
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImpact of porous nanomaterials on inhibiting protein aggregation behaviour†
Munmun Bardhan‡
*a,
Sandip Doluib,
Siddhi Chaudhuric,
Uttam Paul a,
Gaurav Bhattacharjeed,
Manorama Ghosala,
Nakul C. Maitic,
Debashis Mukhopadhyayc and
Dulal Senapati*a
a,
Gaurav Bhattacharjeed,
Manorama Ghosala,
Nakul C. Maitic,
Debashis Mukhopadhyayc and
Dulal Senapati*a
aChemical Sciences Division, Saha Institute of Nuclear Physics, HBNI, 1/AF Bidhannagar, Kolkata 700064, India. E-mail: dulal.senapati@saha.ac.in; Tel: +91-9433139141
bIndian Institute of Chemical Biology, Jadavpur, Kolkata-700032, India
cBiophysics and Structural Genomics Division, Saha Institute of Nuclear Physics, HBNI, 1/AF Bidhannagar, Kolkata 700064, India
dSurface Physics Division, Saha Institute of Nuclear Physics, HBNI, 1/AF Bidhannagar, Kolkata 700064, India
First published on 15th January 2021
Abstract
Aggregation of intrinsically disordered as well as the ordered proteins under certain premises or physiological conditions leads to pathological disorder. Here we have presented a detailed investigation on the effect of a porous metallic (Au) and a non-metallic (Si) nanomaterial on the formation of ordered (fiber-like/amyloid) and disordered (amorphous) aggregates of proteins. Porous nanogold (PNG) was found to reduce the amyloid aggregation of insulin but does not have much impact on the lag phase in the aggregation kinetics, whereas porous nano-silica (PNS) was found both to decrease the amount of aggregation as well as prolong the lag phase of amyloid fiber formation from insulin. On the other hand, both the porous nanoparticles are found to decrease the extent of amorphous aggregation (with slight improvement for PNS) of pathogenic huntingtin (Htt) protein in Huntington's disease cell model. This is a noted direct observation in controlling and understanding protein aggregation diseases which may help us to formulate nanotherapeutic drugs for future clinical applications.
1. Introduction
Aggregation and misfolding of proteins are the major cause of several human neurodegenerative diseases like Huntington's, Alzheimer's, Parkinson's, etc, and one of the leading concerns of present translational healthcare research. The native conformation of globular proteins is generally destabilized by high temperature, low pH, upon addition of denaturants, and truncations or mutations1 which induce protein aggregation on a biochemically suitable timescale. Under the above-mentioned conditions, proteins form toxic aggregates by misfolding into predominantly beta-sheet rich intermediates. Subsequently, it assembles into soluble oligomers and finally to linear matured fibrils.2 Adherence of these fibrils to the cells usually leads to cell degeneration. Fibrillation follows a nucleation-growth pattern, the latter being the main rate-controlling step. This non-specific aggregation via fibrillation alter the physical properties of proteins, which is the main pathological reason behind different diseases like Alzheimer's disease (AD), Parkinson's disease (PD), amyotrophic lateral sclerosis (ALS), Huntington's disease (HD), dementia with Lewy bodies (LBD), temporal diseases (FTD), and several other neurodegenerative diseases.This alteration in conformation leads to the interaction with other proteins and is dependent on age at onset, intensity, and extent of the mutation.3 For the treatment of protein misfolding diseases, therefore, interruption of the amyloid aggregation, either by inhibition or by a delay of the fibrillation process, has been a standard strategy. Currently, there is no successful drug yet which can prevent these diseases in this way. So, the prevention or inhibition of protein aggregation stands as a major issue from the therapeutic point of view. Several approaches have been adopted to delay the fibrillation process including the use of small organic molecules,4 functional polymers,5 and nanoparticles (NPs).6,7 Among these, nanoparticles, materials with a dimension of 1–100 nm, are nowadays considered as the most promising material for its revealing effect on protein fibrillation. In a recent study by Liu et al.,8 MD simulation of the PrP127–147 tetramer with or without carbon NPs suggests that the interactions between prion proteins and carbon NPs reduce the aggregation tendency of PrP127–147 by decreasing the interpeptide interactions and thus inhibiting β-sheet formation. They have explained their result by considering a greater role of aromatic residues from protein chains to the inhibition effects of carbon NPs by a π–π stacking interaction. Designed biocompatible engineered-nanomaterials have the potential to control or cure aggregate related diseases. Among different NPs, porous nanomaterials are the most effective one due to their highly controllable and isotropic nature of large accessible pore size, and easy release of incorporated materials from their pores.9 Of these, porous gold and silica-based nanomaterials are expected to be the most biocompatible and cost-effective ones with a much higher surface to volume (S/V) ratio than the corresponding other non-porous nanomaterials. Though the literature is rich in the therapeutic applications of non-porous inorganic nanoparticles, polymeric particles, and carbon nanomaterials10–22 by modulating amyloid protein aggregation, the role of corresponding porous nanomaterials has not been explored to the same extent. Modulation and inhibition of amyloid protein aggregation by non-porous nanomaterials are mainly governed by the induced electrostatic and magnetic fields offered by the nanomaterials.10–22 Hence, an amyloid aggregation (kinetics) study by porous nanomaterials and exploring the controlling role of various physicochemical parameters is extremely crucial to formulate a successful therapeutic methodology. Aggregation due to unfolding exposes the hydrophobic core of the protein.23 There are reports which document that molecular chaperones interact with non-native proteins through their hydrophobic binding sites to assist the covalent folding or unfolding.24 Similar to molecular chaperones, extent and the type of surface electrostatic potentials, induced magnetic fields, and nanoscopic pH modulation offered by nanoscale materials serve as synthetic nano-chaperones to prevent the aggregation of the protein.15 Porous gold and silica nanoparticles of larger size have been selected and explored here as nano-chaperones for the inhibition of protein aggregation due to their multitude of accessible surface pores, biocompatibility, and easy availability of ingredients for cost-effective synthesis. Applied nanoparticles are also attributed to high stability (against the light, air, and room temperature) and accessible pores on their surface are known to alleviate toxicity. It is also well established that larger porous materials (100–335 nm) show lower toxicity effects relative to smaller ones (14–16 nm) due to their noninvasive nature to the cell cytoplasm.25
Insulin, a 51 residue blood glucose regulatory hormone, can form amyloid fibrils under various conditions in vitro. Amyloid fibrillation in insulin is a major concern during insulin manufacture and long time storage. Patients with insulin-dependent diabetes i.e. type 1 suffers from insulin amyloid fibrillation after insulin infusion and repeated injections. Insulin exists in equilibrium as a mixture of monomer, dimer, hexamer, and possibly higher oligomeric states in solution, but the physiologically predominant storage form is Zn2+-coordinated hexamer which is formed by the association of three insulin dimmers. Under acidic conditions, it is highly prone to form amyloid fibrils in vitro because of the decrease in affinity between insulin and two Zn2+ ions that are coordinated by the imidazole side chains of HisB10 of each monomer equilibrium transfers from hexamers to the lower-order oligomer, and monomeric state. From α to β transition for human insulin appears to happen merely ahead of fibril assembly. The monomeric insulin has the most fibrillation-prone conformation in acidic conditions. The monomeric form of insulin exits as two peptide chains, A and B, having 21 and 30 residues respectively. Under acidic conditions and at high temperatures, insulin undergoes aggregation and forms fibril26,27 which is structurally similar to amyloid. In our experiment, the fibrillogenic conditions of human insulin were selected as 20% acetic acid and pH 1.8 with 100 mM NaCl at 60 °C. Under these conditions, the insulin molecules were present in their monomeric state, and a high temperature was necessary for their partial unfolding, and then allowed insulin to cross the threshold of the fibrillation pathway. It is important to note that physiological conditions such as phosphate-buffered at pH 7.4 were not used in our experiment, as human insulin was not soluble enough under neutral pH conditions. These characteristics of insulin make it a good model system for the study of amyloidal aggregation.
