
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEpitope-imprinted polymers: applications in protein recognition and separation
Tabkrich Khumsap ,
Angelica Corpuz and
Loc Thai Nguyen*
,
Angelica Corpuz and
Loc Thai Nguyen*
Department of Food, Agriculture and Bioresources, School of Environment, Resources and Development, Asian Institute of Technology, 58 Moo 9, Km. 42, Paholyothin Highway, Klong Luang, Pathumthani 12120, Thailand. E-mail: locnguyen@ait.ac.th
First published on 18th March 2021
Abstract
Molecularly imprinted polymers (MIPs) have evolved as promising platforms for specific recognition of proteins. However, molecular imprinting of the whole protein molecule is complicated by its large size, conformational instability, and structural complexity. These inherent limitations can be overcome by using epitope imprinting. Significant breakthroughs in the synthesis and application of epitope-imprinted polymers (EIPs) have been achieved and reported. This review highlights recent advances in epitope imprinting, from the selection of epitope peptide sequences and functional monomers to the methods applied in polymerization and template removal. Technological innovations in detection and extraction of proteins by EIPs are also provided.
1 Introduction
Molecularly imprinted polymers (MIPs) were first reported by Wulff, Sarhan and Zabrocki in 1973.1 In general, MIPs are prepared via controlled polymerization of one or more functional monomers and cross-linking agents in the presence of target templates. Subsequent removal of the templates creates three-dimensional recognition sites, which are complementary to the target molecules in shape, size, and functional groups.2 MIPs are usually referred to as “artificial” or “plastic” antibodies. However, they are significantly simpler and cheaper than antibodies. MIPs also possess superior mechanical properties, and are resistant to extreme pH, temperature, and pressure.3 Various forms of MIPs such as membranes, films, micro- and nano-particles have been synthesized. They have found application in many areas due to their selectivity, stability, and ability to recognize a broad range of target molecules.4 Even though molecular imprinting of low-molecular-weight targets has been established, the imprinting of protein macromolecules remains challenging. The problems are derived from the inherent characteristics of proteins including large molecular size, conformational instability, and structural complexity.5–7 First, the polymerization creates a dense matrix of pores or cavities bearing the template molecules. The large size of protein molecules hinders their complete removal from the polymeric matrix, and eventually impedes the efficient formation of recognition sites. Second, the unstable conformation of proteins is easily affected by the environmental conditions (temperature, pH, organic solvents, etc.) during the imprinting process. Consequently, the affinity of the imprinted cavities towards the native target proteins is diminished. Third, unlike small template molecules, the structurally complex proteins are composed of a large number of functional groups, which are prone to non-specific binding. These interactions reduce the selectivity of MIPs. In addition, the use of protein templates is expensive due to the high purity requirement in MIP synthesis. MIPs undoubtedly play a significant role in protein recognition. However, novel approaches are needed to further improve their synthesis and performance.Epitope is the small active site in the protein structure, which serves as the antigen or target binding site.8 Epitope is normally comprised of nine to fifteen amino acid residues located on the protein surface.9 The principle of epitope imprinting is illustrated in Fig. 1. First, the target protein is analyzed for the desired epitope region. The epitope peptide is then synthesized and used as the template for molecular imprinting. The target protein is recognized when its epitope region binds to the imprinted cavities of EIPs. Epitope-based imprinting overcomes the drawbacks of imprinting the whole protein molecule. Epitope templates are small and can be removed easily from the polymeric matrix. The reduction in structural complexity of the template improves the binding affinity, specificity, and selectivity of EIPs since the synthesis process is analogous to the low-molecular-weight imprinting. The simple structure of epitope is also less sensitive to the environmental conditions. Thus, epitope templates can be used with organic solvents, which broadens the repertoire of functional monomers used for MIP synthesis. Epitope peptides are also cheaper than pure proteins owing to their facile synthesis. This review will focus on recent advances in the synthesis and applications of epitope-imprinted polymers for protein recognition and separation.
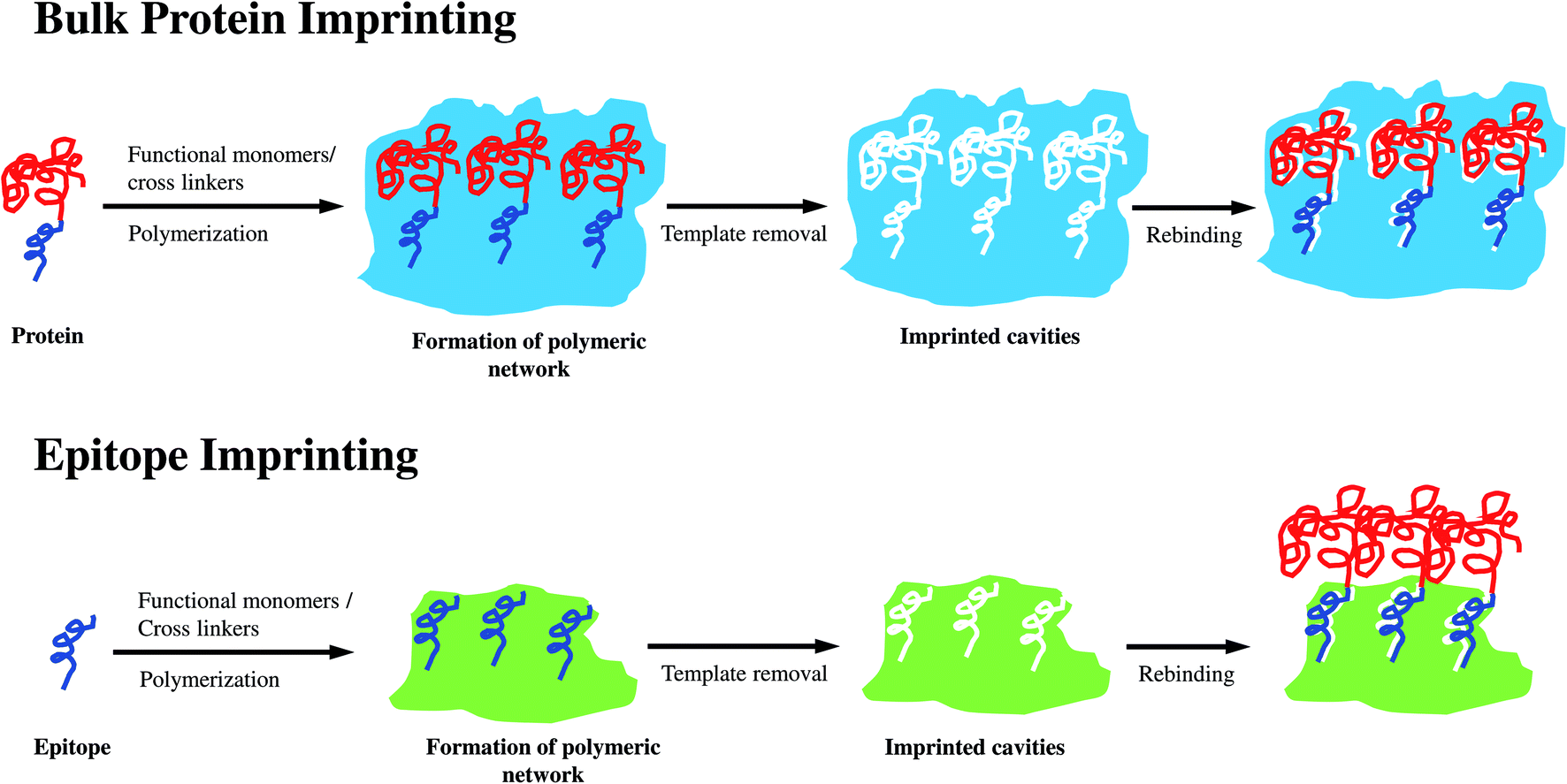 | ||
| Fig. 1 Imprinting approaches used for bulk protein and epitope. | ||
2 Synthesis strategies for epitope-imprinted polymers
2.1 Identification of epitope template
Screening of the suitable epitope in the protein structure for molecular imprinting is of significant importance. Usually, the epitope template is derived from the linear sequence of N- or C-terminus of the target protein. The sequence of other epitopes on proteins can be identified and characterized by crystallography and analysis of antibody binding sites aided by a designated online database. So far, there have been 162![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 529 crystallographic protein structures documented in Protein Databank (PDB).10 The protein sequences are also available in UniProtKB/SwissProt (561911 entries) and UniprotKB/TrEMBL (177754527 entries).11 The screening of epitopes belonging to a protein can be performed by BLAST (Basic Local Alignment Search Tool) software, which helps in comparing protein sequences to databases and in calculating the statistical significance of matches. The method can improve the selectivity of epitope imprinting by removing the epitopes not belonging to the protein target.
