synthons in cross-coupling reactions;6 Thus, they can offer an appealing alternative to the traditional methods.7 In 2014, Feng and Xu's group used one equivalent of AgNO3 to successfully realize the decarboxylative C(sp3)–S cross-coupling reaction of alkyl carboxylic acid with diary disulfides.8 Fu's group reported a visible-light-induced decarboxylative arylthiation using alkyl carboxylic redox-active esters with aryl thiols.9 Recently, Liu and Wang et al. also realized the construction of C(sp3)–S bonds by using alkyl carboxylic redox-active esters mediated by equivalent manganese powder.10 However, the required equivalent of precious metals and oxidant, or stoichiometric chemical premodification of the carboxylate unit, largely limits the scope of these methods in large-scale application. In terms of practicality and scalability, the development of a general and efficient method for transition-metal-free decarboxylative thiolation of stable aliphatic carboxylates would be extremely attractive, albeit challenging. Very recently, Audisio and coworkers developed a carbon isotope exchange procedure on arylacetic acid salts in dimethyl sulfoxide (DMSO) without adding transition metal catalysts.11 Simultaneously, Lundgren's group also demonstrated that isotopic exchange of carboxylic acids with 13CO2 can proceed smoothly; therein, the authors elegantly developed a new decarboxylative carbon–carbon bond-forming reaction.12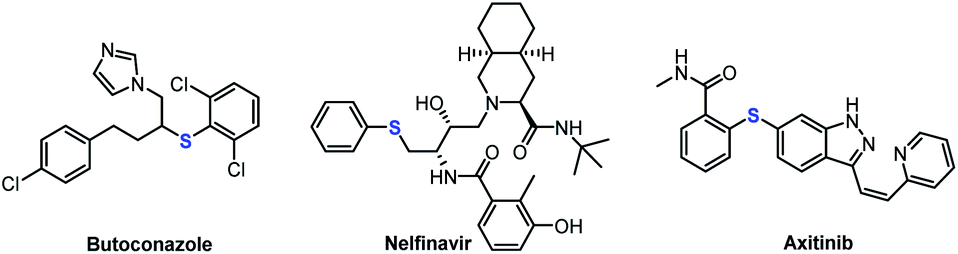 | ||
| Fig. 1 Thioether-containing pharmaceutical agents. | ||
In a continuation of our studies on decarboxylative functionalization,13 and inspired by these seminal works,11,12,14 we wondered whether the alkyl carboxylates I could directly decarboxylate in DMSO or DMF (N,N-dimethylformamide) solution to form the corresponding anion II (Scheme 1A, path a),15 or generate dienolate species IV in situ (path b).14b These intermediates might undergo nucleophilic attack onto aryl disulfides, and subsequently deliver the desired product III (Scheme 1A).
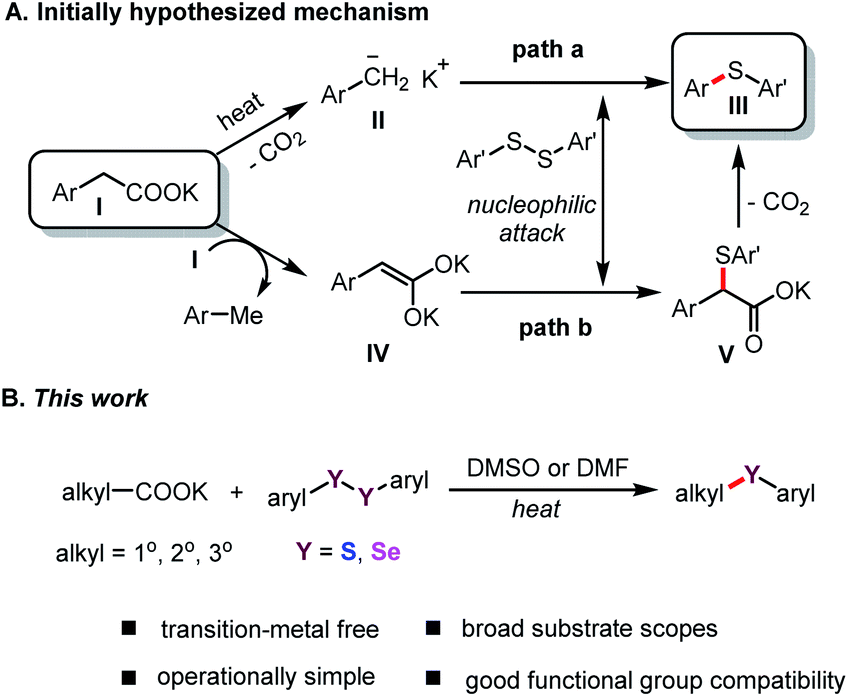 | ||
| Scheme 1 The initially hypothesized mechanism and the focus of this work. | ||
Herein, we report an efficient and operationally simple protocol for the synthesis of aryl alkyl sulfides through transition-metal-free decarboxylative cross-coupling of aryl disulfides. This method has broad substrate scope, (hetero)aryl acetates, and α-CN substituted acetates are amenable, has satisfactory functional group tolerance, and proceeds with good to excellent yields (Scheme 1B). In addition, this protocol was successfully applied to synthesize aryl alkyl selenides, and, to our knowledge, is the first general method for the synthesis of aryl alkyl selenides using stable aliphatic carboxylates without the use of transition-metal catalysts.
Results and discussion
