
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Characterization of NucPNP and NucV involved in the early steps of nucleocidin biosynthesis in Streptomyces calvus†
Utumporn Ngivprom‡
a,
Surayut Kluaiphanngam‡a,
Wenjuan Jic,
Siriwalee Siriwiboola,
Anyanee Kamkaew a,
James R. Ketudat Cairnsab,
Qi Zhang*c and
Rung-Yi Lai*ab
a,
James R. Ketudat Cairnsab,
Qi Zhang*c and
Rung-Yi Lai*ab
aSchool of Chemistry, Institute of Science, Suranaree University of Technology, Nakhon Ratchasima, 30000, Thailand. E-mail: rylai@sut.ac.th
bCenter for Biomolecular Structure, Function and Application, Suranaree University of Technology, Nakhon Ratchasima, 30000, Thailand
cDepartment of Chemistry, Fudan University, Shanghai, 200433, China. E-mail: qizhang@sioc.ac.cn
First published on 15th January 2021
Abstract
Nucleocidin 1 produced by Streptomyces calvus is one of five characterized natural products containing fluorine. It was discovered in 1956, but its biosynthesis is not yet completely resolved. Recently, the biosynthetic gene cluster of 1 was identified. The nucPNP gene, which was initially annotated as orf206 and encodes a putative purine nucleoside phosphorylase, is essential for nucleocidin production. In this study, we performed in vitro assays and showed NucPNP produced adenine 3 from methylthioadenosine (MTA) 2 and adenosine 4. We also showed the downstream enzyme, NucV annotated as adenine phosphoribosyltransferase (APRT), catalyzes AMP formation from adenine 3 and 5-phospho-α-D-ribose-1-diphosphate (PRPP) 5. However, the catalytic efficiency of NucV was much slower than its homolog ScAPRT involved in the biosynthesis of canonical purine nucleoside in the same strain. These results provide new insights in nucleocidin biosynthesis and could guide future research on organofluorine formation.
1. Introduction
Organofluorine compounds play important roles in pharmaceuticals,1 medical imaging,2 materials science,3 agrochemicals,4 and other disciplines. Due to the interest in fluorination, researchers have searched for natural products containing fluorine. Among an estimated more than 130![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 structurally characterized natural products, there were only five unique fluorine-containing metabolites (Fig. 1) found thus far.5 All fluorometabolites, except for nucleocidin, are biosynthesized from 5′-fluorodeoxyadenosine (FDA). The formation of FDA is catalyzed by the only known fluorinase (FldA) in Streptomyces cattleya6,7 or its homologs in other microorganisms.8–10
000 structurally characterized natural products, there were only five unique fluorine-containing metabolites (Fig. 1) found thus far.5 All fluorometabolites, except for nucleocidin, are biosynthesized from 5′-fluorodeoxyadenosine (FDA). The formation of FDA is catalyzed by the only known fluorinase (FldA) in Streptomyces cattleya6,7 or its homologs in other microorganisms.8–10
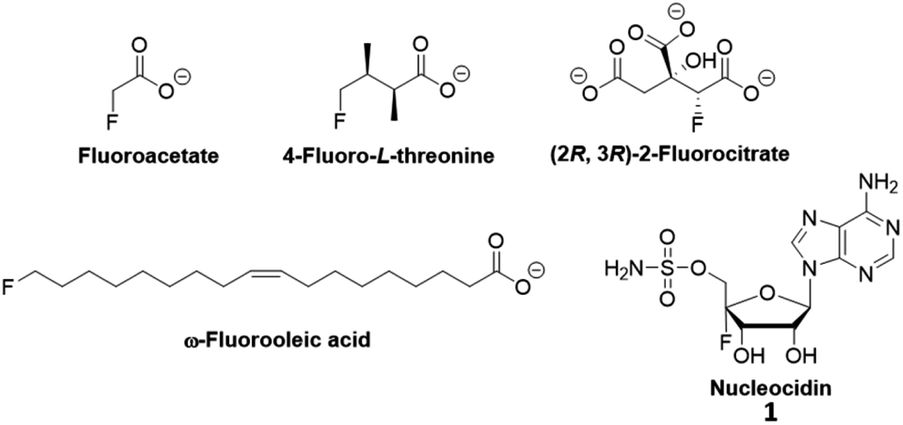 | ||
| Fig. 1 The five characterized natural products containing fluorine. | ||
Nucleocidin was discovered in S. calvus ATCC 13382 in 1956.11 It is an antimicrobial and antitrypyranosomal analogue of adenosine, which contains a fluorine atom at the C-4′ position of the ribose ring.12 Researchers had attempted to investigate its biosynthesis for a few decades without any success. A major reason was that the strain studied could not reproduce nucleocidin. A few years ago, Zechel's group identified a point mutation in the bldA gene that is predicted to encode a misfolded Leu-tRNAUUA molecule.13 The complementation of S. calvus ATCC 13382 with a functional bldA gene restored the production of nucleocidin.14 At the same time, the genome of S. calvus was sequenced and annotated to identify the gene cluster for nucleocidin biosynthesis. Last year, S. asterosporus DSM 41452 was identified to be another nucleocidin producing strain after complementation with a functional adpA gene.15 Its genome sequence revealed a nucleocidin biosynthetic gene cluster nearly identical to that of S. calvus ATCC 13382. However, the fldA gene or its homolog was not identified in either genome, suggesting nucleocidin biosynthesis involves a different fluorinase from FldA. Feeding experiments with deuterated glycerol suggested that the fluorination step was after ribose biosynthesis.16,17 Furthermore, gene knockout experiments for nucleocidin production by O'Hagan's group showed the necessity of nucGT, which encodes a glucosyltransferase.18 The in vitro characterization of NucGT showed that it catalyzed 3′-O-β-glucosylation of adenosine and sulfamoyladenosine using UDP-glucose. Furthermore, NucJ, which is annotated as a radical SAM enzyme, was proposed as a fluorinase candidate to catalyze the C–F formation in the biosynthesis of nucleocidin.