Though there are few in vivo studies28,29 are reported to find out the role of nanomaterials in up- and down-regulation of protein amyloid aggregation, for detailed understanding. In the present study, we have synthesized PNG & PNS and used them further for studying and comparing their efficiency to inhibit protein aggregation. Insulin is used as a model protein to measure the in vitro effects upon the addition of porous NPs. Spectroscopic assays are primarily used for the characterization of protein aggregation upon the addition of NPs. Subsequently, these porous NPs have been used to study the inhibition of protein aggregation in vitro in human neuroblastoma (SH-SY5Y) cell lines where the HTT-83Q mutant gene has been transfected to imitate HD-like pathology. Confocal microscopy has been used to illustrate the impact of reduced aggregation in cells. The purpose of the present work is to investigate the fibrillation kinetics of model protein as well as SH-SY5Y cell lines to reveal the features of the underlying pathway.
2. Materials and methods
2.1. Chemical and reagents
Tetraethyl orthosilicate [TEOS, Si(OC2H5)4], cetyltrimethylammonium chloride (CTAC, CH3–(CH2)14–CH2–N(CH3)3+Cl−), ethanol (EtOH, C2H5OH), cetyltrimethylammonium bromide (CTAB, CH3–(CH2)14–CH2–N(CH3)3+Br−), gold(III) chloride trihydrate (HAuCl4·3H2O), sodium borohydride (NaBH4, 99%), L-ascorbic acid (AA, 99.5+%), mesitylene [C6H3(CH3)3], DMSO, human insulin, acetic acid, sodium chloride (NaCl), all are purchased from Sigma-Aldrich and used without further purification. We used Millipore water (resistivity: 18.2 MΩ) to prepare solutions for all experiments.2.2. Synthesis of porous nano-silica (PNS)
The porous nano-silica (PNS) particles were prepared by a modified general method reported before.30 In brief; 0.05 g of CTAB was completely dissolved in 240 mL water by sonication. Followed by, 1.75 mL of 2 M NaOH and 3.5 mL of mesitylene were added one after another to the above diluted CTAB solution. The resultant mixture was then stirred vigorously at 80 °C for 2 h. Followed by, 2.5 mL of TEOS was added dropwise to the above mixture at a rate of 1 mL min−1. Continued stirring the reaction mixture vigorously at 80 °C for another 2 h. The formation of porous nano-silica is visible from the appearance of turbidity in the clear solution. The resultant precipitate was then isolated by filtration, washed with methanol, and dried under vacuum at 100 °C for 12 h.2.3. Synthesis of porous nanogold (PNG)
Porous nanogold (PNG) was synthesized by a two-step seed-mediated growth method reported before:312.4. Transmission electron microscopy
TEM studies were carried out to measure the particle size and morphology of PNS and PNG. Both samples were mixed in EtOH and sonicated for 15 min to achieve a good dispersion of particles. An FEI, Tecnai™ G2 F30, S-Twin microscope operating at 300 kV is used for all the electron imaging experiments. For all TEM measurements, 300 mesh copper formvar/carbon grids were used as the substrate. By drop-casting a small volume (5–6 μL) of supernatant EtOH solution of nanoparticles on copper grids followed by 24 h drying at room temperature, we made samples for TEM imaging.2.5. Brunauer–Emmett–Teller (BET) surface analysis
We have performed BET analysis of silica nanoparticles to confirm their mesoporous nature and also to determine their pore size as well as the extent of pores to the total volume of the particle. Compared to PNS, since porous gold nanoparticles are stable only in the liquid state, we could not perform their BET analysis in the solid-state. The surface area and porosity were determined from nitrogen adsorption/desorption isotherms with a micromeritics ASAP 2020 physisorption instrument using the BET equation to estimate the overall surface area. The BJH equation yields the surface area in the mesoporous region along with the pore size distribution.2.6. UV-visible and fluorescence spectroscopy
All UV-vis absorption spectra and fluorescence spectra were recorded at room temperature on a V-650 (Made: JASCO) and VARIAN CARY Eclipse fluorescence spectrometer, respectively. Both for absorption and fluorescence spectra we used a quartz cuvette of 1 cm optical path length. For fluorescence spectroscopy, the excitation wavelength was kept fixed at 440 nm with an emission maximum at 485 nm where both the slit widths were fixed at 5 nm.2.7. CD spectroscopy
CD spectra were performed between 190 and 250 nm on a JASCO J-815 spectropolarimeter (JASCO, USA). All the CD measurements were performed at 25 °C. The data acquisition interval (D.I.T.) value was 2 s. Spectra were recorded using a 0.1 mm path length cell with a 50 nm min−1 scan rate. For each sample, three scans were averaged and the buffer background was subtracted in each case.A CD spectrum is usually expressed in terms of mean residue molar ellipticity [MRE] in deg cm2 dmol−1 and can be calculated according to the eqn (1).
