
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dye–catalyst dyads for photoelectrochemical water oxidation based on metal-free sensitizers†
Cristina Decavoli a,
Chiara L. Boldrinia,
Vanira Trifilettib,
Sally Luongb,
Oliver Fenwickb,
Norberto Manfredi*a and
Alessandro Abbotto*a
a,
Chiara L. Boldrinia,
Vanira Trifilettib,
Sally Luongb,
Oliver Fenwickb,
Norberto Manfredi*a and
Alessandro Abbotto*a
aDepartment of Materials Science, INSTM Unit, Solar Energy Research Center MIB-SOLAR, University of Milano – Bicocca, Via R. Cozzi 55, I-20125, Milano, Italy. E-mail: alessandro.abbotto@unimib.it; norberto.manfredi@unimib.it
bSchool of Engineering and Materials Science (SEMS), Queen Mary University of London, Mile End Road, London E1 4NS, UK
First published on 28th January 2021
Abstract
Dye-Sensitized Photoelectrochemical Cells (DS-PECs) have been emerging as promising devices for efficient solar-induced water splitting. In DS-PECs, dyes and catalysts for water oxidation and/or reduction are typically two separate components, thus limiting charge transfer efficiency. A small number of organometallic dyes have been integrated with a catalyst to form an integrated dye–catalyst dyad for photoanodes, but until now no dyads based on metal-free organic dyes have been reported for photoanodes. We herein report the first example of dyad-sensitized photoanodes in DS-PEC water splitting based on metal-free organic dyes and a Ru catalyst. The di-branched donor–π–acceptor dyes carry a donor carbazole moiety which has been functionalized with two different terminal pyridyl ligands in order to coordinate a benchmark Ru complex as a water oxidation catalyst, affording water oxidation dyads. The two dyads have been fully characterized in their optical and electrochemical properties, and XPS has been used to confirm the presence of the catalyst bonded to the dye anchored to the semiconductor anode. The two dyads have been investigated in DS-PEC, showing an excellent faradaic efficiency (88% average across all cells, with a best cell efficiency of 95%), thus triggering new perspectives for the design of efficient molecular dyads based on metal-free dyes for DS-PEC water splitting.
Introduction
In the search for an alternative to fossil fuels, hydrogen is gaining increasing interest in the scientific community and the automotive-aerospace sector, because its combustion generates only water with a zero-carbon footprint.1–3 Hydrogen is today mainly produced from methane via the steam-reforming reaction.4 It is, therefore, critical to develop sustainable and clean processes to produce hydrogen. In this scenario, solar-driven water splitting has been playing an increasing role, with particular reference to the direct conversion from water and sunlight via photocatalysis (PC) or photoelectrochemical cells (PEC).5–7 A PEC is a device composed of two electrodes soaked in a water-based electrolyte, where at least one of them is photoactivated. The photoelectrodes typically consist of a wide bandgap semiconductor (SC), which can be sensitized adequately by a dye to extend the absorbed spectrum into the visible region.8,9 In the case of a DS-PEC with only a photoactive anode, a platinum wire is used as a passive cathode. The sensitizer is adsorbed or chemically bonded onto the SC surface, while the WOC, typically a ruthenium complex, could be either dispersed in aqueous media10,11 or adsorbed onto the SCs surface.12,13 Such arrangements are not optimal for the whole process. In the former case, the device efficiency strongly depends on the diffusion of the catalyst near the electrode. In contrast, in the latter one, the adsorption of the catalyst on the SC surface may compete with the absorption of the dye, limiting the light-harvesting capability. An innovative way out is to design an integrated dye–catalyst supramolecular system, also referred to as a dyad, where the molecular sensitizer and the catalyst are bound together as a single molecular unit. In this way, the drawbacks of the two previous arrangements are circumvented, providing more efficient light-harvesting and a faster charge transfer between the two components. Despite its great potential, only a few examples of molecular dyads for photoanodes have been so far reported in the literature. These examples are limited to organometallic compounds in the dye part of the dyad (such as Ru complexes or porphyrins).14–16 The absence in the literature of organic dyes in dye–WOC dyads for photoanodes is surprising, considering the emerging role of metal-free dyes in the fields of solar energy conversion.8,17–19 Only two examples of dyads containing organic dyes have been so far reported, but both referred to photoactive cathodes.20,21We thus decided to investigate a dyad where a metal-free sensitizer is covalently bonded to a typical WOC, often used as a catalyst benchmark (a derivative of [Ru(bda)(pic)2] (bda = 2,2′-bipyridine-6,6′-dicarboxylate; pic = 4-picoline)).22 As a metal-free sensitizer, we have selected a di-branched donor–acceptor D–(π–A)2 molecule that we have previously investigated, as a class, in the field.10,23,24 The donor moiety was functionalized with a pyridine ligand, which is then used to coordinate the Ru centre of the WOC complex. To study the effect of different spatial arrangements between the dye and the catalyst, two pyridine ligands, with a para and meta relative position between the coordinating nitrogen atom and a methylene linker to the dye, were envisaged, affording the CBZ-4Py and CBZ-3Py precursors, respectively (Fig. 1). The methylene linker of the picolyl functionality was introduced to break the π-conjugation path between the dye and the catalyst. This ensures that the dyad is constituted by two separated π-conjugated entities, thus avoiding perturbation of the optical, electrochemical, and catalytic properties of the two components, and circumventing charge recombination processes.16,25 The two corresponding molecular dyads CBZ-4Py + Ru and CBZ-3Py + Ru were obtained through a two-step procedure directly on the electrode and fully characterized in their optical and electrochemical properties.
