DOI:
10.1039/D0RA10829D
(Paper)
RSC Adv., 2021,
11, 13348-13358
Enhanced photocatalytic properties of a chemically modified blue phosphorene†
Received
25th December 2020
, Accepted 27th March 2021
First published on 9th April 2021
Abstract
It is high time to placate the peak demand for an efficient, economic and green fuel in the form of H2 through photocatalytic water splitting. Several low dimensional materials have been explored for their photocatalytic properties on account of their surface to volume ratio. The present study illustrates the excellent photocatalytic potential of a two-dimensional material, viz. a chemically tempered blue-phosphorene sheet, with single atom thickness and high carrier mobility. Metal-free element, sulphur, is explored as a dopant in a 32-atom blue-phosphorene sheet. The dopant is inserted at three locations viz. central, edge and central edge positions with varying concentrations from 3.125% to 18.75% (corresponding to n = 1 to 6 sulphur atoms within a 32-atom blue-phosphorene sheet, P32−nSn). The cohesive energy studies predict the higher stability of even number S doped sheets as compared to their odd counterparts. Photocatalytic activity is studied in terms of band gap and band alignment for different concentrations of the former. Studies reveal that edge doping demonstrates better water molecule activation independent of S atom concentration. The edge doped systems not only provide the chemical activity to activate water, but also show feasible HER overpotentials of 1.24–1.29 eV at neutral medium. Finally, this work opens up a driving lead of non-corrosive catalysts for water molecule splitting.
Introduction
Low dimensional materials have been a buzzing topic of research in the last two decades due to their characteristic physico-chemical properties. Various quantum phenomena such as phonon–electron interactions, spin interaction, quantum tunnelling effect and quantum confinement within these materials are responsible for their enhanced and yet tuneable electronic, thermal, magnetic and optical properties with far reaching applications in novel devices such as spintronics, photonics, quantum computing and catalytic materials. However, the most widely applied area of these materials is catalysis on account of their higher surface to volume ratio and higher electron transferability making it cost effective.1–6 Among the various low dimensional nano-structures such as quantum dots/nanoparticles (zero dimensional), 1D nanotubes/nanorods and 2D nanofilms/nanosheets, 2D based materials have the upper hand with respect to other dimensional substances as catalysts due to the aforesaid reasons. (a) Firstly, each site on their surface is equally accessible for catalytic process.7 (b) Secondly, these materials offer higher thermal and mechanical stability and thus, operational ability at various temperatures.8 (c) Due to their promising thermal conductivity and large surface space, they also act as efficient heat sinks during exothermic reaction.9 (d) 2D sheet being atomic thick, reduces charge carrier distance from the core to top, thereby reducing electron–hole recombination and enhancing the redox process marginally.10 (e) They offer wide range of options for feasible chemical engagement, thereby enabling charge storage.11 (f) Lastly, quantum confinement and unsaturated surfaces leads to facile movement of electrons along in-plane direction, thus increasing the lifetime of charge carrier.12,13
The journey of 2D materials based catalysts started with graphene, a single atom thick carbon sheet held by sp2 hybrid bonds arranged in hexagonal manner.14 Employment of graphene-based materials, to catalysis quickly has captured the interests of researchers in the last few decades.14,15 However, due to its intrinsic zero band gap and consequently its instantaneous electron–hole recombination, its application in photocatalysis remains challenging.16,17 Although, its oxygenated counterpart is seen to have finite band gap of 2.4 eV to 4.3 eV,18–20 it fails to be within the acceptable slot of 1.6–2.2 eV so as to be activated efficaciously under natural solar light irradiation.21 Several other 2D materials such as silicene,22 germanene,23 borophene,24,25 stanene26,27 and monochalcogenides28 have been explored for their potential application as photocatalysts.28 However, absence of a tunable band gap within the acceptable slot has limited their practical applications.29,30
In this context, phosphorene, a mono-layer/multi-layer sheet of black phosphorous,31 with its tunable band gap ranging between 0.3–2.5 eV has been found to be more attractive as a photocatalyst as compared to other 2D materials.32–35 Following the fact finding journey, many other allotropes/polytypes of phosphorene viz., red, black, violet, green phosphorene have been realized36–39 and some of them have been explored for water splitting application.40–43 More recently, a new member of phosphorene polytype was realized through the synthesis of mono-layered honeycomb buckled blue-phosphorene (B-phosphorene) on an Au (111) sheet using a molecular beam epitaxial method with black-phosphorous as precursor.44 It has been reported that a specific dislocation in black phosphorene induces a new structure with zigzag puckering, called B-phosphorene with a characteristic tunable bandgap upto 2 eV.44 This B-phosphorene is found to be more stable thermodynamically as compared to black-phosphorene,45,46 which is of great importance for an efficient photocatalyst.47–49 These properties inspired the researchers to further examine the photocatalytic properties of nanotube B-phosphorene,50 doped51 and several hetero-structures of B-phosphorene.52–54 Research works also focused on engineering the band gap of B-phosphorene by application of external electric field,55 using stacking effect,56,57 functionalization58–60 and doping.61–63 Doping is the most attractive mode among all the methods for modulating physico-chemical properties64–75 of phosphorenes (or for any given material).76,77 From the experimental point of view, single atomic layered 2D structures can be doped by traditional substitutional doping methods such as implantation,78 diffusion processes79–81 or new techniques such as surface charge transfer method,82 intercalation,83,84 and field effect modulation85 methods.
Non-metal doped B-phosphorene are synthesized and reported in several experimental studies.86,87 Their physicochemical as well as electronic properties are also explored theoretically.88–91 Further, their corresponding application as catalytic materials is also studied by some groups.92–95 In the context of application as photocatalytic material, ample experimental studies of doped B-phosphorene has been reported where N is employed as dopant.96–98 The photocatalytic properties of N, Si, S, doped B-phosphorene have also been studied theoretically.99,100 Moreover, sulphur has also been earlier reported to be a successful catalyst in the context of sensors and opto-elecronics.39,99 Further, S having the same atomic size to that of P atom, plays a vital role in improving its stability.101 The anionic substitutional doping of S results in puckered structure of phosphorene that facilitates anion atoms to get exposed to the outer surface as compared to planar 2D materials like graphene.102 From above studies and available literature reports it is clear that there are very few studies on the photocatalytic activity of sulphur doped B-phosphorene. More importantly, the influence of varying concentration of S on the stability and catalytic activity of B-phosphorene is not known in terms of water molecule activation.103,104 In the present work, using density functional theory, we evaluate the role of S doping, its concentration and its position in modulating the catalytic activity of B-phosphorene in water molecule activation.
With reference to the above we vary the concentration of S from 3.125% to 18.75% in 32 B-phosphorene-sheet. The S atoms are doped at three different ways; (a) all S atoms are placed in the central position (see Fig. 1(a) for elucidation) (b) all S atoms are spread across both edge and central & edge position as shown in Fig. 1(a). Stability of thus doped systems is evaluated in terms of cohesive energies, band gap and band edges. pH is taken into account to ensure that the doped structures qualify to be photocatalyst practically. Finally, the models that qualify the photocatalytic properties are adsorbed with water molecule. Activation of water molecule is evaluated in terms of adsorption energy of water molecule, extent of O–H bond elongation from the optimal O–H bond length in water and the red shift in O–H stretching frequency. The thermostability of the sulphur-doped blue phosphorene systems has also been evaluated through ab initio molecular dynamics (AIMD) simulations at 500 K.
 |
| | Fig. 1 (a) Central (C) and edge (E) atoms in B-phosphorene sheet. (b) Relaxed structure of B-phosphorene from top and side views (a and b are lattice parameters) Wigner–Seitz cells are shown by the shaded area. | |
Computational details
The phosphorene sheets, pristine and doped, considered in this study are modelled using GaussView06 Software.105 Density Functional Theory (DFT) calculations are carried out on these sheets using SIESTA106 (Spanish Initiative for Electronics Simulations with Thousands of Atoms) which uses flexible linear combination of atomic orbital (LCAO) as a basis set. Norm-conserving pseudopotentials are taken up to describe the electron–ion interaction, which was yielded by implementing the Troullier–Martins scheme.107 A Generalized Gradient Approximation-Perdew, Burke, Ernzerhof (GGA-PBE) functional is used for the calculation of correlation and exchange energies108 along with a double-zeta plus (DZP) polarization basis set for all elements. Spin polarized optimization of all the structures are carried out using first-order minimization method, which is known as Conjugated Gradient (CG). All structures are relaxed until the force on each atom is less than 0.01 eV Å−1. In the Self-Consistent Field (SCF) process, the Brillouin Zone (BZ) of the reciprocal lattice is sampled through a 6 × 6 × 1 grid mesh, generating 3 special k-points in the Monkhorst–Pack scheme. For the computation of integrals and the representation of charge densities and potentials, mesh cut-off energy is kept at 300 Ry so as to depict the periodic plane waves in the grid without aliasing. To guarantee better contribution from dopant atoms and save computational time, we have considered a square-size supercell with 32 atoms. In order to prevent interaction between the neighbouring layers, a vacuum space of 20 Å thickness is maintained in the perpendicular direction of the monolayer. Upon optimization, water molecule is introduced and the adsorption energy of water molecule on these structures are computed. To measure the extent of water activation on the phosphorene sheets, the O–H stretching frequency of the adsorbed water molecule is measured using ‘vibra’, a post-processing tool of SIESTA. The AIMD simulations at 500 K, a Nosé–Hoover thermostat has been implemented and the nS-B-phosphorene systems are described with NVT (number, volume, temperature) ensemble.