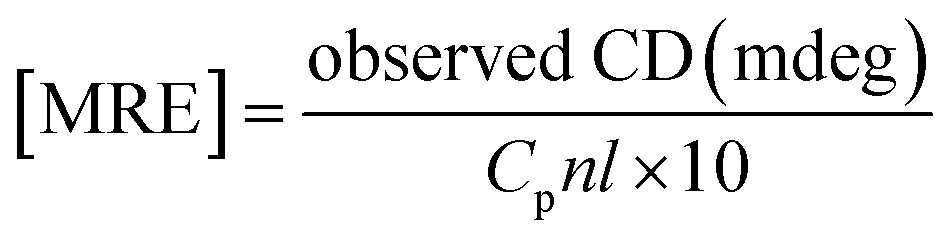 | (1) |
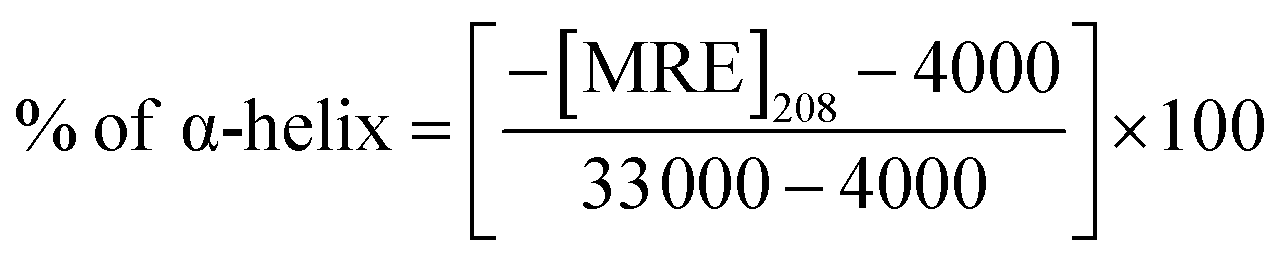 | (2) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 is the MRE value of total α-helix at 208 nm. To estimate the contribution of secondary structural components, data were analyzed using the BeStSel Web server.
000 is the MRE value of total α-helix at 208 nm. To estimate the contribution of secondary structural components, data were analyzed using the BeStSel Web server.
2.8. Sample preparation
2.9. Thioflavin T (ThT) fluorescence spectroscopy assay
The fibrillation kinetics of insulin could be described as a sigmoidal time-dependent curve with sequentially involving three stages, an initial lag phase where no change in the intensity occurs, a subsequent growth phase where intensity increases with time, and the final equilibrium phase where intensity reaches a plateau indicating the end of the fibril formation. A 2 mM stock solution of thioflavin T was prepared in 10 mL double distilled water and filtered through a 0.22 μm membrane filter before we store it at 4 °C. ThT concentration was measured through the absorbance at 412 nm in water by using an extinction coefficient of 36000 M−1 cm−1. ThT assays were performed to investigate the insulin amyloid fibrillation process where 700 μL of the ThT stock solution was taken and 10 μL aliquots of insulin solutions in the presence and absence of PNG (3 μg mL−1) or PNS (3 μg mL−1) were mixed properly at room temperature. The samples were then excited at 440 nm and the emission was recorded at 485 nm on a VARIAN CARY Eclipse fluorescence spectrometer using a quartz cuvette (1 cm path length), where integration time and slit widths were fixed at 1 s and 5 mm, respectively. ThT in buffer without protein was used for baseline correction. All experiments were carried out in 20% (v/v) acetic acid with 100 mM NaCl (pH 1.8) at 60 °C.
2.10. Cell culture
The human neuroblastoma-cell line SH-SY5Y was obtained from The National Cell repository at NCCS, Pune, INDIA by following the licensing protocol and was maintained in F12 medium (F12; HIMEDIA) supplemented with 10% FBS (Sigma), 2% L-glutamine, 1% penicillin, and 1% streptomycin stock solutions. The medium was changed every two days and the cells were passaged by trypsinization before the confluence.2.11. MTT assay
MTT assay was performed with a fresh F12 medium. 5 mg mL−1 of MTT stock was prepared in sterile PBS. 10 μL of the MTT stock solution was added to the cells and incubated for 4 h. DMSO (150 mL) was added to solubilize the formazan before taking the absorbance at 570 nm.2.12. Confocal microscopy
SH-SY5Y cells were seeded in 35 mm cell culture dishes at a seeding density of 5 × 104 cells per well, 24 h before the experiment. Following attachment, the medium was changed and the cells were transiently transfected with the DsRed-HTT-83Q (1.3 μg per mL per well) using Lipofectamine 2000 (Invitrogen) to constitutively express high levels of the protein. After 24 h, the cells were treated with nanoparticles at various concentrations and kept them under nanoparticle treatment for another 24 h before we washed them with PBS buffer and fixed them with 4% formaldehyde for microscopy experiments. The fluorescence of DsRed at an excitation wavelength of 561 nm was used to confirm the transfection. Aggregations were identified from the appeared red puncta inside cells.3. Results and discussion
PNS and PNG were successfully prepared by the reported procedure with slight modifications. A detail of their synthesis is described in the previous section. Though the as-prepared porous silica nanoparticles are spherical, the corresponding gold nanoparticles show a whole walnut kernel shape. Considering both types of particles as overall spherical, their average diameter is comparable and is around 150–200 nm as confirmed by TEM measurement (Fig. 1). | ||
| Fig. 1 TEM images of (a) spherical PNS and (b) whole walnut kernel shaped PNG. | ||
To confirm the porous nature of the synthesized silica nanoparticles, we have performed their BET analysis. Besides confirming their porous nature, BET analysis also provides a quantitative estimation of their pore size as well as the extent of pores to the total volume. From the BET analysis, it is clearly observable that the synthesized PNS are porous in nature with an average pore diameter of 4.55 nm and a total pore volume of 0.136827 cm3 g−1. The obtained summary report from the BET measurement is listed below whereas the associated BET surface area plot and BJH desorption plot are listed as Fig. S1 and S2 respectively in the ESI† section.