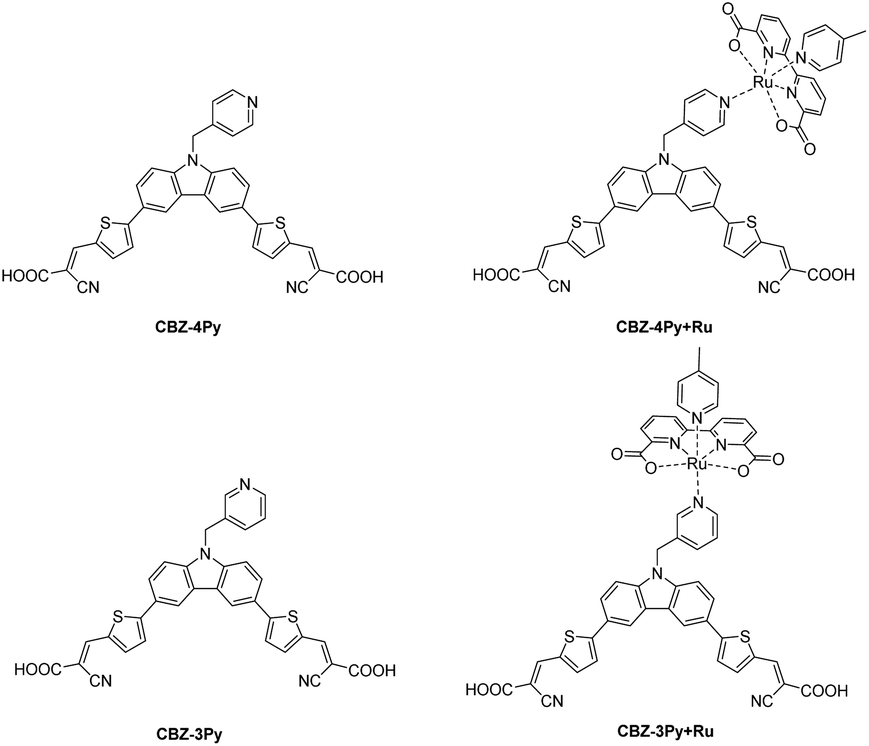 | ||
| Fig. 1 Structures of the investigated dyes and corresponding dye–catalyst dyads. | ||
When used in water splitting PEC, the investigated dyads showed high faradaic efficiencies (FE), with the meta-pyridine dyad CBZ-3Py + Ru showing the highest FE.
As the metal-free D–(π–A)2 component of the dyads, we selected a carbazole (CBZ) derivative, which has been functionalized with pyridine linkers. The CBZ donor building block has been selected as a versatile sulphur-free alternative to the commonly used phenothiazine scaffold.8,18,26 The central pyrrole-like ring ensures the strong electron-donor character.27 The presence of the two benzofused benzene rings can be exploited to easily build the two (π–A) arms carrying the terminal cyanoacrylic acceptor-anchoring groups. Moreover, the NH group of CBZ can be easily functionalised with the pyridine linker needed to couple to the WOC. CBZ-based sensitizes have been successfully employed in PC and PEC water oxidation and hydrogen generation.10,23 As WOC, we selected a close derivative of the ruthenium-based benchmark for DS-PEC, [Ru(bda)(pic)2], which performed well in many literature examples.28
Experimental
General information
FTO plates coated with 3.5 μm transparent TiO2 film, prepared as described above, were treated in a UV-O3 system for 20 min at room temperature, then immersed into a 2 × 10−4 M solution in EtOH of the dye + catalyst precursor for 3 h at room temperature in the dark. The stained substrates were rinsed with EtOH and dried with a stream of dry nitrogen. The UV-vis spectra, CV and LSV were recorded in comparison with a bare 3.5 μm transparent TiO2 film.
| IPCE(λ) = LHE(λ) × APCE(λ) |
In a typical experiment, the working electrodes' currents were recorded for 300 s in the dark, then during 300 s of illumination, and in the dark for an additional 300 s. The procedure allows the diffusion of oxidation products across the solution. The faradaic efficiency for O2 production, ηO2, can be calculated by:
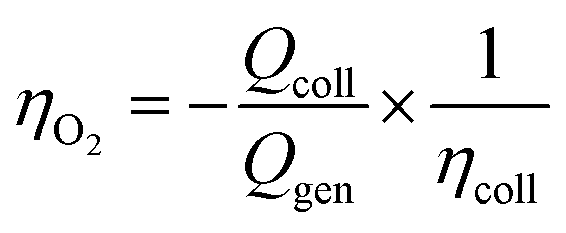
Synthesis
NMR spectra were recorded with a Bruker AMX-500 spectrometer operating at 500.13 MHz (1H) or Bruker Avance Neo spectrometer operating at 400 MHz (1H) and 100 MHz (13C). Coupling constants are given in Hz. ATR FT-IR spectra were recorded with a PerkinElmer Spectrum 100. High resolution mass spectra have been recorded with an Agilent 6230B Time of Flight (TOF) equipped with an electrospray (dual ESI) source. Melting point were recorded with a Büchi Melting Point M-565. Flash chromatography was performed with Merck grade 9385 silica gel 230–400 mesh (60 Å). Reactions performed under inert atmosphere were performed in oven-dried glassware, and a nitrogen atmosphere was generated with Schlenk technique. The conversion was monitored by thin-layer chromatography by using UV light (254 and 365 nm) as a visualizing agent. All reagents were obtained from commercial suppliers at the highest purity grade and used without further purification. Anhydrous solvents were purchased from Acros Organics and used without further purification. Extracts were dried with Na2SO4 and filtered before removal of the solvent by evaporation. Compounds 9H-3,6-dibromocarbazole (1),34 4,4,5,5-tetramethyl-2-[5-(4,4,5,5-tetramethyl-1,3-dioxolan-2-yl)thiophen-2-yl]-1,3,2-dioxaborolane,35 [Ru(bda)(DMSO)(MeCN)(pic)]36 (bda = 2,2′-bipyridine-6,6′-dicarboxylate, pic = 4-picoline) were prepared according to literature.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :AcOEt – 2:1). The desired product was isolated as a white solid (383 mg, 0.65 mmol) with 51% yield. 1H NMR (500 MHz, CDCl3): δ (ppm) = 8.30 (s, 2H), 8.09 (s, 1H), 7.68 (dd, J = 8.4, 1.6 Hz, 2H), 7.41 (d, J = 8.4 Hz, 2H), 7.20 (d, J = 3.7 Hz, 2H), 7.14 (d, J = 3.6 Hz, 2H), 6.22 (s, 2H), 1.39 (s, 12H), 1.34 (s, 12H).:2). The mixture was heated at 50 °C for 2 h and then poured into water. K2CO3 was slowly added until basic pH was reached, and then THF was removed under reduced pressure. The product precipitated was filtered on Hirsh and washed with water.:2). The desired product was isolated as a yellow solid (91 mg, 0.19 mmol) with 91% yield. 1H NMR (400 MHz, DMSO): δ (ppm) = 9.92 (s, J = 0.9 Hz, 2H), 8.91 (dd, J = 1.9, 0.5 Hz, 2H), 8.48 (d, J = 6.1 Hz, 2H), 8.09 (d, J = 4.0 Hz, 2H), 7.96 (dd, J = 8.6, 1.9 Hz, 2H), 7.81 (d, J = 4.0 Hz, 2H), 7.74 (d, J = 8.6 Hz, 2H), 7.10 (d, J = 6.1 Hz, 2H), 5.84 (s, 2H).:2). The desired product was isolated as a yellow solid (290 mg, 0.60 mmol) with 98% yield. 1H NMR (400 MHz, DMSO): δ (ppm) = 9.92 (s, 2H), 8.90 (d, J = 1.8 Hz, 2H), 8.58 (d, J = 1.9 Hz, 1H), 8.46 (dd, J = 4.7, 1.5 Hz, 1H), 8.09 (d, J = 4.0 Hz, 2H), 7.97 (dd, J = 8.6, 1.9 Hz, 2H), 7.85 (d, J = 8.7 Hz, 2H), 7.81 (d, J = 4.0 Hz, 2H), 7.59–7.48 (m, 1H), 7.30 (dd, J = 7.8, 4.8 Hz, 1H), 5.83 (s, 2H).