Before that, Zechel's group reported the necessity of nucPNP (orf206 in the original paper).14 Moreover, the level of mRNA transcript of nucPNP increased by 151 times when bldA is functional to restore nucleocidin production (Table S1†). In comparison, the mRNA level of nucGT only increased by 12 times while that of nucJ increased 85 times. Last but not least, nucV, which encodes an adenine phosphoribosyltransferase (APRT), showed a similarly increasing level of mRNA transcript (110 times) as nucPNP. However, there is a housekeeping APRT in all Streptomyces species. According to the in silico analysis, NucPNP and NucV are both involved in the biosynthesis of adenosine, which is the core structure of nucleocidin.
To investigate the functions of nucPNP and nucV, we heterologously overexpressed these two proteins in E. coli. For comparative analysis, S. calvus APRT was also expressed. All soluble proteins were purified and characterized by in vitro assays. NucPNP converted methylthioadenosine (MTA) 2 or adenosine 4 to adenine 3. NucV catalyzes the synthesis of AMP 6 from adenine 3 and PRPP 5, but its rate was slower than ScAPRT. Based on these findings, the biosynthetic pathway is proposed in Scheme 1, which is similar to the biosynthesis of ascamycin and dealanylascamycin.19 The substrate of NucV is proposed as 4′-F-PRPP 7 from the NucJ reaction. These results provide clues to solve the mystery of C–F formation in nucleocidin biosynthesis.
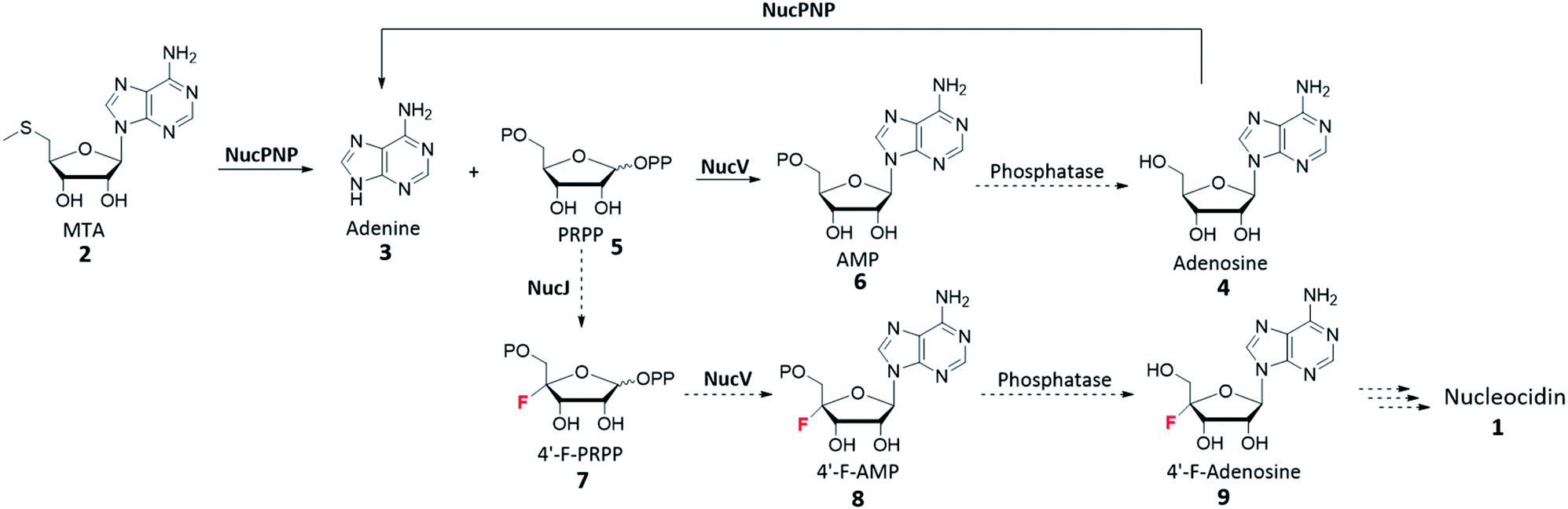 | ||
| Scheme 1 Proposed biosynthetic pathway of nucleocidin 1. The reactions (solid arrow) are studied in this article. | ||
2. Experimental section
2.1 Materials
Chemicals were purchased from either Sigma-Aldrich or TCI Chemicals. Sulfamoyladenosine was synthesized as shown in the ESI.