
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBoosting oxygen evolution reaction activity by tailoring MOF-derived hierarchical Co–Ni alloy nanoparticles encapsulated in nitrogen-doped carbon frameworks†
Xiaobin Liu,
Xudong Zhao and
Li-Zhen Fan *
*
Beijing Advanced Innovation Center for Materials Genome Engineering, Institute of Advanced Materials and Technology, University of Science and Technology Beijing, Beijing 100083, China. E-mail: fanlizhen@ustb.edu.cn
First published on 15th March 2021
Abstract
The growing demand for sustainable energy has led to in-depth research on hydrogen production from electrolyzed water, where the development of electrocatalysts is a top priority. We here report a controllable strategy for preparing the cobalt–nickel alloy nanoparticles encapsulated in nitrogen-doped porous carbon by annealing a bimetal–organic framework. The delicately tailored hierarchical Co2Ni@NC nanoparticles effectively realize abundant synergistic active sites and fast mass transfer for the oxygen evolution reaction (OER). Remarkably, the optimized Co2Ni@NC exhibits a small overpotential of 310 mV to achieve a current density of 10 mA cm−2 and an excellent long-term stability in alkaline electrolyte. Furthermore, the underlying synergistic effect mechanism of the Co–Ni model has been pioneeringly elucidated by density functional theory calculations.
1. Introduction
Moving from fossil fuels to renewable energy is undoubtedly the biggest scientific challenge of our time.1,2 Owing to the pollution free and easy recycling attributes, hydrogen has great potential to be an alternative fuel in the future. Water electrolysis is one of the efficient and promising ways to obtain hydrogen in large quantity.3,4 Nevertheless, the oxygen evolution reaction (OER) process severely limits the efficiency of overall water splitting because of the sluggish kinetics and large overpotential.5 Currently, iridium and ruthenium oxides have been proved to be efficient electrocatalysts for OER, but their limited reserves and high costs are great hindrances.6,7 Despite some progress in recent years, the synthesis of highly-efficient and low-cost non-noble metal alternatives that can compare favorably with noble catalysts remains a challenge.8,9In recent years, some bimetallic alloys (e.g., FeCo, FeNi, and CoNi) have been demonstrated to possess better electrocatalytic activities than their individual entities for OER, because the combination of two metals can form inherent polarity to introduce synergetic effects.10–12 Among them, CoNi alloy stands out on account of their features of low cost and good environmental friendliness.13 According to the previous reports, in an alkaline solution, nickel is more likely to desorb OH− than cobalt, and cobalt is more helpful in increasing the activity of the Tafel step.14 In addition, since the freedom degree of Co–Ni alloy is greater than that of pure metal, more opportunities can be provided to optimize the catalytic activity by regulating the proportion of the metal in the alloy.9,15 However, rational design and controlled synthesis of homogeneous CoNi alloy catalyst with high-activity is still a big challenge: (1) the synthesis process of the porous hierarchical or micro–nano structure is complex and tedious; (2) precise control of multiple phases produced by two metal centers is difficult to achieve; (3) the heteroatoms (N, P, S) are usually doped into carbon to enrich active sites through an additional step, which tends to trigger agglomeration and non-uniform distribution of the active material after the carbonization treatment, resulting in unevenness and inadequate exposure of active sites. To address these problems, the development of a general manufacturing strategy to achieve better catalytic performance is highly desired.
Metal–organic frameworks (MOFs), a family of ordered porous materials consisting of metal center nods and organic ligand linkers, have been proved to be an ideal template to fabricate high-efficiency electrocatalysts.16,17 On one hand, by using the guiding effect of coordination bond, the MOF structure with uniform size and controllable morphology can be constructed efficiently. After pyrolysis in an inert atmosphere, shrinkage and reconstruction are performed based on the MOF precursor to form the hierarchical nanostructures, which provide large surface area and facilitate fast mass transfer. On the other hand, benefiting from the good compatibility of MOF with various metal ions, the synthesis of homogeneous bimetallic MOF precursors is achievable, leading to the highly uniform dispersion of active sites in atomic level.18 Apart from the controllable morphology and compatible metal ions, the as-prepared N-containing bimetal MOFs offer the opportunity to fabricate active materials coated by N-doped carbon (NC) without additional nitrogen source.19 Bearing these in mind, we develop a controllable synthesis strategy for the construction of cobalt–nickel alloy nanoparticles via annealing bimetallic MOF precursors in argon. The optimized Co2Ni@NC integrates the nanostructure engineering and component modulation together, realizing abundant synergistic active sites and fast mass transfer towards high-activity OER. As a result, it requires a small overpotential of 310 mV to achieve a current density of 10 mA cm−2 and exhibits an excellent durability in alkaline electrolyte.