To verify our hypothesis presented in Scheme 1A, we first selected potassium 2-(4-cyanophenyl)acetate (1) and diphenyl disulfide as model substrates. To our delight, after careful optimization of all reaction parameters, optimal conditions were identified in which the desired product 3 was obtained in 97% yield in DMSO solution at 80 °C for 24 h. The salt effect of the counter-cation of the carboxylate on the reaction was investigated (Table 1, row 1). The reaction proceeded smoothly, albeit in lower yield, when the Li+ salt of 1 was used. Na+, Rb+, and Cs+ salts of 1 gave good yields of 3 of 81, 84, and 87%, respectively. The conversion dramatically decreased with divalent metal salts of 1 (Ca2+), and 2-(4-cyanophenyl)acetic acid failed to give the desired product. The nature of the solvent was crucial for the success of the carboxylate decarboxylation.16 The decarboxylative thiolation occurred smoothly in polar aprotic solvents, such as DMF or DMA (Table 1, row 2), only 7% yield of 3 was detected in MeCN solution, and the reaction was completely suppressed in dioxane, glyme, diglyme, or DCE solution. Control experiments showed that changing the ratio of the substrates (1, 0.2 mmol; 2, 0.3 mmol) reduced the yield of product 3 to as low as 69%. The conversion decreased dramatically when the reaction temperature was reduced to 60 °C, delivering the desired product in 52% yield. It should be noted that this protocol was not very sensitive to humidity; 65% yield of the desired product could be obtained in the presence of one equivalent of water.
|
||||||
|---|---|---|---|---|---|---|
| Different cation instead of K+ | ||||||
| Li+ | Na+ | Rb+ | Cs+ | Ca2+ | H+ | |
| a Reaction conditions: 1 (0.3 mmol, 1.5 eq.), 2 (0.2 mmol, 1.0 eq.), DMSO (2.0 mL), 80 °C, 24 h. The yield was determined by GC analysis using diphenylmethane as internal standard.b 0.15 mmol (p-CNC6H5CH2CO2)2Ca instead of 1. | ||||||
| Yield of 3 (%) | 72 | 81 | 84 | 87 | 28b | 0 |

| Different solvent instead of DMSO | |||||||
|---|---|---|---|---|---|---|---|
| DMF | DMA | MeCN | Dioxane | Glyme | Diglyme | DCE | |
| Yield of 3 (%) | 81 | 81 | 7 | 0 | 0 | 0 | 0 |
| Control experiments | |||
|---|---|---|---|
| 1 (0.2 mmol), 2 (0.3 mmol) | Reaction temperature: 60 °C | 0.2 mmol H2O | |
| Yield of 3 (%) | 69 | 52 | 65 |
With the optimized conditions in hand, we further investigated the scope of the decarboxylative thiolation. As shown in Table 2, a myriad of (hetero)aryl acetates and α-CN substituted acetates, including primary, secondary, and even tertiary carboxylates, were readily converted into the corresponding aryl alkyl sulfides in moderate to excellent yields. A broad range of functional groups, such as cyano (3), aryl halides (5, 6, 7), trifluoromethyl (8), trifluoromethoxy (9), sulfonate (10), ether (16), and even nitro (11, 12) groups proved compatible. The aryl halides provided versatile synthetic handles for further functionalization. The steric hindrance of aryl acetates has no clear influence on the reaction (11, 12). Increasing the reaction temperature and addition of 18-crown-6 could promote the efficiency of the reaction (5, 16). Potassium naphthalene acetate was also an amenable substrate (13). Moreover, both monophenyl and diphenyl substituted phenylacetates successfully underwent decarboxylative thioetherification in high yields of 93% (14) and 97% (15), respectively. Notably, various potentially reactive heterocycles, such as pyridines (17 and 18), thiophene (19), pyrimidine (20), isoxazole (21), and benzo[d]thiazole (22) were well tolerated in this protocol.