Results and discussion
Structure, stability and electronic properties of nS-B-phosphorene sheets (n = 0 to 6)
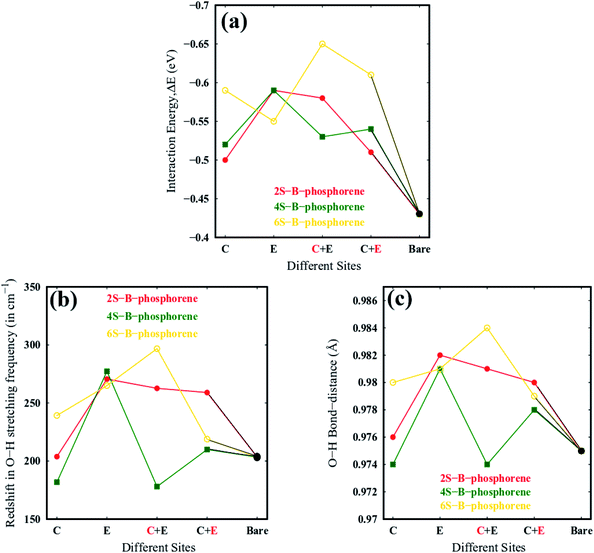
B-phosphorene has a hexagonal primitive unit cell with two atoms per cell109 as shown Fig. 1(b). We obtain an optimized lattice constant (a = b) of 3.27 Å and P–P bond length of 2.28 Å and P–P–P bond angle of 91.6°, which are in satisfactory agreement with previously reported calculations.110 The buckling length (h) of monolayer is found to be 1.24 Å, which is in good accordance with previous report.111 The atoms in the sheet have been further classified as the central (C) and edge (E) atoms as discussed earlier are shown in Fig. 1(a). Accordingly, there are 10 central atoms and 22 edge atoms. In order to initially screen the influence of sulphur concentration on the stability and electronic effects of B-phosphorene, we have replaced phosphorous atoms with sulphur atoms only at the central (C) positions. Concentration of sulphur was varied in this sheet from 3.125% which translates to 1S atom in the sheet. As the concentration of S atom was increased beyond 18.75% the structure was seen to collapse. So, further structural studies are restricted to 18.75% which translates to doping of 1–6 P atoms with S in above sheet. Thus, there are 10 distinct conformations generated for single S atom doped B-phosphorene and are referred to as 1S-B-phosphorene conformations. Each of this conformation was optimized. The six lowest energy 1S-B-phosphorene conformations with their relative energies are shown in ESI Fig. S.F.1.† Next, the number of dopant atoms (S atoms) is increased to 2. The first S atom was placed at the 10 distinct C positions, followed by 9 distinct C positions for the second atom. Thus, totally 10 × 9 distinct conformations were generated. The six lowest energy 2S-B-phosphorene conformations are given in ESI Fig. S.F.2.† Similarly, the number of conformations generated for 3S doped B-phosphorene sheet are 10 × 9 × 8 and so on. The six lowest energy conformation for 3S, 4S, 5S and 6S doped B-phosphorene are shown in ESI Fig. S.F.3–S.F.6,† respectively. In case of doped structures with more than 1S atom, it was noted that defects are formed around the S atoms in the sheet, on account of its lower valency, higher electronegativity and smaller atomic radius as compared to that of P (see ESI Fig. S.F.1–S.F.6,† respectively). This defect formation was also noted in the previous work where blue phosphorene nanoribbons were doped with S atoms.112 These defects are anticipated to play a vital role in enhancing photocatalysis by averting the recombination of photogenerated charges.113
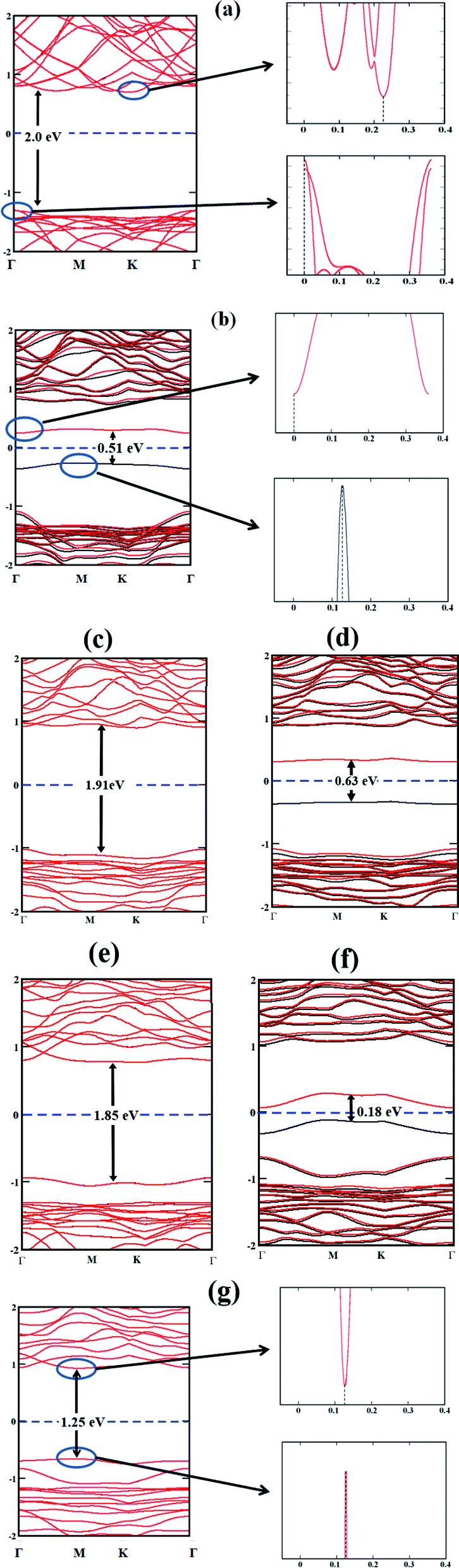
In order to evaluate the potential of a material as a photocatalyst, it is instructive to probe its band structure. Therefore, we analyse the electronic properties of the lowest energy conformation for each nS-B-phosphorene (n = 0–6). Fig. 2 shows the spin resolved band structure for B-phosphorene and nS-B-phosphorene sheets (n = 0, 1, 2, 3, 4, 5, 6). Spin-up (black-lines) and spin-down (red-lines) energy levels are highlighted and follow the representation adopted by Zhu et al.114 It is noted that the first Brillouin zone of hexagonal B-phosphorene is projected between the conduction band minimum and valence band maximum through the high symmetric path Γ–M–K–Γ (see Fig. 2(a)). Our calculations further reveal an indirect band gap of 2 eV for the B-phosphorene monolayer, which is also substantiated by other reported data.115,116 Coming to the discussion on the type of band gap pertinent in the context of visible light driven catalysis, the effective participation of the photogenerated electrons and holes is determined by recombination rate of these pairs. On recombination there is liberation of energy in the form of photon with the excited electrons needing to fulfil the following transition selection rules of momentum conservation117
| | |
ℏkev − ℏkec = ±ℏqphonon
| (2) |
where ℏ is the reduced Planck constant,
kev and
kec are the electron wave vectors at the VBM and CBM, respectively,
qphonon is the wave vector of the assisted phonon,
Eg is the band gap of semiconductor, and
ω photon is angular frequency of the emitted photon. It is seen in case of pristine B-phosphorene, that
kev ≠
kec with an indirect band gap characteristic of semiconductors. Thus, the excited electrons cannot recombine directly and easily with holes, resulting in an increase of the photoexcited electron–hole lifetime, increase in diffusion length and time. Thus, B-phosphorene has better photocatalytic activity than direct band gap semiconductors as reported in a previous study.
118 Analysis of the band structures of the lowest energy
nS-B-phosphorene (
n = 1–6) reveals a dissimilarity in valence orbital occupancy of dopant S with respect to phosphorous (3s
23p
4 as compared 3s
23p
3), bringing in distinct changes in the electronic structure of doped sheets with respect to their pristine counterpart.
nS-B-phosphorene (
n = 1, 2, 3, 4, 5) sheets demonstrate n-type semiconducting characteristics with Conduction Band Minimum (CBM) lying nearer to Fermi level as compared to Valence Band Maximum (VBM). On the other hand, 6S-B-phosphorene is found to have a direct band gap as seen from
Fig. 2(g). The band gap results for pristine and doped B-phosphorene are quantified in
Table 1, from which clearly, we can note that while the pristine and even number S doped (
n = 2, 4, 6) systems have a gap which is higher than the electrolytic potential of water
viz., 1.23 eV, the odd number S doped systems (
n = 1, 3, 5) fails to meet this essential criterion. The even
nS-doped systems with band gaps above 1.23 eV are potential photocatalyst for water splitting and will be explored further. The odd
nS-doped B-phosphorene systems possess a smaller band gap and would inherently show higher carrier mobility and electrical conductivity. However, the adsorption efficiency of H
+ (dissociated from water) decreases eventually leading to lower solar energy to hydrogen evolution efficiency, thereby reducing the photocatalytic efficiency for HER.