2. Experimental
2.1. Chemicals
Nickel(II) nitrate hexahydrate (Ni(NO3)2·6H2O, 98%), cobalt(II) nitrate hexahydrate (Co(NO3)2·6H2O, 98.5%), 1,4-benzenedicarboxylic acid (H2BDC, 99%), 4,4′-bipyridine (4,4′-bipy, 98%), sodium hydroxide (NaOH, 96%) were purchased from Sinopharm Chemical Reagent Co. Ltd (Shanghai, China). Commercial iridium oxide (IrO2, 99.9%) was purchased from Aladdin (Shanghai, China). Nafion (5 wt%) was purchased from Sigma-Aldrich (Beijing, China). The water for experiments was deionized (DI) water (18 MΩ). Other chemicals and solvents used in this work were obtained from commercial suppliers and used without further purification.2.2. Materials synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 1:1 and 1:2, named as Co2Ni-MOF, CoNi-MOF and CoNi2-MOF, respectively.
:1, 1:1 and 1:2, named as Co2Ni-MOF, CoNi-MOF and CoNi2-MOF, respectively.2.3. Materials characterizations
The morphology feature was analyzed by scanning electron microscopy (SEM, JEOL JSM-6330 and QUANTA FEG 450) and transmission electron microscopy (TEM, JEM-2010F). The chemical composition was characterized by energy dispersive spectrometer (EDS) attached to QUANTA FEG 450. The phase composition was characterized by powder X-ray diffraction (PXRD, Rigaku D/max-RB). X-ray photoelectron spectra (XPS) were collected by EscaLab 250Xi Instrument. Raman spectrum was tested by LabRAM HR Evolution. Nitrogen adsorption–desorption measurement was conducted at 77 K with an autosorb iQ automated gas sorption analyzer (Micromeritics ASAP 2460).2.4. Preparation of working electrode
A glassy carbon (GC) electrode (3 mm in diameter, surface area = 0.07065 cm2) was used as the working electrode. Typically, 5 mg sample was dispersed in a solution containing 450 μL ethanol, 500 μL deionized water and 50 μL Nafion solution by sonication for 1 hour to form a homogeneous ink. Then, 5 μL well-dispersed catalysts was loaded onto a GC electrode and dried at room temperature.2.5. Electrochemical measurements
Electrochemical measurements were performed in a three-electrode setup at room temperature using an electrochemical workstation (CHI 660C). A Ag/AgCl (KCl saturated) electrode and a graphite rod were as the reference electrode and the counter electrode, respectively, and 1.0 M KOH was used as the electrolyte. The catalyst was cycled 50 times by CV until a stable CV curve was obtained before testing. Potentials were referenced to a reversible hydrogen electrode (RHE): ERHE = EAg/AgCl + 0.197 + 0.059 pH. The overpotential (η) was calculated according to the following formula: η = ERHE − 1.23 V. Linear sweep voltammetry (LSV) was recorded at a scan rate of 5 mV s−1 to obtain the polarization curves. EIS was performed at a given potential with frequency from 0.1 to 100000 Hz. Long-term stability tests were conducted by continuous cyclic voltammetry scans with a sweep rate of 100 mV s−1, and time dependent current density curves tested by chronopotentiometry method at overpotential of 310 mV vs. RHE. The electrochemically active surface areas (ECSAs) are usually tested from the electrochemical double-layer capacitance (Cdl) by collecting cyclic voltammograms (CVs). The Cdl was estimated from cyclic voltammograms measured in a non-faradaic region at different scan rates (20, 40, 60 and 80 mV s−1) in the potential range from 0.94–1.04 V vs. RHE. Cdl values were evaluated using the equation as follows: Cdl = Ic/ν, where Cdl is the double-layer capacitance (mF cm−2), Ic is the charging current (mA cm−2), ν is the scan rate (mV s−1).
2.6. Computational details
The projector augmented wave (PAW) method20 and Perdew, Burke, and Ernzerhof (PBE) functional21 were adopted in the Vienna ab initio simulation package (VASP).22–24 The energy cutoff was set to 500 eV (8 × 8 × 1) Monkhorst–Pack scheme k-point mesh were generated for (111) surface. The convergence energy threshold for the electronic self-consistent loop was set to 10−5 eV, while that for the force convergence tolerance was set to 0.01 eV Å−1. DFT-D3 method25 was employed to deal with the weak vdW force between adsorbate and (111) surface. Spin polarization was considered in all computations. The free energy diagrams of the OER were calculated according to the method developed by Nørskov:26| ΔG = ΔE + ΔEZPE − TΔS + ΔGU + ΔGpH |
ln10 × pH and pH = 14 was considered. The free energy of gas H2O at 0.035 bar was used as that of the liquid H2O at 298.15 K. The free energy of O2 was obtained from the free energy change of the reaction O2 + 2H2 → 2H2O, which is −4.92 eV at 298.15 K.
For the OER overpotentials, a method developed by Nørskov was applied:27
 ln10 × pH.
ln10 × pH.