529 crystallographic protein structures documented in Protein Databank (PDB).10 The protein sequences are also available in UniProtKB/SwissProt (561911 entries) and UniprotKB/TrEMBL (177754527 entries).11 The screening of epitopes belonging to a protein can be performed by BLAST (Basic Local Alignment Search Tool) software, which helps in comparing protein sequences to databases and in calculating the statistical significance of matches. The method can improve the selectivity of epitope imprinting by removing the epitopes not belonging to the protein target.
Bossi et al. proposed a method to identify distinctive peptide sequences (both internal and terminal peptides) for molecular imprinting.12 The EIP prepared by this method is called fingerprint imprinted polymer (FIP). Briefly, the process consists of six steps: (1) in silico digestion of the desired protein, (2) selection of the length of digested peptide (7–12 residues), (3) alignment of the 7–12 residues with all of the protein sequences stored in UniProtKB using the BLAST software, (4) in silico ranking of the peptides for their uniqueness, (5) selection of the most unique peptide as the template, and (6) synthesis of the FIP. The method was used in preparing FIP for detection of NT-proBNT, the clinical marker of heart failure. Bagan et al. developed a different technique to screen the suitable epitope from proteins via enzyme digestion.5 In this case, the whole target molecules were immobilized on the surface of silica beads and digested by trypsin. The silica beads decorated with remaining peptides were used as the templates for the imprinting process. The silica was eventually dissolved in a solution of 4% hydrogen fluoride in methanol to yield the imprinted polymer particles. The method was successfully used to synthesize hemoglobin-selective polymer particles.
2.2 Functional monomer
Unlike protein macromolecular imprinting, epitope imprinting can be performed in organic solvents. Therefore, the choices of functional monomers are expanded. However, the effect induced by the surface charges of the polymer and target needs to be considered. A positively or negatively charged polymer may repel similarly charged template molecules. Consequently, a smaller number of template–monomer complexes are formed, resulting in lower sensitivity of the EIPs. Obviously, the functional monomers used in imprinting of bulk proteins can also be used for that of epitopes. A detailed description of the monomers and methods used for bulk protein imprinting was reported elsewhere.13 Functional monomers such as acrylate, organometallics, alcohols, and dopamine are usually adopted in epitope imprinting for protein detection. Zinc acrylate (ZnA) is one of the most commonly used acrylate monomers. ZnA can form strong metal chelation and five-membered ring with the amino and hydroxyl groups of the epitopes.14 This metal coordination can overcome the limitations associated to the covalent and non-covalent bonding. Covalent bonding may be too strong for template removal, whereas non-covalent bonding could be too weak for template immobilization. The use of these monomers was found to increase both imprinting sites and factor. Gupta et al. used both 3-sulfopropyl methacrylate potassium salt (3-SPMAP) and benzyl methacrylate (BMA) to develop an EIP for detection of ferric binding (fbp A) protein in Neisseria meningitidis.15 The sulfur in 3-SPMAP enhanced the grafting of polymeric film on the electrode surface while the aromatic ring of BMA facilitated the charge transfer or hydrophobic interactions with the template. The specificity and selectivity of the imprinted sites were reportedly improved due to multipoint non-covalent interactions. Li et al. reported the use of thermoresponsive EIP-coated nanoparticles for recognition of human serum albumin (HSA) with N-isopropylacrylamide (NIPAAm) monomer.16 In this study, the hydrophilic and hydrophobic states of the imprinted polymer can be modulated by the applied temperature. As a result, the capture and release of the target protein can be thermally controlled. Organosilicons, or organometallic compounds, are also used as functional monomers for epitope imprinting. Yang et al. assembled 3-aminopropyltriethoxysilane (APTES) and the peptide epitope of bovine serum albumin (BSA) on the surface of silica nanospheres embedded CdTe quantum dots (QDs).7 APTES formed a complex with the epitope via non-covalent interactions, and then reacted with tetraethoxysilane (TEOS) (cross-linker) to produce the imprinted matrix. The obtained EIP was successfully used to detect and separate BSA from blood samples. In another instance, o-phenylenediamine (o-PD), a derivative of aniline, was used as the functional monomer for epitope imprinting. o-PD can be polymerized over a wide range of pH.17 An o-PD molecule contains two amino groups which can bind with the protein targets through hydrogen bonding or van der Waals forces. The o-PD-based EIP platform was applied in electrochemical detection of BSA18 and insulin.19 Lee et al. developed an EIP from ethylene-co-vinyl alcohol (EVAL) to detect regenerating protein 1 (REG1).20 The use of EVAL as the functional monomer is favored by its low cost and simple imprinting process. In addition, EVAL can form complexes with template molecules by the interactions of its hydroxyl groups with carbonyl groups of the target. These non-covalent bonds allow the templates to be removed without using a strong solvent. EVAL-based EIP was found to have excellent selectivity and affinity towards the target proteins. Recently, dopamine has emerged as a promising monomer for molecular imprinting. Dopamine contains both catechol and amino groups. While the catechol can bind with thiol and amine groups of the protein molecules via Michael addition or Schiff base reactions during protein imprinting, amino can react with hydroxyl and carboxyl groups through hydrogen bonding or van der Waals forces.21 Moreover, polydopamine (PDA) can adhere to different surfaces by self-assembly. PDA can form thin, robust films on the substrate surface over a wide range of pH. Epitope-imprinted PDA was used in surface plasmon resonance (SPR) sensor for detection of troponin T (TnT).22 PDA also found its applications in the imprinting of peptides and proteins such as HIV-1 associated protein,23 human serum albumin (HSA),24 bovine hemoglobin (BHb),25 and hydrophobins.26 Besides dopamine, norepinephrine, a monomer differing from dopamine only for an additional hydroxyl group has been used in epitope-imprinting for the first time by Baldoneschi et al.27 The presence of the hydroxyl group spreading over the polynorepinephrine (PNE) network possibly enhanced the surface hydrophilicity with respect to PDA. This feature helps reduce the non-specific adsorption of proteins and facilitate the selective binding of target molecules. The results showed that the protein adsorption is significantly reduced on the PNE film, up to 87% for BSA, 93% for the antibody and 60% for troponin I–C complex. This epitope-imprinted PNE film was successfully used to detect troponin I from human serum with excellent selectivity, capture capacity, and kinetic binding constant. With the increasing demand of EIPs, the production process should be simple and rapid. The imprinted biopolymers such as PDA and PNE provide great advantages since their syntheses can be conducted in aqueous solutions by a one-step process. The corresponding monomers (dopamine and norepinephrine), hence, have a great potential in the future applications of EIPs.282.3 Polymerization technique