3.1. BET summary report
827 cm3 g−1.3.2. Effect of porous nanomaterials on the aggregation of insulin
To monitor the effect of porous nanomaterials on the aggregation process of insulin, a fluorescence study has been performed by using fluorescence assay based thioflavin T dye. Details of sample preparation which include the preparation of insulin fibrils and porous nanomaterials along with thioflavin T (ThT) fluorescence assay have been described in the Experimental section.† In general, insulin loses efficacy as a result of protein aggregation under high pressure or at low pH above room temperature.32,33 The kinetics of insulin fibrillation has three stages, characterized by the increment of fluorescence intensity of the fibril-specific dye thioflavin T (ThT), which are lag phase, elongation phase (or growth of amyloidal aggregates), and saturation phase.34–36 ThT dye preferentially binds to amyloid deposits as it recognizes the cross β-sheet structure of amyloid fibrils and results in a significant increase in its fluorescence emission intensity. The red curve in Fig. 2 represents the ThT fluorescence of insulin in the absence of nanoparticles, monitoring at different time intervals. Fluorescence measurements were carried out for 300 min at 60 °C. The sigmoidal curve of ThT fluorescence showed in Fig. 2 reveals the nucleation-dependent pathway. Control insulin gave a strong ThT emission with a lag time of 120 min, after that ThT fluorescence increases rapidly (lag phase linked to the elongation and propagation of fibrils) and then finally reached a hill within 250 min (Fig. 2) followed by a prolonged saturation. In the presence of porous gold nanoparticles, the time for the lag phase of insulin changes from 120 min to 150 min with a decrease in fluorescence intensity as shown by the green curve in Fig. 2.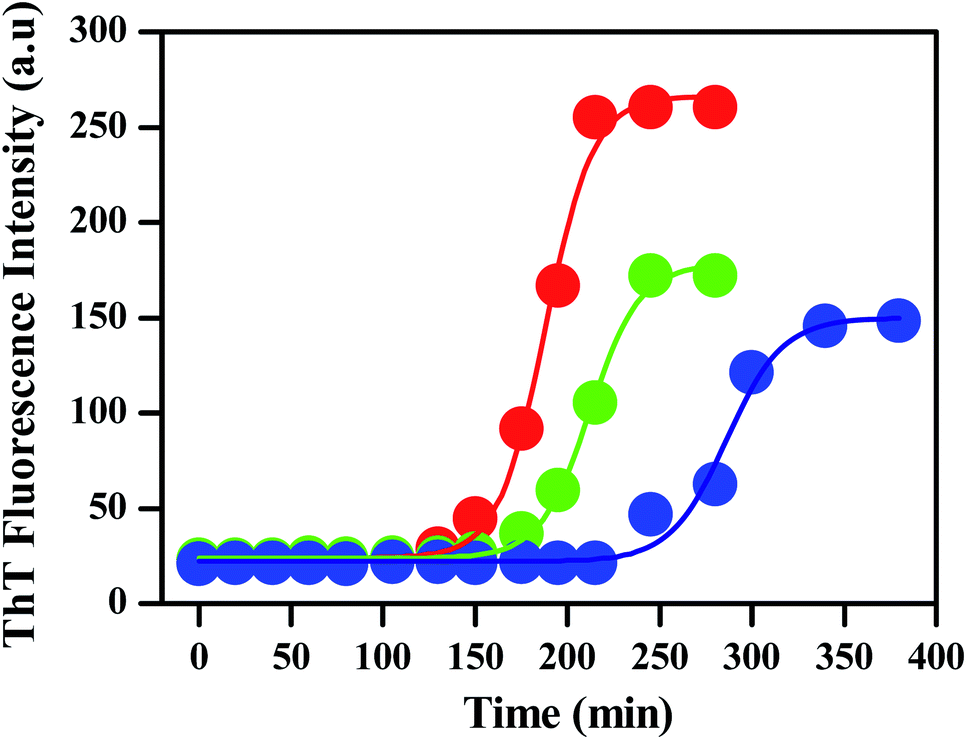 | ||
| Fig. 2 Fibrillation kinetics of human insulin (HI) (concentration: 1.7 mg mL−1) in the absence (red) and presence of PNG (green) and PNS (blue), incubated in a pH of 1.8 at 60 °C, is followed by measuring the ThT fluorescence intensity. | ||
A similar experiment was carried out with PNS as shown by the blue curve in the same figure. It is clearly observable from Fig. 2 that the addition of PNS increases the lag phase significantly from 120 min to 220 min with almost a 50% decrease in fluorescence intensity. The corresponding bar plot for the lag phase of insulin in the absence and presence of PNG and PNS is shown in Fig. S3.†
By comparing the fibrillation kinetics of human insulin (HI) in presence of PNG and PNS, it is clearly observable that though both the nanoparticles inhibit the fibrillation process by increasing their respective lag phases, the effect is strikingly significant for PNS. Similar to the comparison of lag phases, by comparing the extent and rate of the reduction of the ThT fluorescence intensity, we can infer the kinetics of their growth phase. Though both in presence of PNG and PNS the growth phase took ∼100 min to rich to the hill after the lag phase, the extent of reduction in fluorescence intensity in presence of PNS is significantly greater compared to PNG. This indirectly proves the predominant role of PNS in suppressing the fibrillation growth process and highlights its important role in the prevention of HI aggregation. To understand the role of particle charge as an important parameter for understanding the nature of NP–protein interaction and aggregation inhibition, we have measured the pH-dependent zeta potentials of PNS and PNG along with insulin as shown in the Table 1.
| System | Measured zeta potential (mV) at different pH | ||
|---|---|---|---|
| @1.8 | In water | @7.4 | |
| PNS | +8.16 | −41.7 | −31.1 |
| PNG | +54.2 | +38.8 | +31.0 |
| Insulin | +6.12 | +9.14 | −5.81 |
It is clear from the above table that at pH: 1.8, due to the very high +Ve zeta potential of PNG, insulin molecules (which are also positively charged amphiphiles with a zeta potential of + 6.12 mV at pH: 1.8 with 100 mM NaCl) are highly unlikely to be adsorbed either on the surface or inside PNG nanopores to form corona structure37 and hence the insulin peptide chains (A and B) remain as free chains for efficient hydrogen bonding between three central amino acids of attaching peptides to form β-sheets or a reorientation of central Glu side chain of the locking peptide towards the interface between two β-sheets to form fibrils.38 On the other hand, due to the very low +Ve zeta potential of PNS, insulin molecules are highly likely to get adsorbed either on the surface or inside PNS nanopores to form an effective corona structure. As a result of this structural modification of insulin amphiphile chains (inaccessibility of other free insulin molecules inside each pore or presence of big PNS knots on each insulin chain's surface), conformational locking among insulin chains is not so efficient as it is for PNG to form β-sheets or the resultant fibrils.
3.3. Kinetics of human insulin fibril formation from fluorescence study
The kinetics of insulin fibrillation can be described by the sigmoidal curve of thioflavin T fluorescence assay having three characteristic stages34–36 in the absence and presence of two different porous nanomaterials, PNG and PNS. Fitting of the curve is described by the following eqn (3) using sigma plot:39
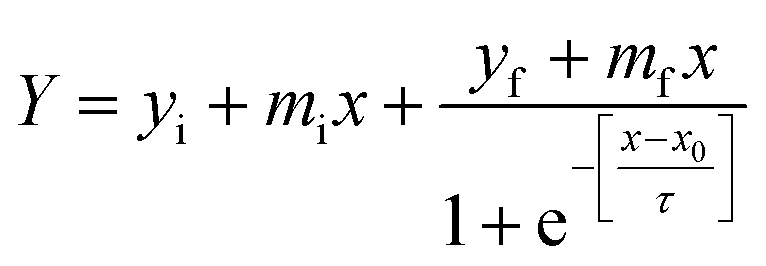 | (3) |
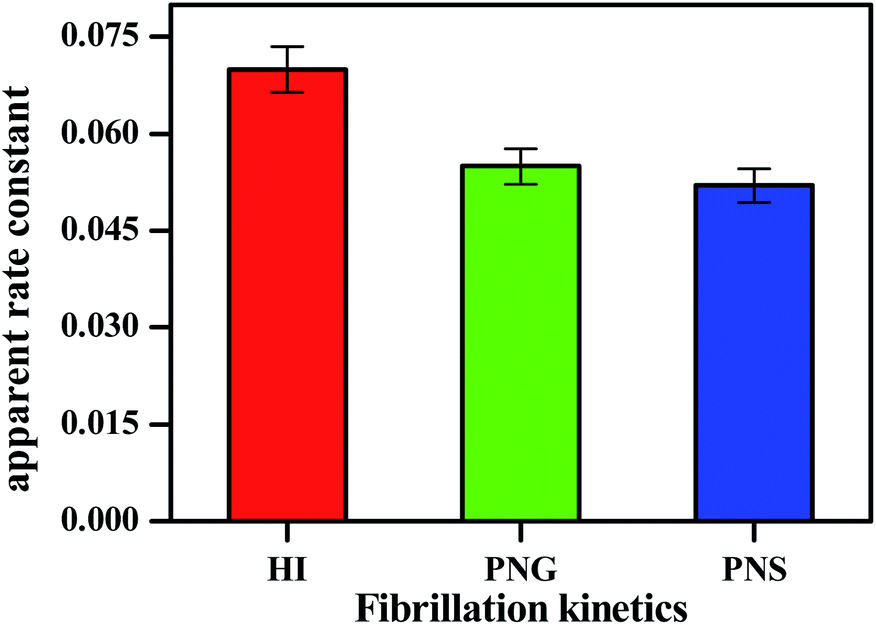 | ||
| Fig. 3 Apparent rate constant (Kapp) during insulin fibrillation in the absence and presence of two different porous nanomaterials PNG and PNS. | ||
From Table 2, the apparent rate constant for PNG and PNS were calculated as 0.055 min−1 and 0.053 min−1. These values of rate constants suggest their involvement in decreasing the fibrillation process along with the delay in the nucleation phase. It is observable from the obtained result that PNS is more effective in slowing down the fibrillation process of model insulin than PNG.