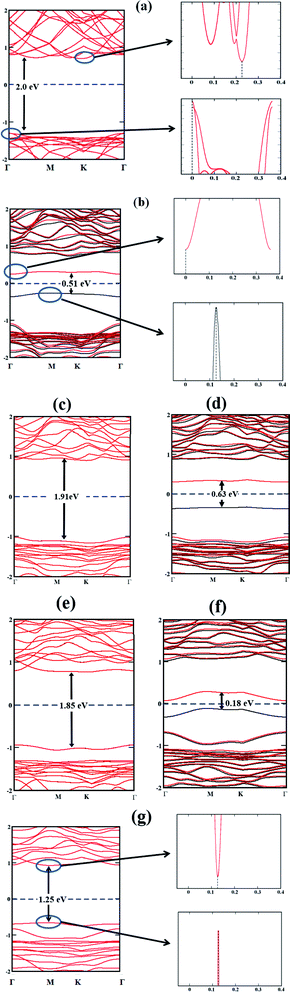 |
| | Fig. 2 Band structures of (a) B-phosphorene; (b) 1S-B-phosphorene (c) 2S-B-phosphorene (d) 3S-B-phosphorene (e) 4S-B-phosphorene (f) 5S-B-phosphorene (g) 6S-B-phosphorene with their respective band gaps indicated with vertical lines. The position of the band edges is indicated by the sharp band lines as shown for pristine B-phosphorene, 1S-B-phosphorene and 6S-B-phosphorene. | |
Table 1 Band gap energy of pristine and various nS-B-phosphorene
| System |
Band gap energy (eV) |
| Pristine phosphorene |
2.00 |
| 1S-B-phosphorene |
0.51 |
| 2S-B-phosphorene |
1.91 |
| 3S-B-phosphorene |
0.63 |
| 4S-B-phosphorene |
1.85 |
| 5S-B-phosphorene |
0.18 |
| 6S-B-phosphorene |
1.25 |
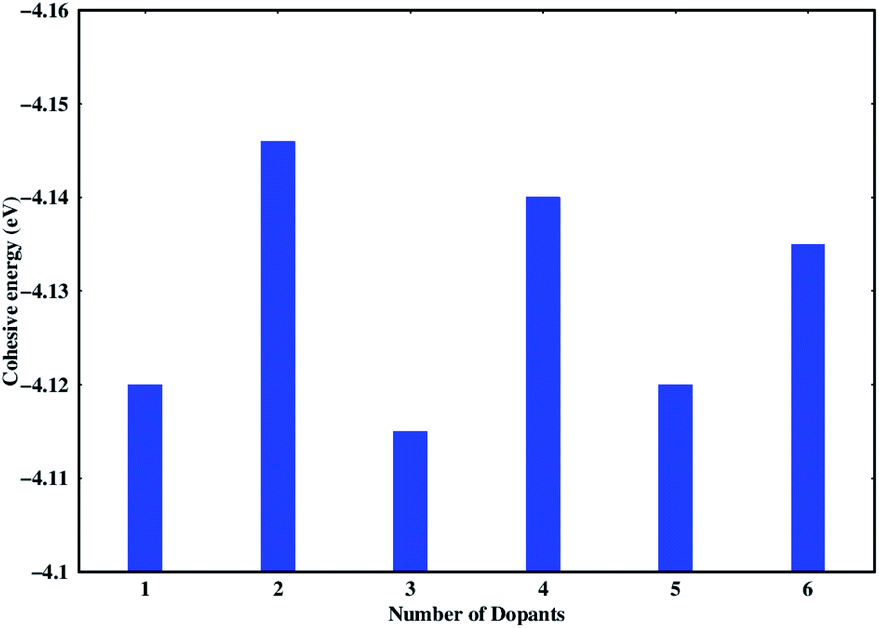
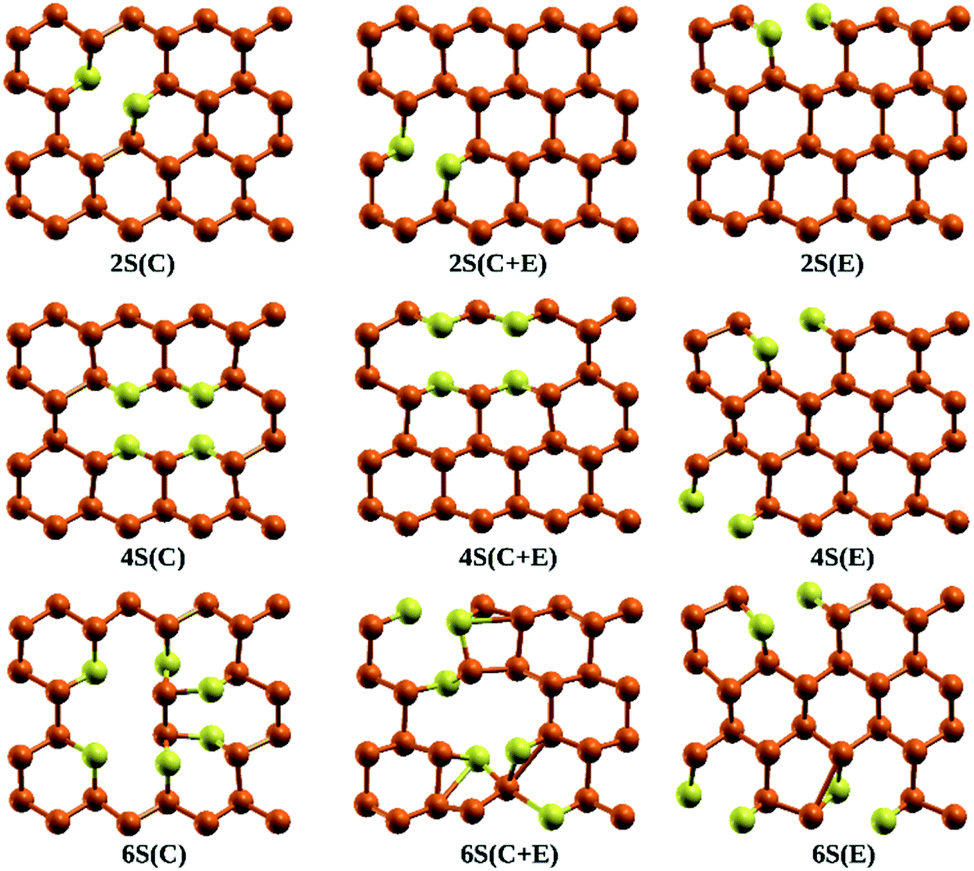
Stability of the generated catalytic systems are studied in terms of their cohesive energies and it is calculated from the total electronic energies.119–121 The cohesive energies of different systems are shown in Fig. 3. Evaluation of cohesive energies suggest that even doped nS-B-phosphorene systems have higher cohesive energies with nearly difference of 0.02 eV than their odd counterparts. This results in better thermal stability of nS-B-phosphorene (n = 2, 4, 6) sheets indicating that a particular concentration of dopants is more favourable (or number of dopant) viz. 6.25%, 12.5% and 18.75%. Based on the cohesive energy results and band gap energy results, we have further restricted our studies to the even doped nS-B-phosphorene (n = 2, 4, 6) sheets. In these dopant concentrations, the most favourable dopant substitution sites are more rigorously explored taking central (C), edge (E) and central & edge (C + E) sites into account. The three sites of S-atom occupation are described as follows; (i) all S atoms occupy central sites of Fig. 1(a) as elaborated in Fig. 4 and this is referred to as nS-B-phosphorene (C). (ii) The S atoms are distributed among various edge and central sites of Fig. 1(a), these conformations are referred to as nS-B-phosphorene (C + E) and representative structure of this is shown in Fig. 4. (iii) All S atoms occupy edge sites of Fig. 1(a) as shown in Fig. 4, and is referred to as nS-B-phosphorene (E).
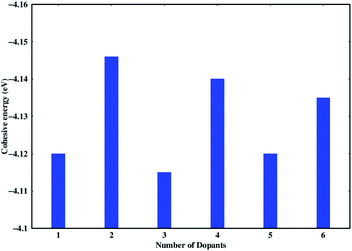 |
| | Fig. 3 Cohesive energies of various studied nS-B-phosphorene systems. | |
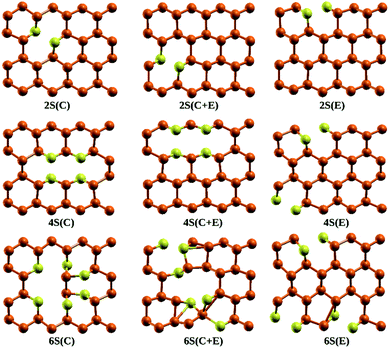 |
| | Fig. 4 Lowest energy conformations of S atom occupation in 2S-B-phosphorene, 4S-B-phosphorene, 6S-B-phosphorene, respectively. | |
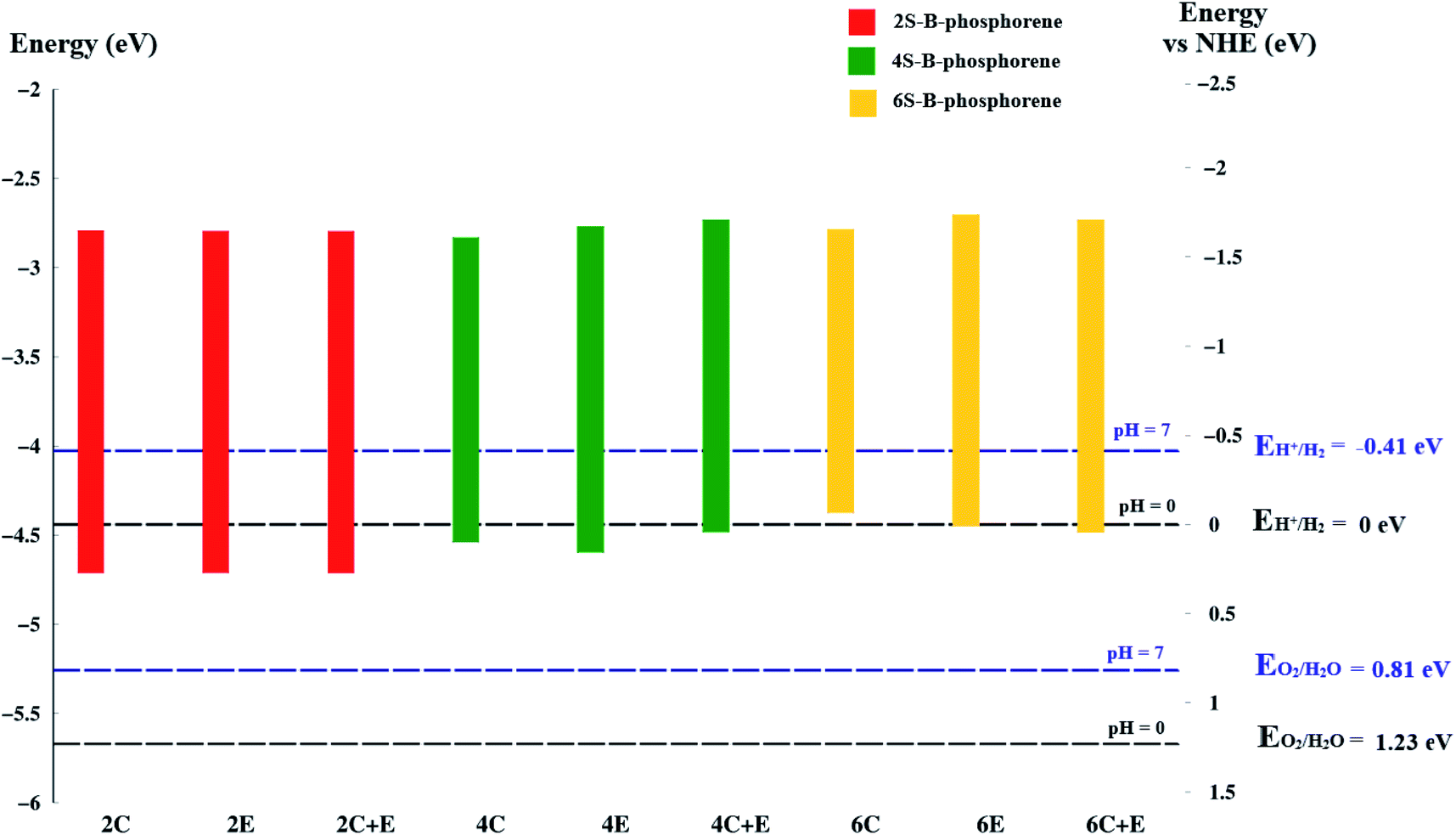
As already discussed, for a material to be an efficient photocatalyst for water molecule activation, band gap must be at least 1.23 eV. Besides, the Conduction Band Minimum (CBM) should be more negative than the redox potential of H+/H2 (0 V vs. NHE) and the Valence Band Maximum (VBM) should be more positive than the redox potential of O2/H2O.120 A material will act as a catalyst for OER when its VBM is more positive than the redox potential of O2/H2O whereas, it will act as a catalyst for HER when its CBM is more negative than the redox potential of H+/H2. The water redox potential energies are also reported to be dependent on the pH of the medium.120 To analyse the potential of the studied materials we first look at the redox potential of water molecule at 0 and 7 pH values using the below standard equation.122–124
| | |
EO2/H2O = −5.67 eV + pH × 0.059 eV
| (3) |
| | |
EH+/H2 = −4.44 eV + pH × 0.059 eV
| (4) |
It is known that the reduction potential of H+/H2 is 0.00 V at pH 0 vs. NHE, however −0.41 V over-potential is required for the evolution of hydrogen in aqueous solutions at pH 7 vs. NHE. Similarly, potential for water oxidation O2/H2O is at +1.23 V vs. NHE at pH 0 (or +0.82 V vs. NHE at pH 7).125 This, reflects as to why a band gap of 1.23 eV is required for a material to drive overall water splitting reaction. To calculate the band edge position at pH = 7 of the medium, the values of CBM and VBM are computed from the data obtained. The Conduction Band (CB) potential of a semiconducting material in aqueous solution usually exhibits a pH dependence explained by ECB = E0CB (pH 0) − 0.059 × pH; where E0CB is the standard reduction potential.126,127 Further, the location of the VBM should be more positive than +0.82 V while the position of the CBM should be more negative than −0.41 V vs. NHE at pH 7, respectively.