† Streptomyces calvus ATCC 13382 was purchased from the American Type Culture Collection. E. coli MG1655 was purchased from NBRP-E. coli at NIG in Japan. The genomic DNA were purified with a BioFact Genomic DNA Prep Kit. Oligonucleotides were purchased from Integrated DNA Technologies. Enzymes for molecular cloning and the E. coli SHuffle T7 Express lysY strain were purchased from New England Biolabs. DNA purification kits were purchased from Vivantis. All plasmids made were constructed by Gibson assembly of PCR products.202.2 Overexpression of MBP-NucPNP, NucV, ScAPRT, and EcAPRT
The gene encoding NucPNP, NucV, and ScAPRT were amplified by PCR from the genomic DNA of S. calvus ATCC 13382. The gene encoding EcAPRT was amplified from the genomic DNA of E. coli MG1655. Every PCR product was assembled into pET28 vectors by Gibson assembly.The NucPNP protein was overexpressed as a fusion with a dual N-terminal 6His/maltose-binding protein (MBP) tag. Soluble MBP-NucPNP was only obtained from expression in E. coli SHuffle T7 Express lysY. MBP-NucPNP was purified by amylose resin followed by Ni-NTA resin.
The NucV, ScAPRT and EcAPRT proteins were overexpressed as a fusion with an N-terminal 6-His tag in E. coli BL21(DE3). All proteins were purified by Ni-NTA resin.
All information on protein expression and purification is shown in the ESI.†
2.3 Enzymatic assays
2.4 Kinetic constant determination
The initial velocity for each substrate concentration was determined by measuring the enzyme activity at different time points in triplicate. The amount of product in the assay was determined by HPLC analysis using the calibration curve of standard, adenine or AMP. The Km and Vmax values were calculated by fitting the rate of product formation and substrate concentration in nonlinear regression of the Michaelis–Menten curves with Grafit 5.0 (Erithacus Software, Horley, Survey, UK) shown in Fig. S3–S6.† The apparent kcat was calculated by dividing Vmax by the final concentration of enzyme.3. Results and discussion
3.1 NucPNP catalysis of phosphorylation of purine nucleoside