The polymerization process in molecular imprinting generally results in the formation of the template–monomer complexes mediated by covalent or non-covalent interactions. Covalent bonds such as those between boronic acid of aminophenylboronic acid (APBA) and cis-diol compounds of glycoprotein targets are rarely used.29,30 Covalent bonding can yield highly-selective EIP due to strong monomer–template interactions. However, this approach can obstruct the removal of templates, the binding and dissociation between template and monomer.2 Non-covalent bonding is more widely used in epitope imprinting due to its simplicity, fast binding kinetics, and ease of template removal.Different polymerization methods including bulk polymerization, precipitation polymerization, electropolymerization, etc. have been applied for epitope-imprinting. In bulk polymerization, the template, functional monomer(s), and crosslinker are all dissolved in an appropriate solvent or porogen. The polymerization is thermally or photochemically carried out and the insoluble polymers obtained are then ground and sieved prior to removal of the templates.31 The process can be carried out at room temperature and does not require sophisticated equipment. However, the grinding step may partially damage the imprinted cavities and adversely affect the sensitivity of EIPs.32 Bulk polymerization was the most commonly applied technique in epitope imprinting.33 Precipitation polymerization is based on the polymerization of monomers in dilute solution (<5% w/v) in near-θ solvents. The particles are formed by the precipitation of nano-gel (seed) particles followed by continuous capture of oligomers from solution.34,35 This technique can produce spherical microparticles with better uniformity and yield than that of bulk polymerization.36,37 The diameter of synthesized particles normally ranges from 0.1 μm to 10μm.38 The size and porosity of EIPs particles can be controlled by the reaction conditions.39 Rossetti et al. reported the use of precipitation polymerization to prepare EIP for detection of ProGastrin releasing peptide (ProGRP),40 a protein biomarker for lung cancer. In this work, the imprinting process was optimized by adjusting the ratio of the functional monomer, cross-linker, and solvent. Sol–gel technique is another attractive method for polymerization, due to its straightforward synthesis path. Moreover, a high degree of cross-linking in the polymeric network helps retain both size and shape of the cavities after template removal. The as-prepared EIPs usually have higher selectivity, surface area, number of binding sites, and can enhance mass transfer rates.41 In addition, this method produces water-soluble polymers which greatly reduces damage to imprinted cavities from swelling.42 Silica-gel materials are usually formed by the condensation of modified silanes in aqueous solution under mild thermal conditions.43 In this connection, the imprinted matrix can be fabricated through the self-assembly of silanes and templates via condensation, and is eventually structured under the aging process. The application of sol–gel method to produce microporous EIP silica scaffolds for determination of lysozyme has been successfully demonstrated.44 Electropolymerization is another method to synthesize in situ EIP layer on the substrate surface. This technique allows for the precise control of the deposited layer thickness. The micro- or nano-scaled film can be readily obtained by adjusting the number of scan cycles or deposition time.45–47 Unlike bulk polymerization, electropolymerization can reduce the structural damages of the protein templates due to thermal or, chemical initiation such as use of strong oxidizing initiators.48,49 Thus, the approach is quite suitable for protein targets.50 Electropolymerization has been used to develop highly sensitive electrochemical sensors, or sensors based on quartz crystal resonator, surface plasmon resonance, and acoustic wave.51 Tchinda et al. has demonstrated the application of an electrosynthesized EIP for molecular recognition of neuron specific enolase (NSE) biomarker.52 Scopoletin was polymerized onto the QCM gold surface by amperometry in the presence of cysteine- or histidine-modified epitope as templates. The thickness of the film was modulated by the applied potential and time. Polymerization can be alternatively performed using “grafting-from” technique, which relies on the photo-initiation of chain growth at the support surface. A precise control of the film thickness could be achieved by adjusting the density of immobilized photoinitiator and UV irradiation time. EIP membrane with affinity toward IgG monoclonal antibody has been prepared using this technique.53
2.4 Template removal
The removal of template is crucial in epitope imprinting as it affects the sensitivity of EIPs. Inappropriate techniques may lead to the breakage, distortion, or shrinkage of the polymeric network.54 Epitope imprinting mainly depends on the non-covalent bonds between the polymer matrix and the template molecules. The templates, thus, can be removed by either inducing the swelling of the polymer to facilitate the migration of the templates or the disruption of the interactions (hydrogen bonds or van der Waals forces) between the templates and the imprinted polymeric network.55 Usually, the templates are removed by incubation in solvent or by Soxhlet extraction. Solvent incubation can be carried out under mild conditions, hence, produces a highly stable imprinted network.56 A wide range of solvents including sodium hydroxide,19,57 sodium dodecyl sulfate,7,20 Tween-20,18 acetic acid,22 and the mixture of methanol and acetic acid9,58,59 solutions have been investigated for template removal. Soxhlet extraction with organic solvent is traditionally used to remove templates in bulk imprinting process. In this method, the quantity and suitability of the solvent, as well as the extraction time, are critical parameters. Soxhlet extraction requires simple, inexpensive equipment, and can be applied for different polymer matrices.60 However, the process is relatively time-consuming (6–24 h) and chemical-intensive (50–300 mL) due to the required circulation of solvent. Moreover, the extreme temperature conditions can cause protein denaturation. These drawbacks thwart the application of Soxhlet extraction in epitope imprinting. So far, the use of Soxhlet extraction for EIP preparation has been scarcely reported.61 The removal of protein template was also studied using electrochemical cleavage. Pirzada et al. immobilized cysteine-modified epitope templates onto the gold electrode surface via thiol linkages.62 After polymerization, the templates were desorbed by anodic treatment, which cleaved the thiol linkages. However, a proper control of the applied potential and time is needed to avoid undesirable cross-linking within the polymer instead of the desired breakage of the bonds.632.5 Solid phase synthesis and template immobilization