3. Results and discussion
The preparation process of hierarchical Co2Ni@NC nanoparticles is displayed in Fig. 1. In a typical experiment, the CoNi-based bimetallic MOF (denoted as CoxNiy-MOF), was synthesized with Co(NO3)2·6H2O, Ni(NO3)2·6H2O, 1,4-benzenedicarboxylic acid (H2BDC) and 4,4′-bipyridine (4,4′-bipy). The similar ionic radius and coordination mode of cobalt and nickel ions enable them to be homogeneously incorporated into the CoxNiy-MOF framework. As shown in Fig. 2a, each metal center contains x Co(II) and y (y = 1 − x) Ni(II) cations, which coordinate with two oxygen atoms from one H2BDC and two nitrogen atoms from two 4,4′-bipy ligands, to form a distorted octahedral geometry. By adjusting the concentrations of cobalt nitrate and nickel nitrate in the reactants, Co/Ni atom molar ratio in CoxNiy-MOF can be accurately controlled. X-ray diffraction (XRD) pattern suggests that the as-prepared CoxNiy-MOFs have very alike diffraction peaks and all can coincide well with the simulated CoNi-MOF (Fig. 2b).28 The obvious color change from brown to green further illustrates the structure transformation of heterogeneous isomorphism from Co-MOF to Ni-MOF.29 It can be clearly observed that the as-synthesized Co2Ni-MOF has micron-sized rod-like framework with well-defined smooth surfaces (Fig. S1†). Subsequently, plentiful N-doped carbon coated nanoparticles were gained based on the original rod-like framework after the annealing treatment, resulting in the well-designed hierarchical Co2Ni@NC architectures (Fig. 2c and d). The SEM energy dispersive spectrometer (EDS) mapping images indicate that the elements of Co, Ni, C and N are uniformly distributed in the Co2Ni@NC (Fig. 2e). Transmission electron microscopy (TEM) images further verify that the Co–Ni alloy nanoparticles with N-doped carbon coating are homogenously inlaid into a rod-like framework (Fig. 2f and g). From the high-resolution transmission electron microscopy (HRTEM) image in Fig. 2h, the clear crystal lattice fringes with a spacing of 0.207 nm correspond to the (111) plane of Co (0.2046 nm, JCPDS card no. 15-0806) and Ni (0.2034 nm, JCPDS card no. 65-2865).13 The selected-area electron diffraction (SAED) pattern displays bright rings corresponding to the (111) and (220) planes of Co and Ni, further proving the polycrystalline character of Co2Ni@NC (Fig. 2i).30 Additionally, Co2Ni@NC-T (T = 600, 700, 900 and 1000 °C) samples derived from Co2Ni-MOF were prepared to investigate the effect of annealing temperature. The SEM images of CoxNiy@NC and Co2Ni@NC-T samples with similar morphology are shown in Fig. S2 and S3.† | ||
| Fig. 1 Schematic illustration of the preparation process of hierarchical Co2Ni@NC nanoparticles derived from bimetal–organic framework with boosting OER activity. | ||
 | ||
| Fig. 2 (a) The crystal structure and (b) XRD patterns of CoxNiy-MOF. (c and d) SEM, (e) EDS element mapping (Co, Ni, C and N), (f and g) TEM, (h) HRTEM images and (i) SAED pattern of Co2Ni@NC. | ||