| System | Lag time (min) | Apparent rate constant Kapp (min−1) |
|---|---|---|
| HI | 120 | 0.072 |
| HI + PNG | 150 | 0.055 |
| HI + PNS | 220 | 0.052 |
The inhibition effect caused by porous nanomaterials was also confirmed from their respective TEM images. Fig. 4 represents different morphologies of aggregated insulin in the absence and presence of nanomaterials which demonstrates the influence of nanomaterials on the aggregation of insulin.
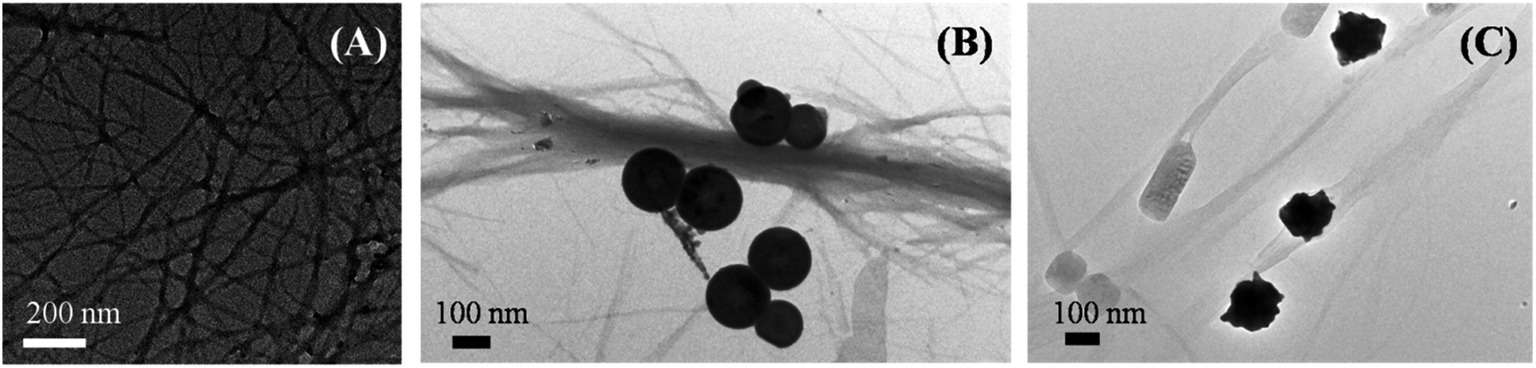 | ||
| Fig. 4 TEM images of aggregated insulin (A) control, in presence of (B) PNS, and in presence of (C) PNG. | ||
3.4. Conformational change of human insulin during fibril formation: a circular dichroism (CD) spectroscopic study
In addition to fluorescence study and electron imaging, we have additionally performed a CD experiment to shows the structural changes of insulin in presence of PNS and PNG at different time intervals. It is well known in the literature that when a normal protein undergoes the fibrillation process, conformation generally changes from α-helix to β-sheet.40 Insulin in its controlled state (i.e. without fibrillation) has the signature of an α-helical structure resulting in two negative ellipses at 208 nm and 222 nm. As it undergoes the aggregated state from 0 min to 220 min, the changes in CD spectra are clearly observable from Fig. 5. Changes in the HI protein secondary structures, in the presence of both nanoparticles (Fig. 5), are analyzed in terms of mean residue molar ellipticity [MRE] in deg cm2 dmol−1 and the subsequent α-helix content of free and bound HI was estimated from the MRE value at 208 nm ([MRE]208) by using eqn (1) and (2) respectively as detailed in the previous section. The changes in α and β fractions as a result of the interaction with porous nanoparticles are carefully calculated and estimated results are enlisted in Table 3.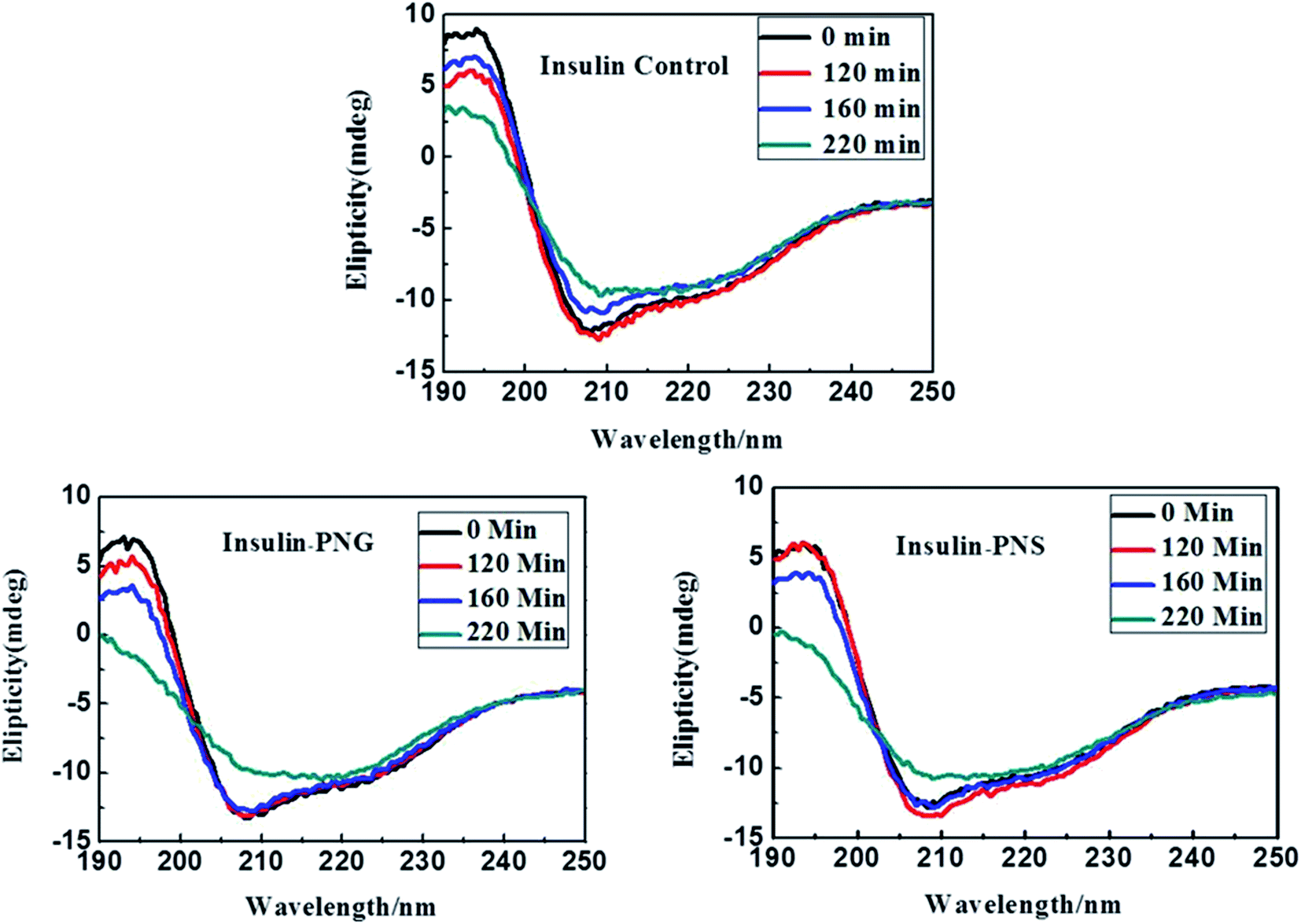 | ||
| Fig. 5 CD spectra of control insulin and insulin in presence of PNG and PNS at different time intervals. | ||
| Time (min) | Species | α-helix (%) | β-sheet (%) | Turn (%) | Random coil (%) |
|---|---|---|---|---|---|
| 0 | Insulin | 45 | 9 | 11 | 30 |
| Insulin + PNS | 43 | 12 | 16 | 29 | |
| Insulin + PNG | 43 | 12 | 15 | 30 | |
| 90 | Insulin | 42 | 13 | 15 | 30 |
| Insulin + PNS | 41 | 13 | 15 | 31 | |
| Insulin + PNG | 40 | 14 | 16 | 30 | |
| 160 | Insulin | 35 | 22 | 15 | 28 |
| Insulin + PNS | 41 | 13 | 15 | 31 | |
| Insulin + PNG | 35 | 22 | 15 | 28 | |
| 220 | Insulin | 20 | 35 | 15 | 30 |
| Insulin + PNS | 39 | 18 | 15 | 28 | |
| Insulin + PNG | 28 | 30 | 14 | 28 |
In the present study, CD spectra of native insulin contain 45% of α-helix, 9% of β-sheet, 11% of turn, and 30% of a random coil configuration at zero time under incubation in a pH 1.8 and 60 °C, as listed in Table 3, which is in good agreement with the earlier report.34 It is also observable from Table 3 that with increasing time, in absence of any nanomaterials, the percentage of α helicity of the same insulin is gradually decreasing and the percentage of β-sheet is increasing, while there is almost no change in the percentage of turns and random coil conformation. But, in presence of porous nanomaterials, both for PNG and PNS, though the difference between the percentage of α-helix and β-sheet formation is not significant at the beginning (at 0 min), it becomes significant as time goes. It is also observable that with time though the relative percentage of α-helix and β-sheet formation for PNS remains almost constant, PNG shows a gradual increment in β-sheet formation.