:AcOEt – 2:1). The desired product was isolated as a white solid (383 mg, 0.65 mmol) with 51% yield. 1H NMR (500 MHz, CDCl3): δ (ppm) = 8.30 (s, 2H), 8.09 (s, 1H), 7.68 (dd, J = 8.4, 1.6 Hz, 2H), 7.41 (d, J = 8.4 Hz, 2H), 7.20 (d, J = 3.7 Hz, 2H), 7.14 (d, J = 3.6 Hz, 2H), 6.22 (s, 2H), 1.39 (s, 12H), 1.34 (s, 12H).:2). The mixture was heated at 50 °C for 2 h and then poured into water. K2CO3 was slowly added until basic pH was reached, and then THF was removed under reduced pressure. The product precipitated was filtered on Hirsh and washed with water.:2). The desired product was isolated as a yellow solid (91 mg, 0.19 mmol) with 91% yield. 1H NMR (400 MHz, DMSO): δ (ppm) = 9.92 (s, J = 0.9 Hz, 2H), 8.91 (dd, J = 1.9, 0.5 Hz, 2H), 8.48 (d, J = 6.1 Hz, 2H), 8.09 (d, J = 4.0 Hz, 2H), 7.96 (dd, J = 8.6, 1.9 Hz, 2H), 7.81 (d, J = 4.0 Hz, 2H), 7.74 (d, J = 8.6 Hz, 2H), 7.10 (d, J = 6.1 Hz, 2H), 5.84 (s, 2H).:2). The desired product was isolated as a yellow solid (290 mg, 0.60 mmol) with 98% yield. 1H NMR (400 MHz, DMSO): δ (ppm) = 9.92 (s, 2H), 8.90 (d, J = 1.8 Hz, 2H), 8.58 (d, J = 1.9 Hz, 1H), 8.46 (dd, J = 4.7, 1.5 Hz, 1H), 8.09 (d, J = 4.0 Hz, 2H), 7.97 (dd, J = 8.6, 1.9 Hz, 2H), 7.85 (d, J = 8.7 Hz, 2H), 7.81 (d, J = 4.0 Hz, 2H), 7.59–7.48 (m, 1H), 7.30 (dd, J = 7.8, 4.8 Hz, 1H), 5.83 (s, 2H).Results and discussion
Synthesis
The synthesis of the dyes CBZ-4Py and CBZ-3Py functionalised with the two pyridine linkers is presented in Scheme 1. The Suzuki–Miyaura cross-coupling between commercially available 9H-3,6-dibromocarbazole and the protected pinacol ester of 5-formyl-2-thienylboronic acid35 affords intermediate 2 with protected formyl groups. The deprotonated derivative is then submitted to alkylation with the proper picolylbromide to give the pyridine derivatives 3a and 3b. Subsequent deprotection of the formyl groups and final Knoevenagel condensation with cyanoacetic acid, in the presence of piperidine, yields the desired sensitizers with the pyridine ligand. The new sensitizers have been fully characterised with 1H and 13C NMR, as well as melting point, FT-IR, and HRMS to check the required structures and purities (see ESI†).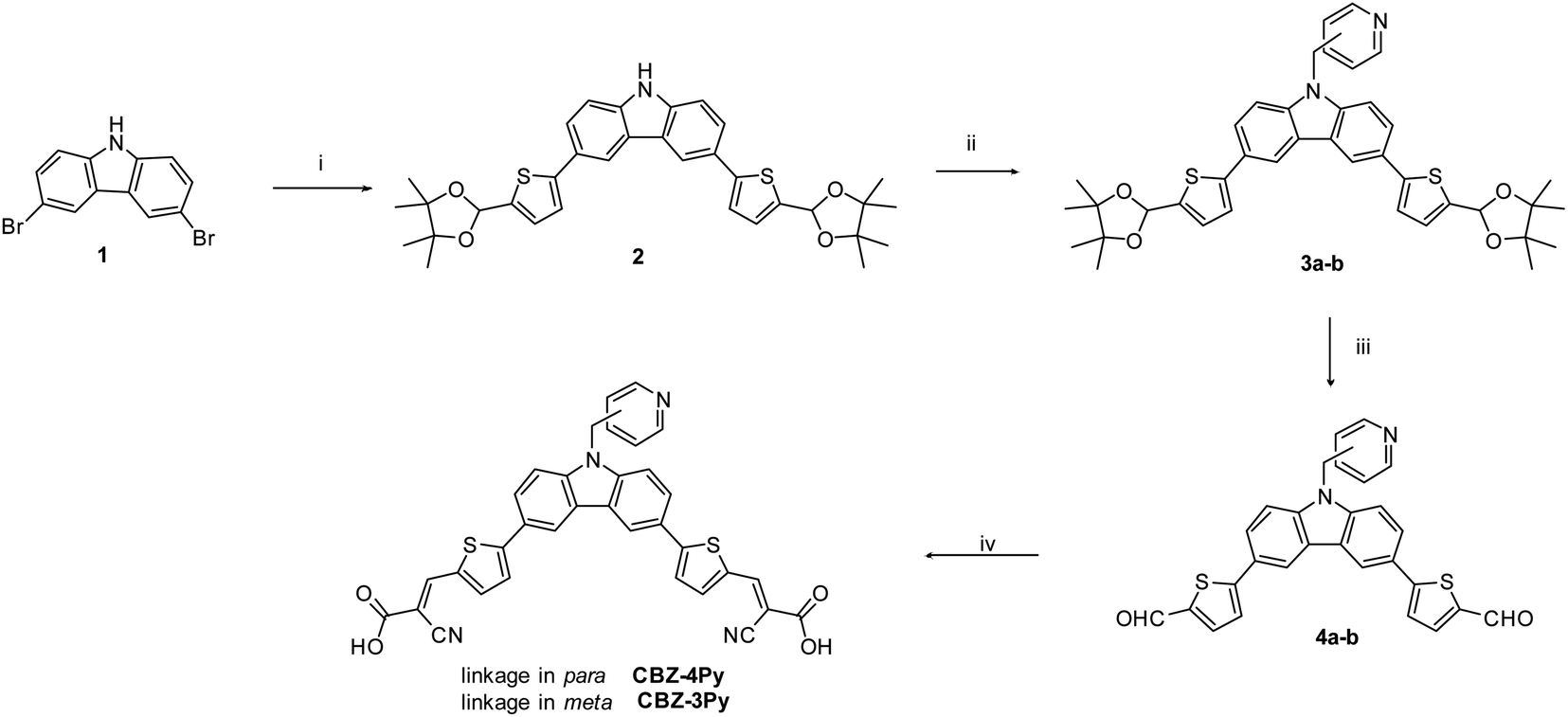 | ||
| Scheme 1 Synthetic pathway for CBZ-4Py and CBZ-3Py. Reagents and conditions: (i) 4,4,5,5-tetramethyl-2-(5-(4,4,5,5-tetramethyl-1,3-dioxolan-2-yl)thiophen-2-yl)-1,3,2-dioxaborolane, Pd(dppf)Cl2·CH2Cl2, K2CO3, DME:MeOH (1:1), microwave 100 °C, 70 W, 60 min; (ii) 4-(bromomethyl)pyridine hydrobromide or 3-(bromomethyl)pyridine hydrobromide, NaH 60%, THF anhydrous, 0 °C, 30 min and then rt, overnight; (iii) HCl 10%:THF (1:2), 50 °C, 2 h; (iv) cyanoacetic acid, piperidine, CHCl3 dry, reflux, 8 h. | ||
For the synthesis of the dyads CBZ-4Py + Ru and CBZ-3Py + Ru, we first attempted to apply the synthetic approach used for heteroleptic [Ru(bda)(pic)(ligand)] complexes (Scheme 2).