Cyano structures are versatile synthetic intermediates and possess biological activity.17 Substituted cyanoacetates have been reported as cyano synthons through decarboxylative coupling under high-temperature conditions using palladium catalyst by Liu and coworkers.18 To our delight, a series of substituted cyanoacetates were also successfully applied to decarboxylative thiolation with diphenyl disulfide without transition-metal catalysts (23–30). Cyanoacetate salts bearing one or two α-substituents were compatible with the reaction conditions, as was a four-membered ring structure with an α-substituent, which provides an efficient and simple practical method for the synthesis of α-CN sulfides. It should be noted that using potassium 1-cyanocyclopropane-1-carboxylate as the substrate, only the ring-opening product of decarboxylative thiolation was obtained (30), which might be due to the effect of ring tension.
Remarkably, some pharmaceutical carboxylates (31–36) were smoothly transformed into sulfides with satisfactory chemoselectivity and functional-group compatibility, demonstrating the potential of this method for post-synthetic-drug modification. Notably, diclofenac (32) and carprofen (35) were also competent substrates in this transformation, with the unprotected N–H structure retained.
We next investigated a diverse set of disulfides to further highlight the versatility of this method (Table 3). A range of aryl disulfides served as effective cross-coupling partners with carboxylates to form compounds bearing C–S bonds in good to excellent yields (57–97%); these compounds contained electron-rich (37, 38), electron-deficient (39), and hindered (40–42) functional groups. In addition, heterocyclic-aryl disulfides 43–45, which have potential physiological activity and are useful in medicinal chemistry, were also amenable substrates and gave the desired products in good yields.

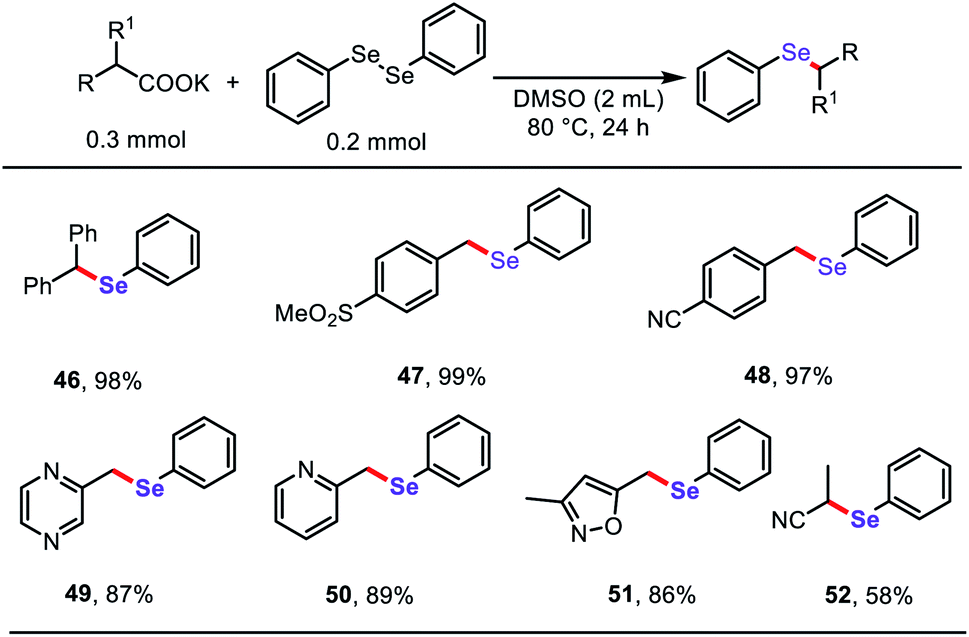
Based on the success of the decarboxylative C–S bond formation in this protocol and the similar reactivity in construction of C–Se bonds,19 we further investigated the reactions of diphenyl diselenide with stable aliphatic carboxylates to generate related aryl selenide scaffolds (Table 4), which are widely applied in antioxidants, fluorescent probes, and functional organic materials.20 To our delight, both (hetero)aryl acetates and α-CN substituted acetates underwent decarboxylative cross-coupling of diphenyl diselenide under the standard reaction conditions described above, providing a simple general approach to synthesize a diverse range of organoselenium compounds.