Though, in all cases, for all samples, the relative percentage of turn and random coil formation remains almost unchanged with time. Gradual increment of β-sheet formation (difference in β-sheet percentage between 0 min and 220 min, ∂β220–0) for control HI (∂β(HI)220–0 = (35 − 9)% = 26%) under the condition of pH 1.8 and 60 °C indicates the most rapid fibrillation whereas in presence of PNG (∂β(PNG)220–0 = (30 − 12)% = 18%) it follows the next and with PNS (∂β(PNS)220–0 = (18 − 12)% = 6%) is the least. From this observation, it can be concluded that Insulin may form corona with the porous nanoparticles or may be buried in the pore of nanoparticles, which inhibits the aggregation process of insulin and PNS is more effective to do this inhibition process than PNG.
3.5. Effect of porous nanomaterials on the protein aggregation in the cellular model
The characteristic images of SH-SY5Y cells with overexpressed DsRed-HTT83Q in presence of different concentrations of PNS and PNG are depicted as red clusters in Fig. 6(i)a–d and 6(i)e–h respectively. The detailed procedure of confocal microscopy is included in the Materials and methods section. Upon treatment with various concentrations (0, 1, 5, and 10 μg mL−1) of PNS and PNG (Fig. 6(i)b–d and 6(i)f–h), the cell cytotoxicity profile of the nanoformulations of less than 40% has been illustrated in Fig. 6(ii). It has been found that both nanomaterials showed effective cell viability, which implies that these porous nanoparticles are safe and sustainable for biotherapeutic applications. A significant reduction in the number of aggregates as well as the size of aggregates was evident (Fig. 6(iii) and (iv)), delineating the effect of the nanoformulations. The Htt gene encodes for a protein, huntingtin, which in its mutated form generates an exaggerated number of glutamine amino acids in its N-terminal region.41 This modification reduces the activity of the protein and is prone to form oligomers and eventually aggregates.42 The size of the Htt protein is about 11 kDa and its approximate dimension is about 2 nm. There are several reports on the effect of nanomaterials on fibrillogenesis, like the effect of fullerene on amyloidogenesis of Aβ peptide by specific binding to its hydrophobic core,43 and fluorinated nanoparticle-induced increased helicity of Aβ peptides preventing aggregation.20 But here in this case we use porous nanomaterials to see the effect on aggregation in SHSY5Y cell and it shows a significant reduction in the aggregation process. The plausible mechanism of the inhibition of protein aggregation by these nanomaterials may be adsorption of the protein in the pore of the nanomaterials. The pores form corona by strong adsorption post interaction with the protein. This would result in an increase in the concentration of the protein on the nanoparticle surface. This process could block the sticky patches on the protein surface that lead to fibril formation and eventually inhibit fibrillation by increasing the lag time. More investigation is needed to get information on the exact mechanism of the process which is on the way.
