
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Revisiting activity of some glucocorticoids as a potential inhibitor of SARS-CoV-2 main protease: theoretical study†
Ayman Abo Elmaatya,
Radwan Alnajjarbc,
Mohammed I. A. Hamedd,
Muhammad Khattab e,
Mohamed M. Khalifaf and
Ahmed A. Al-Karmalawy*g
e,
Mohamed M. Khalifaf and
Ahmed A. Al-Karmalawy*g
aDepartment of Medicinal Chemistry, Faculty of Pharmacy, Port Said University, Port Said 42526, Egypt
bDepartment of Chemistry, Faculty of Science, University of Benghazi, Benghazi, Libya
cDepartment of Chemistry, University of Cape Town, Rondebosch 7701, South Africa
dDepartment of Organic and Medicinal Chemistry, Faculty of Pharmacy, Fayoum University, Fayoum 63514, Egypt
eDepartment of Chemistry of Natural and Microbial Products, Division of Pharmaceutical and Drug Industries, National Research Centre, Cairo, 12622, Egypt
fDepartment of Pharmaceutical Medicinal Chemistry& Drug Design, Faculty of Pharmacy (Boys), Al-Azhar University, Cairo 11884, Egypt
gDepartment of Pharmaceutical Medicinal Chemistry, Faculty of Pharmacy, Horus University-Egypt, New Damietta 34518, Egypt. E-mail: akarmalawy@horus.edu.eg
First published on 9th March 2021
Abstract
The global breakout of COVID-19 and raised death toll has prompted scientists to develop novel drugs capable of inhibiting SARS-CoV-2. Conducting studies on repurposing some FDA-approved glucocorticoids can be a promising prospective for finding a treatment for COVID-19. In addition, the use of anti-inflammatory drugs, such as glucocorticoids, is a pivotal step in the treatment of critical cases of COVID-19, as they can provoke an inflammatory cytokine storm, damaging lungs. In this study, 22 FDA-approved glucocorticoids were identified through in silico (molecular docking) studies as the potential inhibitors of COVID-19's main protease. From tested compounds, ciclesonide 11, dexamethasone 2, betamethasone 1, hydrocortisone 4, fludrocortisone 3, and triamcinolone 8 are suggested as the most potent glucocorticoids active against COVID-19's main protease. Moreover, molecular dynamics simulations followed by the calculations of the binding free energy using MM-GBSA were carried out for the aforementioned promising candidate-screened glucocorticoids. In addition, quantum chemical calculations revealed two electron-rich sites on ciclesonide where binding interactions with the main protease and cleavage of the prodrug to the active metabolite take place. Our results have ramifications for conducting preclinical and clinical studies on promising glucocorticoids to hasten the development of effective therapeutics against COVID-19. Another advantage is that some glucocorticoids can be prioritized over others for the treatment of inflammation accompanying COVID-19.
1. Introduction
In December 2019, a sudden outbreak of a new virus, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), occurred in the Chinese city of Wuhan, causing coronavirus disease 2019 (COVID-19).1 The virus began to spread outside China all over the world within a short time. On March 11, 2020, after the overwhelming global expansion of SARS-CoV-2, the World Health Organization classified COVID-19 as a pandemic.2 The number of confirmed COVID-19 cases on December 12, 2020, was approximately 71![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 797890 patients with a death toll up to 1607590 in over about 216 countries.3 It is a pity that SARS-CoV-2 is highly infectious and might develop some fatal consequences. Some patients diagnosed with COVID-19 may develop septic shock, respiratory failure, and multi-organ dysfunction, leading to a global mortality rate of about 4%.4
797890 patients with a death toll up to 1607590 in over about 216 countries.3 It is a pity that SARS-CoV-2 is highly infectious and might develop some fatal consequences. Some patients diagnosed with COVID-19 may develop septic shock, respiratory failure, and multi-organ dysfunction, leading to a global mortality rate of about 4%.4
Besides, one of the most common features that critical COVID-19 patients may develop is extremely high inflammatory parameters, including C reactive protein and pro-inflammatory cytokines.4,5 Unfortunately, developing a new drug or vaccine that can cure COVID-19 effectively is a big global challenge.
The development of a new drug or vaccine is a long process that requires a lot of time, effort, and money. It may take years starting from the de novo design of drug candidates to the drug being clinically approved and available in markets. Hence this is one of the critical challenges that pharmaceutical industries may experience lately. So, we should shed light on alternative approaches that may allow intellectual intervention, helping our fight against COVID-19 to be accomplished rapidly and effectively. One of these approaches that allow us to overcome drug development boundaries and obstacles is “drug repurposing”, also called drug repositioning, re-profiling, and re-tasking. Drug repurposing is a promising alternative strategy that allows revealing new therapeutic activities for existing approved drugs other than their main original indications.6,7,8 Many pharmaceutical companies pursue this strategy to circumvent expensive conventional drug discovery processes, hence reducing cost and time, which are considered crucial factors in catastrophic circumstances. This is because the safety and pharmacokinetic profiles of repositioned candidates are already established. Approximately one-third of approved drugs are introduced through drug repurposing.9,10
In drug repurposing methods, the other uses of drugs can be revealed using diverse approaches, including computational methods.9,11 Computational approaches play an important role during the drug discovery steps and development trajectory, helping researchers to reveal new promising drug candidates. Data analysis, modeling, and simulation embraced by computational approaches allow scientists to augment their research and advance drug discovery more quickly. An example of these in silico techniques is structure-based virtual screening and molecular docking studies.12 On the other hand, scientists can utilize bioinformatics to detect the main key genes from immense genomic data, and hence druggability will be easier to handle. Virtual screening can then identify new drug candidates based on the chemical properties of these drugs and their target proteins within a short time.13
Due to inflammatory cytokine storms it may provoke, COVID-19 treatment ought to comprise anti-inflammatory drugs to ensure an effective cure.4 Hence, glucocorticosteroids can be used in the treatment of COVID-19, due to their magical anti-inflammatory effect.14 Clinical investigations revealed that COVID-19 patients treated with a daily dose of 6 mg dexamethasone had reduced mortality by 8–26%. This may be particularly useful for short-term severely intubated COVID-19 patients.14,15 Furthermore, there is preliminary non-peer-reviewed evidence suggesting that ciclesonide may have the ability to reduce coronavirus RNA replication owing to its activity against NSP15.16
The main SARS-CoV-2 protease, 3-chymotrypsin-like protease (3CLpro), also known as Mpro, is essential for the virus proteolytic maturation and replication. This main protease is important for the cleavage of the polyproteins to give some essential functional proteins, such as RNA polymerase, endoribonuclease, and exoribonuclease. Furthermore, the human proteases play a key role in the attachment of SARS-CoV-2 to its host cell through the virus spike glycoprotein.17,18
Therefore, in a continuation of our previous work concerning COVID-19,19,20,21 we aimed to utilize the crystal structure of the Mpro (PDB ID: 6LU7) with a small library of the approved glucocorticosteroids (depicted in Fig. 1) by conducting virtual screening, molecular docking, and quantum mechanical calculations.22 Thus, we aimed to investigate the best glucocorticosteroids that might have antiviral activity against COVID-19, or at least to prioritize the best members of the glucocorticosteroids to be used in the short-term treatment of inflammation in COVID-19 patients. In addition, we aimed to investigate the structural and electronic properties of the most promising candidate, ciclesonide.
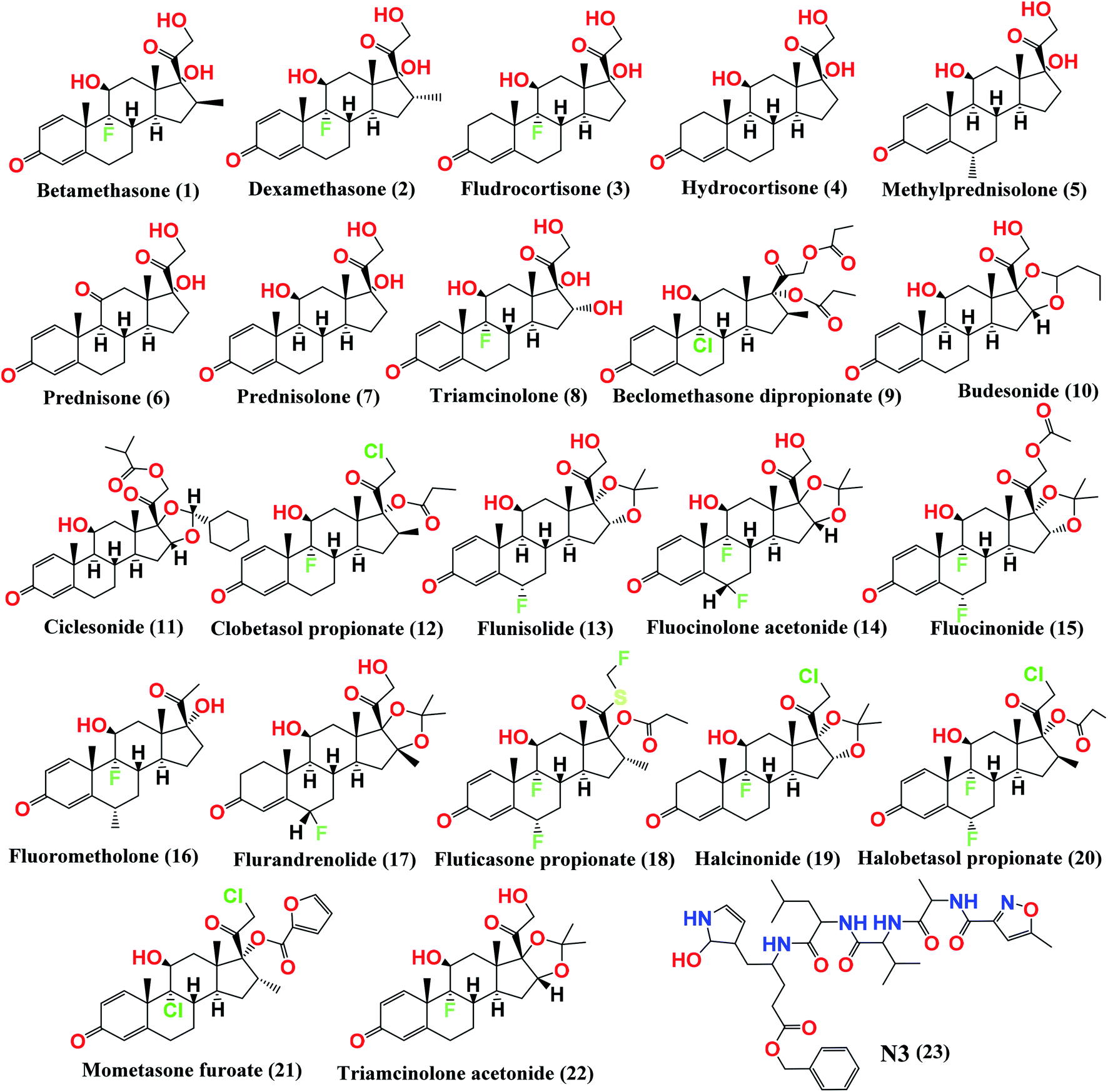 | ||
| Fig. 1 Chemical structures of the screened FDA-approved glucocorticosteroids (1–22) tested against the COVID-19 virus main protease and the co-crystallized inhibitor (N3, 23). | ||
Molecular dynamics (MD) simulations were also carried out on the docked drug–protein complexes to get a deep understanding of the affinity between the ligands and the COVID-19 main protease active site in the explicit solvent model to estimate the stability of the drugs within the active site of the protein. These drug–protein complexes were then subjected to molecular mechanics/generalized Born and surface area (MM/GB-SA) calculations to estimate the corresponding relative binding free energies.