| a Reaction conditions: carboxylates (0.3 mmol), diphenyl diselenide (0.2 mmol), DMSO (2 mL), 80 °C, 24 h, under Ar atmosphere. Isolated yield. |
|---|
 |
To gain more mechanistic insights into this reaction, a series of mechanism experiments and density functional theory (DFT) calculations were carried out (see ESI for detailed data†). Based on these results and on the previous works,11,12,14,15 a plausible mechanism involving dienol formation and proton transfer was proposed (Fig. 2). First, the benzyl carboxylate ion substrate IN1 undergoes benzyl C–H proton migration with the assistance of additional IN1, to deliver an electrostatic-dominated dimer intermediate IN1-dimer. Due to the inhomogeneity of the charge distribution, the migrated proton is easily transferred to the original carboxyl anion with a free-energy decrease of 11.7 kcal mol−1, and the corresponding dienol anion IN3 is obtained. This strategy is more advantageous than the intramolecular proton migration (TS1, 23.2 kcal mol−1 vs. TS2, 44.1 kcal mol−1). Subsequently, the disulfide substrate IN2 electrophilically attacks the dienol anion IN3 to deliver the corresponding sulfurized product IN4. At the same time, the released thiophenol anion binds to the carboxyl O–H bond through electrostatic interaction. After abstracting the proton of the carboxyl group, the generated thiophenol IN6 leaves briefly, and the decarboxylation of the remaining species IN5 becomes achievable (TS5, 18.4 kcal mol−1); this is easier than the direct decarboxylation of benzyl carboxylate ion (TS7, 21.1 kcal mol−1, see ESI for details†). When the new benzyl carbanion intermediate IN7 forms after decarboxylation, the thiophenol IN6 “returns” the previously captured proton to IN7. Thereby, the formation of the product P0 is exergonic by 24.5 kcal mol−1, and the energy barrier of this step is 7.1 kcal mol−1. In addition, we also considered the classical carbanion decarboxylation pathway and a cooperative attack-decarboxylation pathway; the results clearly indicate that this proposal of proton transfer is dominant (see ESI for related calculation details†). Different from previous reports, this transformation might proceed through a novel pathway involving proton migration, electrophilic attack, decarboxylation, and proton exchange, providing great potential and a different perspective for decarboxylative cross-coupling.