The phase characteristics of CoxNiy@NC are determined by XRD patterns (Fig. 3a). Each cobalt or nickel metal atom has a coordination number of twelve, showing a face-centered cubic stack geometry (Fig. S4†). The diffraction peaks of Ni@NC positioned at 44.5, 51.8, and 76.4° are attributed to the (111), (200), and (220) planes of Ni (JCPDS card no. 65-2865), respectively. As the Co/Ni molar ratio increases, the metallic Co (JCPDS card no. 15-0806) is finally obtained.31 During this process, the diffraction peak of the (220) plane shifts from 76.4 to 75.8°, indicating the formation of Co–Ni alloy. Fig. S5† exhibits the XRD patterns of the Co2Ni@NC-T samples, similar diffraction characteristics to those of the Co2Ni@NC can be observed. The porous feature of Co2Ni@NC was estimated by Brunauer–Emmett–Teller (BET) measurement (Fig. S6†). The specific area is 236.6 m2 g−1 and the average pore-size distribution is concentrated at 8.31 nm. The hierarchical porosity of Co2Ni@NC with high surface area guarantees fast electron transfer and affords sufficient active sites for oxygen evolution. X-ray photoelectron spectroscopy (XPS) measurements were used to investigate the chemical constitutions and valence states of the Ni@NC, Co2Ni@NC and Co@NC catalysts. Peaks corresponding to Co, Ni, N, C and O can be clearly observed in the full spectra shown in Fig. S7.†
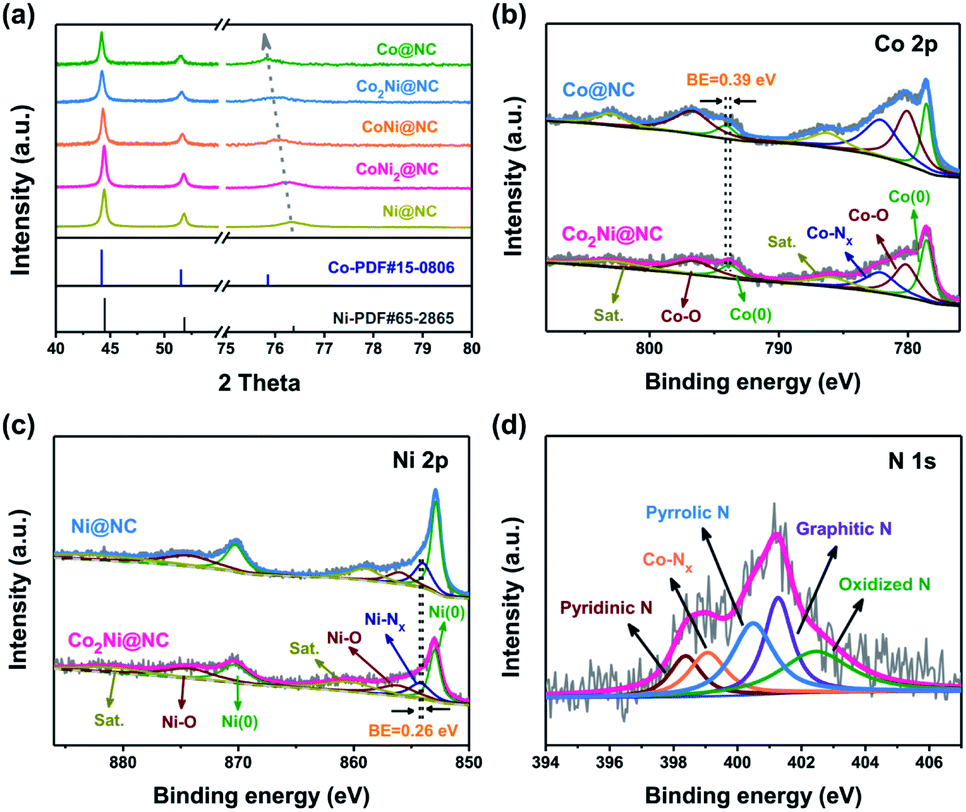 | ||
| Fig. 3 (a) XRD patterns of CoxNiy@NC. High-resolution XPS spectra of (b) Co 2p in Co@NC and Co2Ni@NC, (c) Ni 2p in Ni@NC and Co2Ni@NC, and (d) N 1s in Co2Ni@NC. | ||
High-resolution Co 2p XPS spectrum of Co2Ni@NC is displayed in Fig. 3b. Three important Co-related species including metallic Co, Co–O and Co–Nx are fitted and analyzed. Two pairs of peaks at 778.5/793.8 eV and 780.2/796.6 eV are assigned to the 2p3/2/2p1/2 doublets of metallic Co and Co–O bond, respectively.32 Similar to the Co species, metallic Ni, Ni–O and Ni–Nx signals are characterized in the Ni 2p spectrum of Co2Ni@NC (Fig. 3c). Two pairs of peaks at 853.0/870.4 eV and 856.3/874.4 eV are ascribed to the 2p3/2/2p1/2 doublets of metallic Ni and Ni–O bond, respectively.33 Additionally, two pairs of peaks at 785.9/802.7 and 860.6/880.8 eV are fitted to be satellites due to the shakeup excitation of Co2+ and Ni2+, respectively.32,33 The peaks at 782.2 and 854.3 eV indicate the presence of Co–Nx/Ni–Nx, which has been identified as one of the best active sites for OER.34,35 For Co2Ni@NC, owing to the enhanced electron transfer and strong interactions between Co and Ni, the binding energy of metallic Co exhibits a 0.39 eV positive shift compares to Co@NC, while the binding energy of Ni–Nx exhibits a 0.26 eV negative shift in comparison with Ni@NC.36 N 1s spectrum of Co2Ni@NC can be fitted into five peaks at 398.4, 399.1, 400.5, 401.3 and 402.4 eV, which are attributed to the pyridinic N, Co–Nx, pyrrolic N, graphitic N and oxidized N, respectively (Fig. 3d).19 According to the area percentages of the above five peaks, the content percentages of the five nitrogen types are 11.3%, 14.2%, 26.1%, 24.1% and 24.3%, respectively. It is well known that doping graphitic N atoms can offer p-electrons to the π-conjugated sp2 carbon system, thus enhancing the electronic conductivity of nanostructures. At the same time, the pyridinic-N and pyrrolic-N can create some defects in the nanostructures, promoting the fast mass transfer.37 In addition, the observation of Co–Nx