NucPNP was annotated as purine nucleoside phosphorylase (PNP).18 PNP enzymes catalyze a reversible reaction in purine metabolism,21 converting purine nucleoside and phosphate to purine base and α-D-ribose-1-phosphate. In order to identify the substrate of NucPNP, we tried a few different expression systems and soluble tags to obtain soluble protein. Soluble NucPNP only was overexpressed with a fusion with a dual N-terminal His/MBP tag in E. coli SHuffle T7 Express lysY. It is hereinafter referred to as MBP-NucPNP. Because the nucPNP gene is in the nucleocidin biosynthetic gene cluster, a few nucleoside substrates containing adenine, such as MTA 2, adenosine 4, AMP 6, ADP, ATP, and sulfamoyladenosine, were assayed with MBP-NucPNP in presence of excess phosphate. The reaction mixtures were analyzed by HPLC to monitor the formation of adenine 3, which was confirmed by the standard comigration. Among all testing substrates, MTA 2 and adenosine 4 were shown to generate adenine 3 via phosphorylation compared with the control assays without MBP-NucPNP (Fig. 2 and 3).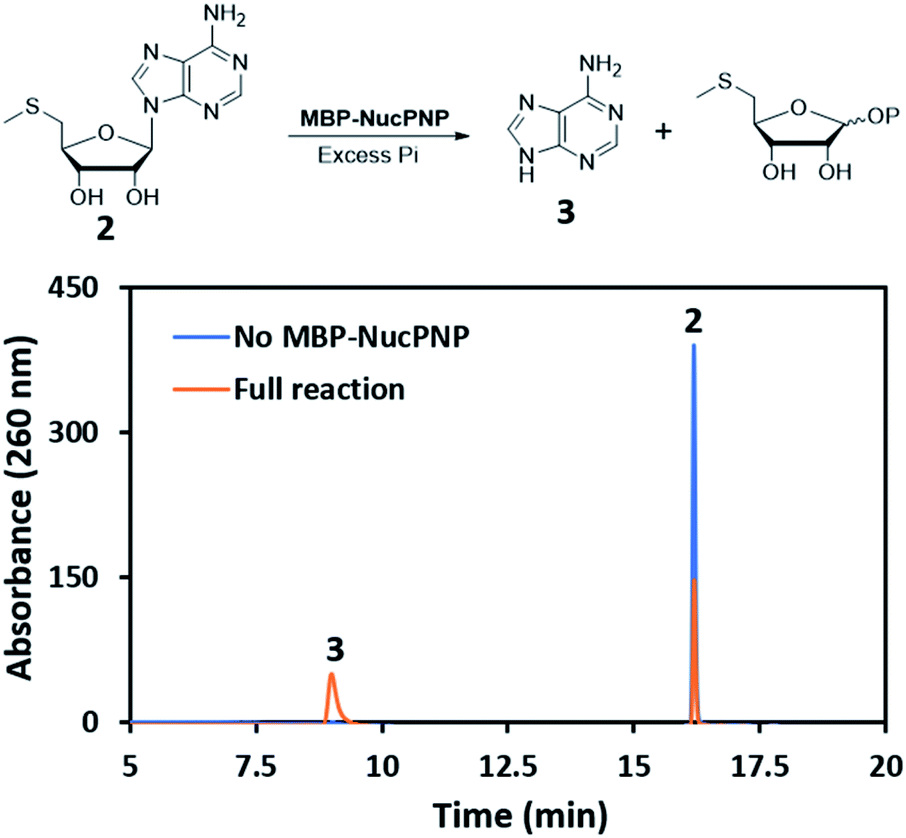 | ||
| Fig. 2 The HPLC analysis of MBP-NucPNP assays to convert MTA 2 to adenine 3. | ||
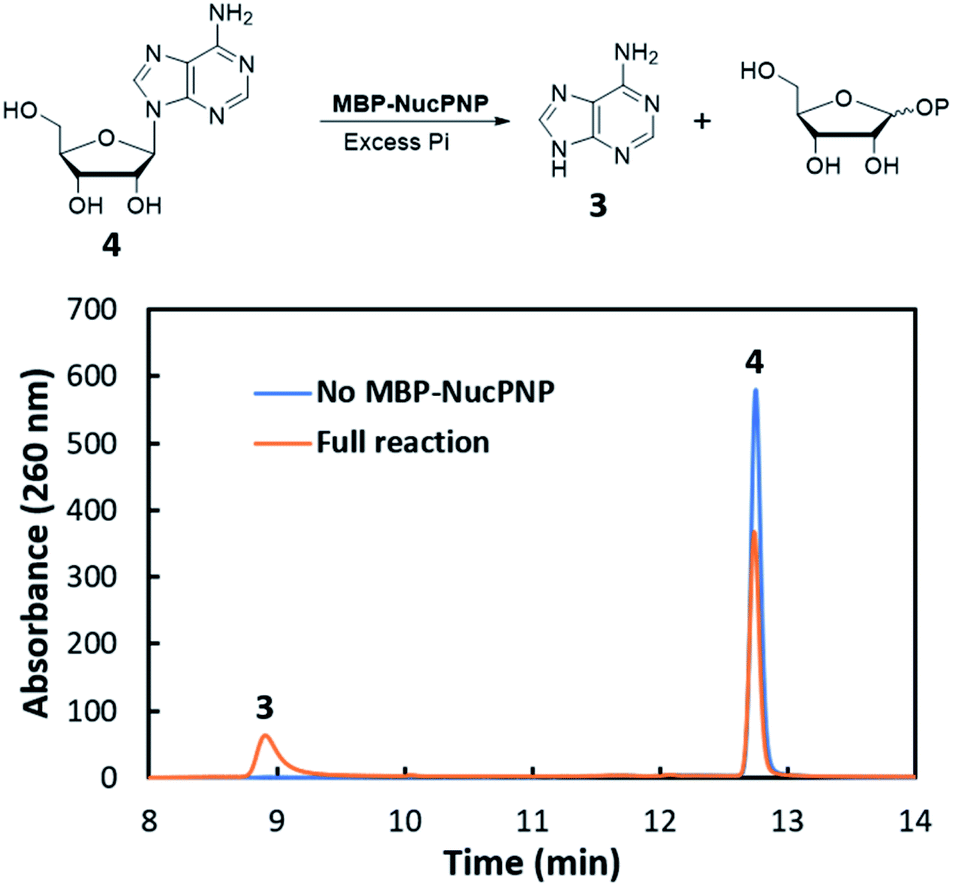 | ||
| Fig. 3 The HPLC analysis of MBP-NucPNP assays to convert adenosine 4 to adenine 3. | ||
Furthermore, the steady-state saturation kinetic constants of MBP-NucPNP for MTA 2 and adenosine 4 were determined by the formation rates of adenine 3 at different concentrations of MTA 2 or adenosine 4 in the presence of 10 mM of phosphate (Table 1). The apparent kcat of MTA 2 is about three-fold lower than adenosine 4 and the apparent Km of MTA 2 is half of adenosine 4. Moreover, the time course experiments were performed at 200 μM adenosine 4 or MTA 2 in the presence of excess phosphate to undergo the phosphorylation at the rate of Vmax. The results showed that the formation of adenine 3 from adenosine 4 was more than MTA 2 in the period of reaction (Fig. 4). Moreover, the amounts of adenine 3 formation from adenosine 4 were about 2.5 folds higher than MTA 2, which was similar to the ratio of their apparent kcat values.