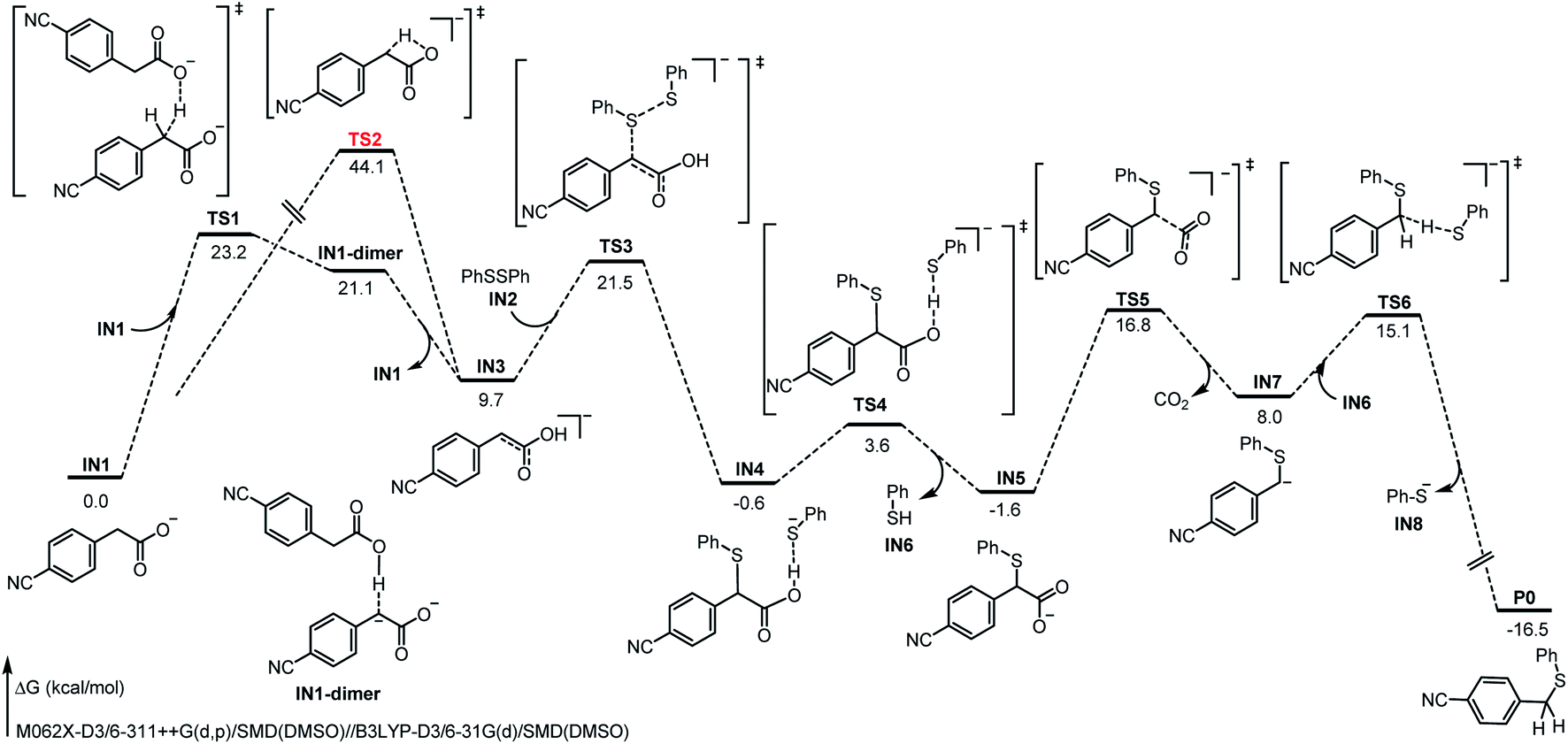 | ||
| Fig. 2 The Gibbs free energies of the feasible mechanism. | ||
Conclusions
We have developed a mild and efficient protocol for the C–S cross-coupling of aryl disulfides with stable aliphatic carboxylates without the use of transition-metal catalysts. A wide range of primary, secondary, and tertiary (hetero)aryl acetates and α-CN substituted acetates underwent the decarboxylative thiolation smoothly in moderate to excellent yields. Moreover, this protocol was also successfully applied to construct C–Se bonds with aryl diselenide. DFT calculations revealed that the pathway involved dienol formation, and proton transfer is dominant in this protocol. The simple and mild conditions of this protocol will allow a wide range of applications for the synthesis of biologically and pharmaceutically active molecules containing aryl sulfur/selenium moieties.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Natural Science Foundation of Anhui (2008085MB40) and the USTC Research Funds of the Double First-Class Initiative (YD2060002003). The authors thank Prof. Rui Shang for helpful discussion on experiment details.Notes and references
- (a) C. Christophersen and U. Anthoni, Sulfur Rep., 1986, 4, 365 CrossRef CAS; (b) M. Feng, B. Tang and S.-H. Liang, Curr. Top. Med. Chem., 2016, 16, 1200 CrossRef CAS; (c) K. A. Scott and J. T. Njardarson, Top. Curr. Chem., 2018, 5, 376 Search PubMed; (d) D. A. Boyd, Angew. Chem., Int. Ed., 2016, 55, 15486 CrossRef CAS.