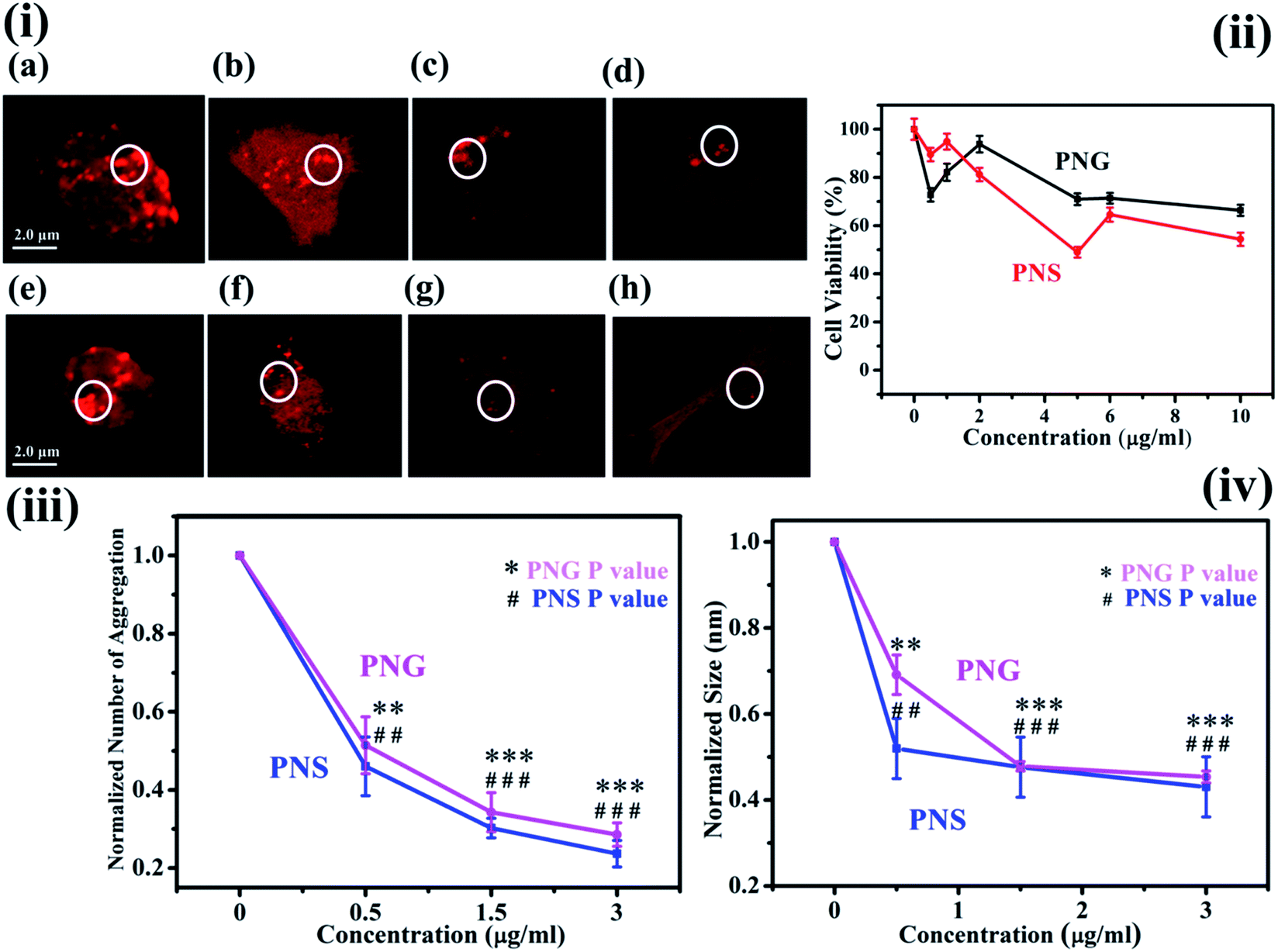 | ||
| Fig. 6 (i) Confocal images of SH-SY5Y cells in presence of different concentrations of PNS (a–d) and PNG (e–h); (ii) MTT assay with PNS and PNG in SH-SY5Y cells; (iii) and (iv) depict the reduction in aggregation number and size of red dots, respectively, in SH-SY5Y cells after the treatment with PNS and PNG, **, ## denote P-value ≤ 0.01 and ***, ### depict P-value ≤ 0.001 for PNG and PNS, respectively. | ||
4. Conclusion
By using ThT-based fluorescence assay, electron microscopy, and circular dichroism-based spectroscopy we have studied the effect of two different porous nanomaterials (PNG and PNS) with similarity in size on protein aggregation of the model system (HI) as well as in vitro. The obtained result shows the significant role of PNS in preventing protein aggregation when we compare it with PNG. Here, insulin may form corona with the porous nanoparticles or may be buried in the pore of nanoparticles, which inhibit the aggregation process of insulin and this conformational change is further supported by the CD data. Further study of these porous materials on huntingtin aggregates in human neuroblastoma cell-lines (SH-SY5Y) shows the same effect as on the in vitro model and proves the significant role of PNS over PNG where both the nanoparticles are nontoxic in nature. These findings pave the way for the use of porous nano-silica (PNS) as a potential therapeutic agent, both in terms of delaying the fibrillation process as well as the extent of fibril formation in comparison to PNG, for inhibition of protein aggregation and augment our understanding of diagnosis and cure of protein misfolding and aggregate associated diseases.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
M. B. would like to express her sincere gratitude to the DST-SERB project (YSS/2015/000589), DST, Govt. of India and UGC, New Delhi for providing Dr D. S. Kothari fellowship. S. C. thanks Science and Engineering Research Board (India) for National Postdoctoral Fellowship (File No: PDF/2016/002789). We thank Mr M. Mahendar for facilities in super-resolution confocal microscopy, SINP.References
- Y. C. Eva, K. Sampathkumar, W. R. Theodore and J. F. Carpenter, Pharm. Res., 2003, 20, 1325–1336 CrossRef.
- D. H. Lopes, A. Meister, A. Gohlke, A. Hauser, A. Blume and R. Winter, Biophys. J., 2007, 93, 3132–3141 CrossRef CAS.
- C. Voisine, J. S. Pedersen and R. I. Morimoto, Neurobiol. Dis., 2010, 40, 12–20 CrossRef CAS.
- M. Li, S. E. Howson, K. Dong, N. Gao, J. Ren, P. Scott and X. Qu, J. Am. Chem. Soc., 2014, 136, 11655–11663 CrossRef CAS.
- S. Palmal, N. R. Jana and N. R. Jana, J. Phys. Chem. C, 2014, 118, 21630–21638 CrossRef CAS.
- J. A. Yang, B. J. Johnson, S. Wu, W. S. Woods, J. M. George and C. J. Murphy, Langmuir, 2013, 29, 4603–4615 CrossRef CAS.