| Substrate | kcat (s−1) | Km (μM) | kcat/Km (μM−1 s−1) |
|---|---|---|---|
| a The kcat, Km, and kcat/Km were determined at a fixed concentration of 10 mM phosphate with varied MTA or adenosine catalyzed by 1 μM of MBP-NucPNP. | |||
| MTA | 0.15 ± 0.01 | 43.5 ± 4.1 | 3.4 × 10−3 |
| Adenosine | 0.49 ± 0.02 | 90.3 ± 8.2 | 5.5 × 10−3 |
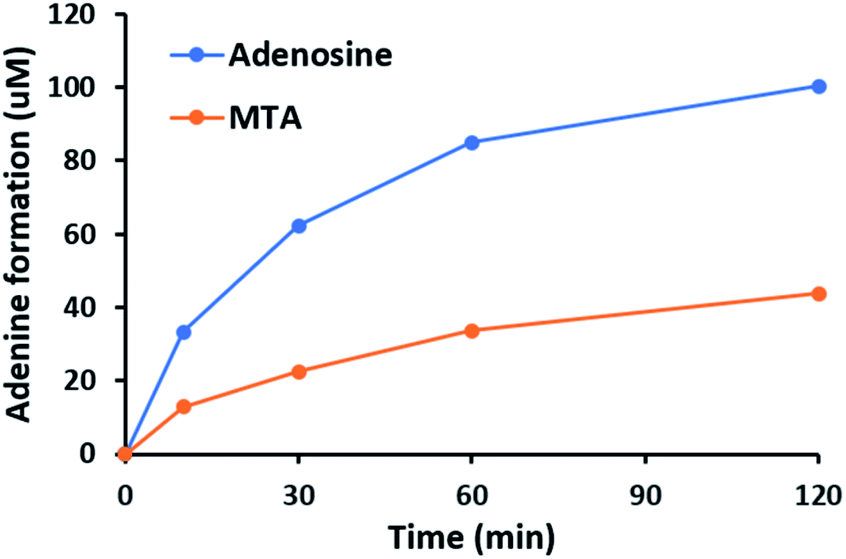 | ||
| Fig. 4 MBP-NucPNP catalyzed the phosphorylation of adenosine 4 to generate adenine 3 faster than MTA 2. | ||
In adenosine metabolism, MTA 2 or adenosine 4 could be metabolized to form adenine 3 by 5′-methylthioadenosine phosphorylase or adenosine phosphorylase, respectively.21 Similarly, adenine 3 could be utilized to biosynthesize nucleocidin 1 (Scheme 1). In natural product biosynthesis, a similar reaction was reported in aristeromycin biosynthesis.22 Its MacI and MacT enzymes were characterized as adenosine phosphorylases. Their functions were proposed to generate adenine and catalyze an irreversible reaction for the biosynthesis of neplanocin A and aristeromycin.
Moreover, to investigate the possible self-resistant function of NucPNP, sulfamoyladenosine, a nucleocidin analogue without fluorine at the C-4′ of ribose, was synthesized and assayed in the phosphorylation condition (Fig. S2†). We did not observe any conversion by HPLC, which suggested that NucPNP does not act on sulfamoyladenosine (and likely nucleocidin).
According to the in vitro characterization, the reported increase in the mRNA level upon complementation to allow nucleocidin synthesis, and the gene knockout experiments,14 NucPNP is proposed to be an essential enzyme to generate adenine for nucleocidin biosynthesis.