bond demonstrates that the Co atoms are indeed doped into carbon structure and bonded with the nitrogen. The C 1s peak of Co2Ni@NC can be deconvoluted into four peaks located at 284.6, 285.2, 286.3, and 289.9 eV, relating to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, CN, C–O, and CO bonds, respectively (Fig. S8a†).38 The presence of CN bond verifies the formation of N-doped carbon coating, which is favorable to improve the inherent electronic conductivity of materials.19 The O element from carboxy group may be also a good source to form O doping in carbon for improving electrocatalytic activity (Fig. S8b†).39 Raman spectrum was used to further confirm the carbon types of Co2Ni@NC (Fig. S9†). Two peaks at 1340 and 1581 cm−1 are ascribed to the sp2-type D band and G band, respectively. The other two peaks locate at 1065 and 1496 cm−1 are indexed to the sp3-type carbon. By calculation, the intensity ratio of the D-band to the G-band (ID/IG) is 0.98, indicating the high degree of graphitization. The intact area ratio of sp3 to sp2 (Asp3/Asp2) is 65.7%, which suggests a high sp2-type carbon content for the high electronic conductivity.40
C, CN, C–O, and CO bonds, respectively (Fig. S8a†).38 The presence of CN bond verifies the formation of N-doped carbon coating, which is favorable to improve the inherent electronic conductivity of materials.19 The O element from carboxy group may be also a good source to form O doping in carbon for improving electrocatalytic activity (Fig. S8b†).39 Raman spectrum was used to further confirm the carbon types of Co2Ni@NC (Fig. S9†). Two peaks at 1340 and 1581 cm−1 are ascribed to the sp2-type D band and G band, respectively. The other two peaks locate at 1065 and 1496 cm−1 are indexed to the sp3-type carbon. By calculation, the intensity ratio of the D-band to the G-band (ID/IG) is 0.98, indicating the high degree of graphitization. The intact area ratio of sp3 to sp2 (Asp3/Asp2) is 65.7%, which suggests a high sp2-type carbon content for the high electronic conductivity.40
The electrocatalytic activities of a series of CoxNiy@NC samples for OER were investigated utilizing a typical three-electrode system in alkaline electrolyte (1.0 M KOH). Fig. 4a exhibits the linear sweep voltammetry (LSV) profiles of CoxNiy@NC. For comparison, the commercial IrO2 catalyst was also tested. It is worth mentioning that Co2Ni@NC reveals the best catalytic performance and the increase or decrease of Co doping content will affect the performance of OER. By precisely adjusting the Co–Ni ratio in the alloy, the charge transfer between the catalyst metal atoms can be accelerated, and a stronger synergy between the two metal centers can be achieved, thereby improving the performance of OER.9,36 As shown in Fig. 4b, the Co2Ni@NC could achieve a current density of 10 mA cm−2 at an overpotential of 310 mV, the overpotential is lower than those of other CoxNiy@NC samples, even better than the commercial IrO2 (340 mV). The electrocatalytic kinetics of CoxNiy@NC for OER were studied by related Tafel plots. In Fig. 4c, the Co2Ni@NC has a much smaller overpotential of 60.8 mV dec−1 than that of IrO2 (93.9 mV dec−1) and those of other CoxNiy@NC samples. These results are superior to that of many reported Co-based OER catalysts in basic condition (as is summarized in Table S1†). In addition, for the purpose of optimizing the annealing temperature, a series of Co2Ni@NC-T catalysts were examined. It is found that the Co2Ni@NC under the 800 °C treatment shows the highest OER activity (Fig. S10†).