2. Materials and methods
Molecular docking studies using the MOE 2014 suite,23 molecular dynamics simulation studies using the Desmond simulation package of Schrödinger LLC,24 and DFT calculations using Gaussian/g09 software25 were carried out to examine and confirm the binding affinities and modes of the 22 selected FDA-approved glucocorticosteroids against the COVID-19 main protease compared to its N3 inhibitor as a reference. The 22 approved glucocorticoid drugs were selected based on their structural similarity except for having different substitutions at the 16- and 17- positions on the glucocorticoid moiety.2.1. Docking studies
Moreover, in the beginning, a validation process was carried out by docking the native N3 inhibitor inside its pocket of the target main protease (Mpro) and a valid performance was indicated (RMSD = 1.23 Å) between the docked and crystal conformations.28,29
2.2. Molecular dynamics simulations
Molecular dynamics simulations were applied using the Desmond simulation package of Schrödinger LLC.24 The NPT ensemble with the temperature of 300 K and pressure of 1 bar was applied in all the runs. The simulation length was 100 ns with a relaxation time of one ps for all the selected ligands. The OPLS3 force-field specifications were utilized in all the simulations.30 The cutoff radius in Coulomb interactions was 9.0 Å. The orthorhombic periodic box boundaries were set 10 Å away from the protein atoms. The water molecules were explicitly described using the transferable intermolecular potential with a three-point (TIP3P) model.31,32 The salt concentration was set at 0.15 M NaCl and established using the system builder utility of Desmond.33 The Martyna–Tuckerman–Klein chain coupling scheme with a coupling constant of 2.0 ps was used for controlling the pressure and the Nosé–Hoover chain coupling scheme was used for the surveillance of the temperature.34,35 Nonbonded forces were calculated using a RESPA integrator and every step of the short-range forces were updated, whereas the long-range forces were updated every three steps. The trajectories were saved at 20 ps intervals for analysis. Analysis of the behavior and interactions between the ligands and protein were performed using the simulation interaction diagram tool implemented in the Desmond MD package. The stability of the MD simulations was kept an eye on by monitoring the RMSD of the ligand and protein atom positions over time.2.3. MD trajectory analysis and prime MM-GBSA calculations
The simulation interactions diagram panel in the Maestro software was utilized to follow the interactions' contributions to the ligand–protein stability. Hence to calculate the ligand binding free energies and ligand strain energies for the docked compounds, the molecular mechanics generalized Born/solvent accessibility (MM-GBSA) was carried out over the 100 ns period with the thermal_mmgbsa.py python script provided by Schrödinger, which takes a Desmond trajectory file, splits it into individual snapshots, runs the MM-GBSA calculations on each frame, and outputs the average computed binding energy along with the standard deviation.2.4. Quantum mechanics calculations details
Becke's three-parameter hybrid exchange–correlation functional (B3LYP)36,37 with different basis sets was employed in the quantum mechanics calculations. The structure of ciclesonide was initially optimized at B3LYP coupled with the 3-21G, 6-31G, 6-311G, and 6-311G* basis sets. The geometry of ciclesonide was finally optimized at B3LYP/6-311+G* where the geometries, charges, and all other structural properties of the ground and excited state structure of ciclesonide were computed using DFT and TD-DFT methods, respectively, using the B3LYP/6-311+G* model. No imaginary frequencies were detected for the optimized structures, indicating that the corresponding geometry was a true local minimum structure. An implicit solvent effect was considered in our calculations to replicate the reported UV-vis absorption spectrum of ciclesonide. Hence, the conductor-like polarizable continuum model (CPCM)38 utilizing the dielectric constant of methanol was used. All the calculations were performed using GAUSSIAN 09 Revision C.01 (ref. 25) on Swinburne supercomputing facilities.3. Results and discussion
3.1. Docking study
Beside a Cys–His catalytic dyad it holds, the COVID-19 main protease substrate-binding pocket is found in a cleft between domains I and II. The N3 inhibitor shows asymmetric units containing only one polypeptide and is stabilized inside the substrate-binding site. Molecular docking of the picked glucocorticoids (1–22) and N3 inhibitor 23 (depicted in Fig. 1) into the Mpro active site was done. They were fitted at the inhibitor-binding pocket by several nonconstant interactions (Table 1). The order of strength according to their binding scores was: N3 inhibitor (23, docked) > ciclesonide (11) > fluorometholone (16) > dexamethasone (2) > betamethasone (1) > halobetasol propionate (20) > hydrocortisone (4) > fludrocortisone (3) > triamcinolone (8) > fluticasone propionate (18) > methylprednisolone (5) > fluocinonide (15) > prednisolone (7) > budesonide (10) > beclomethasone dipropionate (9) > fluocinolone acetonide (14) > triamcinolone acetonide (22) > flurandrenolide (17) > mometasone furoate (21) > halcinonide (19) > flunisolide (13) > prednisone (6) > clobetasol propionate (12).| No. | Glucocorticosteroid | S scorea kcal mol−1 | RMSD_Refineb | Amino acid bond | Distance Å |