3.2 The NucV-catalyzed AMP formation is slower than those by ScAPRT and EcAPRT
Although NucPNP is proposed to have a function to generate adenine 3 from MTA 2 or adenosine 4, adenosine 4 is a precursor to biosynthesize sulfamoyladenosine. It was proposed as an intermediate in the biosynthesis of dealanylascamycin,19 a nucleocidin analogous. In addition, in the purine nucleoside metabolism, adenosine 4 could be synthesized from adenine 3 and PRPP 5 to form AMP 6 in an APRT-catalyzed reaction,23 followed by dephosphorylation. In the gene cluster of nucelocidin, nucV was annotated as an APRT. Therefore, we raised a question why NucPNP could phosphorylase adenosine 4 back to adenine 3. Moreover, the kinetic experiments of MBP-NucPNP also showed that MBP-NucPNP had 1.6-fold higher kcat/Km for adenosine 4 than MTA 2 (Table 1). It suggested that MBP-NucPNP slightly prefers adenosine 4 as the substrate. Therefore, we hypothesized that the possibility of fluorination occurs between the NucPNP and NucV reactions. In the fluorometabolite biosynthesis in S. cattleya, FlB was shown to selectively phosphorylase 5′-fluoro-5′-deoxyadenosine faster than adenosine.24,25 It made us hypothesize that 4′-F-PRPP 7 might be the substrate of NucV. In our scenario, NucV could convert 4′-F-PRPP 7 and adenine 3 to form 4′-F-AMP 8 faster than PRPP 5. Unfortunately, 4′-F-PRPP 7 is not available from any source.Therefore, we used a bioinformatic approach to evaluate our hypothesis. We searched the amino acid sequence of NucV against the genome of S. calvus by BLAST. The results showed that S. calvus has one NucV homolog (hereafter named as ScAPRT) with 79% similarity and 62% identity. Moreover, both amino acid sequences have the PRPP binding site motif.26 We further blasted the ScAPRT amino acid sequence against all Streptomyces species. ScAPRT is a housekeeping APRT for nucleoside biosynthesis. Hence, we compared the reaction rate of ScAPRT and NucV to gain more information. Soluble ScAPRT or NucV with a N-terminal His tag were expressed in E. coli BL21(DE3) and purified by affinity chromatography. Both enzymes were tested to catalyze the formation of AMP 6 from PRPP 5 and adenine 3 in the presence of MgCl2 by HPLC analysis.26,27 Since APRT enzymes were reported to catalyze reversible reactions,27 the backward reactions were tested by incubating AMP 6 and excess pyrophosphate (PPi) with each enzyme. No adenine 3 formation was observed in HPLC and 31P-NMR analysis (data not shown). It suggested that both enzymes favour the forward reaction to generate AMP 6. With the preliminary results of in vitro reconstitution, the saturation kinetic constants of NucV and ScAPRT were determined by the formation rates of AMP 6 at different concentrations of PRPP 5 or adenine 3 with the fixed concentration of excess adenine 3 or PRPP 5, respectively (Table 2).
| PRPPa | |||
|---|---|---|---|
| Enzyme | kcat (s−1) | Km (μM) | kcat/Km (μM−1 s−1) |
| a The constants were determined at the fixed concentration of 500 μM adenine with varied PRPP catalyzed by 1 μM enzyme.b The constants were determined at the fixed concentration of 500 μM PRPP with varied adenine catalyzed by 1 μM enzyme. | |||
| NucV | 1.34 ± 0.07 | 21.4 ± 3.0 | 0.062 |
| ScAPRT | 9.72 ± 0.47 | 9.8 ± 2.0 | 0.987 |
| Adenineb | |||
|---|---|---|---|
| Enzyme | kcat (s−1) | Km (μM) | kcat/Km (μM−1 s−1) |
| NucV | 2.09 ± 0.09 | 6.8 ± 1.4 | 0.309 |
| ScAPRT | 16.59 ± 0.47 | 2.8 ± 0.6 | 5.863 |
In the Table 2, the apparent kcat(PRPP) and kcat(adenine) of NucV are both much lower than those of ScAPRT. The apparent Km(PRPP) and Km(adenine) of NucV are both higher than those of ScAPRT. These results suggested that NucV is far less efficient in AMP synthesis compared to ScAPRT. Then, we conducted time course experiments to monitor the rate of AMP formation by both enzymes in the Vmax condition by HPLC analysis. In the designed condition, ScAPRT completed the reaction in 5 min, while NucV took about 1 hour (Fig. 5A). To confirm these results, 31P-NMR was utilized to monitor the formation of PPi (δ = −4.75 ppm) and the disappearance of PRPP 5 (δ = 4.09, −5.17, and −10.95 ppm) in the assays (Fig. 5B and C). The NMR analysis showed the same results. In order to confirm that the rapid AMP synthesis is a characteristic of APRT, E. coli APRT was also analyzed in the same condition, which is its Vmax condition according to its kinetic constants (Table S2†). The amino acid sequence of EcAPRT has 70% similarity and 51% identity with ScAPRT. In the 31P-NMR analysis, EcAPRT also completed the reaction in 5 min (Fig. S7†). All experiments suggested that NucV catalyzed slower AMP formation than ScAPRT.