- C. Cabaleiro-Lago, F. Quinlan-Pluck, I. Lynch, S. Lindman, A. M. Minogue, E. Thulin, D. M. Walsh, K. A. Dawson and S. Linse, J. Am. Chem. Soc., 2008, 130, 15437–15443 CrossRef CAS.
- S. Zhou, Y. Zhu, X. Yao and H. Liu, J. Chem. Inf. Model., 2019, 59, 1909–1918 CrossRef CAS.
- S. Jafari, H. Derakhshankhah, L. Alaei, A. Fattahi, B. S. Varnamkhasti and A. A. Saboury, Biomed. Pharmacother., 2019, 109, 1100–1111 CrossRef CAS.
- C. Wang, M. Zhang, X. Mao, Y. Yu, C. X. Wang and Y. L. Yang, Adv. Mater., 2013, 25, 3780–3801 CrossRef.
- M. Mahmoudi, H. R. Kalhor, S. Laurent and I. Lynch, Nanoscale, 2013, 5, 2570–2588 RSC.
- L. Fei and S. Perrett, Int. J. Mol. Sci., 2009, 10, 646–655 CrossRef CAS.
- M. Zaman, E. Ahmad, A. Qadeer, G. Rabbani and R. H. Khan, Int. J. Nanomedicine, 2014, 9, 899–912 Search PubMed.
- M. A. Busquets, R. Sabaté and J. Estelrich, Nanoscale Res. Lett., 2014, 9, 538 CrossRef.
- Y. H. Liao, Y. J. Chang, Y. Yoshiike, Y. C. Chang and Y. R. Chen, Small, 2012, 8, 3631–3639 CrossRef CAS.
- M. Mahmoudi, F. Quinlan-Pluck, M. P. Monopoli, S. Sheibani, H. Vali, K. A. Dawson and I. Lynch, ACS Chem. Neurosci., 2013, 4, 475–485 CrossRef CAS.
- A. M. Saraiva, I. Cardoso, M. J. Saraiva, K. Tauer, M. C. Pereira, M. A. N. Coelho, H. Möhwald and G. Brezesinski, Biosci, 2010, 10, 1152–1163 CAS.
- A. M. Saraiva, I. Cardoso, M. C. Pereira, M. A. N. Coelho, M. J. Saraiva, H. Möhwald and G. Brezesinski, ChemBioChem, 2010, 11, 1905–1913 CrossRef CAS.
- C. Cabaleiro-Lago, F. Quinlan-Pluck, I. Lynch, K. A. Dawson and S. Linse, ACS Chem. Neurosci., 2010, 1, 279–287 CrossRef CAS.
- S. Rocha, A. F. Thünemann, M. D. C. Pereira, M. Coelho, H. Möhwald and G. Brezesinski, Biophys. Chem., 2008, 137, 35–42 CrossRef CAS.
- S. I. Yoo, M. Yang, J. R. Brender, V. Subramanian, K. Sun, N. E. Joo, S. H. Jeong, A. Ramamoorthy and N. A. Kotov, Angew. Chem. Int Ed., 2011, 50, 5110–5115 CrossRef CAS.
- S. Mirsadeghi, S. Shanehsazzadeh, F. Atyabi and R. Dinarvand, Mater. Sci. Eng. C., 2016, 59390–59397 Search PubMed.
- J. I. Clark and P. J. Muchowski, Curr. Opin. Struct. Biol., 2000, 10, 52–59 CrossRef CAS.
- M. Bhattacharyya, S. Ray, S. Bhattacharya and A. Chakrabarti, J. Biol. Chem., 2004, 279, 55080–55088 CrossRef CAS.
- S. Sharifi, S. Behzadi, S. Laurent, M. L. Forrest, P. Stroeve and M. Mahmoudi, Chem. Soc. Rev., 2016, 41, 2323–2343 RSC.
- D. F. Waugh, J. Am. Chem. Soc., 1944, 66, 663 CrossRef CAS.
- D. F. Waugh, J. Am. Chem. Soc., 1946, 68, 247–250 CrossRef CAS.
- B. Wang, E. H. Pilkington, Y. Sun, T. P. Davis, P. C. Ke and F. Ding, Environ. Sci. Nano., 2017, 4, 1772–1783 RSC.
- I. Javed, T. Yu, G. Peng, A. Sanchez-Ferrer, A. Faridi, A. Kakinen, M. Zhao, R. Mezzenga, T. P. Davis, S. Lin and P. C. Ke, Nano Lett., 2018, 18, 5797–5804 CrossRef CAS.
- I. I. Slowing, B. G. Trewyn and V. S.-Y. Lin, J. Am. Chem. Soc., 2007, 129, 8845–8849 CrossRef CAS.
- Q. Zhang, N. Large, P. Nordlander and H. Wang, J. Phys. Chem. Lett., 2014, 5, 370–374 CrossRef CAS.
- W. Dzwolak, R. Ravindra, J. Lendermann and R. Winter, Biochemistry, 2003, 42, 11347–11355 CrossRef CAS.
- J. Haas, E. Vöhringer-Martinez, A. Bögehold, D. Matthes, U. Hensen, A. Pelah, B. Abel and H. Grubmüller, ChemBioChem, 2009, 10, 1816–1822 CrossRef CAS.
- Y. Song, P.-N. Cheng, L. Zhu, E. G. Moore and J. S. Moore, J. Am. Chem. Soc., 2014, 136, 5233–5236 CrossRef CAS.
- Q.-X. Hua and M. A. Weiss, J. Biol. Chem., 2004, 279, 21449–21460 CrossRef CAS.
- V. Sluzky, A. M. Klibanov and R. Langer, Biotechnol. Bioeng., 1992, 40, 895–903 CrossRef CAS.
- S. Zhang, K. Saito, H. Matsumoto, M. Minagawa and A. Tanioka, Polymer Journal, 2008, 40, 837–841 CrossRef CAS.
- M. Schor, J. Vreede and P. G. Bolhuis, Biophys. J., 2012, 103, 1296–1304 CrossRef CAS.
- S. Roy Chowdhury, S. Mondal and P. K. Iyer, ACS Biomater. Sci. Eng., 2018, 12, 4076–4083 CrossRef.
- C. M. Dobson, Trends Biochem. Sci., 1999, 24, 329–332 CrossRef CAS.
- E. Zanforlin, G. Zagotto and G. Ribaudo, ACS Chem. Neurosci., 2017, 8, 2356–2368 CrossRef CAS.
- M. Sinha, J. Ghose and N. P. Bhattarcharyya, RNA Biol., 2011, 8, 1005–1021 CrossRef CAS.
- J. E. Kim and M. Lee, Biochem. Biophys. Res. Commun., 2003, 303, 576–579 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra10927d |
| ‡ Current address: Department of Physics, Jadavpur University, Kolkata-700032, India, E-mail: bardhanm@gmail.com, +91-9434414318. |
| This journal is © The Royal Society of Chemistry 2021 |