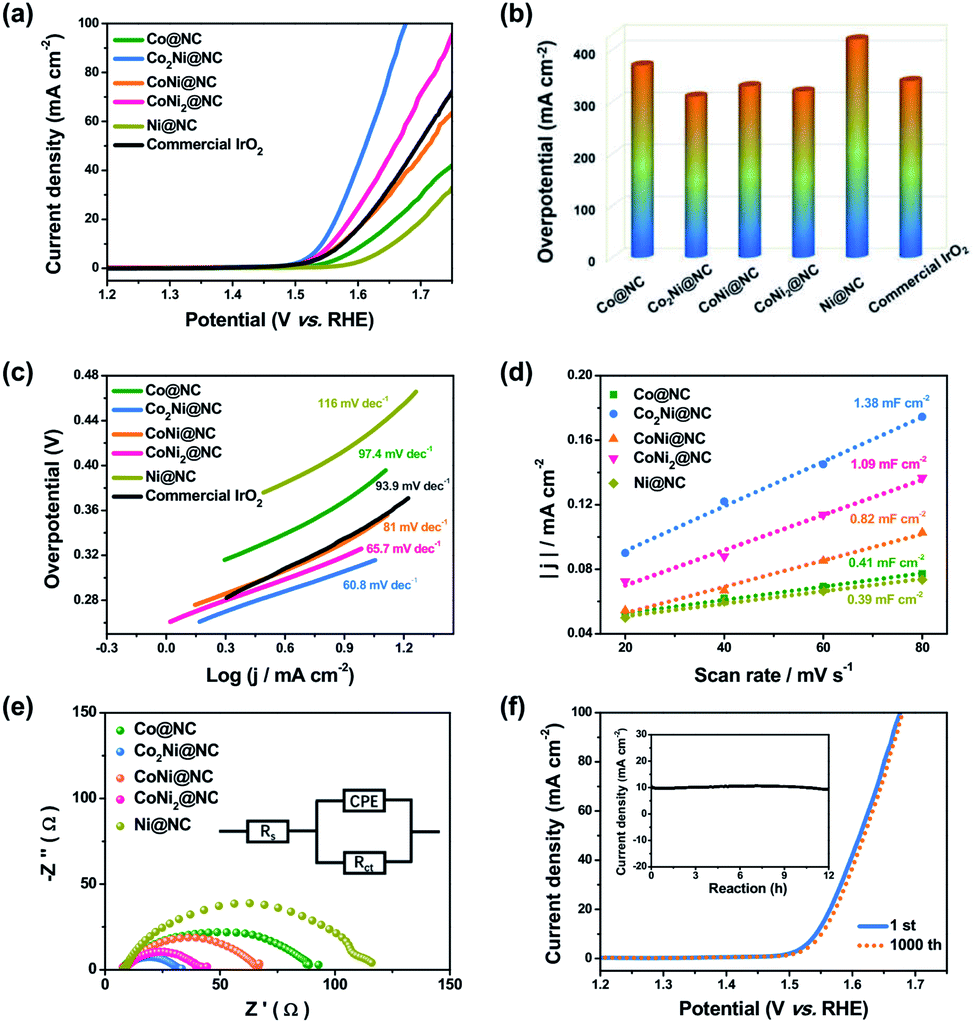 | ||
| Fig. 4 (a) The OER LSV curves, (b) overpotentials, and (c) Tafel plots of Co@NC, Co2Ni@NC, CoNi@NC, CoNi2@NC, Ni@NC and commercial IrO2. (d) The capacitive currents as a function of scan rates, (e) EIS spectra of Co@NC, Co2Ni@NC, CoNi@NC, CoNi2@NC and Ni@NC. (f) The LSV curves of Co2Ni@NC at the initial state and after 1000 CV cycles. The inset was the chronoamperometric curve. | ||
To deeply realize the intrinsic high activity of Co2Ni@NC for OER, we conducted the electrochemical active surface area (ECSA) tests. The electrochemical area of Co2Ni@NC (1.38 mF cm−2) examined by cyclic voltammogram (CV) (Fig. S11†) is larger than those of other samples (Fig. 4d). The larger electrochemical area is associated with more active sites at the solid–liquid interface. Besides, the electrochemical impedance spectroscopy (EIS) measurements show that the Co2Ni@NC has the smallest charge transfer resistance (Rct = 32.8 Ω) in comparison with other catalysts, indicating a fast electron transfer and the rapid OER kinetics at the catalyst/electrolyte interface (Fig. 4e). Additionally, we calculated the turn over frequencies (TOFs) per surface site at different potentials for these catalysts to gain an insight into the intrinsic catalytic activity. Fig. S12† shows the TOF value on the Co2Ni@NC (0.043 s−1) at an overpotential of 450 mV, which is about 4.1 and 6.8 times of the pure Ni@NC and Co@NC catalysts, respectively, indicating that the introduction of the Co–Ni alloy increases the OER catalytic activity of Co2Ni@NC.
Apart from the outstanding OER activity, Co2Ni@NC also displays an excellent long-term stability. After 1000 consecutive CV cycles at a scan rate of 100 mV s−1 in 1.0 M KOH, Co2Ni@NC only shows a slight increase of potential (Fig. 4f). What's more, the stable time-dependent current over 12 h further demonstrates the good stability of Co2Ni@NC for OER, which should be associated with the distinctive architecture of Co–Ni alloy encapsulated in nitrogen-doped porous carbon, protecting active materials from agglomeration and pulverization. XRD and XPS characterizations were conducted to verify the catalyst composition after the durability test. The XRD pattern shows that the phase characteristic of the Co2Ni@NC catalyst is not changed (Fig. S13†). In addition, from the high-resolution Co 2p and Ni 2p XPS spectra after the OER stability test, the signals of Co3+ (779.1 eV) and Ni3+ (853.7 eV) are emerged on the surface of the sample, which is beneficial for enhancing the OER activity (Fig. S14†).13
To further understand the relationship between Co–Ni alloy composition and OER performance, first-principles simulations based on density functional theory (DFT) were performed via Vienna ab initio simulation package (VASP).22–24 The calculations were made for models in a different regime of that expected for the experimental particles. As is well-known, OER follows a four-electron transfer process in alkaline environment and could be written as:
| OH− + * → HO* + e− | (1) |
| OH* + OH− → H2O + O* + e− | (2) |
| O* + OH− → HOO* + e− | (3) |
 | (4) |
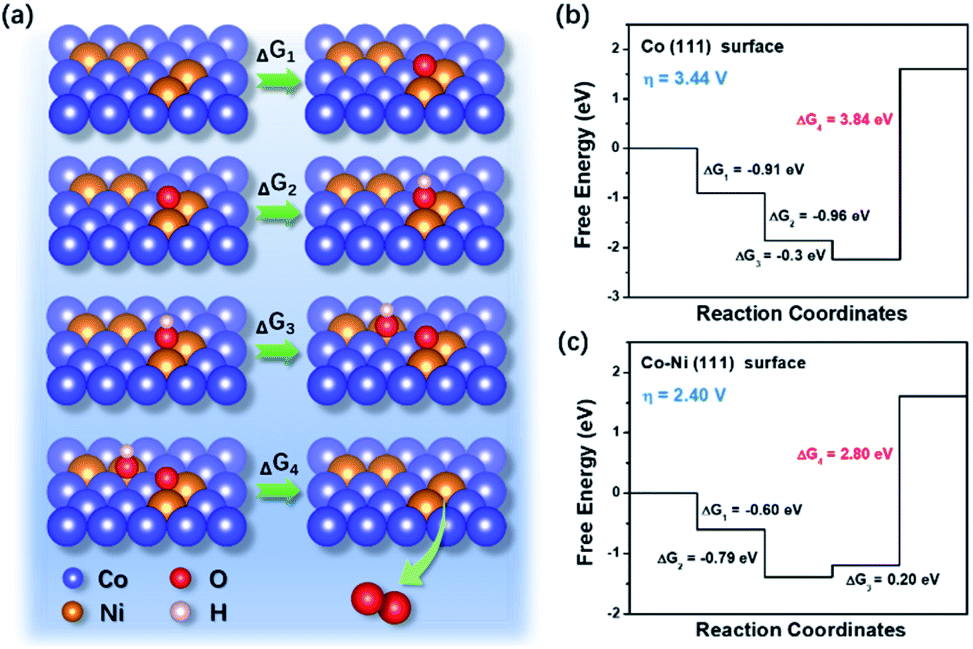 | ||
| Fig. 5 (a) The schematic illustration of OER process on the Co–Ni surfaces. Free energy diagrams for the OER of (b) Co (111) and (c) Co–Ni (111) surfaces. | ||
The outstanding OER activity of the Co2Ni@NC could be ascribed to the following aspects: firstly, the well-designed hierarchical structures with large surface area effectively increase the contact area between electrode and electrolyte. Second, the homogeneous CoNi alloy with an optimized component can efficiently adjust the electronic structure and significantly improve the electrocatalytic performance owing to their additional synergistic effects. Third, the in situ nitrogen doped carbon coating not only enhances the electrical conductivity, but also induces a charge rearrangement on the carbon materials to create new catalytic centers. What's more, the carbon coating protects active materials from agglomeration and pulverization, resulting in an improved cycling stability.
4. Conclusions
In summary, we have developed a controllable strategy to assemble the cobalt–nickel alloy nanoparticles encapsulated in nitrogen-doped porous carbon frameworks by annealing a bimetal–organic framework. Benefiting from the porous hierarchical nanostructures and component modulation, the optimized Co2Ni@NC catalyst realizes abundant synergistic active sites and fast mass transfer for OER, showing superiorly high electrocatalytic activities (310 mV, 10 mA cm−2) and excellent durability in alkaline solution. The outstanding electrochemical performance along with the clear explanation of the reaction mechanism, provides a deep understanding and great prospects for the MOF-derived Co2Ni@NC as a high-performance OER electrocatalyst.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from National Natural Science Foundation of China (51872027) is gratefully acknowledged.References
- N. Armaroli and V. Balzani, Angew. Chem., Int. Ed., 2007, 46, 52–66 CrossRef CAS PubMed.
- M. S. Dresselhaus and I. L. Thomas, Nature, 2001, 414, 332–337 CrossRef CAS PubMed.
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- C. Wang, J. Jiang, T. Ding, G. Chen, W. Xu and Q. Yang, Adv. Mater. Interfaces, 2016, 3, 1500454 CrossRef.
- M. Pramanik, C. Li, M. Imura, V. Malgras, Y. M. Kang and Y. Yamauchi, Small, 2016, 12, 1709 CrossRef CAS PubMed.
- K. C. Neyerlin, G. Bugosh, R. Forgie, Z. Liu and P. Strasser, J. Electrochem. Soc., 2009, 156, 363–369 CrossRef.
- T. Reier, M. Oezaslan and P. Strasser, ACS Catal., 2012, 2, 1765–1772 CrossRef CAS.
- Y. Hou, M. R. Lohe, J. Zhang, S. H. Liu, X. D. Zhuang and X. L. Feng, Energy Environ. Sci., 2016, 9, 478–483 RSC.
- Y. Yang, Z. Y. Lin, S. Q. Gao, J. W. Su, Z. Y. Lun, G. L. Xia, J. T. Chen, R. R. Zhang and Q. W. Chen, ACS Catal., 2017, 7, 469–479 CrossRef CAS.
- M. Li, T. T. Liu, X. J. Bo, M. Zhou and L. P. Guo, J. Mater. Chem. A, 2017, 5, 5413–5425 RSC.
- X. Zhang, H. Xu, X. Li, Y. Li, T. Yang and Y. Liang, ACS Catal., 2016, 6, 580–588 CrossRef CAS.