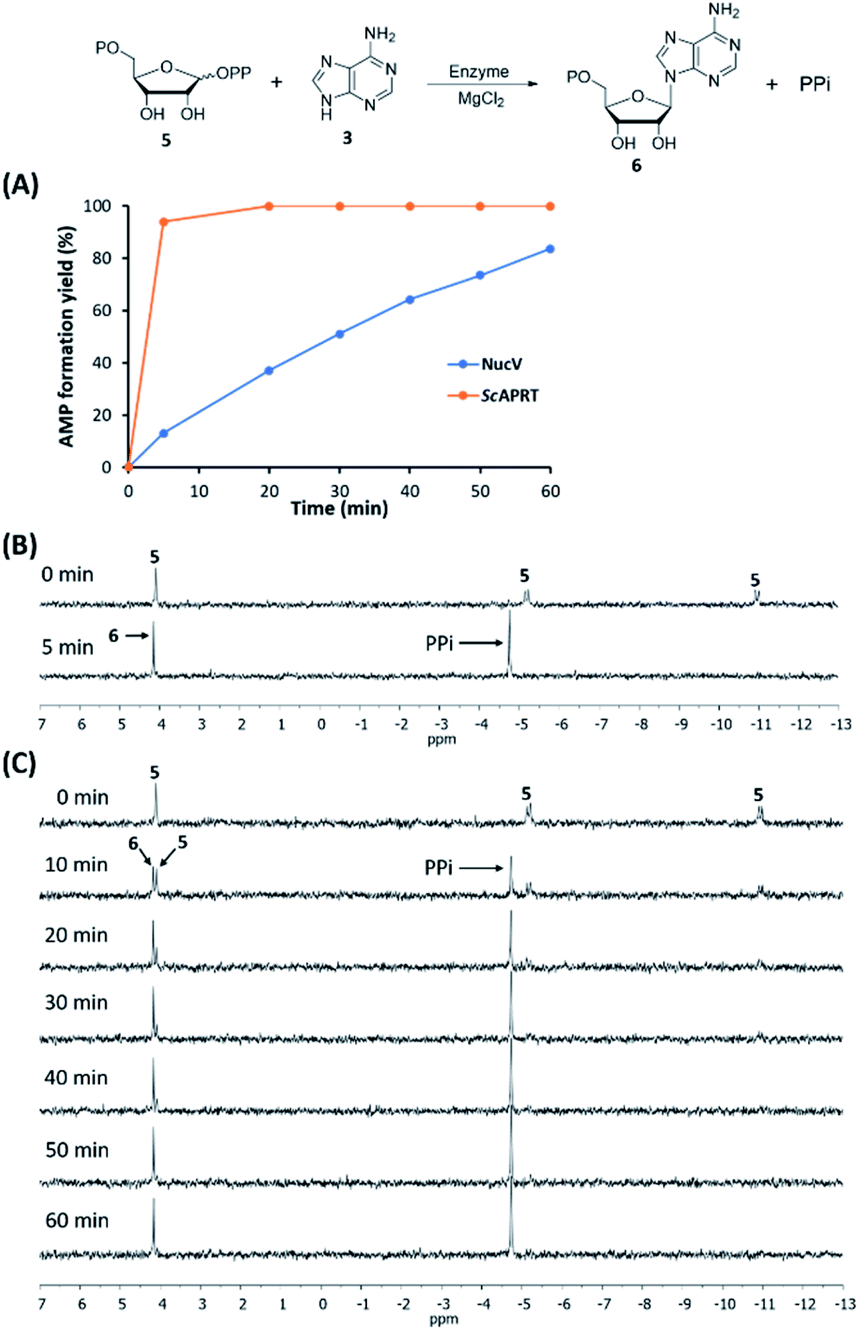 | ||
| Fig. 5 NucV catalyzed the reaction slower than ScAPRT under the same condition. The relative rate of AMP 6 formation was analyzed by HPLC (A). PPi formation and PRPP 5 consumption by ScAPRT (B) and NucV (C) were monitored by 31P-NMR. | ||
Although we can not show a direct evidence that NucV could catalyze 4′-F-PRPP 7 faster than PRPP 5, our indirect experiments showing that NucV acts relatively slowly on PRPP 5 support our hypothesis (Scheme 1). Additionally, the mRNA level of nucV transcript was reported to increase by 110 times with bldA complementation to rescue nucleocidin synthesis, suggesting the importance of NucV for nucleocidin production.
4. Conclusions and perspective
In this research, soluble MBP-NucPNP and NucV were successfully expressed and purified. The in vitro characterization suggested that NucPNP acts on MTA 2 or adenosine 4 to generate adenine 3 in presence of phosphate. Furthermore, NucV catalyzes slower AMP 6 formation from PRPP 5 and adenine 3 compared with ScAPRT, suggesting PRPP may not be its natural substrate.Taking all information in account, the proposed nucleocidin biosynthetic pathway is shown in Scheme 1. NucPNP catalyzes adenine 3 formation from MTA 2. NucV could catalyze 4′-F-AMP 8 from 3 and 4′-F-PRPP 7. However, NucV also catalyzes AMP 6 formation. After dephosphorylation, adenosine 4 could be recycled to form adenine 3.
For nucleocidin biosynthesis, C–F formation is the most attractive step to investigate, but is still mysterious. NucJ has been proposed as a fluorinase candidate.18 We would suggest to test PRPP 5 for in vitro reconstitution of NucJ or any candidate proteins. We hope that our results and hypothesis can help researchers to solve the enigma of C–F formation in nucleocidin biosynthesis.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research is supported by the Thailand Research Fund (TRF) and Office of the Higher Education Commission (OHEC) Grant (No. MRG6280142), and by National Natural Science Foundation of China (21822703).References
- J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. Del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432–2506 CrossRef CAS.
- S. M. Ametamey, M. Honer and P. A. Schubiger, Chem. Rev., 2008, 108, 1501–1516 CrossRef CAS.
- R. Berger, G. Resnati, P. Metrangolo, E. Weber and J. Hulliger, Chem. Soc. Rev., 2011, 40, 3496–3508 RSC.