Coming to the present work, we plot the band edge locations for the lowest energy nS-B-phosphorene (n = 2, 4, 6) where dopants positions are nS-B-phosphorene (C), nS-B-phosphorene (E) and nS-B-phosphorene (C + E), as shown in Fig. 5 with the redox potentials of hydrogen evolution (H+/H2) and oxygen evolution (O2/H2O) at pH 0 and 7. The band width of all the systems surpasses the required band gap of 1.23 eV and are favourable for photocatalytic water splitting at acidic (pH = 0) as well as neutral medium (pH = 7) without any application of an external potential. The redox potential of oxygen evolution (O2/H2O) is found to be more positive than the VBM of all nS-B-phosphorene systems, see Fig. 5. Thus, these systems will fail to catalyse the oxygen evolution reaction (OER). However, the CBM is seen to be more negative than the redox potential of hydrogen evolution (H+/H2) at both pH values; thereby all nS-B-phosphorene systems can behave as efficient catalysts for HER. An acidic medium can lead to etching or degradation of the material; therefore, a neutral medium is always preferred for carrying out experiments. At neutral medium, i.e., pH = 7, all 2S-B-phosphorene structures show an overpotential of 1.24 eV for HER in all three sites (i.e. C, E and C + E). 4S-B-phosphorene shows varying overpotentials ranging from 1.2 eV in C-site, 1.26 eV in E-site and 1.3 eV in C + E site; while the overpotentials in 6S-B-phosphorene sites range from 1.24 eV in C-site, 1.29 eV in E-site to 1.32 eV in C + E site. The nS-B-phosphorene (C) systems show a lower overpotential compared to systems with dopants at other sites. Also, these nS-B-phosphorene (C) systems with lower overpotentials also possess a lower band gap compared to nS-B-phosphorene (E) or nS-B-phosphorene (C + E) systems. Therefore, all nS-B-phosphorene (C) systems will show higher photocatalytic activity towards HER; in particular, the 4S-B-phosphorene (C) with overpotential of 1.2 eV will show highest activity at pH = 7. Interestingly, the even nS-B-phosphorene systems are also found to show high thermostability without any distortion in their geometry at temperature as high as 500 K. AIMD simulations of the nS-B-phosphorene (E) systems carried out for 10 ps and the geometries generated for every 1 fs is as shown in Fig. 7. Considering the band edge alignment at ambient pH condition and the thermostability at 500 K, the nS-B-phosphorene (E) systems potential photocatalysts that can be pragmatically implemented at room temperature and neutral pH.
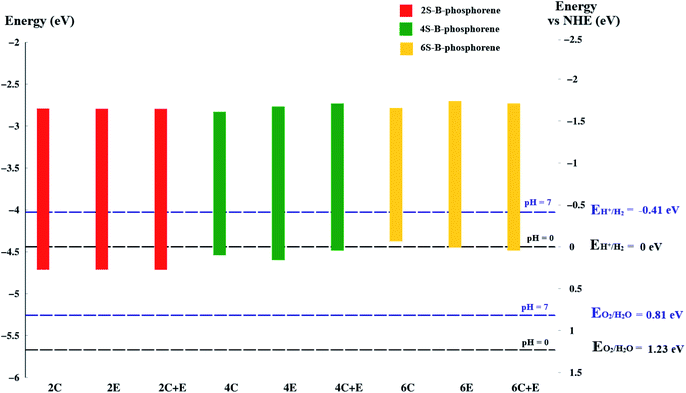 |
| | Fig. 5 Band edge positions of nS-B-phosphorene (n = 2, 4, 6), at (C), (E), (C + E) locations in vacuum energy and NHE energy scale. The horizontal black dashed line is the redox potential of water splitting at pH = 0 and the blue dashed line is the redox potential of water splitting at pH = 7. | |
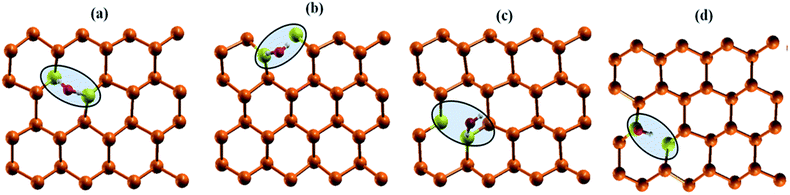 |
| | Fig. 6 Interaction of water molecule at various dopant sites (a) nS-B-phosphorene (C) (b) nS-B-phosphorene (E) (c) nS-B-phosphorene ( ) (d) nS-B-phosphorene ( ) (d) nS-B-phosphorene (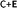 ). The highlighted and red coloured letter in C + E corresponds to the site on which water molecule is adsorbed. ). The highlighted and red coloured letter in C + E corresponds to the site on which water molecule is adsorbed. | |
 |
| | Fig. 7 AIMD simulations of (a) 2S-B-phosphorene (E) (b) 4S-B-phosphorene (E) and (c) 6S-B-phosphorene systems for 10 ps at 500 K. Relaxed geometries of nS-B-phosphorene (E) systems are as shown inset. | |
Activation of water molecule on nS-B-phosphorene sheet (n = 2, 4, 6)
The electronic properties of nS-B-phosphorene (m), n = 2, 4, 6 and m = C, E, C + E is further corroborated by explicit adsorption and activation of water molecule on the sheet. The water molecule in all cases is adsorbed on the dopant site, viz.; S atom. The activation is analysed in terms of adsorption energy of water molecule and activation of O–H bond through analysis of its red shift and O–H bond elongation. Adsorption energy is indicative of chemical interaction the nS-B-phosphorene and the water molecules and is calculated by using the following equation128| | |
Eads = Ewater+system − (Ewater + Esystem)
| (5) |
In the above equation, Eads is the adsorption energy, Ewater+system is the electronic energy of water molecule adsorbed B-phosphorene, Ewater is the electronic energy of a water molecule and Esystem is the electronic energy of B-phosphorene sheet.