|---|---|---|---|---|---|
| a S: the score of a compound positioned into the binding pocket of the protein utilizing the London ΔG scoring function.b RMSD_Refine: the root-mean-squared-deviation (RMSD) between the refined predicted pose and those of the unrefined crystal structure. | |||||
| 1 | Betamethasone | −16.88 | 0.69 | Glu166/H-acceptor | 1.70 |
| Glu166/H-donor | 1.60 | ||||
| Gln189/H-acceptor | 2.00 | ||||
| Thr26/H-donor | 2.20 | ||||
| 2 | Dexamethasone | −17.26 | 1.02 | Glu166/H-donor | 1.80 |
| Gln189/H-acceptor | 1.70 | ||||
| Gln189/H-acceptor | 1.60 | ||||
| 3 | Fludrocortisone | −16.30 | 1.33 | Glu166/H-donor | 1.70 |
| Glu166/H-acceptor | 1.50 | ||||
| 4 | Hydrocortisone | −16.70 | 0.98 | Glu166/H-acceptor | 1.50 |
| Ser144/H-donor | 2.90 | ||||
| His163/H-donor | 1.90 | ||||
| 5 | Methylprednisolone | −15.50 | 1.66 | Glu166/H-acceptor | 2.70 |
| Asn142/H-acceptor | 2.00 | ||||
| 6 | Prednisone | −11.45 | 1.12 | Glu166/H-acceptor | 1.30 |
| Glu166/H-donor | 2.00 | ||||
| 7 | Prednisolone | −14.91 | 1.10 | Thr26/H-donor | 2.00 |
| Thr26/H-acceptor | 2.00 | ||||
| Asn142/H-acceptor | 1.70 | ||||
| 8 | Triamcinolone | −16.26 | 1.21 | Gln189/H-acceptor | 1.70 |
| Glu166/H-acceptor | 1.40 | ||||
| Phe140/H-acceptor | 2.10 | ||||
| 9 | Beclomethasone dipropionate | −13.98 | 1.47 | Glu166/H-donor | 1.80 |
| Gln189/H-acceptor | 2.60 | ||||
| Thr26/H-donor | 1.90 | ||||
| 10 | Budesonide | −14.06 | 0.51 | Glu166/H-donor | 2.60 |
| His163/H-acceptor | 2.10 | ||||
| Gly143/H-donor | 2.40 | ||||
| Ser46/H-donor | 2.60 | ||||
| 11 | Ciclesonide | −18.88 | 1.56 | Ser46/H-donor | 1.80 |
| 12 | Clobetasol propionate | −9.57 | 1.78 | — | — |
| 13 | Flunisolide | −12.66 | 1.00 | Thr26/H-acceptor | 1.60 |
| Thr26/H-donor | 3.10 | ||||
| Gly143/H-donor | 2.50 | ||||
| Asn142/H-acceptor | 2.0 | ||||
| 14 | Fluocinolone acetonide | −13.98 | 1.77 | Asn142/H-acceptor | 1.80 |
| 15 | Fluocinonide | −15.38 | 1.62 | — | — |
| 16 | Fluorometholone | −17.45 | 1.68 | Asn142/H-acceptor | 1.50 |
| 17 | Flurandrenolide | −13.15 | 1.50 | Gln189/H-acceptor | 1.70 |
| His164/H-acceptor | 1.60 | ||||
| 18 | Fluticasone propionate | −15.88 | 1.09 | Gln189/H-acceptor | 1.90 |
| His41/H-donor | 3.20 | ||||
| 19 | Halcinonide | −12.81 | 1.77 | Glu166/H-donor | 2.00 |
| 20 | Halobetasol propionate | −16.80 | 1.43 | Asn142/H-acceptor | 1.80 |
| 21 | Mometasone furoate | −13.02 | 1.55 | His163/H-donor | 2.20 |
| Asn142/H-donor | 1.90 | ||||
| 22 | Triamcinolone acetonide | −13.21 | 1.68 | Glu166/H-donor | 2.70 |
| Glu166/H-acceptor | 1.30 | ||||
| 23 | N3 | −22.71 | 1.65 | Glu166/H-acceptor | 1.62 |
| Glu166/H-acceptor | 1.88 | ||||
| Glu166/H-donor | 1.52 | ||||
| Gln189/H-acceptor | 1.50 | ||||
| Thr190/H-acceptor | 2.10 | ||||
| Phe140/H-acceptor | 1.91 | ||||
| His164/H-acceptor | 1.62 | ||||
| His41/H-donor | 2.01 | ||||
Although many poses were gained for each selected compound inside the receptor pocket with even better binding modes and/or interactions, the poses with the best scores (indicating the stability of the pose) and rmsd_refine values (indicating the proximity of the elected pose to the position of the authentic ligand inside the receptor pocket) were selected. The scores, RMSD values, and different binding interactions with the COVID-19 Mpro pocket amino acids are presented in Table 1 and their detailed figures are included in ESI data 1 (Fig. S1†).
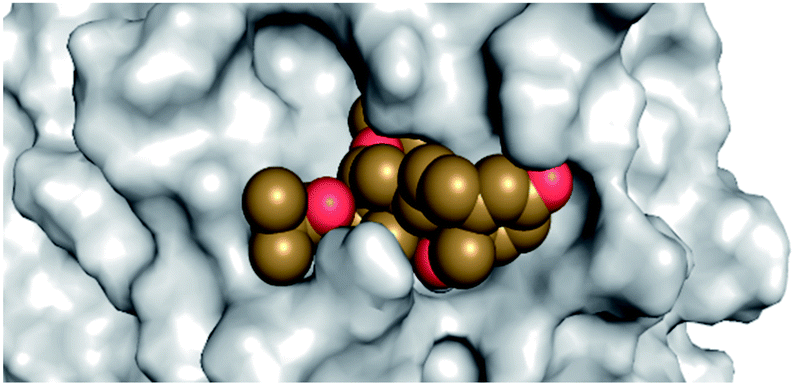
By analyzing the docking results of the selected glucocorticoids, it was found that most of the selected compounds manifested very close binding scores and modes compared to the co-crystallized inhibitor (N3) at the COVID-19 Mpro target receptor. Ciclesonide 11, dexamethasone 2, betamethasone 1, hydrocortisone 4, fludrocortisone 3, and triamcinolone 8 were found to have the best binding affinities and modes against COVID-19 protease with binding scores of −18.88, −17.26, −16.88, −16.70, −16.30, and −16.26 kcal mol−1, respectively (Table 1). These energy values were very close to that of the docked N3 inhibitor (binding energy = −22.71 kcal mol−1). The detailed binding modes of the docked N3 23 and all of the tested glucocorticoids (1–22) are presented in Table 1. Furthermore, all of their 3D binding interactions, surfaces and maps, and 3D positioning inside the protein pocket can be found in ESI data 1 (Fig. S1–S3†). Also, the 3D binding interactions and 3D protein positioning of the best selected six glucocorticoids are presented in Table 2.
| Drug | 3D interaction | 3D protein positioning |
|---|---|---|
| Ciclesonide 11 | 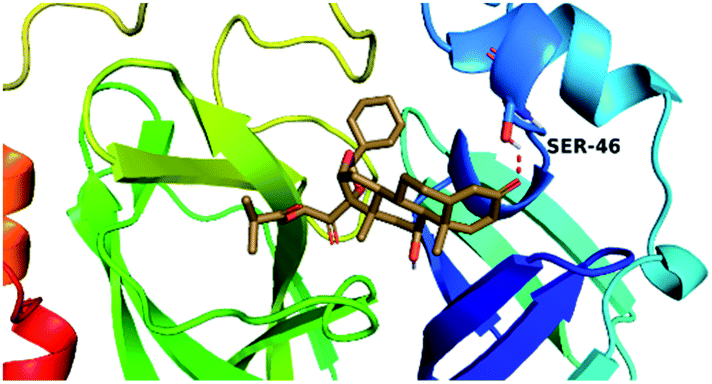 |
 |
| Dexamethasone 2 | 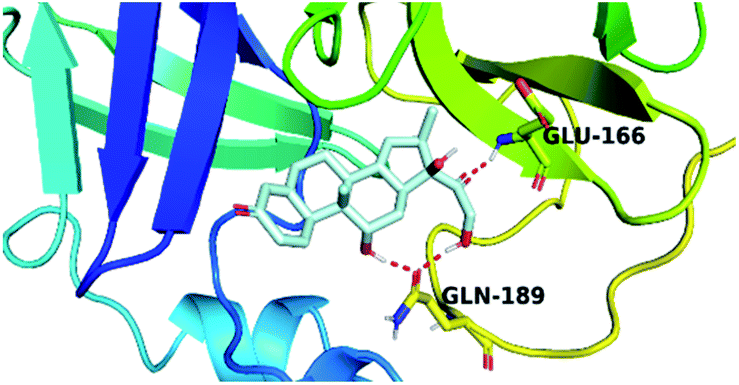 |
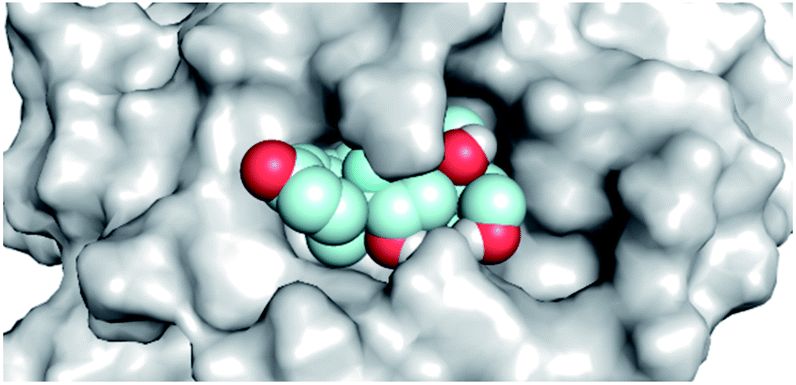 |
| Betamethasone 1 | 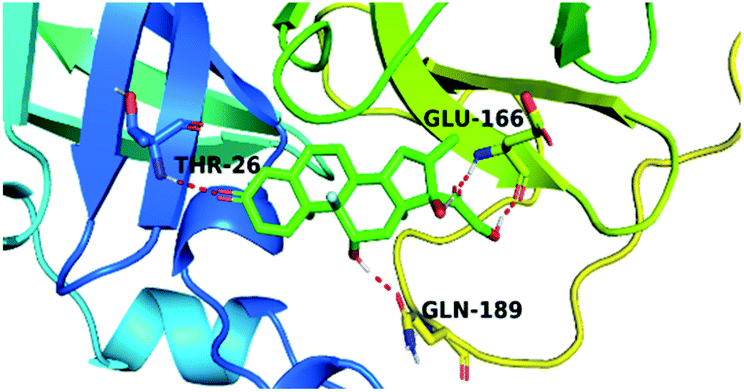 |
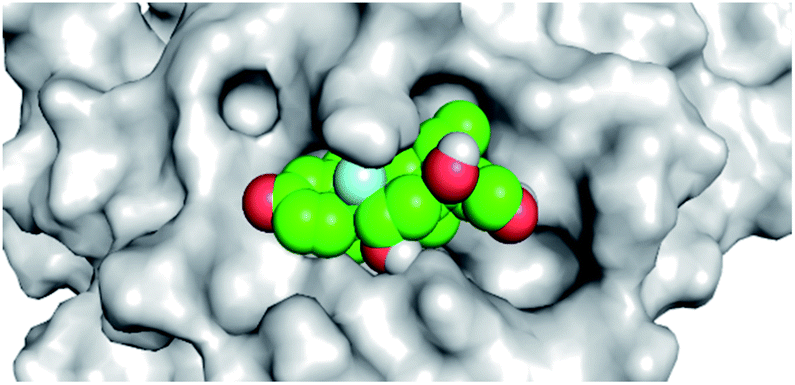 |