- T. Fujiwara and D. O'Hagan, J. Fluorine Chem., 2014, 167, 16–29 CrossRef CAS.
- D. O'Hagan and H. Deng, Chem. Rev., 2015, 115, 634–649 CrossRef.
- D. O'Hagan, C. Schaffrath, S. L. Cobb, J. T. G. Hamilton and C. D. Murphy, Nature, 2002, 416, 279 CrossRef.
- C. Schaffrath, H. Deng and D. O'Hagan, FEBS Lett., 2003, 547, 111–114 CrossRef CAS.
- H. Deng, L. Ma, N. Bandaranayaka, Z. Qin, G. Mann, K. Kyeremeh, Y. Yu, T. Shepherd, J. H. Naismith and D. O'Hagan, ChemBioChem, 2014, 15, 364–368 CrossRef CAS.
- Y. Wang, Z. Deng and X. Qu, F1000Research, 2014, 3, 61 Search PubMed.
- S. Huang, L. Ma, M. H. Tong, Y. Yu, D. O'Hagan and H. Deng, Org. Biomol. Chem., 2014, 12, 4828–4831 RSC.
- S. O. Thomas, V. L. Singleton, J. A. Lowery, R. W. Sharpe, L. M. Pruess, J. N. Porter, J. H. Mowat and N. Bohonos, Antibiot. Annu., 1956, 716–721 Search PubMed.
- G. O. Morton, J. E. Lancaster, G. E. Van Lear, W. Fulmor and W. E. Meyer, J. Am. Chem. Soc., 1969, 91, 1535–1537 CrossRef CAS.
- L. Kalan, A. Gessner, M. N. Thaker, N. Waglechner, X. Zhu, A. Szawiola, A. Bechthold, G. D. Wright and D. L. Zechel, Chem. Biol., 2013, 20, 1214–1224 CrossRef CAS.
- X. M. Zhu, S. Hackl, M. N. Thaker, L. Kalan, C. Weber, D. S. Urgast, E. M. Krupp, A. Brewer, S. Vanner, A. Szawiola, G. Yim, J. Feldmann, A. Bechthold, G. D. Wright and D. L. Zechel, ChemBioChem, 2015, 16, 2498–2506 CrossRef CAS.
- S. Zhang, D. Klementz, J. Zhu, R. Makitrynskyy, A. R. Ola Pasternak, S. Günther, D. L. Zechel and A. Bechthold, J. Biotechnol., 2019, 292, 23–31 CrossRef CAS.
- A. Bartholomé, J. E. Janso, U. Reilly and D. O'Hagan, Org. Biomol. Chem., 2017, 15, 61–64 RSC.
- X. Feng, N. Al Maharik, A. Bartholomé, J. E. Janso, U. Reilly and D. O'Hagan, Org. Biomol. Chem., 2017, 15, 8006–8008 RSC.
- X. Feng, D. Bello, P. T. Lowe, J. Clark and D. O’hagan, Chem. Sci., 2019, 10, 9501–9505 RSC.
- C. Zhao, J. Qi, W. Tao, L. He, W. Xu, J. Chan and Z. Deng, PLoS One, 2014, 9, e114722 CrossRef.
- D. G. Gibson, L. Young, R. Y. Chuang, J. C. Venter, C. A. Hutchison and H. O. Smith, Nat. Methods, 2009, 6, 343–345 CrossRef CAS.
- K. Buckoreelall, L. Wilson and W. B. Parker, J. Bacteriol., 2011, 193, 5668–5674 CrossRef CAS.
- G. Xu, L. Kong, R. Gong, L. Xu, Y. Gao, M. Jiang, Y.-S. Cai, K. Hong, Y. Hu, P. Liu, Z. Deng, N. P. J. Price and W. Chen, Appl. Environ. Microbiol., 2018, 84, e01860 CAS.
- B. Hove-Jensen, K. R. Andersen, M. Kilstrup, J. Martinussen, R. L. Switzer and M. Willemoës, Microbiol. Mol. Biol. Rev., 2017, 81, e00040 CrossRef CAS.
- S. L. Cobb, H. Deng, J. T. G. Hamilton, R. P. McGlinchey and D. O'Hagan, Chem. Commun., 2004, 592–593 RSC.
- F. Huang, S. F. Haydock, D. Spiteller, T. Mironenko, T. L. Li, D. O'Hagan, P. F. Leadlay and J. B. Spencer, Chem. Biol., 2006, 13, 475–484 CrossRef CAS.
- J. Del Arco, M. Martinez, M. Donday, V. J. Clemente-Suarez and J. Fernández-Lucas, Biocatal. Biotransform., 2018, 36, 216–223 CrossRef CAS.
- C. Bashor, J. M. Denu, R. G. Brennan and B. Ullman, Biochemistry, 2002, 41, 4020–4031 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra10878b |
| ‡ These authors contributed equally to this article. |
| This journal is © The Royal Society of Chemistry 2021 |