- (a) B. Liu, C.-H. Lim and G. M. Miyake, J. Am. Chem. Soc., 2017, 139, 13616 CrossRef CAS; (b) D. Liu, H.-X. Ma, P. Fang and T.-S. Mei, Angew. Chem., Int. Ed., 2019, 58, 5033 CrossRef CAS; (c) F. Sandfort, T. Knecht, T. Pinkert, C. G. Daniliuc and F. Glorius, J. Am. Chem. Soc., 2020, 142, 6913 CrossRef CAS.
- (a) T. Kondo and T. Mitsudo, Chem. Rev., 2000, 100, 3205 CrossRef CAS; (b) S. V. Ley and A. W. Thomas, Angew. Chem., Int. Ed., 2003, 42, 5400 CrossRef CAS; (c) M. S. Oderinde, M. Frenette, D. W. Robbins, B. Aquila and J. W. Johannes, J. Am. Chem. Soc., 2016, 138, 1760 CrossRef CAS; (d) J. Lou, Q.-N. Wang, P. Wu, H.-M. Wang, Y.-G. Zhou and Z.-K. Yu, Chem. Soc. Rev., 2020, 49, 4307 RSC.
- (a) D. Baranano and J. F. Hartwig, J. Am. Chem. Soc., 1995, 117, 2937 CrossRef CAS; (b) G. Mann, D. Baranano, J. F. Hartwig, A. L. Rheingold and I. A. Guzei, J. Am. Chem. Soc., 1998, 120, 9205 CrossRef CAS; (c) M. A. Fernández-Rodríguez, Q.-L. Shen and J. F. Hartwig, J. Am. Chem. Soc., 2006, 128, 2180 CrossRef; (d) E. Alvaro and J. F. Hartwig, J. Am. Chem. Soc., 2009, 131, 7858 CrossRef CAS.
- (a) C. Uyeda, Y.-C. Tan, G. C. Fu and J. C. Peters, J. Am. Chem. Soc., 2013, 135, 9548 CrossRef CAS; (b) M. W. Johnson, K. I. Hannoun, Y.-C. Tan, G. C. Fu and J. C. Peters, Chem. Sci., 2016, 7, 4091 RSC.
- (a) L. J. Gooßen, G.-J. Deng and L. M. Levy, Science, 2006, 313, 662 CrossRef; (b) R. Shang and L. Liu, Sci. China Chem., 2011, 54, 1670 CrossRef CAS; (c) D. Kong, P. J. Moon, O. Bsharat and R. J. Lundgren, Angew. Chem., Int. Ed., 2020, 59, 1313 CrossRef CAS; (d) Á. M. Martínez, D. Hayrapetyan, T. van Lingen, M. Dyga and L. J. Gooßen, Nat. Commun., 2020, 11, 4407 CrossRef.
- (a) C. Shen, P.-F. Zhang, Q. Sun, S.-Q. Bai, T. S. Andy Hor and X.-G. Liu, Chem. Soc. Rev., 2015, 44, 291 RSC; (b) W.-J. Wang, X.-J. Liu and X. Wang, ChemistrySelect, 2017, 2, 1140 CrossRef CAS; (c) A. Hosseinian, P. D. K. Nezhad, S. Ahmadi, Z. Rahmani and A. Monfared, J. Sulfur Chem., 2019, 40, 88 CrossRef CAS; (d) T.-P. Cao, T.-X. Xu, R.-T. Xu, X.-L. Shu and S.-H. Liao, Nat. Commun., 2020, 11, 5340 CrossRef CAS; (e) F.-H. Cui, J. Chen, S.-X. Su, Y.-l. Xu, H.-S. Wang and Y.-M. Pan, Adv. Synth. Catal., 2017, 359, 3950 CrossRef CAS.