| Hydrocortisone 4 | 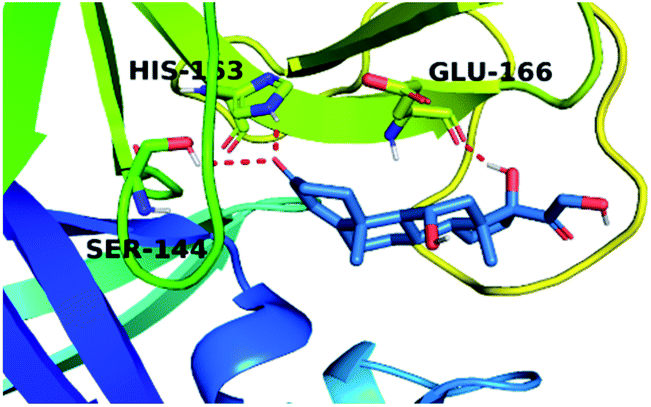 |
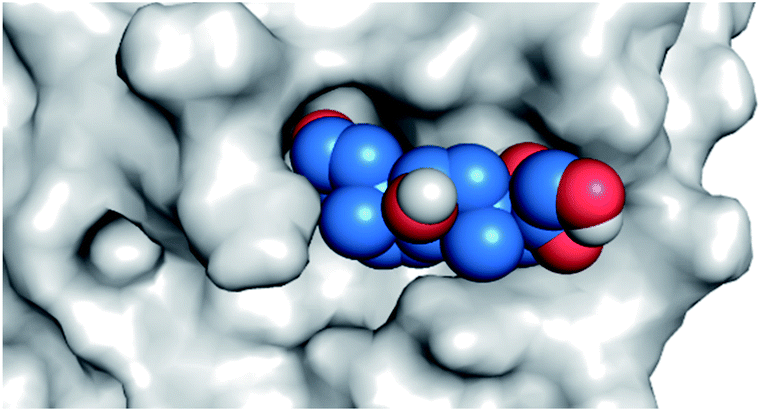 |
| Fludrocortisone 3 | 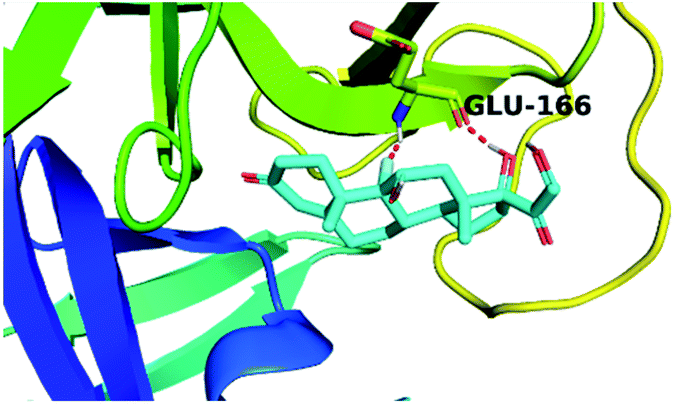 |
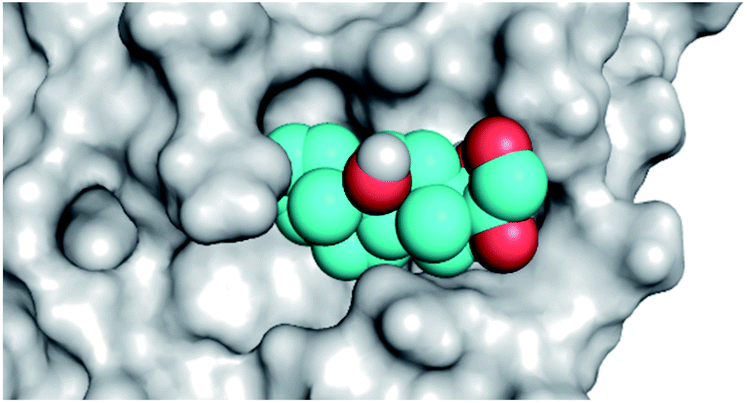 |
| Triamcinolone 8 | 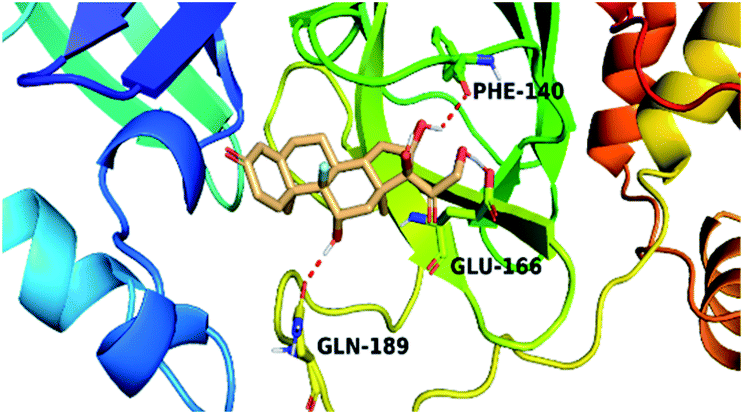 |
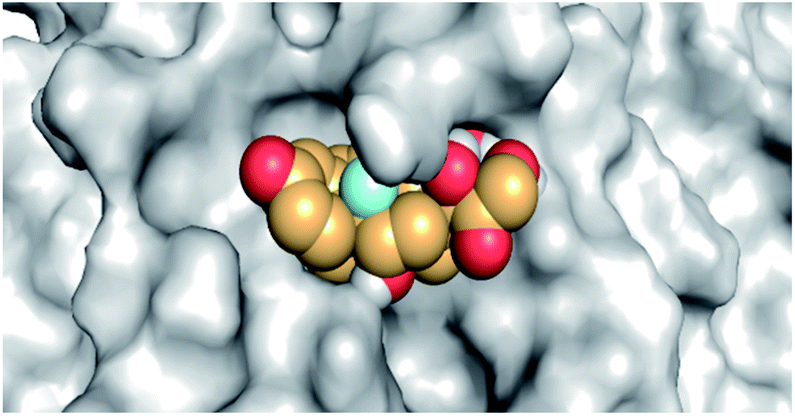 |
| N3 23 | 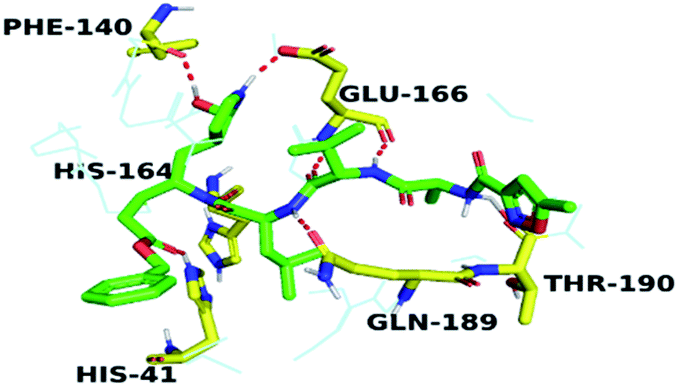 |
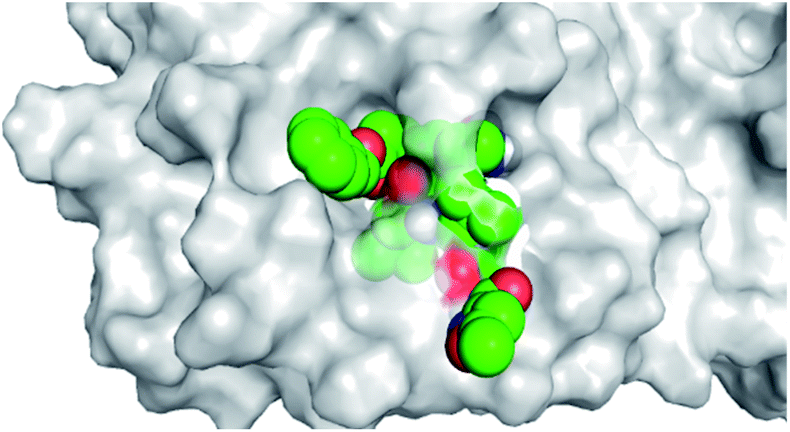 |
3.2. Molecular dynamics (MD) simulation
Despite their usual fast and approximate utility, docking protocols lack protein flexibility, which may be related to the thoroughness of the resulting ligand–protein complexes. Therefore, docking is often accompanied by the more computationally expensive but more accurate molecular dynamic (MD) simulations techniques to provide a better complementary result. In summary, MD simulations can be used to study the macromolecule features, but it counts on classical mechanics and the use of Newton's equation of motion to calculate the position of and speed of each atom of the studied system.Therefore, we can say that MD can perform a more rigorous conformational search than docking does, thus providing a more accurate representation of protein motion. Hence, MD simulations were carried out utilizing the Desmond package on the ligand–potential complex to imitate the interaction of the best selected six candidates (ciclesonide 11, dexamethasone 2, betamethasone 1, hydrocortisone 4, fludrocortisone 3, and triamcinolone 8) selected from the docking study with the COVID-19 main protease active site for 100 ns.
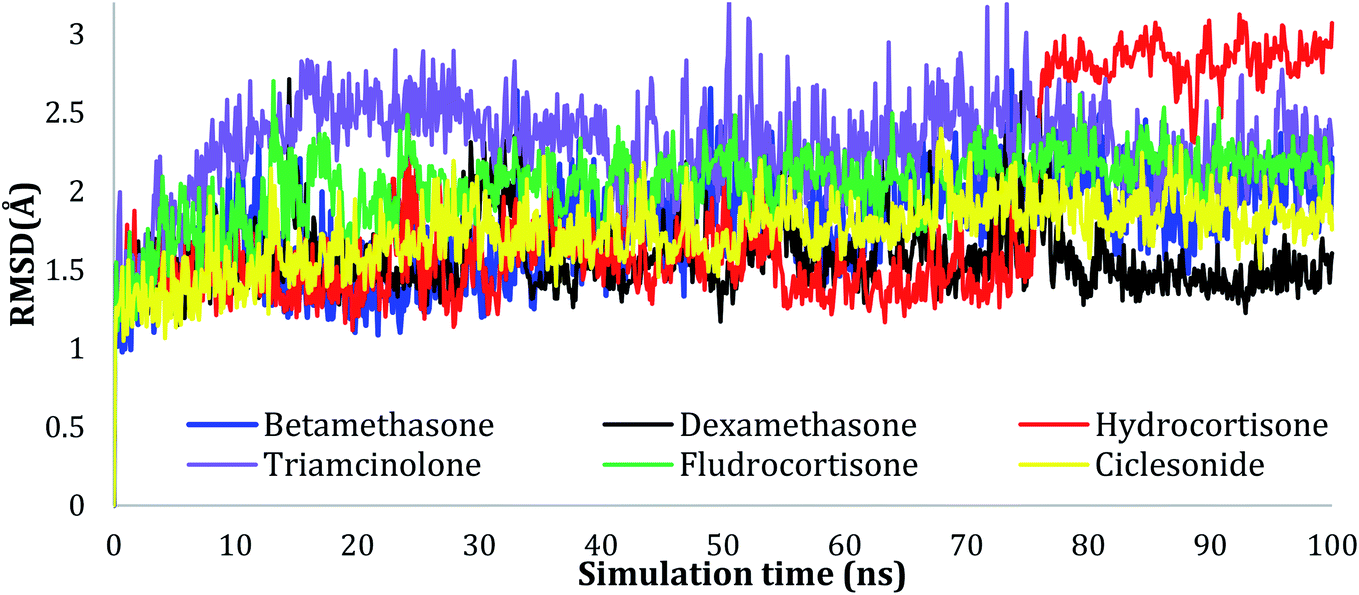 | ||
| Fig. 2 Plots of RMSD for Cα atoms (Å) for the initial structure vs. the simulation time (ns) for all the complexes. | ||
The RMSD values of the ligands were also plotted as a function of simulation time to show the RMSD of a ligand aligned and measured just on its reference confirmation within the active site, and as can be seen from Fig. 3, betamethasone 1, dexamethasone 2, and triamcinolone 8 were stable within the active site of the protein and reached equilibrium in the first 20 ns. Their average RMSD values over the last 50 ns were 3.74, 3.75, and 4.43 Å respectively. Ciclesonide 11 and fludrocortisone 3 showed equilibrium stability at about 60 ns and 70 ns, with RMSD values in the last 50 ns found to be 4.68 Å and 5.10 Å, respectively. Hydrocortisone 4 was the most fluctuate ligand among others, it only reached equilibrium at 80 ns with an RMSD of 7.70 Å. The RMSD of all the ligands over all the simulation times and the last 50 ns are reported in Table 1SI, ESI data 2.†
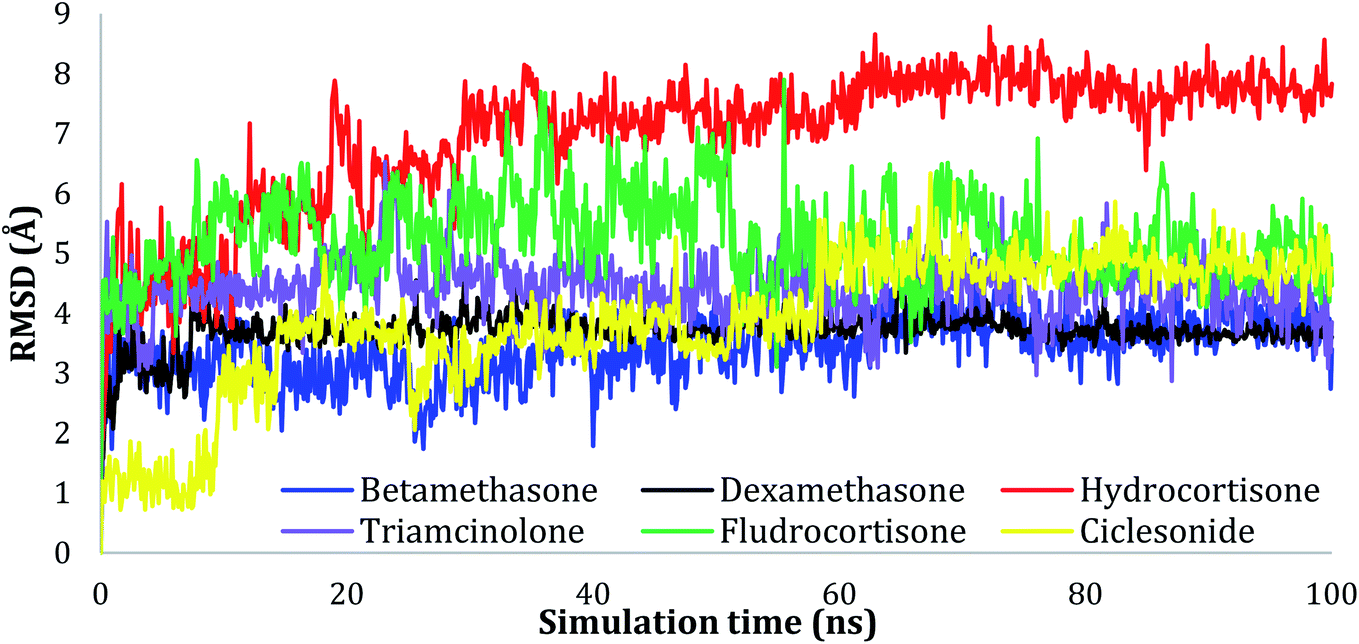 | ||
| Fig. 3 Plots of RMSD for ligand atoms (Å) concerning the initial structure vs. simulation time (ns) for all the complexes. | ||
The active site contains the following polar amino acids (threonine (Thr26), asparagine (Asn142), and glutamine (Gln189, Gln192)), a nonpolar amino acid (alanine (Ala193)), and negatively charged amino acids (glutamic (Glu166) and aspartic acid (Asp187)). Since ciclesonide 11 and betamethasone 1 showed the highest binding score and highest MM-GBSA energy (Tables 1 and 3), hence their interactions will be discussed in detail. As can be seen from Fig. 4, the histogram explains the contacts that occur during the simulations between the ligands and protein, which were generated with simulation interactions, through a diagram panel implemented in Maestro software. For ciclesonide 11, hydrogen bonding between the Thr26 and Glu166 residues and the ligand were maintained during most of the time, either directly or through water bridging H-bonds as can be seen in Fig. 5. Other residues such as Asn142 and Gln189 were able to develop an H-bond for almost 50% of the time, again, directly or through a water bridge. Hydrophobic interactions with Met49 and Met165 were very weak and could be nongalactic (Fig. 4a). Betamethasone 1, was able to form a donor–acceptor hydrogen bond with Glu166, leading to 180% of the time, 90% of the time as donor and 90% as acceptor, while other hydrogen bonds were formed with His41 and Gln189 during 50% and 40% of the simulation time, respectively. Bridging hydrogen bonds through water were formed with Thr24, Thr25, Asp187, and Gln192 between 40–75% of the simulation time as can be seen in Fig. 4b, other selected drug interactions histograms and figures are presented in the ESI data 2 file.†
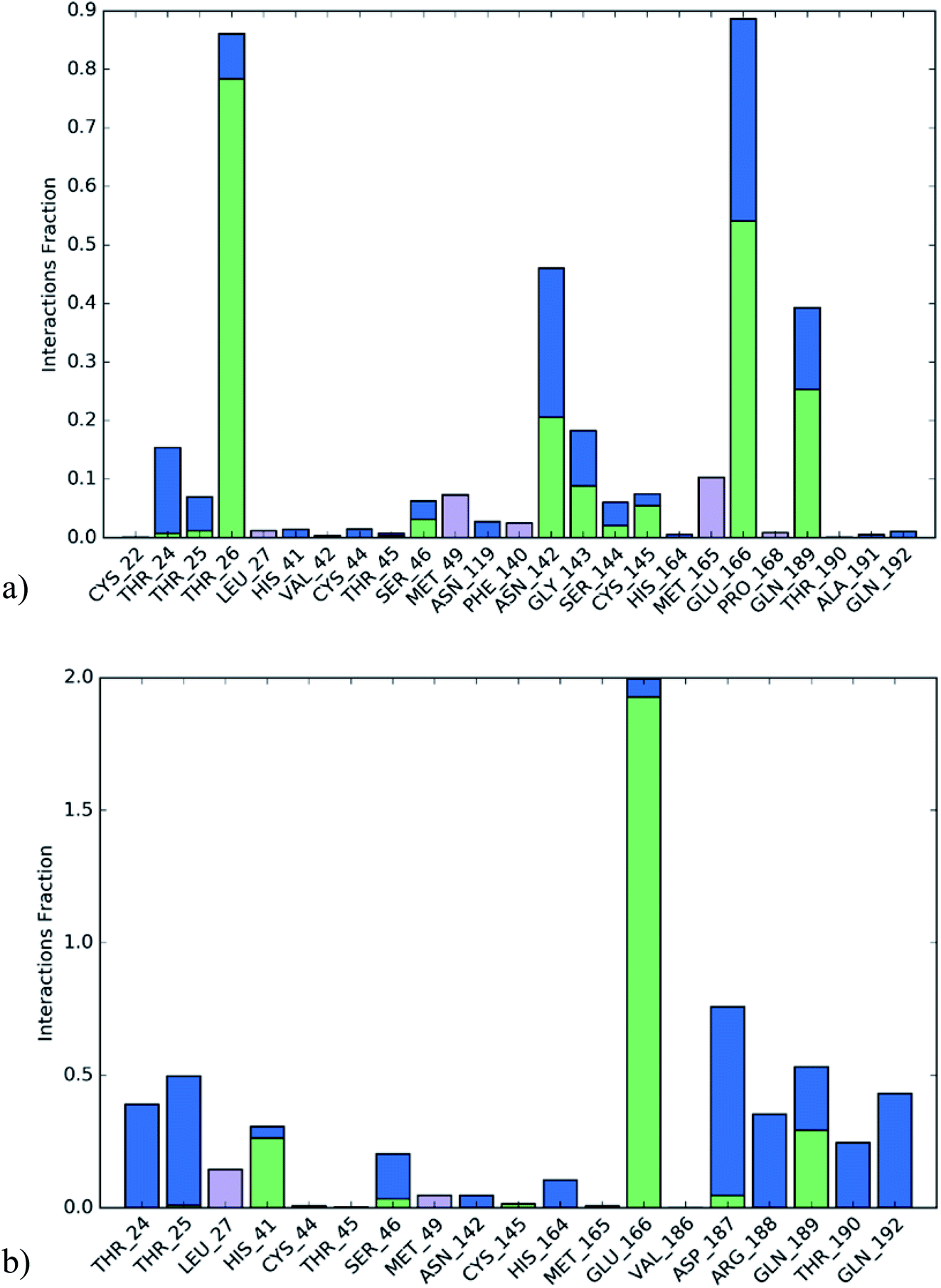 | ||
| Fig. 4 The histogram of ligand-6LU7 contact throughout the trajectory. (a) Ciclesonide, (b) Betamethasone. | ||
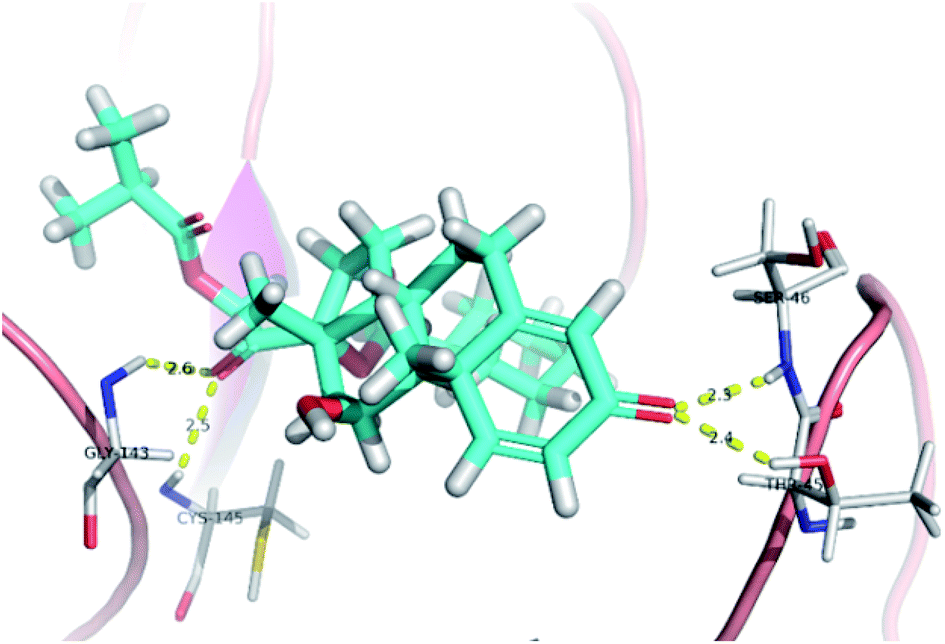 | ||
| Fig. 5 Snapshot of ciclesonide interactions with the protein residues (hydrogen bonds are shown as yellow dashed) at 100 ns. | ||
To monitor the protein–ciclesonide interactions during the simulation, a plot of active site residues was plotted against trajectories frames (Fig. 6). Notably, Thr26 and Glu166, Asn142, and Gln129 were in contact with ciclesonide during most of the time. While, for example, Gly143 had a strong interaction with ciclesonide at the beginning of the simulation time, and then it was lost at about 10 ns.