Solid-phase synthesis approach was developed to overcome the inherent limitations of MIPs prepared by traditional methods, including the binding site heterogeneity (“polyclonal”), presence of residual template, and complicated production process.64 These issues are partly derived from the rotational and translational motion of the soluble templates during the formation of binding sites. In addition, the use of free templates has low imprinting efficiency due to the tendency of being buried within the polymer matrix. In solid phase approach, the template is covalently immobilized on the surface of a suitable solid support such as glass or silica beads. The polymerization is carried out under controlled conditions to promote the formation of imprinted nanoparticles.65 The solid support also acts as an affinity medium to purify the high-affinity polymer particles. Solid-phase method has been successfully used to synthesize EIPs for protein separation and detection. For example, Xing et al. developed a new process based on controllable oriented surface imprinting.66 Briefly, a glycated C-terminus nonapeptide epitope was used as the template and was anchored onto a boronic acid-functionalized substrate. Then, the polycondensation was conducted using multiple silylating reagents, including aminopropyltriethoxysilane (APTES), 3-ureidopropyl-triethoxysilane (UPTES), isobutyltriethoxysilane (IBTES), and tetraethyl orthosilicate (TEOS). The approach provided strong affinity and, high specificity towards β2-microglobulin (B2M). EIPs are also prepared by solid-phase synthesis on the porous silica and glass beads. Poma et al. adopted a different method in which the peptide template was immobilized onto the glass beads by 3-aminopropyltrimethyloxysilane (APTMS).64 After polymerization, the high-affinity nanosized EIP particles were eluted from the glass substrate and collected. The apparent dissociation constant (Kd) of the obtained EIP for a model peptide (TATTSVLG-NH2) was estimated to be 4.8 × 10−8 M. Additional advantages of this method are ease of automation and multiple reuse of the templates. Unlike the monoliths, membranes, films or beads, the as-prepared EIP nanoparticles can be readily integrated into various diagnostic test and assay platforms. Using a similar technique, Gómez-Arribas et al. used microporous silica beads as the substrate to synthesize EIP nanoparticles for recognition of FLAG tag,67 a short peptide usually used for the purification of recombinant proteins. In this study, the epitope template, a pentapeptide (DYKDC), was first covalently immobilized on the surface of microporous silica beads previously pre-functionalized with aminosilanes. The polymerization was then performed using N-(2-aminoethyl)methacrylamide hydrochloride (EAMA) as functional monomer, ethylene glycol dimethacrylate (EDMA) as cross-linker, dimethylsulfoxide (DMSO) as porogen and 2,2-azobis(2,4-dimethylvaleronitrile) (ABDV) as initiator. The silica was eventually dissolved by ammonium hydrogen difluoride to produce imprinted polymer particles. The unique features of the solid phase synthesis make it a promising approach to produce EIPs for practical applications.3 Advances in protein sensing and separation
Epitope imprinting has attracted an increasing interest in the development of platforms for protein recognition. Epitope-imprinted polymers have been used in optical, electrochemical, and piezoelectric sensors for detection of various protein targets. A summary of the latest EIP-based sensors is provided in Table 1. Other applications of EIPs include the extraction and separation of proteins by magnetic extraction, solid phase extraction, and chromatography.| Sensor type | Detection technique | Target | Polymerization method | Template | Length (aa) | Monomer | Cross-linker | Solvent | Eluent | LOD | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a OC: optical, EC: electrochemical, PE: piezoelectric, HAc: acetic acid, NaAc: sodium acetate, TnT: Troponin T, TnI: Troponin I, Cyt c: cytochrome c, BSA: bovine serum albumin, APTES: 3-aminopropyltriethoxysilane, TEOS: tetraethoxysilane, ZnA: zinc(II) acrylate, EGDMA: ethylene glycol dimethacrylate, DMF: dimethylformamide, CV: cyclic voltammetry, DPV: differential pulse voltammetry, SWV: square wave voltammetry, ELP: electropolymerization, SA: self-assembly, o-PD: o-phenylenediamine, NSE: neuron specific enolase, CME: cysteine modified epitope, HME: histidine modified epitope, TCEP: tris(2-carboxyethyl)phosphine, AV treatment: anodic potential treatment, REG 1: regenerating protein 1, PBS: phosphate buffer solution, n/a: not available, DMF: dimethylformamide, QCM: quartz crystal microbalance, 3-SPMAP: 3-sulfo propyl methacrylate potassium salt, BMA: benzyl methacrylate, NIPAM: N-isopropylacrylamide, MBA: methylene bisacrylamide, TCEP: tris(2-carboxyethyl)phosphine, SPS: solid phase synthesis, TBAM: N-tert-butylacrylamide. | |||||||||||
| OC | SPR | Cardiac TnT | SA | TnT-derived peptide | 10–15 | Dopamine | — | Tris–HCl | HAc | 15.4 nM | 22 |
| SPR | TnI | SA | N-terminus | 13 | Norepinephrine | — | Tris–HCl | HAc | 7.1 nM | 27 | |
| Fluorescent | BSA | Precipitation | C-terminus | 9 | APTES | TEOS | ACN/MeOH/H2O | EtOH | 0.11 μM | 7 | |
| Fluorescent | Cyt c | Precipitation | C-terminus | 9 | ZnA | EGDMA | DMF | MeOH/HAc | 0.32 μM | 59 | |
| N-terminus | 9 | ||||||||||
| Fluorescent | Cyt c | Co-polymerization | C-terminus | 9 | ZnA | EGDMA | DMF | MeOH/HAc | 0.11 μM | 69 | |
| N-terminus | 9 | ||||||||||
| Fluorescent | HSA | Bulk polymerization | C-terminus | 12 | NIPAM | MBA | NaHTe | ACN/H2O | 44.3 pM | 70 | |
| EC | DPV | BSA | ELP | C-terminus | 9 | o-PD | — | HAc–NaAc | Tween-20 | 0.30 pM | 18 |
| DPV | NSE biomarker | ELP | CME | 12 | Scopoletin | — | TCEP | AV treatment | 15.6 nM | 52 | |
| HME | 12 | NaOH | 128 μM | ||||||||
| DPV | Insulin | ELP | C-terminus | n/a | o-PD | — | PBS | NaOH | 7.24 fM | 19 | |
| DPV | Insulin | SPS | Computationally derived epitope | n/a | NIPAM | MBA, TBAM | DMSO | EtOH | 26 fM | 72 | |
| CV | REG 1 | Co-precipitation | REG1-derived peptide | 13–18 | EVAL | — | DMSO | SDS | n/a | 20 | |
| CV | α-Synuclein | ELP | Derived peptides | 13–15 | Aniline | — | DI water | EtOH | n/a | 73 | |
| SWV | NSE biomarker | ELP | Cys-modified epitopes (2) | 11, 13 | Scopoletin | — | PBS-TCEP | Anodic treatment | 2.56 pM | 62 | |
| SWV | NSE biomarker | ELP | Cys-modified epitopes (2) | 10 | Scopoletin | — | NaCl | Anodic treatment | 0.25 μM | 71 | |
| PE | QCM | HSA | Sol–gel | C-terminus | 9 | ZnA | EGDMA | DMF | MeOH/HAc | 0.39 nM | 9 |
| QCM | fbp A | SA | Computationally derived peptide | 14 | 3-SPMAP and BMA | NNMBAA | ACN | PBS | 38.87 pM | 15 | |
3.1 Optical sensors
So far, EIPs have been mainly employed in optical sensors based on fluorescence quenching and surface plasmon resonance. Fluorescence quenching sensors are usually fabricated from quantum dots (QDs) due to their high photoluminescence capability, wide absorption spectrum, narrow emission band (30–50 nm), and high quantum yield.68 A novel strategy for preparing EIP-coated quantum dots to determine and separate BSA from bovine blood sample was described by Yang et al.7 In this study, the surface-exposed C-terminus of BSA (residues 599–607) was selected as the template and APTES served as the functional monomer. The developed approach produced EIP with higher imprinting factors (4.80) and binding capacity (66.97 mg g−1) than that of bulk protein imprinted polymer. The coated QDs had excellent selectivity, which can discriminate against one mismatched amino acid in the epitope sequence. Zhang et al. prepared a fluorescent composite SiNP@SiO2@EIP using dual epitope imprinting and metal-chelating method for detection of cytochrome c (Cyt c).59 The templates were composed of both C-terminal nonapeptide (AYLKKATNE) and N-terminal nonapeptide (GDVEKGKKI). The chelation between Zn(II) of ZnA monomer and the amino or hydroxy groups of the nonapeptide template was responsible for recognition and capture of Cyt c. Dual epitope imprinting exhibited better selectivity than single epitope approach. The obtained detection limit (LOD) and imprinting factor (IF) were 0.32 μM and 2.43, respectively. The recovery of the target Cyt c from human serum ranged from 94.0% to 107.5%. Yan et al. used a similar approach to synthesize the nitrogen-doped graphene quantum dots MIP for recognition and detection