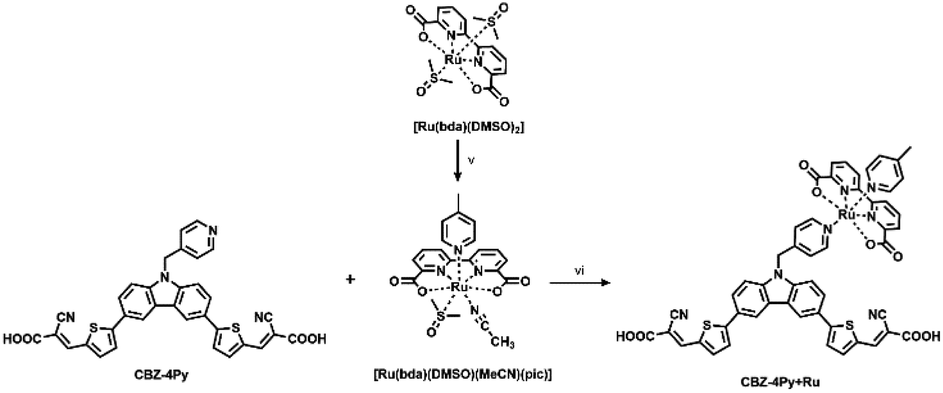 | ||
| Scheme 2 General procedure for the synthesis of the dyads in solution. Reagents and conditions: (v) 4-picoline, MeCN, reflux, 1 h; (vi) MeOH, DMSO, reflux, 1 day. | ||
A stoichiometric amount of the dye was added to [Ru(bda)(DMSO)(MeCN)(pic)],14,16 but, unfortunately, the desired product could not be characterized due to good solubility only in coordinating solvents, thus decomposing the Ru complex of the dyad. To overcome this issue, the dyad was completed directly on the electrode surface, starting from the synthetic precursors. A TiO2 electrode was soaked into a 2 × 10−4 M EtOH/DMSO (9:1) dye solution in the presence of a stoichiometric amount of the Ru precursor. To eliminate the unreacted Ru precursor, the photoelectrode was then rinsed with EtOH. Since the Ru precursor does not carry any anchoring group, the final step ensures that the presence of Ru in the rinsed electrode can only be due to the formation of the desired complex with the functionalized dye, in turn, anchored to the SC via the acceptor-carboxylic functionality.
Optical and electrochemical properties
The absorption spectra (molar absorption coefficient vs. wavelength) of the dyes CBZ-4Py and CBZ-3Py in solution (10−5 M in DMSO) are shown in Fig. 2 and the main optical parameters are listed in Table 1. As expected, the spectra of the two dyes are almost identical, since the picolyl functionality does not affect the π-framework of the D–(π–A)2 dyes. The UV-vis spectra in solution exhibited the main intramolecular charge transfer (ICT) transition at ca. 450 nm. The UV-vis analysis of the dyes adsorbed onto a 1 μm transparent TiO2 film presented a similar absorption intensity for the ICT band, with the peak at ca. 30 nm lower wavelengths. The optical band gaps were estimated using the method of Tauc plot37 and listed in Table 1. The normalized spectra of the TiO2 films sensitized with the dyads CBZ-4Py + Ru and CBZ-3Py + Ru are shown in Fig. S1 (ESI).† They are in good agreement with the dyes' absorption spectra: the shape in the visible range is dominated by the ICT band of the dye portion. The electrochemical properties of the dyes in solution have been investigated, and the main electrochemical parameters are collected in Table 1.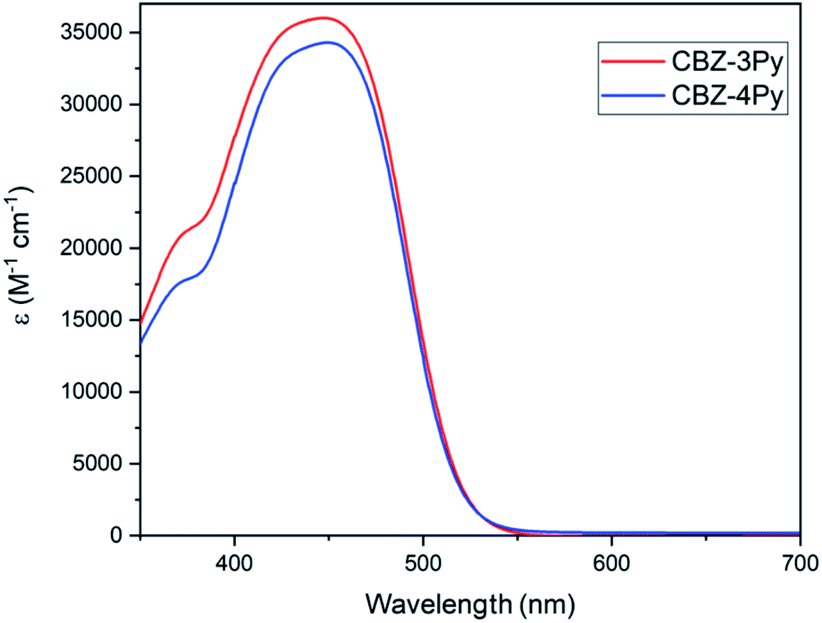 | ||
| Fig. 2 Absorption spectra of dyes in solution. | ||
| Dye | λmaxa,b (nm) | εa (M−1 cm−1) | Dye loading (nmol cm−2) | Voxa,b (V vs. Fc/Fc+) ± 10 mV | HOMOa,b,c (eV) ± 0.05 eV | Eoptgapa,b (eV) | LUMOa,b,c (eV) ± 0.05 eV | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Soln | Film | Soln | Film | Soln | Film | Soln | Film | Soln | Film | |||
| a Dye solution 10−5 M in DMSO.b 1 μm transparent TiO2 photoanode.c Fc+/Fc = −5.23 eV vs. vacuum. | ||||||||||||
| CBZ-4Py | 448 | 417 | 34300 ± 100 |
37.1 | 0.83 | 0.59 | −6.06 | −5.82 | 2.26 | 2.12 | −3.80 | −3.70 |
| CBZ-3Py | 447 | 413 | 36000 ± 2000 |
55.8 | 0.85 | 0.78 | −6.08 | −6.01 | 2.26 | 2.14 | −3.82 | −3.87 |
Cyclic voltammetry (CV) profiles (Fig. S2, ESI†) showed irreversible behaviour for the oxidation processes of CBZ-3Py and CBZ-4Py. Differential Pulsed Voltammetry (DPV) (Fig. S3, ESI†) was thus used to determine the oxidation potential from the current peak38 and then calculate the HOMO energy levels, which were estimated to be around −6 eV. This value is lower than the electron-donating level of the WOC (−5.8 eV),39 and therefore dye regeneration can take place. The LUMO levels have been derived from electrochemical HOMO values and optical bandgaps, calculated as described above. The so calculated LUMO energies are also very similar, and both are at higher energy than that of the CB of TiO2 (−4.0 eV vs. vacuum), thus ensuring efficient electron injection from the excited dye to the SC. Therefore, the electrochemical study guarantees proper energy levels alignments for electron–hole donation among the dye and other components of the cell.