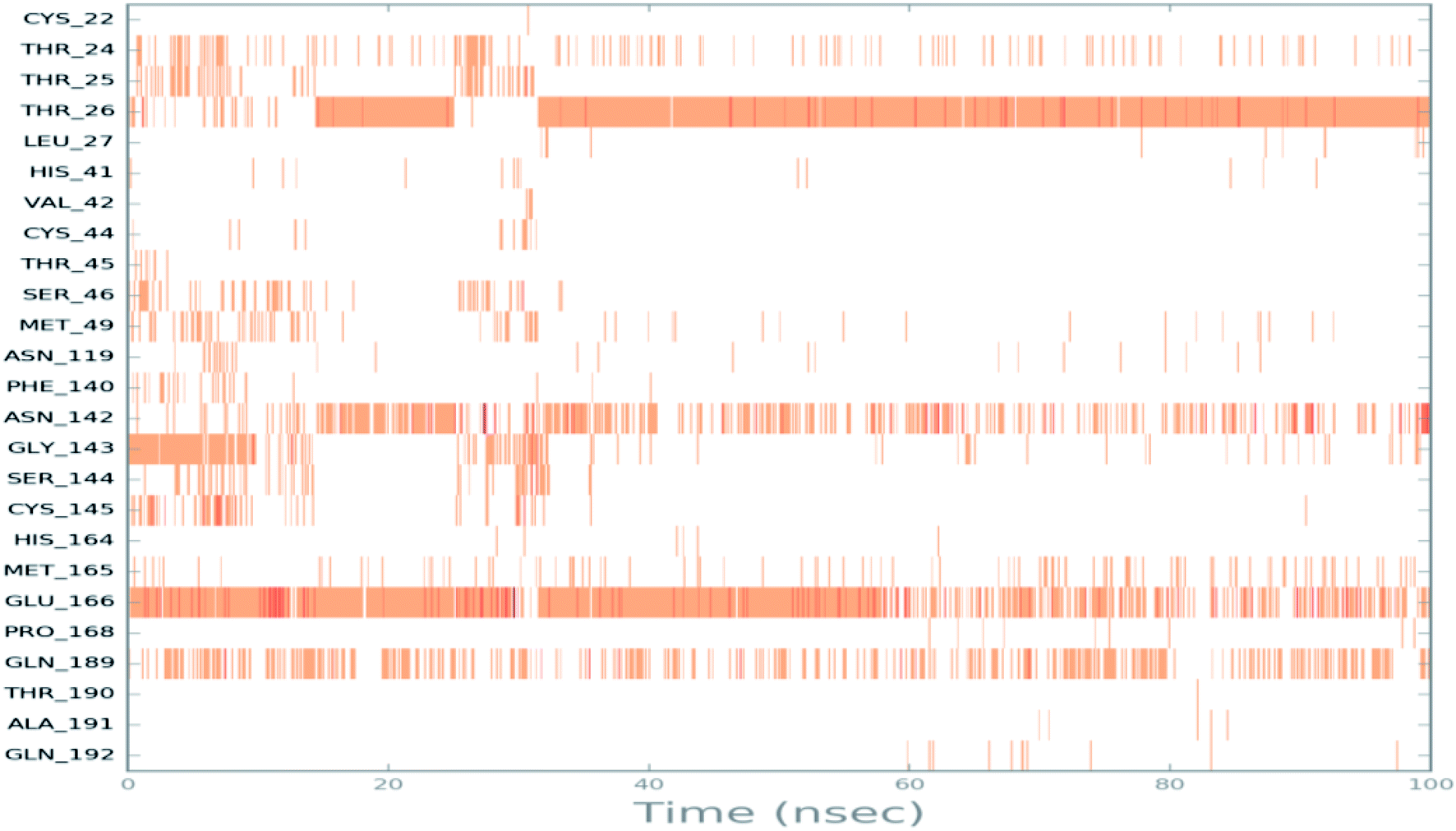 | ||
| Fig. 6 Ciclesonide–6LU7 interactions shown by the active site amino acids in each trajectory frame, white refers to zero interaction while the deep color indicates more interactions. | ||
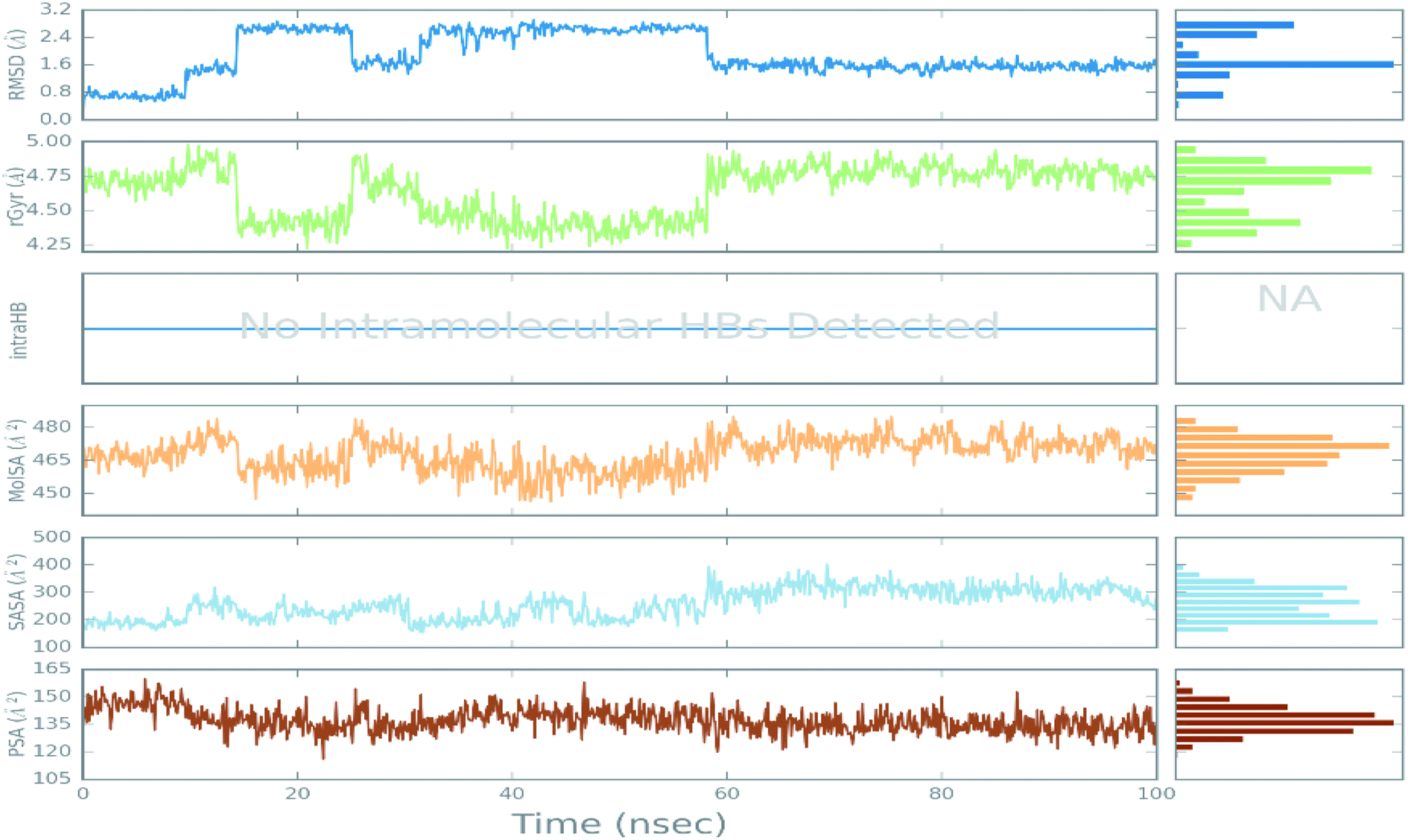 | ||
| Fig. 7 The ligand property trajectory of the ciclesonide–6LU7 complex during the 100 ns simulation. | ||
3.3. MM-GBSA study
Here, 200 snapshots were selected for further analysis taken within a 50 ps interval after calculating the average binding energy for the equilibrated MD trajectory. The binding energy was calculated using the previously mentioned equations.39 The average MM-GBSA binding energy was created using the thermal_mmgbsa.py python script provided by Schrödinger, which also produces the lipophilic energy, Coulomb energy, van der Waals energy, generalized Born electrostatic solvation energy, covalent binding energy, and hydrogen-bonding energy. All the obtained data are listed in Table 3.From the MM-GBSA calculations, the most favored binding was exerted by both betamethasone 1 and ciclesonide 11 compared to the N3 ligand, with ciclesonide 11 showing highly favored van der Waals interactions and lipophilic energy compared to 1 (Table 3), while betamethasone 1 showed a more favorable Columbo energy. Since all the drugs awere aliphatic, no π– π interaction energy was reported.
| ΔG Binding | Coulomb | Covalent | H-bond | Lipo | Solv_GB | vdW | St. Dev. | |
|---|---|---|---|---|---|---|---|---|
| a Coulomb: Coulomb energy; covalent: covalent binding energy; vdW: Van der Waals energy; Lipo: lipophilic energy; Solv_GB: generalized Born electrostatic solvation energy; H-bond: hydrogen-bonding energy; St. Dev.: standard deviation. | ||||||||
| Ciclesonide 11 | −51.24 | −11.00 | 1.57 | −1.16 | −14.32 | 19.33 | −45.67 | 8.67 |
| Betamethasone 1 | −51.05 | −19.48 | 0.86 | −1.58 | −12.53 | 21.71 | −40.03 | 4.35 |
| Hydrocortisone 4 | −49.00 | −15.59 | 2.19 | −1.01 | −14.05 | 20.84 | −41.38 | 9.07 |
| Fludrocortisone 3 | −48.48 | −15.46 | 1.58 | −0.91 | −12.98 | 18.66 | −39.36 | 7.32 |
| Triamcinolone 8 | −47.53 | −21.96 | 1.86 | −1.49 | −11.77 | 19.86 | −34.03 | 9.56 |
| Dexamethasone 2 | −37.37 | −15.83 | 1.27 | −0.82 | −10.36 | 22.99 | −34.62 | 4.23 |
| N3 23 | −80.00 | −43.59 | 4.67 | −3.45 | −16.27 | 45.02 | −64.12 | 6.62 |
3.3. Quantum mechanics studies
Since the molecular docking results revealed that ciclesonide 11 was the most promising candidate exhibiting COVID-19 main protease inhibitory activity, we therefore studied its structural and electronic configurations in detail. The chemical structures (2D and 3D) and nomenclature of ciclesonide (CAS 126544-47-6) are given in Fig. 8. It is obvious that ciclesonide possessed some chiral centers and rotatable single bonds, rendering it a flexible medium-size molecule (MW = 540.69 Da). In addition, ciclesonide contained a small system of conjugated double bonds found in one ring (labeled as ring A), see Fig. 8.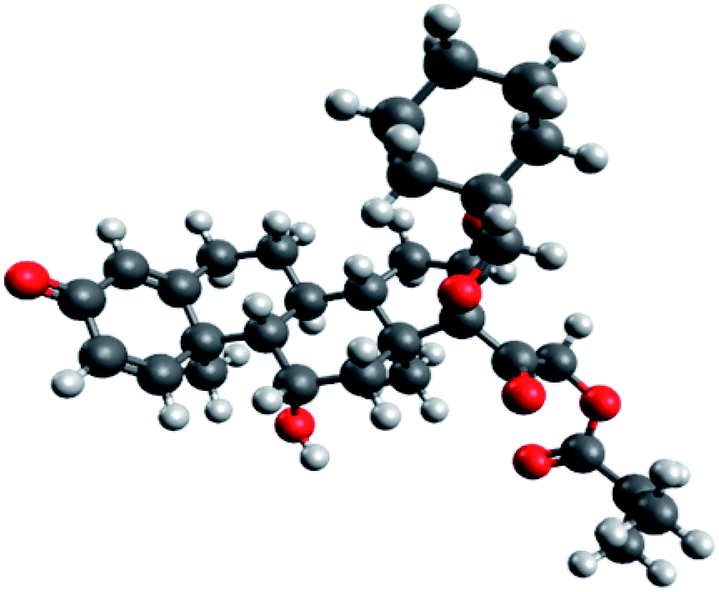 | ||
| Fig. 8 Optimized structure of ciclesonide obtained at the B3LYP/6-311G* level. | ||
Ciclesonide's structure was energetically optimized at different levels of theory as described in the computational methods. By reviewing the literature, B3LYP coupled with different types of basis sets have been predominantly utilized for describing the glucocorticoid systems.40,41 Therefore, studies on ciclesonide 11 using B3LYP/6-311G* and B3LYP/6-311+G* are herein reported in our study. Geometries of the ciclesonide structure obtained at B3LYP/6-311G* and B3LYP/6-311+G* are deposited in the ESI data 1.† Some of the spatial and electronic parameters of ciclesonide are listed in Table 4.
| B3LYP/6-311G* | B3LYP/6-311+G* | ||
|---|---|---|---|
| Rot. Const. | A (GHz) | 0.1080486 | 0.1080863 |
| B (GHz) | 0.0614718 | 0.0608067 | |
| C (GHz) | 0.0442127 | 0.0438208 | |
| Spatial moment r2 (a.u.) | 5216651.8603 | 5216696.0275 | |
| Eh (a.u.) | −1772.881464 | −1772.909008 | |
| ZPE (a.u.) | 0.719611 | 0.718603 | |
| Eh + ZPE (a.u.) | −1772.161853 | −1772.190406 | |
| Polarizability (a.u.) | 477.742460 | 512.314524 | |
| μ (D) | 9.2967 | 10.1645 | |
| HOMO–LUMO gap (eV) | 5.0764 | 5.1115 | |
| Entropy (cal mol−1 Kelvin) | 224.240 | 225.821 | |
The rotational constants parameter is a geometrical descriptor of the molecular size at vibrational equilibrium. The rotational constants at the A, B, and C states of ciclesonide were obtained at 0.1080486, 0.0614718, and 0.0442127 GHz (B3LYP/6-311G*), whereas nearly similar values were calculated at 0.1080863, 0.0608067, and 0.0438208 GHz using the B3LYP/6-311+G* method. The electronic spatial extent r2 (also called the spatial moment) conveys roughly the molecular size (volume) of a molecule. The values obtained by using either B3LYP/6-311G* or B3LYP/6-311+G* were nearly similar. Other parameters, such as the total energy (Eh), zero-point energy (ZPE), polarizability, dipole moment (μ), HOMO–LUMO energy gap, and entropy, did not show significant discrepancies in the computed values, as can be seen from Table 4.
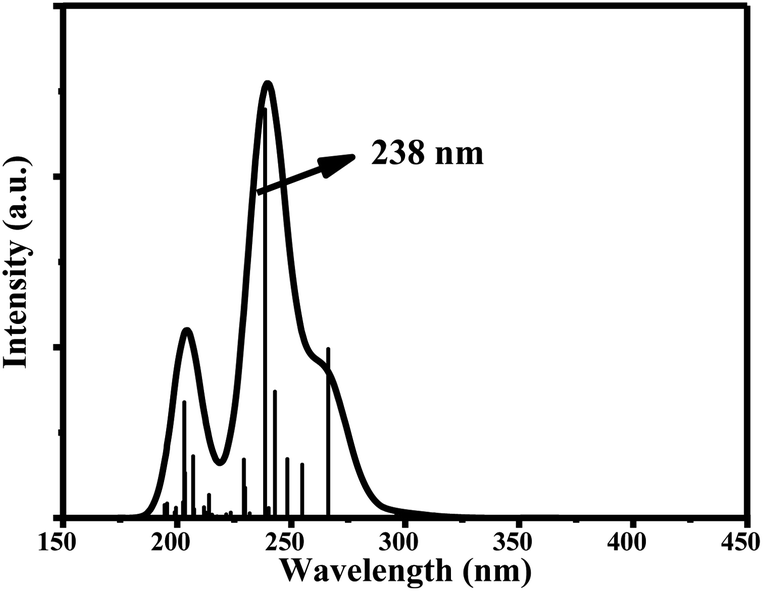
The main UV-vis absorption peak of ciclesonide was reported at 242 nm.42,43 The UV-vis peak using the B3LYP/6-311+G* model was calculated at 238 nm, showing a discrepancy from the reported value by only 4 nm; unlike for the B3LYP/6-311G* model, where the absorption peak was computed at 233 nm with a 9 nm difference from the reported value. This result suggests that the B3LYP/6-311+G* model is—most probably—the best model to describe the ground and transition states of ciclesonide. The computed absorption (excitation) energy, oscillator strength, and major electronic contributions of ciclesonide are listed in Table 5. The calculated UV-vis spectrum results are also shown in Table 6.