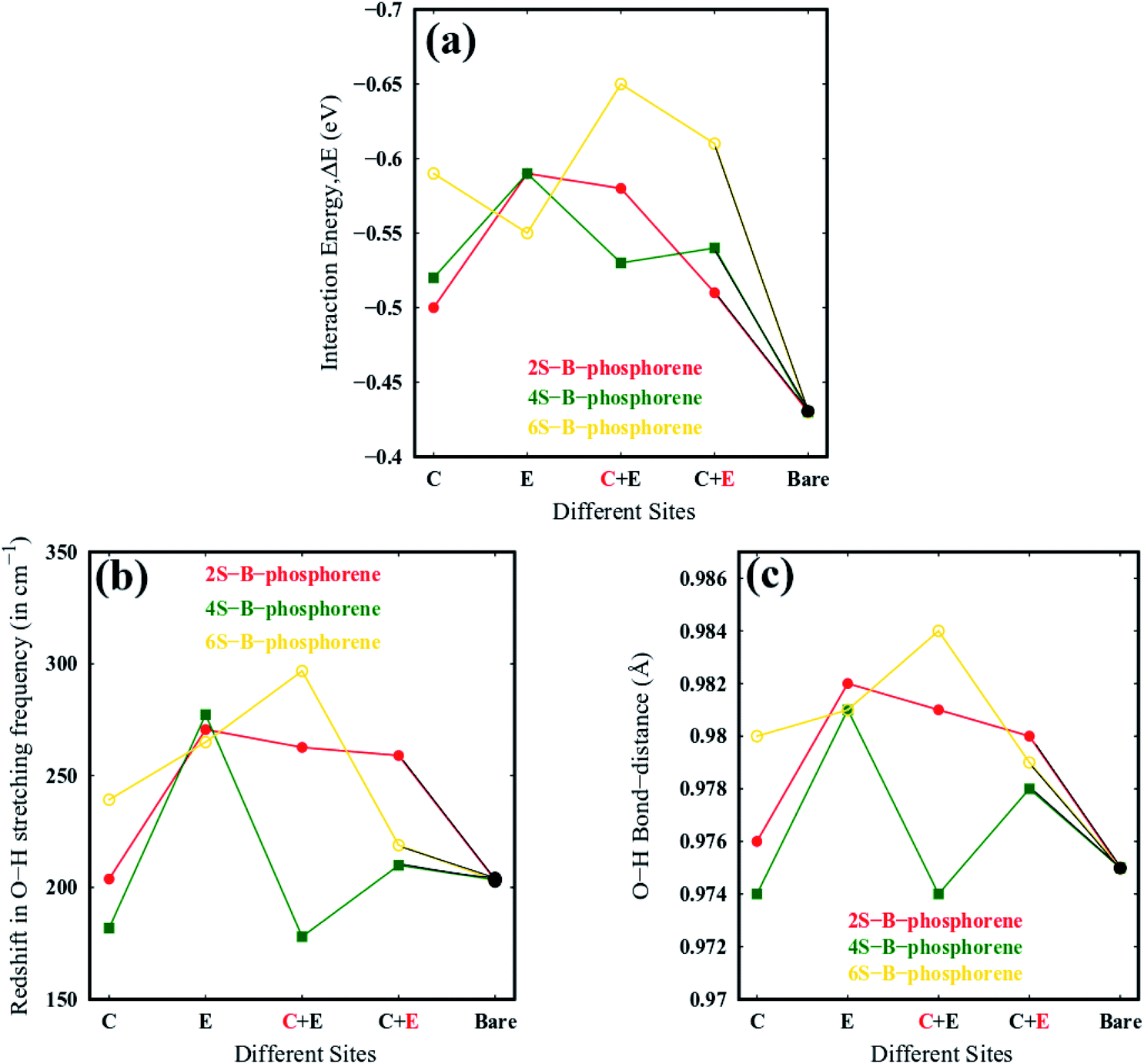
As mentioned earlier, water molecule is adsorbed on the dopant sites (S atom) of lowest energy conformation of nS-B-phosphorene (C), nS-B-phosphorene (E) and nS-B-phosphorene (C + E), n = 2, 4, 6. In case of nS-B-phosphorene (C + E), water molecule can be adsorbed on centrally placed S atom or edge S atom giving rise to nS-B-phosphorene ( ) and nS-B-phosphorene (
) and nS-B-phosphorene ( ), respectively (see Fig. 6(c) and (d)). The adsorption energies, OH stretching frequencies of the activated OH bond and activated OH bond length is given in Fig. 8. The absolute values for the same are given in ESI Table S.T 2.† The adsorption energy of water over B-phosphorene is noted to be 0.43 eV. All doped structures have higher interaction energies with the water. In particular, 6S-B-phosphorene (
), respectively (see Fig. 6(c) and (d)). The adsorption energies, OH stretching frequencies of the activated OH bond and activated OH bond length is given in Fig. 8. The absolute values for the same are given in ESI Table S.T 2.† The adsorption energy of water over B-phosphorene is noted to be 0.43 eV. All doped structures have higher interaction energies with the water. In particular, 6S-B-phosphorene (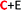 ) shows a higher adsorption energy (0.65 eV) in comparison to other nS-B-phosphorene structures. As we know experimentally, water splitting results in formation of adsorbed hydroxyl ions and hydrogen, respectively, on the catalyst interface. The obtained adsorption energy trends are further evaluated in terms of O–H bond elongation and red shift in O–H stretching frequencies. Red shifts in O–H stretching frequencies are obtained by taking the difference between O–H stretching frequency of free water molecule and that of water adsorbed on nS-B-phosphorene sheets; the values are as shown in Fig. 8(b). This shift in O–H stretching frequency is taken as a measure of O–H bond activation and the highest O–H bond activation is seen in the case of 6S-B-phosphorene (
) shows a higher adsorption energy (0.65 eV) in comparison to other nS-B-phosphorene structures. As we know experimentally, water splitting results in formation of adsorbed hydroxyl ions and hydrogen, respectively, on the catalyst interface. The obtained adsorption energy trends are further evaluated in terms of O–H bond elongation and red shift in O–H stretching frequencies. Red shifts in O–H stretching frequencies are obtained by taking the difference between O–H stretching frequency of free water molecule and that of water adsorbed on nS-B-phosphorene sheets; the values are as shown in Fig. 8(b). This shift in O–H stretching frequency is taken as a measure of O–H bond activation and the highest O–H bond activation is seen in the case of 6S-B-phosphorene ( ) system with a red shift of 296.82 cm−1. This is followed by nS-B-phosphorene (E) systems with their red shifts approximately at 270 cm−1. The observed red shifts in O–H stretching frequencies are also corroborated through the analysis of the activated O–H bond lengths as shown in Fig. 8(c). O–H bond length in free water moiety is 0.96 Å however, the O–H bond length for 6S-B-phosphorene corresponds to 0.985 Å indicating that it gets activated upon adsorption on nS-B-phosphorene sheets resulting in higher interaction energy and maximum red shift in O–H stretching frequency.
) system with a red shift of 296.82 cm−1. This is followed by nS-B-phosphorene (E) systems with their red shifts approximately at 270 cm−1. The observed red shifts in O–H stretching frequencies are also corroborated through the analysis of the activated O–H bond lengths as shown in Fig. 8(c). O–H bond length in free water moiety is 0.96 Å however, the O–H bond length for 6S-B-phosphorene corresponds to 0.985 Å indicating that it gets activated upon adsorption on nS-B-phosphorene sheets resulting in higher interaction energy and maximum red shift in O–H stretching frequency.
 |
| | Fig. 8 (a) Water molecule adsorption energies, magnitude of the Ead values are plotted (b) O–H stretching frequency of the activated bond in the water molecule, (c) activated O–H bond length of water molecule on various nS-B-phosphorene (n = 2, 4, 6). C, E, C + E on the x axis represents the doping sites of S atom viz; namely, centre, edge, centre + edge. The highlighted and red coloured letter in C + E corresponds to the site on which water molecule is adsorbed. | |
While the 6S-B-phosphorene system shows a higher activation of water molecule when compared to rest of the systems; interestingly, all nS-B-phosphorene (E) systems show a relatively efficient activation of water molecule with the red shift in O–H red stretching frequency approximately at 270 cm−1. Also, in several other 2D materials the edge sites have been reported to show higher catalytic activity when compared to the basal plane and our results are in correspondence to several such results. The overpotentials required for photocatalytic HER in these nS-B-phosphorene (E) systems range from 1.24 eV to 1.29 eV from our previous discussion on band edge alignment. When compared to the HER overpotential of 1.2 eV in 4S-B-phosphorene, the HER overpotentials in nS-B-phosphorene (E) systems are slightly higher but the chemically active edge sites available on these systems are an advantage when it comes to photocatalytic water splitting. These active sites bridge the energy required to activate the water molecule which the incident photons from solar light photons cannot provide sufficiently. Therefore, the nS-B-phosphorene (E) systems with catalytically active edge sites and HER overpotentials of 1.24–1.29 eV are efficient photocatalysts that can be experimentally explored.
Conclusions
In this work, the photocatalytic reactivity of B-phosphorene and chemically modified B-phosphorene monolayer is analysed and explored in terms of band structure, S-dopant concentration at best probable atomistic positions, band alignment with respect to pH. Cohesive energy demonstrates a higher stability of even nS-doped B-phosphorene monolayer at specific S-dopant concentration, i.e.; 6.25%, 12.5% and 18.75%. Further, band alignment studies suggest that even doped nS-B-phosphorene (n = 2, 4, 6) fulfils the band gap criteria and indirect band gap with an exception of 6S-B-phosphorene. Moreover, all nS-B-phosphorene (at all doping sites), respectively meet the redox potential level of H2 evolution only at pH = 0 and 7, therefore they will behave as efficient photocatalysts for HER. Aside from an overpotential of ∼1.24–1.29 eV for hydrogen evolution; these even nS-B-phosphorene systems show high feasibility to adsorb water molecule and activate the O–H bond and show high thermal stability at 500 K. The chemical activity of these systems are higher on the 6S-B-phosphorene (C + E) system followed by the nS-B-phosphorene (E) systems. Taking the photochemical activity i.e.; overpotentials required for HER at neutral medium, thermostability at 500 K as well as chemical activity towards water activation, the nS-B-phosphorene (E) systems are found to be effective photocatalysts. This computational investigation can be used to design and improvise phosphorene photocatalyst experimentally for water splitting.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
S. K. D., P. P. S. and S. K. acknowledges the Centre of Excellence in Scientific Computing (CoESC) at CSIR-NCL Pune for providing access to their High-Performance Computing Facility. A. M. acknowledges CSIR for funding of the JRF (Junior Research Fellowship).
Notes and references
- C. X. Lin, R. Li, M. Lu, C. Chen, D. Li, Y. Zhang and L. Jiang, Fuel, 2015, 162, 271–280 CrossRef.
- M. M. J. Li, C. Chin, T. Ayval, H. Suo, J. Zheng, I. F. Teixeira, L. Ye, H. Zou, D. Here and S. C. E. Tsang, ACS Catal., 2018, 8, 4390–4401 CrossRef CAS.
- S. Iguchi, K. Teramura, S. Hosokawa and T. Tanaka, Phys. Chem. Chem. Phys., 2015, 17, 17995–18003 RSC.
- Z. Zhao, L. Zhang and Z. Xia, J. Phys. Chem. C, 2016, 120(4), 2166–2175 CrossRef CAS.
- T. Oshima, T. Ichibha, K. S. Qin, K. Muraoka, J. J. M. Vequizo, K. Hibino, R. Kuriki, S. Yamashita, K. Hongo, T. Uchiyama, K. Fujii, D. Lu, R. Maezono, A. Yamakata, H. Kato, K. Kimoto, M. Yashima, Y. Uchimoto, M. Kakihana, O. Ishitani, H. Kageyama and K. Maeda, Angew. Chem., Int. Ed., 2018, 57, 8154–8158 CrossRef CAS PubMed.
- Q. Shao, P. Wang, T. Zhu and X. Huang, Acc. Chem. Res., 2019, 52, 3384–3396 CrossRef CAS PubMed.
- H. K. Chae, D. Y. Siberio-Pérez, J. Kim, Y. Go, M. Eddaoudi, A. J. Matzger, M. O'Keeffe and O. M. Yaghi, Nature, 2004, 427, 523–527 CrossRef CAS PubMed.
- I. W. Frank, D. M. Tanenbaum, A. M. Van der Zande and P. L. McEuen, J. Vac. Sci. Technol., B: Microelectron. Nanometer Struct.--Process., Meas., Phenom., 2007, 25, 2558–2561 CrossRef CAS.
- Y. Fu, J. Hansson, Y. Liu, S. Chen, A. Zehri, M. K. Samani, N. Wang, Y. Ni, Y. Zhang, Z. B. Zhang, Q. Wang, M. Li, H. Lu, M. Sledzinska, C. M. S. Torres, S. Volz, A. A. Balandin, X. Xu and J. Liu, 2D Mater, 2020, 7(012001), 1–42 CAS.