Characterisation of the photoanodes
The electrochemical properties of the dyad-sensitized anodes have been investigated by CV and compared with those of the dye and the reference catalyst [Ru(bda)(pic)2] (Fig. S4 and S5, ESI†). The CV profile of the CBZ-4Py + Ru-sensitized electrode presented a redox peak at E1/2 = +0.70 V vs. NHE, which well compares with the reported value for the reference Ru complex (+0.65 V vs. NHE).39 The CBZ-3Py + Ru-sensitized electrode showed a less intense, but still reversible, redox peak at E1/2 = +0.77 V vs. NHE. It is slightly higher than the reported potential for the redox benchmark, but still comparable to it and other similar systems.13,25 In contrast, the dye-sensitized electrode evidenced only the beginning process of the dye oxidation, in agreement with the CV profile of the corresponding N-alkylcarbazole derivative10 not carrying the terminal pyridine functionality (Fig. S6, ESI†). The electrochemical study of the dyad-sensitized electrodes thus supports the presence of a [Ru(bda)(pic)(ligand)] complex anchored, through the dye, to the SC. X-ray photoelectron spectroscopy (XPS) analysis was performed to confirm the formation of the anchored dyads. The XPS survey spectra (Fig. S7, ESI†) show the presence of sulphur and nitrogen peaks on the TiO2 surface, thus supporting the presence of the dye portion. Ruthenium peaks are not clearly resolved in the survey spectra due to the proximity to the carbon peak. The core-electron binding energy of C 1s and Ru 3d5/2 are plotted in Fig. 3: C 1s peaks for adventitious carbon are visible at 285.4 eV (C–C/C–H), 286.9 eV (C–O) and 289.5 eV (O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O).40 After adsorption of the dyads, the C–O peak is reduced in intensity compared to C–C/C–H and O–CO. This is likely to be due to the presence of O–CO groups on the dyads at the binding positions. The XPS spectra of the ruthenium complexes show the peak for Ru 3d5/2 with a binding energy of 281.7 eV, thus supporting the presence of the Ru complex in the dyad. Meanwhile, Ru 3d3/2 peaks are hidden by the C 1s peak.16,41 The presence of the Ru complex is further supported by the N 1s peak shift to higher binding energy (core-level shown in Fig. S8, ESI†), which is more pronounced in the CBZ-3Py + Ru dyad (0.3 eV). Finally, the valence-band XPS (Fig. S9, ESI†) contains the overlapping O 2p and Ru 4d peaks. The intensity increase in the Ru 4d region (<3.5 eV) further confirms the presence of the coordination complex.
O).40 After adsorption of the dyads, the C–O peak is reduced in intensity compared to C–C/C–H and O–CO. This is likely to be due to the presence of O–CO groups on the dyads at the binding positions. The XPS spectra of the ruthenium complexes show the peak for Ru 3d5/2 with a binding energy of 281.7 eV, thus supporting the presence of the Ru complex in the dyad. Meanwhile, Ru 3d3/2 peaks are hidden by the C 1s peak.16,41 The presence of the Ru complex is further supported by the N 1s peak shift to higher binding energy (core-level shown in Fig. S8, ESI†), which is more pronounced in the CBZ-3Py + Ru dyad (0.3 eV). Finally, the valence-band XPS (Fig. S9, ESI†) contains the overlapping O 2p and Ru 4d peaks. The intensity increase in the Ru 4d region (<3.5 eV) further confirms the presence of the coordination complex.