- W. J. Wan, X. J. Liu, H. Y. Li, X. Y. Peng, D. S. Xi and J. Luo, Appl. Catal., B, 2019, 240, 193–200 CrossRef CAS.
- Y. Fu, H. Y. Yu, C. Jiang, T. H. Zhang, R. Zhan, X. W. Li, J. F. Li, J. H. Tian and R. Z. Yang, Adv. Funct. Mater., 2018, 1705094 CrossRef.
- L. T. Yan, L. Cao, P. C. Dai, X. Gu, D. D. Liu, L. J. Li, Y. Wang and X. B. Zhao, Adv. Funct. Mater., 2017, 1703455 CrossRef.
- N. N. Du, C. M. Wang, X. J. Wang, Y. Lin, J. Jiang and Y. J. Xiong, Adv. Mater., 2016, 28, 2077–2084 CrossRef CAS PubMed.
- X. B. Liu, Y. C. Liu and L. Z. Fan, J. Mater. Chem. A, 2017, 5, 15310–15314 RSC.
- Y. Xu, W. G. Tu, B. W. Zhang, S. M. Yin, Y. Z. Huang, M. Kraft and R. Xu, Adv. Mater., 2017, 29, 1605957 CrossRef PubMed.
- Y. D. Ma, X. P. Dai, M. Z. Liu, J. X. Yong, H. Y. Qiao, A. X. Jin, Z. Z. Li, X. L. Huang, H. Wang and X. Zhang, ACS Appl. Mater. Interfaces, 2016, 8, 34396–34404 CrossRef CAS PubMed.
- Y. Z. Chen, C. M. Wang, Z. Y. Wu, Y. J. Xiong, Q. Xu, S. H. Yu and H. L. Jiang, Adv. Mater., 2015, 27, 5010–5016 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B, 1999, 59, 1758–1775 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B, 1993, 47, 558–5611 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B, 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jonsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- I. C. Man, H. Y. Su, F. Calle-Vallejo, H. A. Hansen, J. I. Martínez, N. G. Inoglu, J. Kitchin, T. F. Jaramillo, J. K. Nørskov and J. Rossmeisl, ChemCatChem, 2011, 3, 1159–1165 CrossRef CAS.
- A. Abbasi, M. Soleimani, M. Najafi and S. Geranmayeh, J. Mol. Struct., 2017, 1133, 458–463 CrossRef CAS.
- X. F. Xiao, C. T. He, S. L. Zhao, J. Li, W. S. Lin, Z. K. Yuan, Q. Zhang, S. Y. Wang, L. M. Dai and D. S. Yu, Energy Environ. Sci., 2017, 10, 893–899 RSC.
- Y. Hou, S. M. Cui, Z. H. Wen, X. R. Guo, X. L. Feng and J. H. Chen, Small, 2015, 44, 5940–5948 CrossRef PubMed.
- X. T. Yuan, M. S. Riaz, X. Wang, C. L. Dong, Z. Zhang and F. Q. Huang, Chem.–Eur. J., 2018, 24, 3707–3711 CrossRef CAS PubMed.
- Z. H. Xue, H. Su, Q. Y. Yu, B. Zhang, H. H. Wang, X. H. Li and J. S. Chen, Adv. Energy Mater., 2017, 7, 1602355 CrossRef.
- Y. X. Zhang, L. Sun, L. Q. Bai, H. C. Si, Y. Zhang and Y. H. Zhang, Nano Res., 2019, 12, 607–618 CrossRef CAS.
- J. S. Zhang, C. X. Wu, M. H. Huang, Y. Zhao, J. X. Li and L. H. Guan, ChemCatChem, 2018, 10, 1336–1343 CrossRef CAS.
- S. L. Yang, T. R. Zhang, G. C. Li, L. Q. Yang and J. Y. Lee, Energy Storage Mater., 2017, 6, 140–148 CrossRef.
- X. G. Feng, X. J. Bo and L. P. Guo, J. Power Sources, 2018, 389, 249–259 CrossRef CAS.
- Y. Li, H. X. Li, K. Z. Cao, T. Jin, X. J. Wang, H. M. Sun, J. X. Ning, Y. J. Wang and L. F. Jiao, Energy Storage Mater., 2018, 12, 44–53 CrossRef.
- W. P. Kang, Y. Zhang, L. L. Fan, L. L. Zhang, F. N. Dai, R. M. Wang and D. F. Sun, ACS Appl. Mater. Interfaces, 2017, 9, 10602–10609 CrossRef CAS PubMed.
- X. F. Lu, L. F. Gu, J. W. Wang, J. X. Wu, P. Q. Liao and G. R. Li, Adv. Mater., 2017, 29, 1604437 CrossRef PubMed.
- Z. Q. Zhu, F. Y. Cheng and J. Chen, J. Mater. Chem. A, 2013, 1, 9484–9490 RSC.
- X. Zhang, A. Chen, Z. H. Zhang, M. G. Jiao and Z. Zhou, J. Mater. Chem. A, 2018, 6, 11446–11452 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra10713a |
| This journal is © The Royal Society of Chemistry 2021 |