- L. Wang, W. Chen, D. Zhang, Y. Du, R. Amal, S. Qiao, J. Wu and Z. Yin, Chem. Soc. Rev., 2019, 48, 5310–5349 RSC.
- R. Sahoo, A. Pal and T. Pal, Chem. Commun., 2016, 52, 13528–13542 RSC.
- B. Luo, G. Liu and L. Wang, Nanoscale, 2016, 6904–6920 RSC.
- D. Deng, K. S. Novoselov, Q. Fu, N. Zheng, Z. Tian and X. Bao, Nat. Nanotechnol., 2016, 11, 218–230 CrossRef CAS PubMed.
- D. Dreyer, P. Jia and C. Bielawski, Angew. Chem., Int. Ed. Engl., 2010, 49, 6813–6816 CAS.
- H. Jia, D. Dreyer and C. Bielawski, Tetrahedron, 2011, 67, 4431–4434 CrossRef CAS.
- Z. H. Sheng, H. L. Gao, W. J. Bao, F. B. Wang and X. H. Xia, J. Mater. Chem., 2012, 22, 390–395 RSC.
- M. Burghard, H. Klauk and K. Kern, Adv. Mater., 2009, 21, 2586–2600 CrossRef CAS.
- D. R. Dreyer, R. S. Ruoff and C. W. Bielawski, Angew. Chem., Int. Ed., 2010, 49, 9336–9344 CrossRef CAS PubMed.
- J. Albero, D. Mateo and H. García, Molecules, 2019, 24(906), 1–21 Search PubMed.
- T. F. Yeh, J. M. Syu, C. Cheng, T. H. Chang and H. Teng, Adv. Funct. Mater., 2010, 20, 2255–2262 CrossRef CAS.
- L. Clarizia, D. Russo, I. D. Somma, R. Andreozzi and R. Marotta, Energies, 2017, 10(1624), 1–21 Search PubMed.
- P. Vogt, P. D. Padova, C. Quaresima, J. Avila, E. Frantzeskakis, M. C. Asensio, A. Resta, B. Ealet and G. L. Lay, Phys. Rev. Lett., 2012, 108, 155501–155505 CrossRef PubMed.
- A. Acun, L. Zhang, P. Bampoulis, M. Farmanbar, A. Houselt, A. N. Rudenko, M. Lingenfelder, G. Brocks, B. Poelsema, M. I. Katsnelson and H. J. W. Zandvliet, J. Phys.: Condens. Matter, 2015, 27, 443002–443012 CrossRef CAS PubMed.
- L. Shi, C. Ling, Y. Ouyang and J. Wang, Nanoscale, 2017, 8, 533–537 RSC.
- S. H. Mir, S. Chakraborty, P. C. Jha, J. Wärnå, H. Soni, P. K. Jha and R. Ahuja, Appl. Phys. Lett., 2016, 109, 53903–53907 CrossRef.
- P. Garg, I. Choudhuri, A. Mahata and B. Pathak, Phys. Chem. Chem. Phys., 2017, 19, 3660–3669 RSC.
- H. Tada, T. Mitsui, T. Kiyonaga, T. Akita and K. Tanaka, Nat. Mater., 2006, 5, 782–786 CrossRef CAS PubMed.
- C. J. Rupp, S. Chakraborty, J. Anversa, R. J. Baierle and R. Ahuja, ACS Appl. Mater. Interfaces, 2016, 8, 1536–1544 CrossRef CAS PubMed.
- C. Chowdhury, S. Karmakar and A. Datta, J. Phys. Chem. C, 2017, 121, 7615–7624 CrossRef.
- R. Jain, R. Narayan, S. P. Sasikala, K. E. Lee, H. J. Jung and S. O. Kim, 2D Mater, 2017, 4, 042006–042089 CrossRef.
- H. Liu, A. T. Neal, Z. Zhu, X. Xu, D. Tomanek, P. D. Ye and Z. Luo, ACS Nano, 2014, 8, 4033–4041 CrossRef CAS PubMed.
- V. Tran, R. Soklaski, Y. Liang and L. Yang, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 235319 CrossRef.
- L. Li, J. Kim, C. Jin, G. J. Ye, D. Y. Qiu, F. H. D. Jornada, Z. Shi, L. Chen, Z. Zhang, F. Yang, K. Watanabe, T. Taniguchi, W. Ren, S. G. Louie, X. H. Chen, Y. Zhang and F. Wang, Nat. Nanotechnol., 2017, 12(1), 21–25 CrossRef CAS PubMed.
- S. Larentis, B. Fallahazad and E. Tutuc, Appl. Phys. Lett., 2012, 101, 223104 CrossRef.
- H. Liu, A. T. Neal, Z. Zhu, D. Tomanek and P. D. Ye, ACS Nano, 2014, 8, 4033–4041 CrossRef CAS PubMed.
- Y. L. Lu, S. Dong, J. Li, Y. Wu, L. Wang and H. Zhao, Phys. Chem. Chem. Phys., 2020, 22, 13713–13720 RSC.
- K. Zhang, B. Jin, C. Park, Y. Cho, X. Song, X. Shi, S. Zhang, W. Kim, H. Zeng and J. H. Park, Nat. Commun., 2019, 10, 1–10 CrossRef PubMed.
- L. Zhang, H. Huang, B. Zhang, M. Gu, D. Zhao, X. Zhao, L. Li and Y. Cheng, Angew. Chem., Int. Ed., 2020, 132(3), 1090–1096 CrossRef.
- T. Kaewmaraya, L. Ngamwongwan, P. Moontragoon, W. Jaremboon, D. Singh, R. Ahuja, A. Karton and T. Hussain, J. Hazard. Mater., 2021, 401, 123340 CrossRef CAS PubMed.
- M. Rahman, C. Kwong, K. Davey and S. Qiao, Energy Environ. Sci., 2016, 9, 709–728 RSC.
- S. Kaur, A. Kumar, S. Srivastava, K. Tankeshwar and R. Pandey, J. Phys. Chem. C, 2018, 122, 26032–26038 CrossRef CAS.
- N. Jiao, P. Zhou, L. Xue, C. He and L. Sun, J. Phys.: Condens. Matter, 2019, 31, 075702 CrossRef PubMed.
- Y. Lu, S. Dong, W. Zhou, S. Dai, B. Zhou, H. Zhao and P. Wu, Phys. Chem. Chem. Phys., 2018, 20, 11967–11975 RSC.
- J. L. Zhang, S. Zhao, C. Han, Z. Wang, S. Zhong, S. Sun, R. Guo, X. Zhou, C. D. Gu and K. D. Yuan, Nano Lett., 2016, 16, 4903–4908 CrossRef CAS PubMed.
- J. Zeng, P. Cui and Z. Zhang, Phys. Rev. Lett., 2017, 118, 046101 CrossRef PubMed.
- Z. Zhu and D. Tománek, Phys. Rev. Lett., 2014, 112, 176802 CrossRef PubMed.
- J. Li, X. Sun, C. Xu, X. Zhang, Y. Pan, M. Ye, Z. Song, R. Quhe, Y. Wang, H. Zhang, Y. Guo, J. Yang, F. Pan and J. Lu, Nano Res., 2018, 11, 1834–1849 CrossRef CAS.
- E. Montes and U. Schwingenschlög, J. Mater. Chem. C, 2017, 5, 5365–5371 RSC.
- F. Safari, M. Moradinasab, M. Fathipour and H. Kosina, Appl. Surf. Sci., 2018, 464, 153–161 CrossRef.
- L. Ju, Y. Dai, W. Wei, Y. Liang and B. Huang, J. Mater. Chem. A, 2018, 6, 21087 RSC.
- B. Wang, X. Li, X. Cai, W. Yu, L. Zhang, R. Zhao and S. Ke, J. Phys. Chem. C, 2018, 122, 7075–7080 CrossRef CAS.
- Y. Cheng, Y. Song and Y. Zhang, Phys. Chem. Chem. Phys., 2019, 21, 24449 RSC.
- C. Li, Y. Xu, W. Sheng, W. Yin, G. Nie and Z. Ao, Phys. Chem. Chem. Phys., 2020, 22, 615 RSC.
- B. Wang, X. Li, R. Zhao, X. Cai, W. Yu, W. Li, Z. Liu, L. Zhang and S. Ke, J. Mater. Chem. A, 2018, 6, 8923 RSC.
- B. Ghosh, S. Nahas, S. Bhowmick and A. Agarwal, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 91, 115433 CrossRef.
- Y. Mogulkoc, M. Modarresi, A. Mogulkoc and Y. O. Ciftci, Comput. Mater. Sci., 2016, 124, 23–29 CrossRef CAS.
- R. B. Pontes, R. H. Miwa, A. J. R. da Silva, A. Fazzio and J. E. Padilha, Phys. Rev. B, 2018, 97, 235419 CrossRef CAS.
- L. Zhu, S. Wang, S. Guan, Y. Liu, T. Zhang, G. Chen and S. A. Yang, Nano Lett., 2016, 16, 6548–6554 CrossRef CAS PubMed.
- G. Yang, Z. Xu, Z. Liu, S. Jin, H. Zhang and Z. Ding, J. Phys. Chem. C, 2017, 121(23), 12945–12952 CrossRef CAS.
- M. Sun, S. Wang, J. Yu and W. Tang, Appl. Surf. Sci., 2017, 392, 46–50 CrossRef CAS.
- M. Sun, W. Tang, Q. Ren, S.-K. Wang, J. Yu and Y. Du, Appl. Surf. Sci., 2015, 356, 110–114 CrossRef CAS.
- M. Sun, Y. Hao, Q. Ren, Y. Zhao, Y. Du and W. Tang, Solid State Commun., 2016, 242, 36–40 CrossRef CAS.
- W. X. Zhang, J. W. Zhao, W. H. He, L. J. Luan and C. He, Chem. Phys. Lett., 2017, 675, 20–26 CrossRef CAS.
- H. Schmidt, F. Giustiniano and G. Eda, Chem. Soc. Rev., 2015, 44, 7715–7736 RSC.
- Y. D. Zhao, K. Xu, F. Pan, C. J. Zhou, F. C. Zhou and Y. Chai, Adv. Funct. Mater., 2017, 27, 1603484 CrossRef.
- F. Wang, Z. X. Wang, C. Jiang, L. Yin, R. Q. Cheng, X. Y. Zhan, K. Xu, F. M. Wang, Y. Zhang and J. He, Small, 2017, 13, 1604298 CrossRef PubMed.
- B. Li, T. Xing, M. Z. Zhong, L. Huang, N. Lei, J. Zhang, J. B. Li and Z. M. A. Wei, Nat. Commun., 2017, 8, 1958 CrossRef PubMed.
- V. Kochat, A. Apte, J. A. Hachtel, H. Kumazoe, A. Krishnamoorthy, S. Susarla, J. C. Idrobo, F. Shimojo, P. Vashishta, R. Kalia, A. Nakano, C. S. Tiwary and P. M. Ajayan, Adv. Mater., 2017, 29, 1703754 CrossRef PubMed.
- Z. Chen, K. Leng, X. Zhao, S. Malkhandi, W. Tang, B. Tian, L. Dong, L. Zheng, M. Lin, B. S. Yeo and K. P. Loh, Nat. Commun., 2017, 8, 14548 CrossRef CAS PubMed.
- G. L. Liu, A. W. Robertson, M. M. J. Li, W. C. H. Kuo, M. T. Darby, M. H. Muhieddine, Y. C. Lin, K. Suenaga, M. Stamatakis, J. H. Warner and S. C. E. Tsang, Nat. Chem., 2017, 9, 810–816 CrossRef CAS PubMed.
- Y. J. Yu, F. Y. Yang, X. F. Lu, Y. J. Yan, Y. H. Cho, L. G. Ma, X. H. Niu, S. Kim, Y. W. Son, D. L. Feng, S. Y. Li, S. W. Cheong, X. H. Chen and Y. B. Zhang, Nat. Nanotechnol., 2015, 10, 270–276 CrossRef CAS PubMed.
- J. R. Schaibley, H. Yu, G. Clark, P. Rivera, J. S. Ross, K. L. Seyler, W. Yao and X. Xu, Nat. Rev. Mater., 2016, 1, 16055 CrossRef CAS.
- V. V. Kulish, O. I. Malyi, C. Persson and P. Wu, Phys. Chem. Chem. Phys., 2015, 17, 992–1000 RSC.
- R. Zhang, B. Li and J. Yang, J. Phys. Chem. C, 2015, 119(5), 2871–2878 CrossRef CAS.
- G. Wang, R. Pandey and S. P. Karna, Appl. Phys. Lett., 2015, 106, 173104 CrossRef.
- W. Yu, Z. Zhu, C. Y. Niu, C. Li, J. H. Cho and Y. Jia, Phys. Chem. Chem. Phys., 2015, 17, 16351–16358 RSC.
- G. Wang, R. Pandey and S. P. Karna, Nanoscale, 2015, 7, 524–531 RSC.
- M. A. Hughes, Y. Fedorenko, B. Gholipour, J. Yao, T. H. Lee, R. M. Gwilliam, K. P. Homewood, S. Hinder, D. W. Hewak, S. R. Elliott and R. J. Curry, Nat. Commun., 2014, 5, 5346 CrossRef CAS PubMed.
- V. P. Pham and G. Y. Yeom, Adv. Mater., 2016, 28, 9024–9059 CrossRef CAS PubMed.
- J. Suh, T. L. Tan, W. J. Zhao, J. Park, D. Y. Lin, T. E. Park, J. Kim, C. H. Jin, N. Saigal, S. Ghosh, Z. M. Wong, Y. B. Chen, F. Wang, W. Walukiewicz, G. Eda and J. Q. Wu, Nat. Commun., 2018, 9, 199 CrossRef PubMed.
- J. Gao, Y. D. Kim, L. B. Liang, J. C. Idrobo, P. Chow, J. W. Tan, B. C. Li, L. Li, B. G. Sumpter, T. M. Lu, V. Meunier, J. Hone and N. Koratkar, Adv. Mater., 2016, 28, 9735–9743 CrossRef CAS PubMed.
- X. Zhang, Z. Shao, X. Zhang, Y. He and J. Jie, Adv. Mater., 2016, 28, 10409–10442 CrossRef CAS PubMed.
- J. Y. Wan, S. D. Lacey, J. Q. Dai, W. Z. Bao, M. S. Fuhrer and L. B. Hu, Chem. Soc. Rev., 2016, 45, 6742–6765 RSC.
- Y. Jung, Y. Zhou and J. J. Cha, Inorg. Chem. Front., 2016, 3, 452–463 RSC.
- J. Wang, H. Fang, X. Wang, X. Chen, W. Lu and W. Hu, Small, 2017, 13, 1700894 CrossRef PubMed.
- M. O. Valappil, M. Ahlawat, K. V. Pillai and S. Alwarappan, Chem. Commun., 2018, 54, 11733–11736 RSC.
- S. Mukherjee, L. Kavalsky, K. Chattopadhyay and C. V. Singh, Nanoscale, 2018, 10, 21335–21352 RSC.
- J. Wang, S. Li, F. Yun, X. Zhang and Q. Li, Int. J. Quantum Chem., 2020, 13, e26230 Search PubMed.
- F. Safari, M. Fathipour and A. Y. Goharrizi, J. Comput. Electron., 2018, 17, 499–513 CrossRef CAS.
- J. D. Correa, Superlattices Microstruct., 2019, 30, 401–408 CrossRef.
- H. Li, L. Zhang, X. Cai, X. Li, B. Wang, W. Yu and R. Zhao, Mater. Res. Express, 2018, 5, 055007 CrossRef.
- B. Meshgingalam and J. Baryestani, Appl. Surf. Sci., 2020, 526, 146692 CrossRef.
- E. A. Z. Hernandez, E. Florez, L. Dorkis, E. M. Mora-Ramos and J. E. Correa, Int. J. Quantum Chem., 2020, 120, e26075 Search PubMed.
- Y. Xiao, J. Wang, Y. Wang and W. Zhang, Appl. Surf. Sci., 2019, 488, 620–628 CrossRef CAS.
- K. Liu, J. Fu, L. Zhu, X. Zhang, H. Li, H. Liu, J. Hu and M. Liu, Nanoscale, 2020, 12, 4903–4908 RSC.
- Y. Cheng, Y. Song and Y. Zhang, Phys. Chem. Chem. Phys., 2019, 21, 24449–24457 RSC.
- W. Zhang, J. Wang and Y. Xiao, Appl. Surf. Sci., 2019, 488, 620–628 CrossRef.
- L. Ju, Y. Dai, W. Wei, Y. Liang and B. Huang, J. Mater. Chem. A, 2018, 6, 21087–21097 RSC.
- B. J. Wang, X. H. Li, R. Zhao, X. L. Cai, W. Y. Yu, W. B. Li, Z. S. Liu, L. W. Zhang and S. H. Ke, J. Mater. Chem. A, 2018, 6, 8923–8929 RSC.
- J. D. Correa, Superlattices Microstruct., 2019, 130, 401–408 CrossRef CAS.
- W. Lv, B. Yang, B. Wang, W. Wan, Y. Ge, R. Yang, C. Hao, J. Xiang, B. Zhang, Z. Zeng and Z. Liu, ACS Appl. Mater. Interfaces, 2018, 10, 9663–9668 CrossRef CAS PubMed.
- P. Luo, F. Zhuge, Q. Zhang, Y. Chen, L. Lv, Y. Huang, H. Li and T. Zhai, Nanoscale Horiz., 2019, 4, 26–51 RSC.
- M. Z. Rahman, C. Kwong, K. Davey and S. Z. Qiao, Energy Environ. Sci., 2016, 9, 709–728 RSC.
- A. Singh, H. Bae, S. Lee, K. Shabbiri, T. Hussain and H. Lee, Appl. Surf. Sci., 2020, 512, 145641 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- J. M. Soler, E. Artacho, J. D. Gale, A. García, J. Junquera, P. Ordejón and D. Sanchez-Portal, J. Phys.: Condens. Matter, 2002, 14, 2745–2779 CrossRef CAS.
- N. Troullier and J. L. Martins, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 43, 1993–2006 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- J. Guan, Z. Zhu and D. Tomanek, Phys. Rev. Lett., 2014, 113, 046804 CrossRef CAS PubMed.
- J. Bao, L. Zhu, H. Wang, S. Han, Y. Jin, G. Zhao, Y. Zhu, X. Guo, J. Hou, H. Yin and J. Tian, J. Phys. Chem. C, 2018, 122(41), 23329–23335 CrossRef CAS.
- B. Ghosh, S. Nahas, S. Bhowmick and A. Agarwal, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 91, 115433 CrossRef.
- J. D. Correa, Superlattices Microstruct., 2019, 130, 401–408 CrossRef CAS.
- Z. Q. Guo, J. P. Zhou, J. Z. Wang, Q. U. Hassan, J. Yang and Y. Ma, APL Mater., 2017, 5, 026104 CrossRef.
- Z. Zhu and D. Tománek, Phys. Rev. Lett., 2014, 112, 176802 CrossRef PubMed.
- J. Zhang, P. Zhou, J. Liu and J. Yu, Phys. Chem. Chem. Phys., 2014, 16, 20382–20386 RSC.
- M. Sun, W. Tang, Q. Ren, S. K. Wang, J. Yu and Y. Du, Appl. Surf. Sci., 2015, 356, 110–114 CrossRef CAS.
- J. Li, Q. Pei, R. Wang, Y. Zhou, Z. Zhang, Q. Cao, D. Wang, W. Mi and Y. Du, ACS Nano, 2018, 12(4), 3351–3359 CrossRef CAS PubMed.
- W. Gao, J. Lu, S. Zhang, X. Zhang, Z. Wang, W. Qin, J. Wang, W. Zhou, H. Liu and Y. Sang, Adv. Sci., 2019, 6, 1901244 CrossRef CAS PubMed.
- C. Chen, B. Huang and J. Wu, AIP Adv., 2018, 8, 105105 CrossRef.
- C. Kittel, Introduction to Solid State Physics, John Wiley & Sons, New York, 8th edn, 2005 Search PubMed.
- P. Bhatia, R. Swaroop and A. Kumar, RSC Adv., 2016, 6, 101835–101845 RSC.
- U. Gupta and C. N. R. Rao, Nano Energy, 2017, 41, 49–65 CrossRef CAS.
- V. Chakrapani, J. C. Angus, A. B. Anderson, S. D. Wolter, B. R. Stoner and G. U. Sumanasekera, Science, 2007, 318, 1424–1430 CrossRef CAS PubMed.