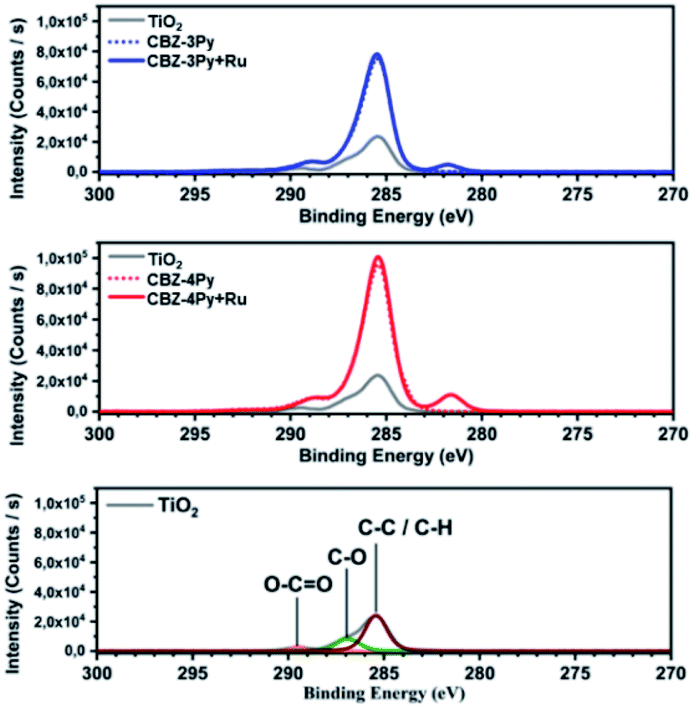 | ||
| Fig. 3 XPS survey spectra of the high-resolution core level for C 1s and Ru 3d region of the investigated dyes and corresponding dyads, compared to the bare TiO2. | ||
Photoelectrochemical properties
The ability of the dyad-sensitized PECs to perform the photoinduced water splitting reaction was measured through photoelectrochemical measurements under white light illumination (200 W Xe lamp, 420 < λ < 800 nm). The bias applied was chosen according to the linear sweep voltammogram (LSV) of the sensitized TiO2 films (Fig. S10†) by selecting the potential at which the best light/dark ratio was recorded. The obtained photocurrents are shown in Fig. S11 and S12 (ESI),† and Fig. 4 (inset) for CBZ-4Py + Ru and CBZ-3Py + Ru sensitized electrodes, respectively. Each dyad-sensitized electrode has been checked against dye-sensitized electrodes (without WOC). In all cases, the dye-sensitized electrode was much less efficient compared to the dyad-sensitized electrode (Fig. S11 and S12, ESI,† and inset of Fig. 4). The CBZ-3Py dye-sensitized electrode had a negligible photocurrent of 3.5 μA cm−2, while the corresponding dyad CBZ-3Py + Ru, after a 10 min chronoamperometry, showed 25 μA cm−2, corresponding to an 8-fold increase. In the case of CBZ-4Py (8.6 μA cm−2) and the corresponding dyad CBZ-4Py + Ru (30 μA cm−2), a performance increase for the latter was observed, but was less pronounced (Fig. S12, ESI†). These values well match the behaviour of other organic sensitizers reported in the literature.16,42,43 Long-term measurements were performed to evaluate the stability of the dyad sensitized electrodes and are shown in ESI (Fig. S13 and S14†).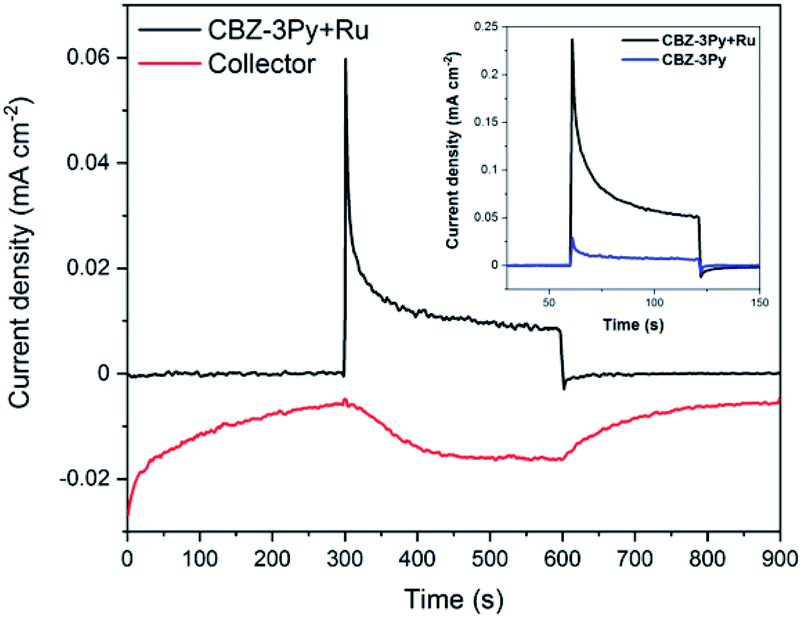 | ||
| Fig. 4 Collector–generator plot of a CBZ-3Py + Ru sensitized electrode. Black line: current–time trace at illuminated (200 W Xe lamp; 420 < λ < 800 nm) CBZ-3Py + Ru dyad on TiO2 in 0.1 M Na2SO4 at pH 5.8 with an applied bias of ∼0.5 V versus NHE. Red line: current–time traces at an FTO collector electrode, 400 μm from the photoanode at an applied bias of ∼−0.6 V vs. NHE measured concurrently with the photoelectrochemical–time trace (FE of shown measurement = 95%). Inset: 60 s chronoamperometry of CBZ-3Py (blue) and CBZ-3Py + Ru (black) in the same conditions of C-G measures. | ||
The O2 evolution was measured through the collector–generator technique,38 as recently reported by many studies on DS-PEC water splitting.8,12,14,32,44 In this technique, a second FTO working electrode (the collector) is positioned at a fixed distance (ca. 400 μm) from the photoanode (the generator) and kept at a reducing potential (−0.8 V vs. Ag/AgCl, ∼−0.6 V vs. NHE). In this way, the collector reduces the oxygen gas as soon as it is formed, thus transforming a very-difficult-to-evaluate variable (affected, among other things, by leakage and air contamination) into a simple electric signal. The direct quantification of oxygen production FE is possible from the ratio between the charge flowing through the collector and the one through the generator. The FE for CBZ-4Py + Ru and CBZ-3Py + Ru sensitized electrodes were 58 ± 12% and 88 ± 9% (average of 4 measurements on different samples), respectively (Fig. S15† and 4).