of Cyt c.69 The C- and N-terminal nonapeptides of Cyt c served as the double templates, which were fixed by zinc acrylate through metal chelation and steady six-membered ring. In this study, the LOD and IF were 0.11 μM and 3.06, respectively. The recovery of Cyt c from urine sample by the material was between 99.3–114.0%. EIP synthesized for detection of HSA in human serum sample via fluorescence quenching was reported by Wang et al.70 The EIP was prepared by one-pot polymerization of NIPAAm in the presence of CdTe QDs and a twelve amino acid-length epitope of HSA. The linear trend was in the range of 0.25–5.0 mM with LOD of 44.3 μM. Palladino et al. developed an EIP-based SPR sensor for detecting TnT using TnT-derived peptides as templates.22 The effectiveness of C-terminus peptide of TnT (GKAKVTGRWK) in producing the imprinted film was attributed to flexibility and basicity of this template. Troponin I (TnI) from human serum, on the other hand, was detected by epitope-imprinted PNE coupled with a SPR transducer.27 The sensor could detect TnI in PBS (pH 7.4) and human plasma with LOD of 460 pM and 8.9 nM, respectively. The sensitivity of the sensor can be improved to LOD as low as 193 pM and 7.1 nM by using alkaline phosphatase-labeled anti-TnC antibody for signal amplification.3.2 Electrochemical sensors
EIPs have been embedded in various electrochemical sensors for the recognition of protein targets. Epitope imprinted o-PD using C-terminus nonapeptide (VVSTQTALA, residues 599–607) as the template molecules was successfully applied to determination of BSA.18 The sensitivity of the sensor was also enhanced by enzyme amplification strategy. The underlying principle is based on the competitive reaction between the horseradish peroxidase-labeled nonapeptide (HRP-nonapeptide) and BSA at the recognition cavities of the EIP. The enzymatic reaction derived from the labeled HRP resulted in amplified current signal. The sensor can detect BSA in the range of 15.04 pM to 2.26 nM with a LOD of 0.30 pM, and recovery rate of 98.3–102.5%. Tchinda et al. reported an EIP for recognition of NSE biomarker based on the imprinting of two different epitopes.52 As mentioned earlier, the peptide epitope derived from the NSE was synthesized and modified with either cysteine or histidine and scopoletin served as the monomer. The histidine modification was found to exhibit better sensitivity, selectivity, and specificity than the other method attributed to template–monomer compatibility. Similarly, Pirzada et al. applied EIP for electrochemical detection of NSE.62 The cysteine-modified epitopes of NSE (templates) were self-assembled on the surface of gold electrode via thiol linkages. Then, the electrode was coated with electropolymerized scopoletin and AuNPs. The incorporation of AuNPs in the EIP film has helped in signal amplification and sensitivity enhancement of the sensor. Using this method, NSE was detected in the concentration range of 0.32–51.28 pM with the LOD of 2.56 pM. The schematic representation of the sensor is shown in Fig. 2.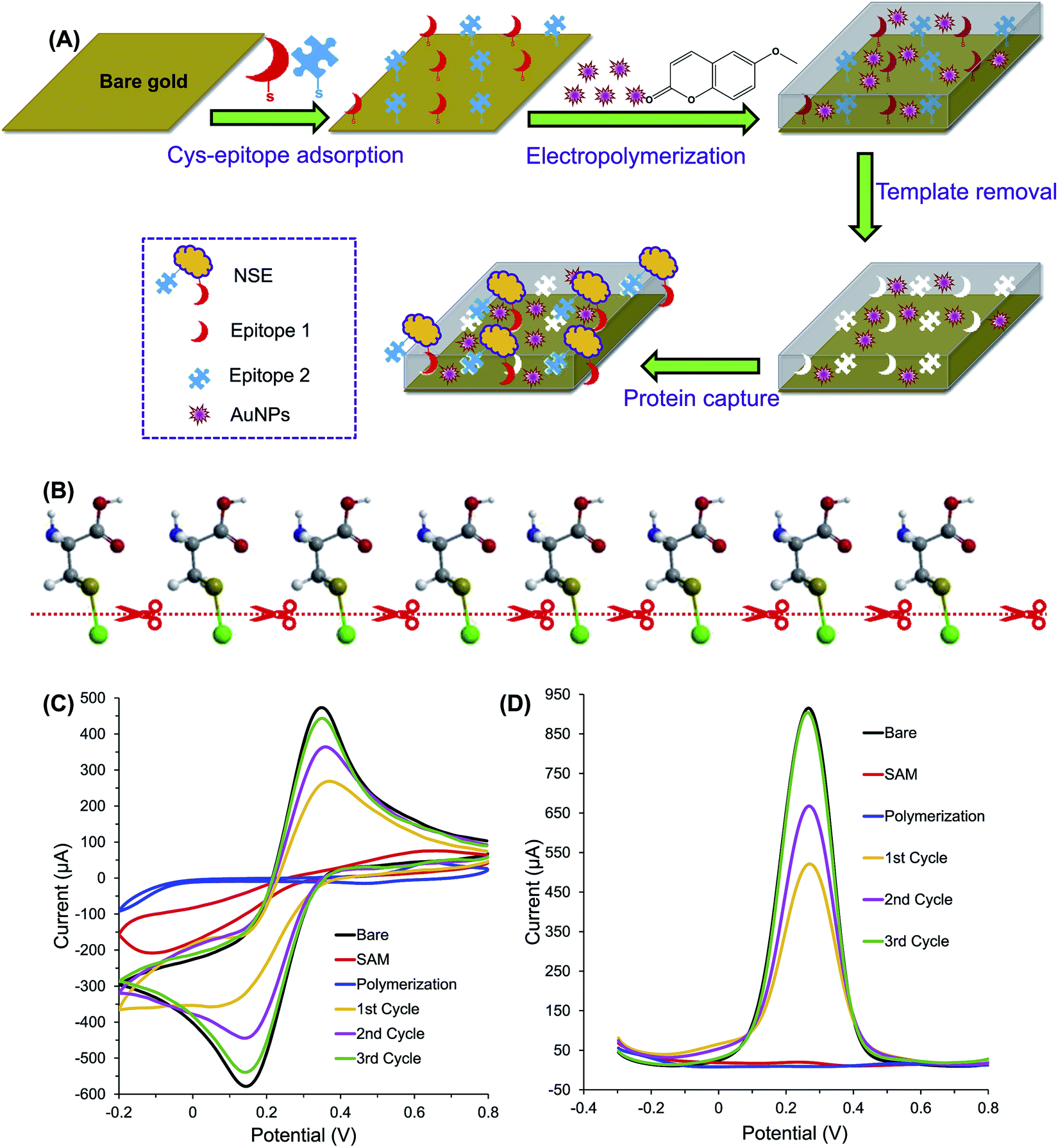 | ||
| Fig. 2 (A) The principle of protein detection using the AuNP-decorated hybrid MIP sensor, which was fabricated by electropolymerization technique in the presence of two different NSE epitopes and AuNPs. (B) Illustration of electrolytic cleavage of thiol bonds of chemisorbed cysteine monolayer and gold (green). The sulfur, carbon, oxygen, nitrogen and hydrogen atoms are represented by golden, dark grey, red, blue and white balls, respectively. (C) CV and (D) SWV optimization of template removal for MIP film. Cycles from 1 to 3 indicate the sequential template removal process using anodic potential application at 1.4 V for 30 s per cycle (this figure has been reproduced from ref. 62 with permission from ELSEVIER, copyright 2020). | ||
Drzazgowska et al. used a different approach to develop an EIP-based electrochemical sensor for detection of NSE.71 The double-cysteine-modified peptide template was assembled on the gold surface via thiol groups of cysteine amino acids on both terminals of the epitope, followed by polymerization. This method has helped form stable epitope template bridges on the electrode surface. The obtained sensor exhibited an enhanced performance within the linear range of 0.125–10 μM, and has LOD of 0.25 μM. In another instance, an electrochemical sensor was fabricated from the EIP prepared with C-terminal polypeptide of insulin as the template and o-PD as the functional monomer.19 The small molecular size of C-insulin polypeptide template reduced the steric hindrance in the recognition process and simplified the elution of the template. The insulin could be detected in the concentration range of 1.0 × 10−14 M to 5.0 × 10−13 M by the sensor with LOD of 7.24 × 10−15 M. Electrochemical detection of insulin was also reported by Cruz et al. However, the need of mediator in conventional electrochemical sensors was eliminated by incorporating ferrocene as a redox probe in the imprinted polymer. EIP was prepared using solid phase synthesis with N-(3-aminopropyl) methacrylamide hydrochloride (NAPMA) as the functional monomer, NIPAAM, N,N′-methylene-bis-acrylamide (MBA), and N-tert-butylacrylamide (TBAM) as the cross-linkers. The sensor could detect insulin in the concentration range of 50 pM to 2000 pM. The LOD of 26 and 81 fM was obtained in buffer and human plasma, respectively.72 Selection of the right peptide epitope can significantly improve the sensor performance. Lee et al. compared three epitopes for preparing EIP targeting alpha-synuclein, the biomarker of the Parkinson's disease. The results showed that AEAAGKTKEGVLY (P3 epitope) yielded the highest imprinting effectiveness.73 On the other hand, the length and hydrophobicity of the peptide templates have a profound effect on the selectivity of EIPs. Lee et al. screened seven different peptides with different ethylene mole fractions during the synthesis of the EIP for regenerating protein 1 (REG1).20 When the peptide templates contained fewer aromatic and hydrophobic amino acids, better EIP was formed. It was also observed that the imprinted films recognized the target peptides well. However, they showed different affinity to the parent protein. Therefore, in the imprinting process, the response of the EIP to target proteins also needs to be optimized.
3.3 Piezoelectric sensors
EIPs were reported to improve the selectivity of the quartz crystal microbalance sensors. Ma et al. employed an EIP-based QCM sensor for detection of HSA.9 This study proposed a simple method by which EIP in ethanol solution was dropped onto the QCM chip and dried to form the sensing layer. The sensitivity towards HSA was within the linear range of 0.75 nM to 7.5 nM with LOD of 0.39 nM. The imprinting factor of EIP in QCM sensor can be improved by the synergistic effects of multiple monomers. Gupta et al. used 3-sulfopropyl methacrylate potassium-salt and benzyl methacrylate to synthesize the EIP for recognition of fbp A.15 The sensor exhibited high binding capacity and specific recognition towards the target with calculated LOD and IF of 38.87 pM and 12.27, respectively.3.4 Applications of EIPs in protein separation