| Model | Absorption energy | Oscillator strength | Major contributions |
|---|---|---|---|
| 6-311G* | 233 nm | 0.1998 | H-5 → LUMO (43%) |
| H-4 → LUMO (13%) | |||
| H-2 → LUMO (20%) | |||
| 6-311+G* | 238 nm | 0.2393 | H-5 → LUMO (53%) |
| H-4 → LUMO (12%) | |||
| H-2 → LUMO (18%) |
| B3LYP/6-311G* | B3LYP/6-311+G* | |
|---|---|---|
| UV-vis | 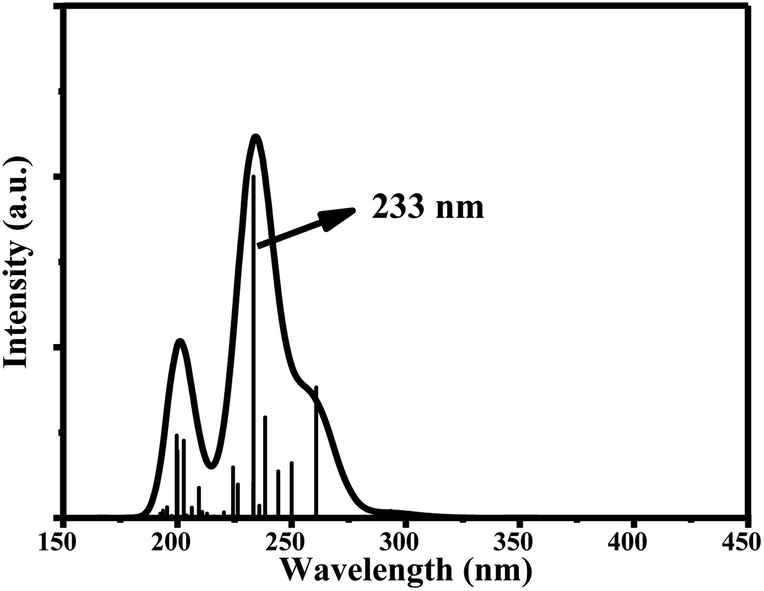 |
 |
| ECD | 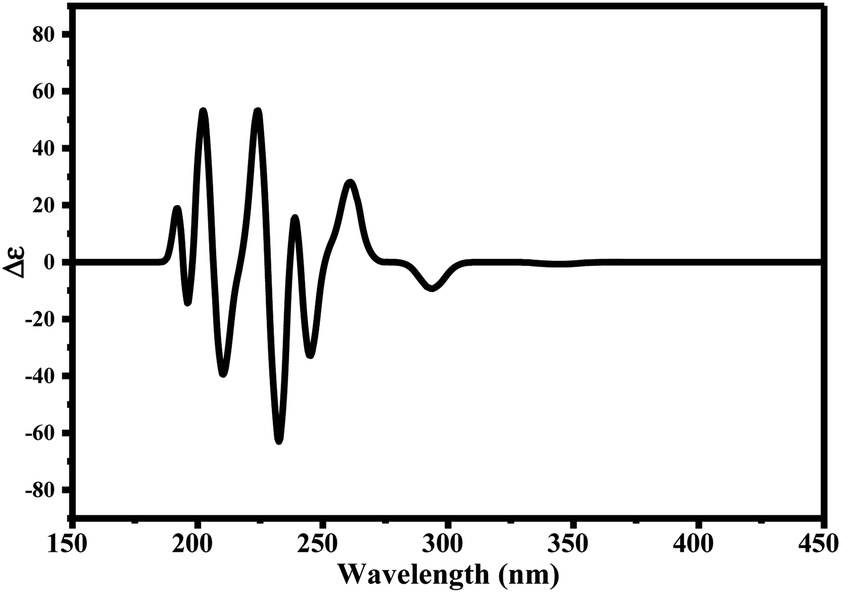 |
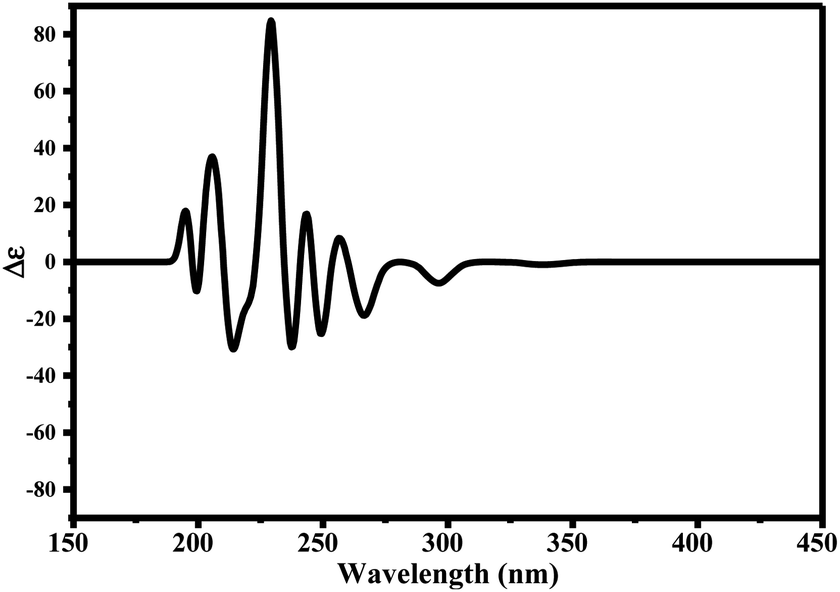 |
Since ciclesonide 11 possessed various chiral centers, it was important to calculate the electronic circular dichroism spectrum (ECD), which has not been reported in the literature to the best of our knowledge. The ECD spectrum measures the difference in absorbance of right- and left-circularly polarized light by a molecule rather than the commonly used absorbance of isotropic light as in UV-vis measurements.44 The ECD spectra of ciclesonide 11 calculated at the B3LYP/6-311G* and B3LYP/6-311+G* levels are reported for the first time, as depicted in Table 6.
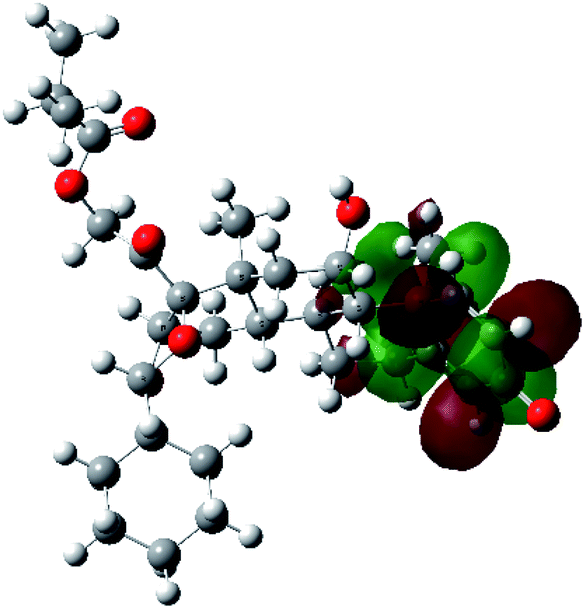
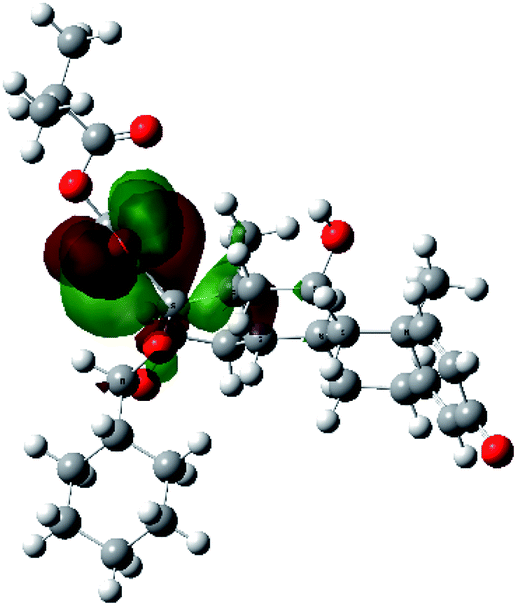
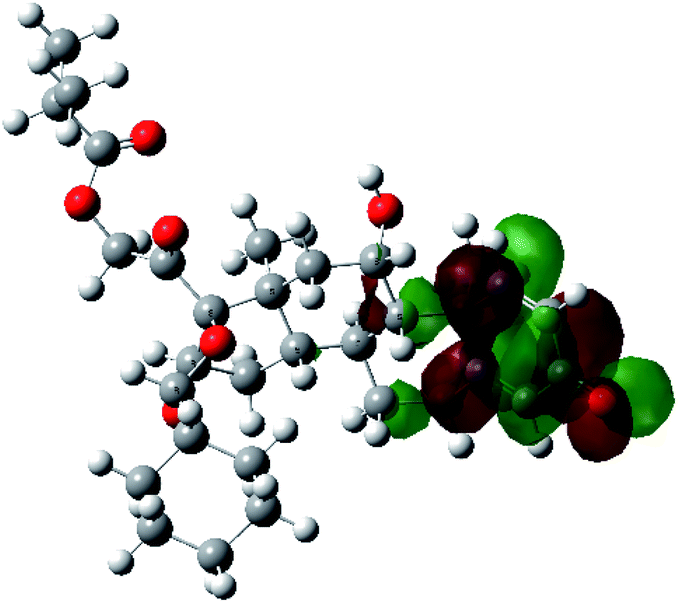
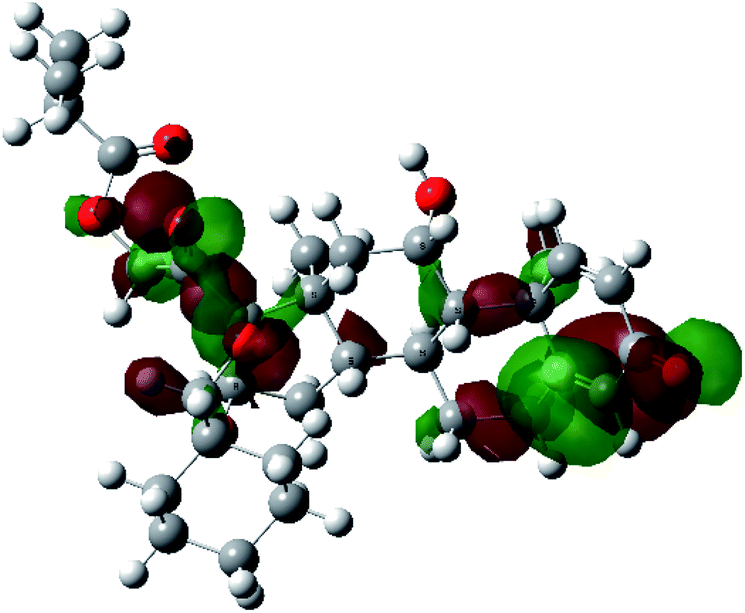
It is known that the most energetically stable structure of a drug is not necessary the most biologically active form ref. 45, which can be attributed to the combined importance of the electronic configuration besides the geometrical configuration in determination of the binding interactions between a drug and its target protein. Therefore, the electrons distribution over some molecular orbitals were computed. The electron charge density of the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO), besides the two outermost molecular orbitals (occupied and virtual) of ciclesonide 11, are depicted in Table 7.