- P.-F. Wang, X.-Q. Wang, J.-J. Dai, Y.-S. Feng and H.-J. Xu, Org. Lett., 2014, 16, 4586 CrossRef CAS.
- Y.-H. Jin, H.-J. Yang and H. Fu, Chem. Commun., 2016, 52, 12909 RSC.
- Z. Li, K.-F. Wang, X. Zhao, H.-H. Ti, X.-G. Liu and H.-G. Wang, Nat. Commun., 2020, 11, 5036 CrossRef CAS.
- G. Destro, K. Horkka, O. Loreau, D.-A. Buisson, L. Kingston, A. Del Vecchio, M. Schou, C. S. Elmore, F. Taran, T. Cantat and D. Audisio, Angew. Chem., Int. Ed., 2020, 59, 13490 CrossRef CAS.
- D.-Y. Kong, P. J. Moon, E. K. J. Lui, O. Bsharat and R. J. Lundgren, Science, 2020, 369, 557 CrossRef CAS.
- (a) M.-C. Fu, R. Shang, B. Zhao, B. Wang and Y. Fu, Science, 2019, 363, 1429 CrossRef CAS; (b) M.-C. Fu, J.-X. Wang and R. Shang, Org. Lett., 2020, 22, 8572 CrossRef CAS.
- (a) S. C. Sha, J. Zhang and P. J. Walsh, Org. Lett., 2015, 17, 410 CrossRef CAS; (b) D.-Y. Kong, P. J. Moon, W.-Y. Qian and R. J. Lundgren, Chem. Commun., 2018, 54, 6835 RSC; (c) D. Kong, P. J. Moon, O. Bsharat and R. J. Lundgren, Angew. Chem., Int. Ed., 2020, 59, 1313 CrossRef CAS.
- P. J. Moon and R. J. Lundgren, ACS Catal., 2020, 10, 1742 CrossRef CAS.
- P. J. Moon, A. Fahandej-Sadi, W. Qian and R. J. Lundgren, Angew. Chem., Int. Ed., 2018, 57, 4612 CrossRef CAS.
- L.-Y. Wu and J. F. Hartwig, J. Am. Chem. Soc., 2005, 127, 15824 CrossRef CAS.
- (a) R. Shang, D. -S. Ji, L. Chu, Y. Fu and L. Liu, Angew. Chem., Int. Ed., 2011, 50, 4470 CrossRef CAS; (b) R. Shang, Z. Huang, X. Xiao, X. Lu, Y. Fu and L. Liu, Adv. Synth. Catal., 2012, 354, 2465 CAS.
- (a) I. P. Beletskaya and V. P. Ananikov, Chem. Rev., 2011, 111, 1596 CrossRef CAS; (b) V. P. Ananikov, N. V. Orlov, S. S. Zalesskiy, I. P. Beletskaya, V. N. Khrustalev, K. Morokuma and D. G. Musaev, J. Am. Chem. Soc., 2012, 134, 6637 CrossRef CAS.
- (a) S. Patai and Z. Rappoport, The Chemistry of Organic Selenium and Tellurium Compounds, John Wiley & Sons, Chichester, 1986, vol. 1 CrossRef; (b) V. P. Reddy, A. V. Kumar, K. Swapna and K. R. Rao, Org. Lett., 2009, 11, 951 CrossRef CAS; (c) H. Xu, Y. Gu, S. Zhang, H. Xiong, F. Ma, F. Lu, Q. Ji, L. Liu, P. Ma, W. Hou, G. Yang and R. A. Lerner, Angew. Chem., Int. Ed., 2020, 59, 13273 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental protocols and spectral data. See DOI: 10.1039/d1ra00063b |
| This journal is © The Royal Society of Chemistry 2021 |