To gain a better insight on the performances of the dyads to convert photons to electrons the incident photon-to-current efficiency (IPCE) was also measured (Fig. 5). The recorded IPCE well matched the trend of the measured faradaic efficiency. The intensity of the signal, related to the produced current density, was higher for the CBZ-3Py + Ru sensitized electrode, with a maximum efficiency of ∼16% at 450 nm. The IPCE of the TiO2 films sensitized with the dyads were compared with those of the corresponding films sensitized with the dye only (without WOC, Fig. S16 and S17†). The light harvesting efficiency (LHE) of both dyads sensitized electrodes were also measured to calculate the absorbed photon-to-current efficiency (APCE), shown in Fig. S18 and S19 (ESI).†
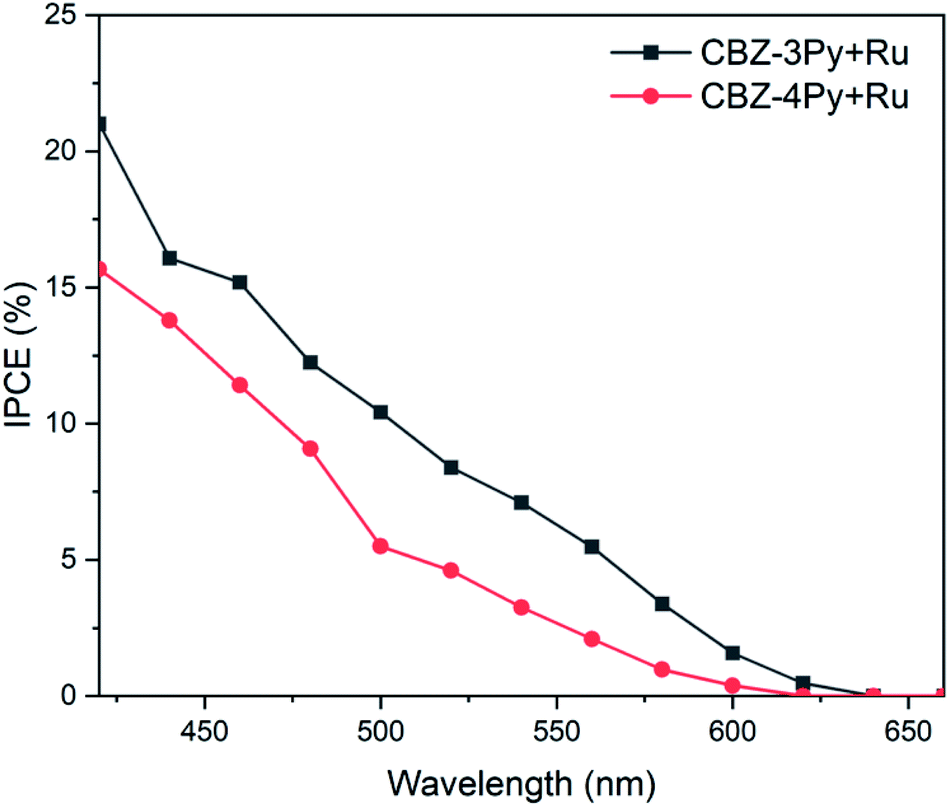 | ||
| Fig. 5 Incident photon-to-current efficiency (IPCE) of dyads sensitized TiO2 films in 0.1 M Na2SO4 at pH 5.8 with an applied bias of ∼0.5 V vs. NHE under monochromatic illumination. | ||
It is worth noting, as an addition evidence of the formation of the dyads, that the IPCE of the devices sensitized with the dye by itself (e.g., without the integrated catalyst) showed a negligible efficiency, in agreement with the hereabove described photocurrent experiments (see Fig. S11 and S12 in ESI† and inset of Fig. 4).
The notably higher efficiency of the 3Py derivative, compared with the 4Py analogue, can be ascribed to the fact that in the former dyad the rotation about the single bond (between the methylene bridge and the quaternary carbon atom of pyridine) allows the catalyst portion to come closer to the dye. Therefore, a more efficient charge transfer between the dye and the catalyst fragment of the dyad is ensured (see the simulated movie in ESI†).
Conclusions
In conclusion, a new series of metal-free organic dyes, bearing a coordinating pyridine functionality, has been synthesized, fully characterised and used to sensitize photoanodes for photoelectrochemical water oxidation. These sensitizers have been used to coordinate a proper Ru precursor to yield the first example ever of a molecular dyad based on a metal-free organic dye integrated to a benchmark Ru complex for water oxidation. Optical, electrochemical, and XPS experiments confirmed the formation of the dyad and, in particular, the presence of the Ru catalyst anchored to the TiO2 photoanode via the dye linker. Both dyad-sensitized photoanodes showed state-of-the-art FE in the O2 generation process,8,45–47 with the CBZ-3Py + Ru dyad reaching an average efficiency of nearly 90% and a best cell efficiency of 95%. Moreover, IPCE measurements confirmed the validity of the approach showing efficiencies among the best recorded for similar compounds and noticeably supported the evidence of the formation of dyads when compared to isolated dyes. Further studies, including a detailed computational investigation, are currently underway to further support and extend the potential of the herein introduced molecular dyad approach to water splitting.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors CD, CLB, NM and AA wish to acknowledge financial support from MIUR (grant Dipartimenti di Eccellenza – 2017 “Materials for Energy”) and University of Milano-Bicocca (Fondo di Ateneo – Quota Competitiva 2017 and 2019). VT acknowledges the European Union's Horizon 2020 Research and Innovation Programme under the Marie Skłodowska-Curie grant agreement, grant number 798271, for funding. OF acknowledges the Royal Society University Research Fellowship UF/40372 for financial support. We are also thanks Dr R. Brambilla for her contribution.Notes and references
- C. Acar, Y. Bicer, M. E. Demir and I. Dincer, Int. J. Hydrogen Energy, 2019, 44, 25347–25364 CrossRef CAS.
- N. Armaroli and V. Balzani, Energy Environ. Sci., 2011, 4, 3193 RSC.
- N. Armaroli and V. Balzani, Chem.–Eur. J., 2016, 22, 32–57 CrossRef CAS PubMed.
- N. Armaroli and V. Balzani, ChemSusChem, 2011, 4, 21–36 CrossRef CAS PubMed.
- S. W. Boettcher, ACS Energy Lett., 2020, 5, 70–71 CrossRef CAS.
- Photoelectrochemical Solar Fuel Production: From Basic Principles to Advanced Devices, ed. S. Giménez and J. Bisquert, Springer International Publishing, Switzerland, 2016 Search PubMed.
- F. J. Niu, D. G. Wang, F. Li, Y. M. Liu, S. H. Shen and T. J. Meyer, Adv. Energy Mater., 2020, 10, 1900399 CrossRef.
- C. Decavoli, C. L. Boldrini, N. Manfredi and A. Abbotto, Eur. J. Inorg. Chem., 2020, 978–999 CrossRef CAS.
- N. Vlachopoulos and A. Hagfeldt, Chimia, 2019, 73, 894–905 CrossRef CAS PubMed.
- N. Manfredi, C. L. Boldrini and A. Abbotto, ChemElectroChem, 2018, 5, 2395–2402 CrossRef CAS.
- Y. Tsubonouchi, S. Lin, A. R. Parent, G. W. Brudvig and K. Sakai, Chem. Commun., 2016, 52, 8018–8021 RSC.
- K.-R. Wee, B. D. Sherman, M. K. Brennaman, M. V. Sheridan, A. Nayak, L. Alibabaei and T. J. Meyer, J. Mater. Chem. A, 2016, 4, 2969–2975 RSC.
- O. Suryani, Y. Higashino, J. Y. Mulyana, M. Kaneko, T. Hoshi, K. Shigaki and Y. Kubo, Chem. Commun., 2017, 53, 6784–6787 RSC.
- B. D. Sherman, Y. Xie, M. V. Sheridan, D. Wang, D. W. Shaffer, T. J. Meyer and J. J. Concepcion, ACS Energy Lett., 2017, 2, 124–128 CrossRef CAS.