| B3LYP/6-311G* | B3LYP/6-311+G* | |
|---|---|---|
| LUMO+2 | 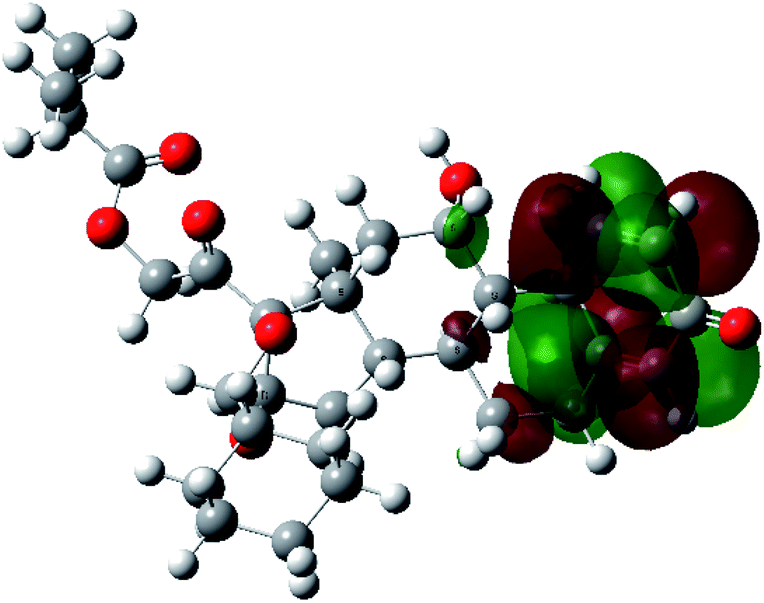 |
 |
| LUMO+1 | 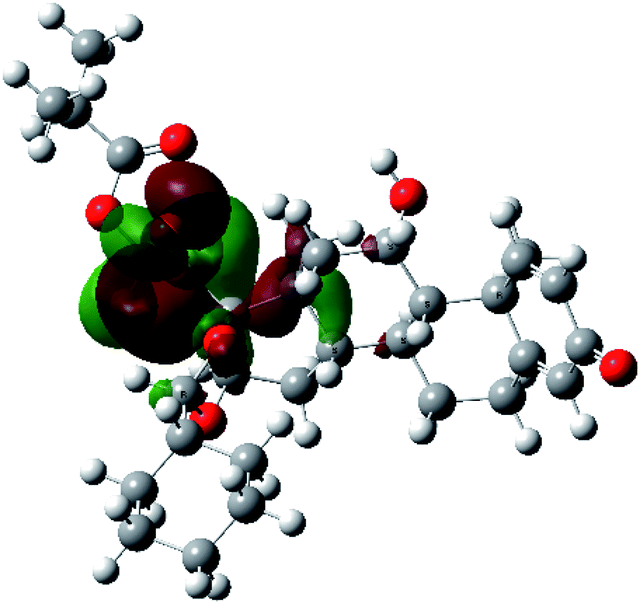 |
 |
| LUMO | 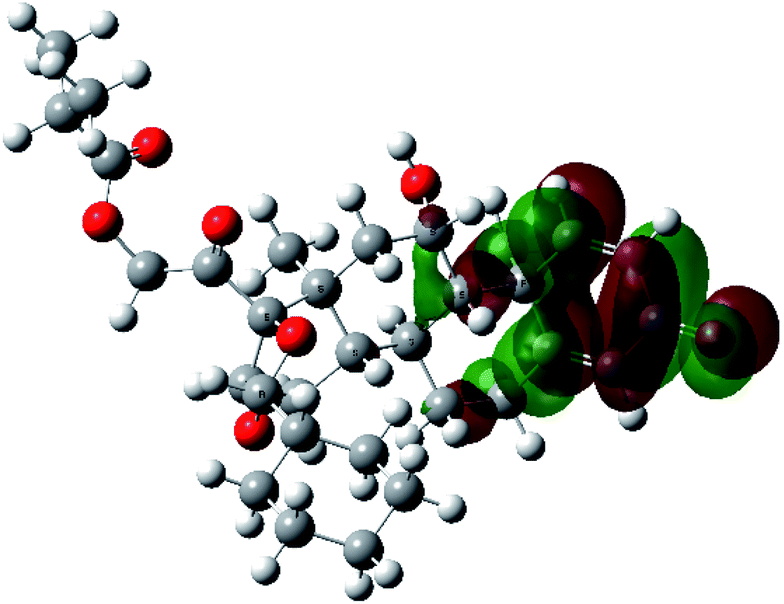 |
 |
| HOMO | 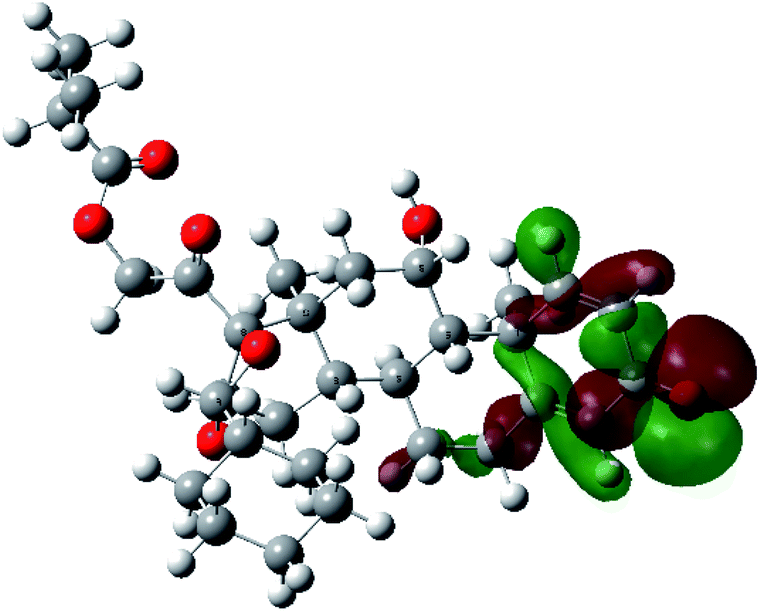 |
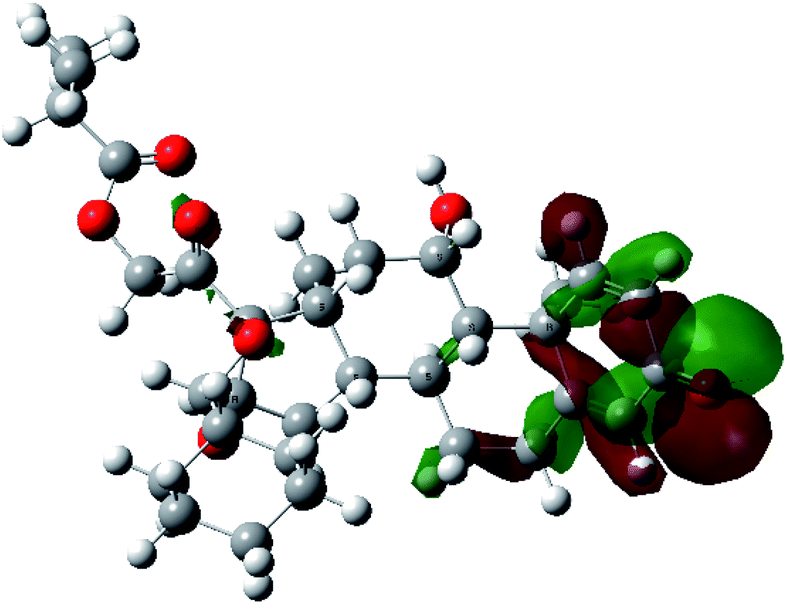 |
| HOMO−1 | 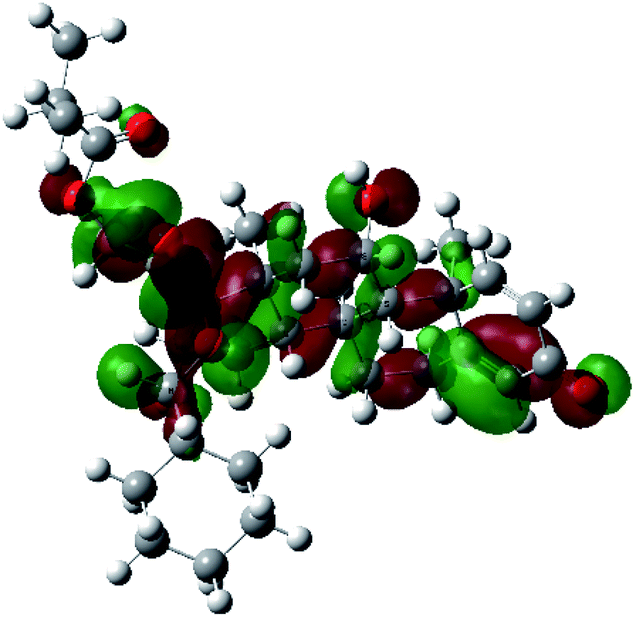 |
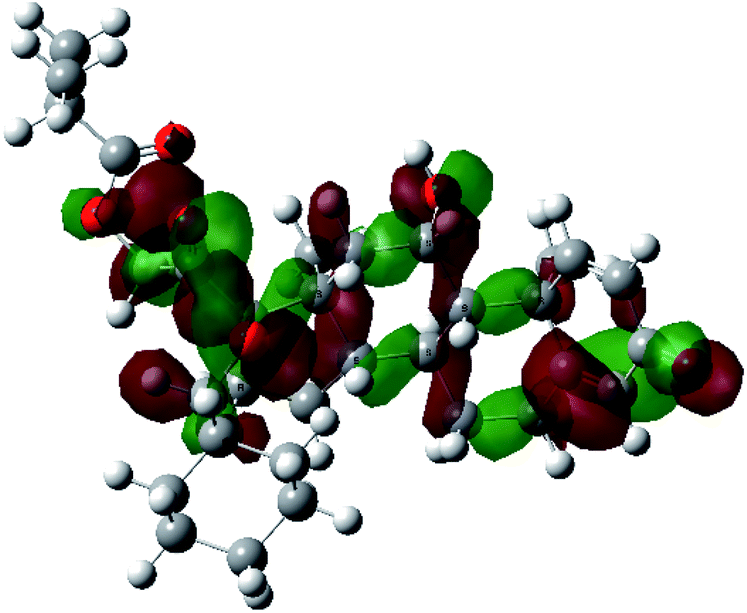 |
| HOMO−2 | 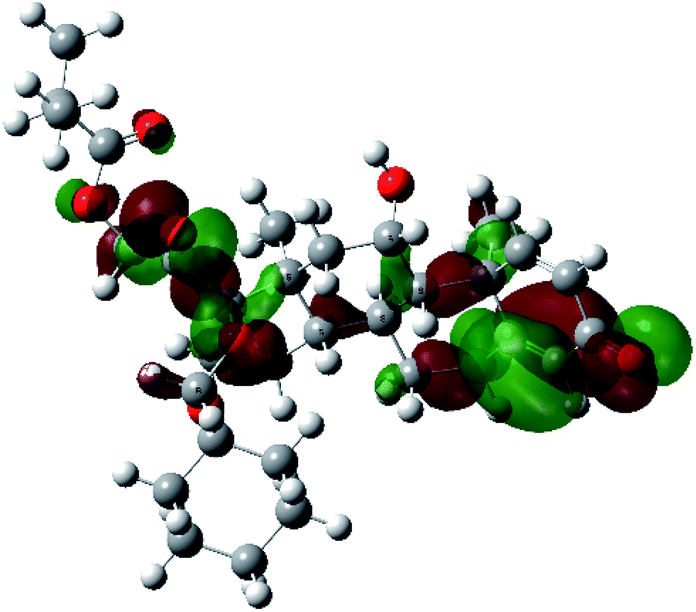 |
 |
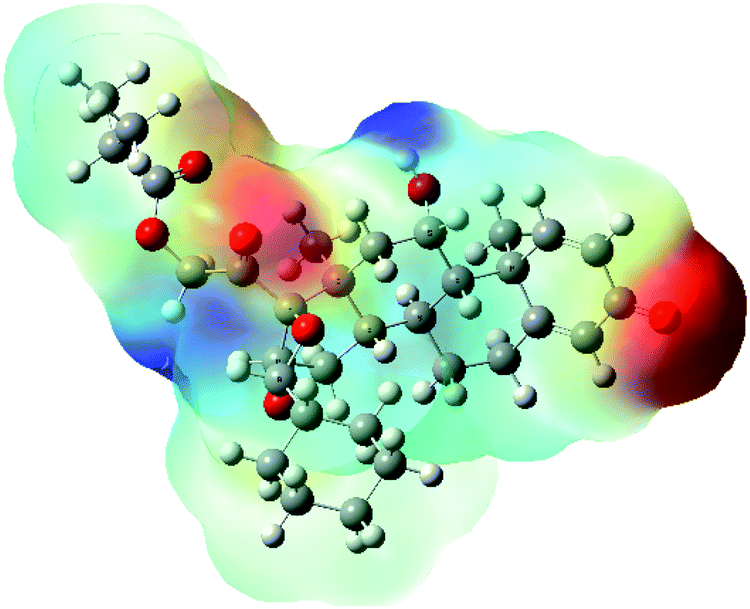
| MEP | 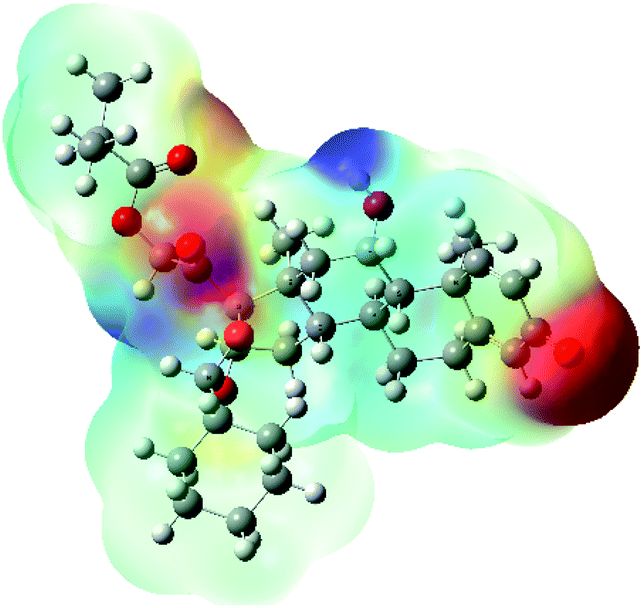 |
 |
It was noted that the electron density is mainly localized on ring A at the HOMO of ciclesonide 11, while the electron density was slightly spread to the adjacent ring in the case of the LUMO. Since the HOMO and LUMO are the main contributors to the binding interactions between a drug and the target receptor, the obtained calculations postulate the occurrence of electron transfer from ring A to the adjacent ring. Further studies should be conducted to confirm the occurrence of such a charge transfer process, which is beyond the scope of our study.
The molecular electrostatic potential (MEP) map quantifies the electronic density distribution at a molecular level. MEP maps of ciclesonide 11 are depicted in Table 7. The electronic density distribution changes from the more electronegative (red) to the more electropositive (blue). It was found that the carbonyl group on ring A was electron rich and therefore a hydrogen bond with Ser46 was formed, as demonstrated by the molecular docking calculations. Another electronegative site was observed on the carboxylic group connecting an isobutyryl moiety to the main nucleus of ciclesonide. The observed electronegativity may account for the metabolic attack for cleavage of the isobutyryl group, resulting in formation of the active metabolite desisobutyryl–ciclesonide. It is noteworthy that the MD simulation revealed a transient binding interaction between that carboxylic group and Gly143. In addition to the two electron-rich sites, two electron-deficient sites were observed, which can act as hydrogen bond donor sites.
Overall, the conducted quantum mechanical calculations emphasized that the use of a proper computational model is crucial for obtaining an accurate description of the structural and electronic configurations of ciclesonide 11. In addition, QM calculations can provide us with quantitative analysis of physicochemical properties that cannot be experimentally measured, such as the electron density of molecular orbitals.
3.4. Structure–activity relationship
The study of the structure–activity relationship of the tested glucocorticoids (1–22) according to their binding affinities to the SARS-CoV-2 main protease showed the following interesting results.The cyclization at C16 and C17 of the steroidal nucleus with a cyclohexyl methylene dioxy moiety and C20 substitution with an isobutyrate ester with an extra double bond between C1 and C2 (compound 11) showed the most favorable activity against the SARS-CoV-2 main protease. Besides, the studied SAR revealed that the addition of an α-fluoro group at C9 experienced better activity. Moreover, the substitution of C16 of the steroidal nucleus with a methyl group showed enhanced activity against the virus main protease, yet the α orientation of the methyl group was more favorable than the β one for better activity and that may be attributed to the steric hindrance with β substituents at C17 and the β angular methyl of C18. In addition, the studied SAR let us observe the favorable extra double bond added between C1 and C2 and its effect for enhancing the activity. On the other hand, SAR revealed that substitution at C6 with an alkyl group had little effect on the activity against the SARS-CoV-2 main protease (Fig. 9).