The extraction and purification of proteins is usually required prior to their analysis. Traditional methods mainly depend on solid phase extraction (SPE) and magnetic extraction (ME). However, these methods are still time-consuming, require large volume of solvent, and have poor selectivity.74 EIPs have been explored to improve the efficiency and selectivity of protein separation processes. A summary of EIPs applied for protein separation and extraction is provided in Table 2. Nishino et al.75 developed an EIP film from acrylamide, N,N-ethylene-bis-acrylamide, and polyethylene glycol 200-diacrylate for capturing BSA, Cyt c, and alcohol dehydrogenase (ADH). The film exhibited high selectivity to the target. The amount of Cyt c bound to the EIP film was 22.4 pmol cm−2, corresponding to about 75% coverage of 1 cm2 of the EIP film surface. EIPs have been used in SPE column (MIPSPE) to increase the selectivity of protein extraction. Yang et al. employed EIP-coated silica beads as the sorbents in SPE.76 The column was applied for the analysis of beta 2-microglobulin (β2M) with a recovery of more than 83%. The LOD and LOQ were estimated to be 0.058 mg L−1 and 0.195 mg L−1, respectively. Similarly, EIP was embedded in a micropipette tip by in situ polymerization of MAA for the separation of enkephalins in human cerebrospinal fluid (CSF).77 The EIP, prepared from a tetrapeptide (YGGF) template, had high extraction efficiency for enkephalins. The technique was coupled with MISPME/HPLC-UV to quantify Met-enkephalin and Leu-enkephalin in CSF with LOD ranging from 0.05 nM to 0.08 nM. Gómez-Arribas et al. developed a hierarchically imprinted polymer for separation of FLAG tag via solid-phase synthesis.67 The obtained EIP particles can be used as high-affinity sorbents for solid phase extraction of the FLAG peptide with recovery percentage up to 87.4%. Protein extraction can be facilitated by magnetic nanoparticles (MNPs). MNPs are known for their superparamagnetism, high surface area, large surface-to-volume ratio, and ease of separation under external magnetic fields.75 The incorporation of EIP on MNP support materials can enhance the selectivity and efficiency of protein separation. Zhao et al. developed an EIP synthesized on the surface of core–shell magnetic nanoparticles (Fe3O4@EMIPs) for the recognition of BSA.78 The surface of the MNPs was functionalized with polyvinylpyrrolidone (PVP), and then imprinted with APTES in the presence of C-terminal nonapeptide (VVSTQTALA) of BSA. The adsorption capacity and imprinting factor were determined to be 20.25 mg g−1 and 3.21, respectively. Li et al.79 demonstrated the applications of EIP-coated MNPs for recognition of HSA. The synthesis involved the immobilization of the His-tag modified C-terminal nonapeptide template of HSA on the surface of functionalized Fe3O4 nanoparticles. The polymeric layer was then formed by self-polymerization of dopamine under weakly alkaline conditions. This technique addressed the difficulty of template removal and accessibility of cavities by target proteins, usually encountered when the epitopes were blended with monomer mixture in imprinting process. Moreover, the improved hydrophilicity of the His-tag-anchored epitope can be exploited for imprinting of epitopes with various polarities. In another work, EIP was prepared from cyclodextrin-based ionic liquid as the functional monomer to improve the separation and enrichment of Cyt c.80 The charged monomer cyclodextrins (CDs) (mono-6A-deoxy-6-(1-vinylimidazolium)-β-cyclodextrin tosylate) was found to exhibit good adsorption and recognition of Cyt c. The improved performance was ascribed to the multiple molecular interaction forces such as hydrogen bonds, π–π stacking, electrostatic attraction, hydrophobic, and steric effects between CDs and the template (Fig. 3).| Sensor type | Target | Polymerization method | Template | Length (aa) | Monomer | Cross-linker | Solvent | Eluent | Adsorption capacity or recoveries rate | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| a HAc: acetic acid, Cyt c: cytochrome c, APTES: 3-aminopropyltriethoxysilane, TEOS: tetraethoxysilane, ZnA: zinc(II) acrylate, EGDMA: ethylene glycol dimethacrylate, PBS: phosphate buffer solution, n/a: not available, MBA: methylene bisacrylamide, ME: magnetic extraction, AA: acrylic acid, DMAPMA: N-[3-(dimethylamino)propyl]methacrylamide, EBA: N,N-ethylenebis(acrylamide), HHb: human hemoglobin, MAA: methacrylic acid, ADH: alcohol dehydrogenase, BSA: bovine serum albumin, TFA: trifluoroacetic acid. | ||||||||||
| SPE | PSA | Distillation-precipitation | C-terminus, N-terminus | n/a | ZnA | EGDMA | ACN | MeOH/HAc | 45.05 mg g−1 | 58 |
| FLAG tag | Bulk polymerization | C-terminus | 5 | EAMA | EDMA | n/a | MeOH/TFA | 87.4% | 67 | |
| Beta 2-microglobulin | Bulk polymerization | N-terminus | 9 | APTES | EBA | DMF | CH3CN/HAc | > 83% | 76 | |
| Enkephalins | Bulk polymerization | Enkephalins derived peptide | 4 | MAA | EGDMA | CH3CN/MeOH | CH3CN/H2O | 1.20 mg g−1 | 77 | |
| ME | Cyt c | Precipitation | C-terminus | 9 | Cyclodextrin | MBA | PBS | MeOH/HAc | 67.6 mg g−1 | 80 |
| HSA | n/a | His-tag HSA derived peptide | 9 | AA, DMAPMA | MBA | n/a | EDTA | 52.4 mg g−1 | 6 | |
| HSA | Sol–gel | His-tag HSA C-terminus | 9 | Dopamine | — | PBS | EDTA | 3.11 mg g−1 | 24 | |
| Others | IgG | Photoinitiation | C-terminus | 10 | MAA | EGDMA | H2O | EtOH | 3.9 mg g−1 | 53 |
| HHb | Bulk polymerization | Enzyme-digested peptides | — | MAA | EGDMA | H2O | HAc/SDS | ∼80% | 5 | |
| Cyt c | Photoinitiation | C-terminus | 9 | Acrylamide | Polyethylene glycol 200-diacrylate | PBS | PBS | 22.4 pmol cm−2 | 75 | |
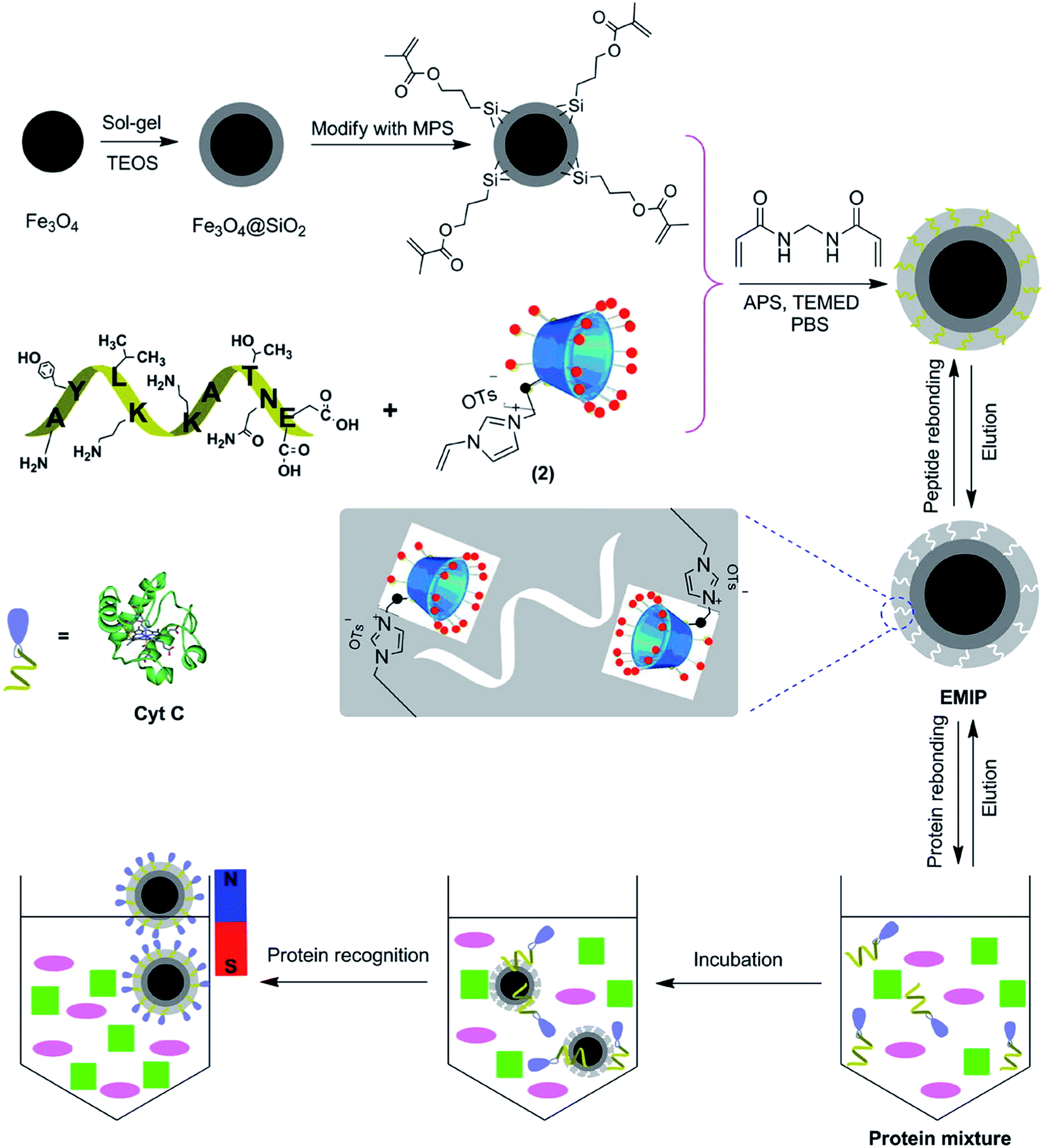 | ||
| Fig. 3 Schematic illustration of synthetic procedure for the Fe3O4@EMIPs and the selective process for the enrichment of Cyt c using Fe3O4@EMIPs microspheres and magnetic separation (this figure has been reproduced from ref. 80 with permission from ELSEVIER, copyright 2020). | ||
4 Conclusions
Epitope imprinted polymers play an increasingly important role in the analysis and separation of protein molecules in biological samples. The field has witnessed significant advances in the development of these materials. However, there remain technological challenges and gaps that should be overcome to facilitate the synthesis as well as the application of EIPs. Molecular modeling should be exploited more effectively to reduce the time required for screening the epitope templates, to optimize the production process and to enhance the sensitivity and selectivity of the EIPs. In addition, the modeling can shed light on the imprinting mechanisms, especially the interaction between the template, monomers, oligomers, and the polymeric matrix. New functional monomers such as NE with straightforward synthetic protocols and improved performance need to be further explored. The reporting capability of the EIPs, such as their electro-activity, should be considered to promote their sensing application. In some medical diagnoses, a single target protein may provide limited information. Therefore, more emphasis should be directed toward the preparation of EIPs for simultaneous detection of protein molecules or biomarkers. With the increasing applications of EIPs, new methods for large-scale synthesis of the materials are required. Solid phase synthesis can be a viable option. However, the current processes need to be automated and simplified for industrial production. The incorporation of EIPs in the state-of-art sensing platforms such as microfluidic or lab-on-a-chip devices should be promoted for better point-of-care analysis.Author contributions
Tabkrich Khumsap: conceptualized and selected the review areas; drafted the original manuscript. Angelica Corpuz: drafted and edited the manuscript. Dr Loc Thai Nguyen: conceptualized the review areas; edited and supervised the preparation of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support for the research was partially provided by Asian Institute of Technology (AIT).References
- G. Wulff, A. Sarhan and K. Zabrocki, Tetrahedron Lett., 1973, 14, 4329–4332 CrossRef.