- M. K. Brennaman, M. K. Gish, L. Alibabaei, M. R. Norris, R. A. Binstead, A. Nayak, A. M. Lapides, W. Song, R. J. Brown, J. J. Concepcion, J. L. Templeton, J. M. Papanikolas and T. J. Meyer, J. Phys. Chem. C, 2018, 122, 13017–13026 CrossRef CAS.
- M. Yamamoto, L. Wang, F. Li, T. Fukushima, K. Tanaka, L. Sun and H. Imahori, Chem. Sci., 2016, 7, 1430–1439 RSC.
- V. Rondán-Gómez, I. Montoya De Los Santos, D. Seuret-Jiménez, F. Ayala-Mató, A. Zamudio-Lara, T. Robles-Bonilla and M. Courel, Appl. Phys. A, 2019, 125, 836 CrossRef.
- B. Cecconi, N. Manfredi, T. Montini, P. Fornasiero and A. Abbotto, Eur. J. Org. Chem., 2016, 5194–5215 CrossRef CAS.
- J.-F. Huang, Y. Lei, T. Luo and J.-M. Liu, ChemSusChem, 2020, 13, 5863–5895 CrossRef CAS PubMed.
- N. Kaeffer, J. Massin, C. Lebrun, O. Renault, M. Chavarot-Kerlidou and V. Artero, J. Am. Chem. Soc., 2016, 138, 12308–12311 CrossRef CAS PubMed.
- P. B. Pati, L. Zhang, B. Philippe, R. Fernández-Terán, S. Ahmadi, L. Tian, H. Rensmo, L. Hammarström and H. Tian, ChemSusChem, 2017, 10, 2480–2495 CrossRef CAS PubMed.
- L. Duan, A. Fischer, Y. Xu and L. Sun, J. Am. Chem. Soc., 2009, 131, 10397–10399 CrossRef CAS PubMed.
- N. Manfredi, M. Monai, T. Montini, M. Salamone, R. Ruffo, P. Fornasiero and A. Abbotto, Sustainable Energy Fuels, 2017, 1, 694–698 RSC.
- N. Manfredi, B. Cecconi and A. Abbotto, Eur. J. Org. Chem., 2014, 7069–7086 CrossRef CAS.
- Y. Gao, L. Duan, Z. Yu, X. Ding and L. Sun, Faraday Discuss., 2014, 176, 225–232 RSC.
- C. Decavoli, C. L. Boldrini, N. Manfredi and A. Abbotto, Rend. Lincei Sci. Fis. Nat., 2019, 30, 469–483 CrossRef.
- A. R. Katritzky and C. W. Rees, Comprehensive Heterocyclic Chemistry, Pergamon, Oxford, 1984 Search PubMed.
- B. Zhang and L. Sun, J. Am. Chem. Soc., 2019, 141, 5565–5580 CrossRef CAS PubMed.
- V. V. Pavlishchuk and A. W. Addison, Inorg. Chim. Acta, 2000, 298, 97–102 CrossRef CAS.
- J. O. M. Bockris, A. K. N. Reddy and M. Gamboa-Aldeco, Modern Electrochemistry 2A: Fundamentals of Electrodics, Springer US, Boston, MA, 2000 Search PubMed.
- S. Ito, T. N. Murakami, P. Comte, P. Liska, C. Grätzel, M. K. Nazeeruddin and M. Grätzel, Thin Solid Films, 2008, 516, 4613–4619 CrossRef CAS.
- B. D. Sherman, M. V. Sheridan, C. J. Dares and T. J. Meyer, Anal. Chem., 2016, 88, 7076–7082 CrossRef CAS PubMed.
- J. T. Kirner and R. G. Finke, ACS Appl. Mater. Interfaces, 2017, 9, 27625–27637 CrossRef CAS PubMed.
- M. Sailer, A. W. Franz and T. J. J. Müller, Chem.–Eur. J., 2008, 14, 2602–2614 CrossRef CAS PubMed.
- V. Leandri, R. Ruffo, V. Trifiletti and A. Abbotto, Eur. J. Org. Chem., 2013, 6793–6801 CrossRef CAS.
- Y. Jiang, F. Li, B. Zhang, X. Li, X. Wang, F. Huang and L. Sun, Angew. Chem., Int. Ed., 2013, 52, 3398–3401 CrossRef CAS PubMed.
- J. Tauc, Mater. Res. Bull., 1968, 3, 37–46 CrossRef CAS.
- K. Izutsu, in Electrochemistry in Nonaqueous Solutions, Wiley-VCH Verlag GmbH & Co. KGaA, 2009, DOI:10.1002/9783527629152.fmatter.
- Y. Gao, L. Zhang, X. Ding and L. Sun, Phys. Chem. Chem. Phys., 2014, 16, 12008–12013 RSC.
- G. Greczynski and L. Hultman, ChemPhysChem, 2017, 18, 1507–1512 CrossRef CAS PubMed.
- H. Rensmo, K. Westermark, S. Södergren, O. Kohle, P. Persson, S. Lunell and H. Siegbahn, J. Chem. Phys., 1999, 111, 2744–2750 CrossRef CAS.
- M. Yamamoto, Y. Nishizawa, P. Chabera, F. Li, T. Pascher, V. Sundstrom, L. Sun and H. Imahori, Chem. Commun., 2016, 52, 13702–13705 RSC.
- Y. K. Eom, L. Nhon, G. Leem, B. D. Sherman, D. Wang, L. Troian-Gautier, S. Kim, J. Kim, T. J. Meyer, J. R. Reynolds and K. S. Schanze, ACS Energy Lett., 2018, 3, 2114–2119 CrossRef CAS.
- G. A. Volpato, M. Marasi, T. Gobbato, F. Valentini, F. Sabuzi, V. Gagliardi, A. Bonetto, A. Marcomini, S. Berardi, V. Conte, M. Bonchio, S. Caramori, P. Galloni and A. Sartorel, Chem. Commun., 2020, 56, 2248–2251 RSC.
- B. Zhang and L. Sun, Chem. Soc. Rev., 2019, 48, 2216–2264 RSC.
- M.-N. Collomb, D. V. Morales, C. N. Astudillo, B. Dautreppe and J. Fortage, Sustainable Energy Fuels, 2020, 4, 31–49 RSC.
- X. Ding, L. Zhang, Y. Wang, A. Liu and Y. Gao, Coord. Chem. Rev., 2018, 357, 130–143 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Optical and electrochemical properties (CV and DPV of dyes in solution and CV of dyes and dyads on TiO2 films), XPS, collector–generator plot, simulated movies of dyad conformers, 1H, 13C NMR and HRMS spectra. See DOI: 10.1039/d0ra10971a |
| This journal is © The Royal Society of Chemistry 2021 |