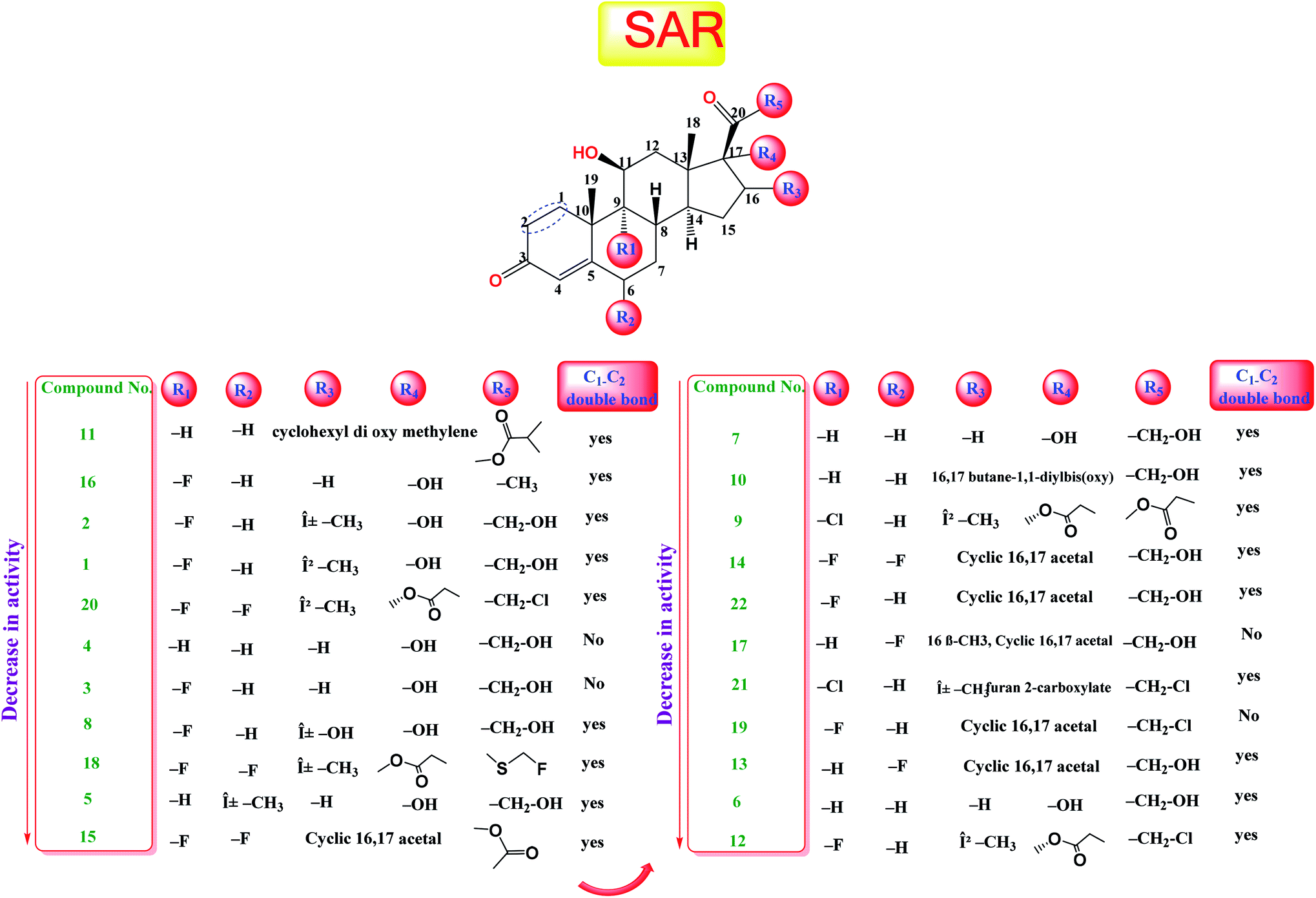 | ||
| Fig. 9 Structure–activity relationship (SAR) analysis for the studied 22 glucocorticoids as promising SARS-CoV-2 main protease inhibitors. | ||
4. Conclusion
Our study revealed the potential of repurposing glucocorticoid drugs to bind in the active site of the SARS-CoV-2 main protease. Six of the screened drugs (betamethasone 1, dexamethasone 2, fludrocortisone 3, hydrocortisone 4, triamcinolone 8, and ciclesonide 11) showed better binding through molecular docking simulation in the enzyme active site. Furthermore, the molecular dynamic simulations showed good interactions between the selected drugs and the COVID-19 main protease active site. It was noted that the drugs did not affect the structure of the protein, as the RMSD was less than 3 Å. For the MM-GBSA frontier analysis, ciclesonide 11 and betamethasone 1 were found to be the most active candidates among the other drugs. MD simulations also showed that Glu166 residue to be critical to the active site interaction and this might help in the rational designing of new molecules or modification of the current drugs. Quantum mechanics calculations were consistent with the molecular docking studies as the carbonyl group on ring A and the carboxylic group connecting isobutyryl moiety to the main nucleus of ciclesonide 11 were found to be electron-rich sites and involved in interactions with biological targets. Based on our study, the screened FDA-approved drugs—especially ciclesonide 11—could undergo preclinical and clinical trials for further evaluation of their activity against COVID-19 and to ensure their safe use. Besides, glucocorticoids may be used as lead compounds for the development of potent SARS-CoV-2 (Mpro) inhibitors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors acknowledge Swinburne University of Technology for using Swinburne's supercomputing facilities.References
- S. N. Mali, The Rise of New Coronavirus Infection-(COVID-19): A Recent Update, Eurasian J. Med. Oncol., 2020, 4(1), 35–41 Search PubMed.
- WHO, Archived: WHO Timeline – COVID-19, WHO, 2020, p. 2020 Search PubMed.
- Worldometer, Coronavirus Update (Live): Cases and Deaths from COVID-19 Virus Pandemic, Worldometers, 2020, p. 1 Search PubMed.
- W. Zhang, et al., The use of anti-inflammatory drugs in the treatment of people with severe coronavirus disease 2019 (COVID-19): The experience of clinical immunologists from China, Clin. Immunol., 2020, 214, 108393 CrossRef CAS PubMed.
- J. Chen, et al., Protection against COVID-19 injury by qingfei paidu decoction via anti-viral, anti-inflammatory activity and metabolic programming, Biomed. Pharmacother., 2020, 129, 110281 CrossRef CAS.
- S. Pushpakom, et al., Drug repurposing: Progress, challenges and recommendations, Nat. Rev. Drug Discovery, 2018, 18(1), 41–58 CrossRef.
- S. G. Eliaa, A. A. Al-karmalawy, R. M. Saleh and M. F. Elshal, Empagliflozin and Doxorubicin Synergistically Inhibit the Survival of Triple-Negative Breast Cancer Cells via Interfering with the mTOR Pathway and Inhibition of Calmodulin: In Vitro and Molecular Docking Studies, ACS Pharmacol. Transl. Sci., 2020, 3(6), 1330–1338 CrossRef CAS PubMed.
- A. Ghanem, et al., Tanshinone IIA synergistically enhances the antitumor activity of doxorubicin by interfering with the PI3K/AKT/mTOR pathway and inhibition of topoisomerase II: in vitro and molecular docking studies, New J. Chem., 2020, 44, 17374–17381 RSC.
- A. Talevi and C. L. Bellera, Challenges and opportunities with drug repurposing: finding strategies to find alternative uses of therapeutics, Expert Opin. Drug Discov., 2020, 15(4), 397–401 CrossRef PubMed.
- S. M. Corsello, et al., Information Resource, Nat. Med., 2017, 23(4), 405–408 CrossRef CAS PubMed.
- Y. Masoudi-Sobhanzadeh, Y. Omidi, M. Amanlou and A. Masoudi-Nejad, Drug databases and their contributions to drug repurposing, Genomics, 2020, 112(2), 1087–1095 CrossRef CAS PubMed.
- S. Brogi, Computational approaches for drug discovery, Molecules, 2019, 24(17), 1–6 CrossRef.
- X. Lin, X. Li and X. Lin, A review on applications of computational methods in drug screening and design, Molecules, 2020, 25(6), 1–17 CrossRef.
- R. M. Johnson and J. M. Vinetz, Dexamethasone in the management of covid -19, BMJ, 2020, 370, 1–2 Search PubMed.
- T. C. Theoharides and P. Conti, Dexamethasone for COVID-19? Not so fast, J. Biol. Regul. Homeost. Agents, 2020, 34(4), 1241–1243 CAS.
- S. Matsuyama, et al., The inhaled corticosteroid ciclesonide blocks coronavirus RNA replication by targeting viral NSP15, J. Biomol. Struct. Dyn., 2020, 1–13 Search PubMed.
- A. D. Elmezayen, A. Al-Obaidi, A. T. Şahin and K. Yelekçi, Drug repurposing for coronavirus (COVID-19): in silico screening of known drugs against coronavirus 3CL hydrolase and protease enzymes, J. Biomol. Struct. Dyn., 2020, 1–12 CrossRef PubMed.
- H. Li, S. M. Liu, X. H. Yu, S. L. Tang and C. K. Tang, Coronavirus disease 2019 (COVID-19): current status and future perspectives, Int. J. Antimicrob. Agents, 2020, 55(5), 105951 CrossRef CAS PubMed.
- R. Alnajjar, A. Mostafa, A. Kandeil and A. A. Al-karmalawy, Molecular docking, molecular dynamics, and in vitro studies reveal the potential of angiotensin II receptor blockers to inhibit the COVID-19 main protease, Heliyon, 2020, 6, e05641 CrossRef.
- A. A. Al-Karmalawy, R. Alnajjar, M. A. Dahab, A. M. Metwaly and I. H. Eissa, Molecular docking and dynamics simulations reveal the potential of anti-HCV drugs to inhibit COVID-19 main protease, Pharm Sci., 2021 DOI:10.34172/PS.2021.3.
- A. A. Zaki and A. A. Al-karmalawy, Molecular docking reveals the potential of Cleome amblyocarpa isolated compounds to inhibit COVID-19 virus main protease, New J. Chem., 2020, 44, 16752–16758 RSC.
- Z. Jin, X. Du, Y. Xu, Y. Deng, M. Liu, Y. Zhao, B. Zhang, X. Li, L. Zhang, C. Peng, Y. Duan, Y. Jing, L. Wang, K. Yang, F. Liu, R. Jiang, X. Yang, Y. Tian, X. Liu, X. Yang and F. Bai, Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors, Nature, 2020, 582, 289–293 CrossRef CAS PubMed.
- C. C. G. Inc., Molecular operating environment (MOE), Chemical Computing Group Inc, 1010 Sherbooke St. West, Suite# 910, Montreal, 2016 Search PubMed.
- Schrödinger Release, 3: Desmond molecular dynamics system, DE Shaw research, New York, NY, 2017 Search PubMed; Maestro-Desmond Interoperability Tools, Schrödinger, New York, NY, 2017 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, et al., Gaussian 16, revision C. 01, 2016 Search PubMed.
- A. A. Al-karmalawy and M. Khattab, Molecular modelling of mebendazole polymorphs as a potential colchicine binding site inhibitor, 2020 Search PubMed.
- W. L. Delano and D. Ph, PyMOL: An Open-Source Molecular Graphics Tool, 2002 Search PubMed.
- I. W. Davis and D. Baker, RosettaLigand Docking with Full Ligand and Receptor Flexibility, J. Mol. Biol., 2009, 385(2), 381–392 CrossRef CAS PubMed.
- B. J. McConkey, V. Sobolev and M. Edelman, The performance of current methods in ligand-protein docking, Curr. Sci., 2002, 83(7), 845–856 CAS.
- E. Harder, et al., OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins, 2016 Search PubMed.
- W. L. Jorgensen, et al., Comparison of simple potential functions for simulating liquid water, J. Chem. Phys., 1983, 79, 926 CrossRef CAS.
- E. Neria, S. Fischer and M. Karplus, Simulation of activation free energies in molecular systems, J. Chem. Phys., 1996, 105, 1902 CrossRef CAS.
- S. Release, 4. Desmond Molecular Dynamics System, Maest. Interoperability Tools, DE Shaw Research, 2016 Search PubMed.
- G. J. Martyna, M. L. Klein and M. Tuckerman, Nosé – Hoover chains: The canonical ensemble via continuous dynamics Nose-Hoover chains: The canonical ensemble via continuous dynamics, J. Chem. Phys., 2011, 97, 2635 CrossRef.
- G. J. Martyna, D. J. Tobias and M. L. Klein, Constant pressure molecular dynamics algorithms, J. Chem. Phys., 1994, 6323 Search PubMed.
- A. D. Becke, A new mixing of Hartree–Fock and local density-functional theories, J. Chem. Phys., 1993, 1372 CrossRef CAS.
- A. D. Becke, Density-functional thermochemistry. III . The role of exact exchange, J. Chem. Phys., 1992, 5648 Search PubMed.
- M. Cossi, et al., Molecules in Solution with the C-PCM Solvation Model, J. Comput. Chem., 2003, 24(6), 669–681 CrossRef CAS PubMed.
- R. Alnajjar, N. Mohamed and N. Kawafi, Bicyclo[1.1.1]Pentane as Phenyl Substituent in Atorvastatin Drug to improve Physicochemical Properties: Drug-likeness, DFT, Pharmacokinetics, Docking, and Molecular Dynamic Simulation, J. Mol. Struct., 2020, 129628 Search PubMed.
- A. Rex, R. David, K. Jeremy, N. John and P. Chandra, Crystal and Molecular Structure and DFT Calculations of the Steroidal Oxime 6 E -Hydroximino-androst-4-ene-3 , 17-dione - ( C 19 H 25 NO 3 ) a Molecule with Antiproliferative Activity, J. Chem. Crystallogr., 2019, 49, 29–36 CrossRef.
- P. Manickam, J. E. Soc, P. Manickam, F. Arizaleta and M. Gurusamy, Theoretical Studies of Cortisol-Imprinted Prepolymerization Mixtures: Structural Insights into Improving the Selectivity of Affinity Sensors, J. Electrochem. Soc., 2017, 164, B3077 CrossRef CAS.
- N. A. H. Cabral-marques, Obtention and characterization of a ciclesonide: methyl-beta cyclodextrin complex, J. Inclusion Phenom. Macrocyclic Chem., 2014, 125–132 Search PubMed.
- P. Prajapati, and V. Vaghela, Densitometric Measurement for Estimation of Ciclesonide in Bulk and Its Dosage Form ( Rotacap ) by H. T. Chromatography, 26, 435–439, 2013.
- M. Khattab, Theoretical study of the geometric and electronic characterization of carbendazim-based drug (Nocodazole), Heliyon, 2020, 6(6), e04055 CrossRef PubMed.
- M. Khattab, S. Chatterjee, A. H. A. Clayton and F. Wang, Absorption spectroscopy, New J. Chem., 2016, 8296–8304 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra10674g |
| This journal is © The Royal Society of Chemistry 2021 |