- L. Chen, X. Wang, W. Lu, X. Wu and J. Li, Chem. Soc. Rev., 2016, 45, 2137–2211 RSC.
- A. Poma, A. P. F. Turner and S. A. Piletsky, Trends Biotechnol., 2010, 28, 629–637 CrossRef CAS PubMed.
- J. Wackerlig and P. A. Lieberzeit, Sens. Actuators, B, 2015, 207, 144–157 CrossRef CAS.
- H. Bagan, RSC Adv., 2017, 7, 41705–41712 RSC.
- S. Li, K. Yang, N. Deng, Y. Min, L. Liu, L. Zhang and Y. Zhang, ACS Appl. Mater. Interfaces, 2016, 8, 5747–5751 CrossRef CAS PubMed.
- Y. Q. Yang, X. W. He, Y. Z. Wang, W. Y. Li and Y. K. Zhang, Biosens. Bioelectron., 2014, 54, 266–272 CrossRef CAS PubMed.
- B. Alberts, A. Johnson, J. Lewis, M. Raff, K. Roberts and P. Walter, Molecular Biology of the Cell, Meiosis, 4th edn, Garland Science, New York, 2002 Search PubMed.
- X. T. Ma, X. W. He, W. Y. Li and Y. K. Zhang, Sens. Actuators, B, 2017, 246, 879–886 CrossRef CAS.
- H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov and P. E. Bourne, Nucleic Acids Res., 2000, 28, 235–242 CrossRef CAS PubMed.
- A. Bateman, M. J. Martin, C. O'Donovan, M. Magrane, E. Alpi, R. Antunes, B. Bely, M. Bingley, C. Bonilla, R. Britto, B. Bursteinas, H. Bye-AJee, A. Cowley, A. Da Silva, M. De Giorgi, T. Dogan, F. Fazzini, L. G. Castro, L. Figueira, P. Garmiri, G. Georghiou, D. Gonzalez, E. Hatton-Ellis, W. Li, W. Liu, R. Lopez, J. Luo, Y. Lussi, A. MacDougall, A. Nightingale, B. Palka, K. Pichler, D. Poggioli, S. Pundir, L. Pureza, G. Qi, S. Rosanoff, R. Saidi, T. Sawford, A. Shypitsyna, E. Speretta, E. Turner, N. Tyagi, V. Volynkin, T. Wardell, K. Warner, X. Watkins, R. Zaru, H. Zellner, I. Xenarios, L. Bougueleret, A. Bridge, S. Poux, N. Redaschi, L. Aimo, G. ArgoudPuy, A. Auchincloss, K. Axelsen, P. Bansal, D. Baratin, M. C. Blatter, B. Boeckmann, J. Bolleman, E. Boutet, L. Breuza, C. Casal-Casas, E. De Castro, E. Coudert, B. Cuche, M. Doche, D. Dornevil, S. Duvaud, A. Estreicher, L. Famiglietti, M. Feuermann, E. Gasteiger, S. Gehant, V. Gerritsen, A. Gos, N. Gruaz-Gumowski, U. Hinz, C. Hulo, F. Jungo, G. Keller, V. Lara, P. Lemercier, D. Lieberherr, T. Lombardot, X. Martin, P. Masson, A. Morgat, T. Neto, N. Nouspikel, S. Paesano, I. Pedruzzi, S. Pilbout, M. Pozzato, M. Pruess, C. Rivoire, B. Roechert, M. Schneider, C. Sigrist, K. Sonesson, S. Staehli, A. Stutz, S. Sundaram, M. Tognolli, L. Verbregue, A. L. Veuthey, C. H. Wu, C. N. Arighi, L. Arminski, C. Chen, Y. Chen, J. S. Garavelli, H. Huang, K. Laiho, P. McGarvey, D. A. Natale, K. Ross, C. R. Vinayaka, Q. Wang, Y. Wang, L. S. Yeh and J. Zhang, Nucleic Acids Res., 2017, 45, 158–169 Search PubMed.
- A. M. Bossi, P. S. Sharma, L. Montana, G. Zoccatelli, O. Laub and R. Levi, Anal. Chem., 2007, 84, 4036–4041 CrossRef PubMed.
- A. Bossi, F. Bonini, A. P. F. Turner and S. A. Piletsky, Biosens. Bioelectron., 2007, 22, 1131–1137 CrossRef CAS PubMed.
- Y. P. Qin, D. Y. Li, X. W. He, W. Y. Li and Y. K. Zhang, ACS Appl. Mater. Interfaces, 2016, 8, 10155–10163 CrossRef CAS PubMed.
- N. Gupta, R. S. Singh, K. Shah, R. Prasad and M. Singh, J. Mol. Recognit., 2018, 31, 1–9 CrossRef PubMed.
- S. Li, K. Yang, N. Deng, Y. Min, L. Liu, L. Zhang and Y. Zhang, ACS Appl. Mater. Interfaces, 2016, 8, 5747–5751 CrossRef CAS PubMed.
- I. Losito, F. Palmisano and P. G. Zambonin, Anal. Chem., 2003, 75, 4988–4995 CrossRef CAS.
- M. X. Li, X. H. Wang, L. M. Zhang and X. P. Wei, Anal. Biochem., 2017, 530, 68–74 CrossRef CAS PubMed.
- Z. Cheng-jun, M. A. Xiong-hui and L. I. Jian-ping, Chin. J. Anal. Chem., 2017, 45, 1360–1366 Search PubMed.
- M. H. Lee, J. L. Thomas, C. L. Liao, S. Jurcevic, T. Crnogorac-Jurcevic and H. Y. Lin, Sep. Purif. Technol., 2018, 192, 213–219 CrossRef CAS.
- M. J. LaVoie, B. L. Ostaszewski, A. Weihofen, M. G. Schlossmacher and D. J. Selkoe, Nat. Med., 2005, 11, 1214–1221 CrossRef CAS PubMed.
- P. Palladino, M. Minunni and S. Scarano, Biosens. Bioelectron., 2018, 106, 93–98 CrossRef CAS PubMed.
- C. H. Lu, Y. Zhang, S. F. Tang, Z. Bin Fang, H. H. Yang, X. Chen and G. N. Chen, Biosens. Bioelectron., 2012, 31, 439–444 CrossRef CAS PubMed.
- S. Li, K. Yang, J. Liu, B. Jiang, L. Zhang and Y. Zhang, Anal. Chem., 2015, 87, 4617–4620 CrossRef CAS PubMed.
- Y. Li, L. Huang, X. Wang and Y. Chen, Anal. Sci., 2017, 33, 1105–1110 CrossRef CAS PubMed.
- D. Riveros, G. K. Cordova, C. Michiels, H. Verachtert and G. Derdelinckx, Talanta, 2016, 160, 761–767 CrossRef PubMed.
- V. Baldoneschi, P. Palladino, M. Banchini, M. Minunni and S. Scarano, Biosens. Bioelectron., 2020, 157, 112161 CrossRef CAS PubMed.
- Y. H. Ding, M. Floren and W. Tan, Biosurf. Biotribol., 2016, 2, 121–136 CrossRef CAS PubMed.
- L. Jiang, R. Lu and L. Ye, Polymers, 2019, 11, 173 CrossRef PubMed.
- R. Xing, Y. Ma, Y. Wang, Y. Wen and Z. Liu, Chem. Sci., 2019, 10, 1831–1835 RSC.
- A. Beltran, F. Borrull, R. M. Marcé and P. A. G. Cormack, TrAC, Trends Anal. Chem., 2010, 29, 1363–1375 CrossRef CAS.
- D. R. Kryscio and N. A. Peppas, Acta Biomater., 2012, 8, 461–473 CrossRef CAS PubMed.
- A. Rachkov and N. Minoura, J. Chromatogr. A, 2000, 889, 111–118 CrossRef CAS.