- B. Sa, Y.-L. Li, J. Qi, R. Ahuja and Z. Sun, J. Phys. Chem. C, 2014, 118, 26560–26568 CrossRef CAS.
- P. Kumar, R. Boukherroub and K. Shankar, J. Mater. Chem. A, 2018, 6, 12876–12931 RSC.
- S. N. Habisreutinger, L. Schmidt-Mende and J. K. Stolarczyk, Angew. Chem., Int. Ed., 2013, 52, 7372–7408 CrossRef CAS PubMed.
- G. Rothenberger, D. Fitzmaurice and M. Graetzel, J. Phys. Chem., 1992, 96, 5983–5986 CrossRef CAS.
- W. Hu, Z. Li and J. Yang, Nano Res., 2017, 10, 2223–2233 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra10829d |
| ‡ Equal contribution to the work. |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Sawan Kumar Das‡a,
Pragnya Paramita Samalab and
Sailaja Krishnamurty
ab,
Sawan Kumar Das‡a,
Pragnya Paramita Samalab and
Sailaja Krishnamurty
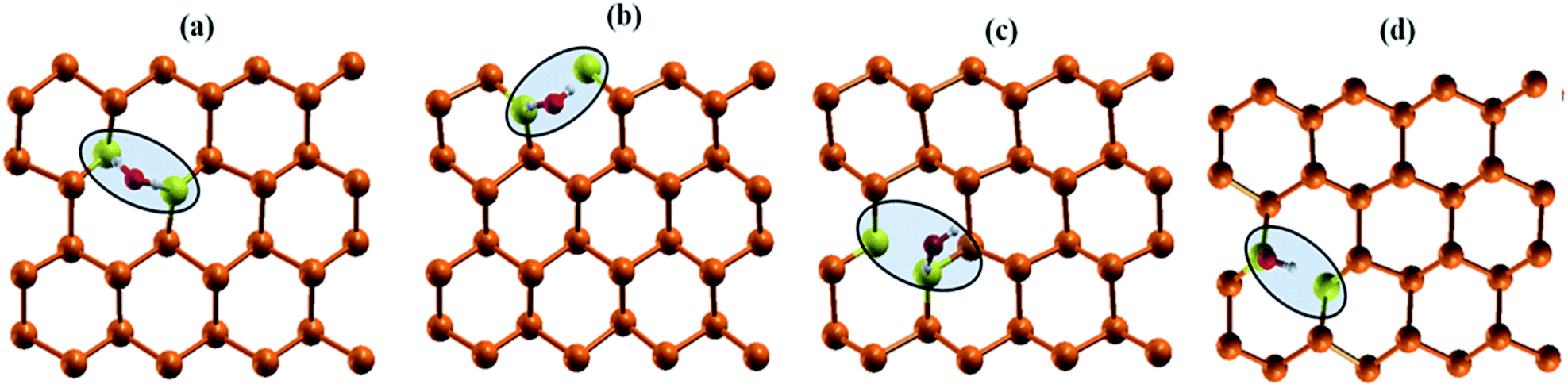
 ) (d) nS-B-phosphorene (
) (d) nS-B-phosphorene ( ). The highlighted and red coloured letter in C + E corresponds to the site on which water molecule is adsorbed.
). The highlighted and red coloured letter in C + E corresponds to the site on which water molecule is adsorbed.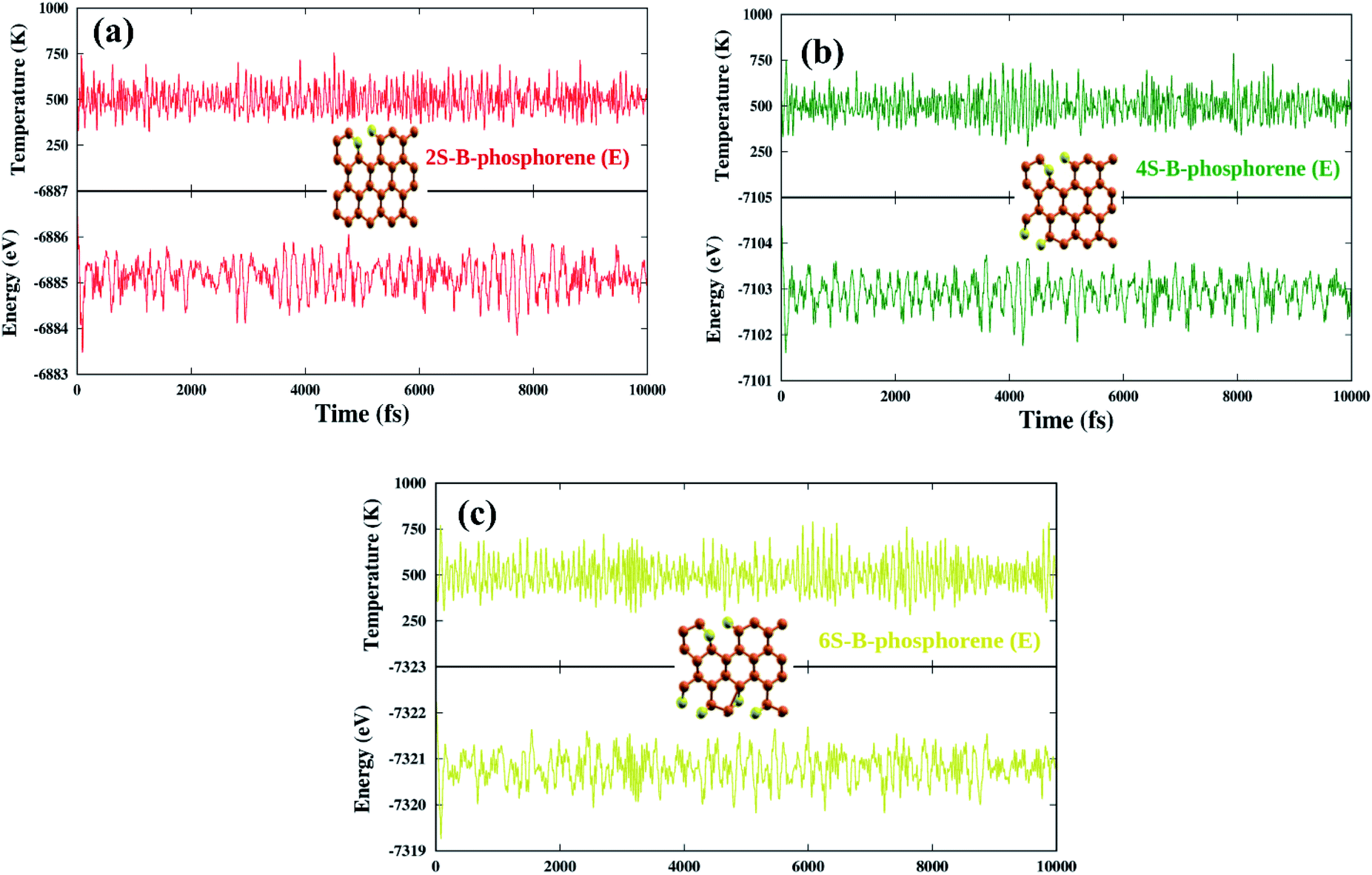
 ) and nS-B-phosphorene (
) and nS-B-phosphorene (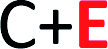 ), respectively (see Fig. 6(c) and (d)). The adsorption energies, OH stretching frequencies of the activated OH bond and activated OH bond length is given in Fig. 8. The absolute values for the same are given in ESI Table S.T 2.† The adsorption energy of water over B-phosphorene is noted to be 0.43 eV. All doped structures have higher interaction energies with the water. In particular, 6S-B-phosphorene (
), respectively (see Fig. 6(c) and (d)). The adsorption energies, OH stretching frequencies of the activated OH bond and activated OH bond length is given in Fig. 8. The absolute values for the same are given in ESI Table S.T 2.† The adsorption energy of water over B-phosphorene is noted to be 0.43 eV. All doped structures have higher interaction energies with the water. In particular, 6S-B-phosphorene ( ) shows a higher adsorption energy (0.65 eV) in comparison to other nS-B-phosphorene structures. As we know experimentally, water splitting results in formation of adsorbed hydroxyl ions and hydrogen, respectively, on the catalyst interface. The obtained adsorption energy trends are further evaluated in terms of O–H bond elongation and red shift in O–H stretching frequencies. Red shifts in O–H stretching frequencies are obtained by taking the difference between O–H stretching frequency of free water molecule and that of water adsorbed on nS-B-phosphorene sheets; the values are as shown in Fig. 8(b). This shift in O–H stretching frequency is taken as a measure of O–H bond activation and the highest O–H bond activation is seen in the case of 6S-B-phosphorene (
) shows a higher adsorption energy (0.65 eV) in comparison to other nS-B-phosphorene structures. As we know experimentally, water splitting results in formation of adsorbed hydroxyl ions and hydrogen, respectively, on the catalyst interface. The obtained adsorption energy trends are further evaluated in terms of O–H bond elongation and red shift in O–H stretching frequencies. Red shifts in O–H stretching frequencies are obtained by taking the difference between O–H stretching frequency of free water molecule and that of water adsorbed on nS-B-phosphorene sheets; the values are as shown in Fig. 8(b). This shift in O–H stretching frequency is taken as a measure of O–H bond activation and the highest O–H bond activation is seen in the case of 6S-B-phosphorene ( ) system with a red shift of 296.82 cm−1. This is followed by nS-B-phosphorene (E) systems with their red shifts approximately at 270 cm−1. The observed red shifts in O–H stretching frequencies are also corroborated through the analysis of the activated O–H bond lengths as shown in Fig. 8(c). O–H bond length in free water moiety is 0.96 Å however, the O–H bond length for 6S-B-phosphorene corresponds to 0.985 Å indicating that it gets activated upon adsorption on nS-B-phosphorene sheets resulting in higher interaction energy and maximum red shift in O–H stretching frequency.
) system with a red shift of 296.82 cm−1. This is followed by nS-B-phosphorene (E) systems with their red shifts approximately at 270 cm−1. The observed red shifts in O–H stretching frequencies are also corroborated through the analysis of the activated O–H bond lengths as shown in Fig. 8(c). O–H bond length in free water moiety is 0.96 Å however, the O–H bond length for 6S-B-phosphorene corresponds to 0.985 Å indicating that it gets activated upon adsorption on nS-B-phosphorene sheets resulting in higher interaction energy and maximum red shift in O–H stretching frequency.