- K. Li and H. D. H. Stöver, J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 3257–3263 CrossRef CAS.
- J. Wang, P. A. G. Cormack, D. C. Sherrington and E. Khoshdel, Angew. Chem., Int. Ed., 2003, 42, 5336–5338 CrossRef CAS PubMed.
- F. G. Tamayo, E. Turiel and A. Martín-Esteban, J. Chromatogr. A, 2007, 1152, 32–40 CrossRef CAS PubMed.
- P. Li, F. Rong and C. Yuan, Polym. Int., 2003, 52, 1799–1806 CrossRef CAS.
- J. Wang, P. A. G. Cormack, D. C. Sherrington and E. Khoshdel, Pure Appl. Chem., 2007, 79, 1505–1519 CAS.
- K. Yoshimatsu, K. Reimhult, A. Krozer, K. Mosbach, K. Sode and L. Ye, Anal. Chim. Acta, 2007, 584, 112–121 CrossRef CAS PubMed.
- C. Rossetti, M. A. Switnicka-Plak, T. Grønhaug Halvorsen, P. A. G. Cormack, B. Sellergren and L. Reubsaet, Sci. Rep., 2017, 7, 44298 CrossRef CAS PubMed.
- X. Jiang, W. Tian, C. Zhao, H. Zhang and M. Liu, Talanta, 2007, 72, 119–125 CrossRef CAS PubMed.
- H. Bagheri and H. Piri-Moghadam, Anal. Bioanal. Chem., 2012, 404, 1597–1602 CrossRef CAS PubMed.
- A. Fernández-González, R. Badía-Laíño and M. E. Díaz-García, Microchim. Acta, 2011, 172, 351–356 CrossRef.
- M. E. Brown and D. A. Puleo, Chem. Eng. J., 2011, 172, 351–356 Search PubMed.
- K. J. Jetzschmann, G. Jágerszki, D. Dechtrirat, A. Yarman, N. Gajovic-Eichelmann, H. D. Gilsing, B. Schulz, R. E. Gyurcsányi and F. W. Scheller, Adv. Funct. Mater., 2015, 25, 5178–5183 CrossRef CAS.
- M. Bosserdt, J. Erdossy, G. Lautner, J. Witt, K. Köhler, N. Gajovic-Eichelmann, A. Yarman, G. Wittstock, F. W. Scheller and R. E. Gyurcsányi, Biosens. Bioelectron., 2015, 73, 123–129 CrossRef CAS PubMed.
- G. Ceolin, Á. Orbán, V. Kocsis, R. E. Gyurcsányi, I. Kézsmárki and V. Horváth, J. Mater. Sci., 2013, 45, 5209–5218 CrossRef.
- M. Teresa, S. Petersen and G. Prakash, in Molecular Photochemistry – Various Aspects, 2012 Search PubMed.
- M. J. Davies, Biochem. J., 2016, 473, 805–825 CrossRef CAS PubMed.
- J. Erdossy, V. Horváth, A. Yarman, F. W. Scheller and R. E. Gyurcsányi, TrAC, Trends Anal. Chem., 2016, 79, 179–190 CrossRef CAS.
- P. S. Sharma, A. Pietrzyk-Le, F. D'Souza and W. Kutner, Anal. Bioanal. Chem., 2012, 402, 3177–3204 CrossRef CAS PubMed.
- R. Tchinda, A. Tutsch, B. Schmid, R. D. Süssmuth and Z. Altintas, Biosens. Bioelectron., 2019, 123, 260–268 CrossRef CAS PubMed.
- S. Schwark, W. Sun, J. Stute, D. Lütkemeyer, M. Ulbricht and B. Sellergren, RSC Adv., 2016, 6, 53162–53169 RSC.
- R. A. Lorenzo, A. M. Carro, C. Alvarez-Lorenzo and A. Concheiro, Int. J. Mol. Sci., 2011, 12, 4327–4347 CrossRef CAS PubMed.
- S. Wu, J. Brandrup, E. H. Immergut, E. A. Grulke, A. Abe and D. R. Bloch, Polymer Handbook, 2005, vol. 7 Search PubMed.
- A. L. Hillberga, K. R. Braina and C. J. Allendera, J. Mol. Recognit., 2009, 22, 223–231 CrossRef PubMed.
- B. Schmid, Z. Altintas, R. Tchinda, R. D. Süssmuth and A. Tutsch, Biosens. Bioelectron., 2018, 123, 260–268 Search PubMed.
- Y. P. Qin, H. Y. Wang, X. W. He, W. Y. Li and Y. K. Zhang, Talanta, 2018, 185, 620–627 CrossRef CAS PubMed.
- X. M. Zhang, Y. P. Qin, H. L. Ye, X. T. Ma, X. W. He, W. Y. Li and Y. K. Zhang, Microchim. Acta, 2018, 185, 1–9 CrossRef CAS PubMed.
- M. D. Luque de Castro and F. Priego-Capote, J. Chromatogr. A, 2010, 1217, 2383–2389 CrossRef CAS PubMed.
- M. Yang and Y. Mine, in Egg Bioscience and Biotechnology, 2007, pp. 239–288 Search PubMed.
- M. Pirzada, E. Sehit and Z. Altintas, Biosens. Bioelectron., 2020, 166, 112464 CrossRef CAS PubMed.
- A. Kowalczyk, E. Matysiak-Brynda and A. M. Nowicka, Curr. Opin. Electrochem., 2019, 14, 44–52 CrossRef CAS.
- A. Poma, A. Guerreiro, M. J. Whitcombe, E. V. Piletska, A. P. F. Turner and S. A. Piletsky, Adv. Funct. Mater., 2013, 23, 2821–2827 CrossRef CAS PubMed.
- F. Canfarotta, A. Poma, A. Guerreiro and S. Piletsky, Nat. Protoc., 2016, 11, 443–455 CrossRef CAS PubMed.
- R. Xing, S. Wang, Z. Bie, H. He and Z. Liu, Nat. Protoc., 2017, 12, 964–987 CrossRef CAS PubMed.
- L. N. Gómez-Arribas, M. D. M. Darder, N. García, Y. Rodriguez, J. L. Urraca and M. C. Moreno-Bondi, ACS Appl. Mater. Interfaces, 2020, 12, 49111–49121 CrossRef.
- J. M. Klostranec and W. C. W. Chan, Adv. Mater., 2006, 18, 1953–1964 CrossRef CAS.
- Y. J. Yan, X. W. He, W. Y. Li and Y. K. Zhang, Biosens. Bioelectron., 2017, 91, 253–261 CrossRef CAS PubMed.
- Y. Z. Wang, D. Y. Li, X. W. He, W. Y. Li and Y. K. Zhang, Microchim. Acta, 2015, 182, 1465–1472 CrossRef CAS.
- J. Drzazgowska, B. Schmid, R. D. Sussmuth and Z. Altintas, Anal. Chem., 2020, 92, 4798–4806 CrossRef CAS PubMed.
- A. Garcia Cruz, I. Haq, T. Cowen, S. Di Masi, S. Trivedi, K. Alanazi, E. Piletska, A. Mujahid and S. A. Piletsky, Biosens. Bioelectron., 2020, 169, 112536 CrossRef CAS.
- M. H. Lee, J. L. Thomas, Z. L. Su, W. K. Yeh, A. S. Monzel, S. Bolognin, J. C. Schwamborn, C. H. Yang and H. Y. Lin, Biosens. Bioelectron., 2021, 175, 112852 CrossRef CAS.
- A. Speltini, A. Scalabrini, F. Maraschi, M. Sturini and A. Profumo, Anal. Chim. Acta, 2017, 974, 1–26 CrossRef CAS PubMed.
- H. Nishino, C. S. Huang and K. J. Shea, Angew. Chem., Int. Ed., 2006, 45, 2392–2396 CrossRef CAS PubMed.
- F. Yang, D. Deng, X. Dong and S. Lin, J. Chromatogr. A, 2017, 1494, 18–26 CrossRef CAS.
- H. Li and D. Li, J. Pharm. Biomed. Anal., 2015, 115, 330–338 CrossRef CAS PubMed.
- X. L. Zhao, D. Y. Li, X. W. He, W. Y. Li and Y. K. Zhang, J. Mater. Chem. B, 2014, 2, 7575–7582 RSC.
- S. Li, K. Yang, J. Liu, B. Jiang, L. Zhang and Y. Zhang, Anal. Chem., 2015, 87, 4617–4620 CrossRef CAS PubMed.
- X. Zhang, N. Zhang, C. Du, P. Guan, X. Gao, C. Wang, Y. Du, S. Ding and X. Hu, Chem. Eng. J., 2017, 317, 988–998 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |