
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn search of therapeutic candidates for HIV/AIDS: rational approaches, design strategies, structure–activity relationship and mechanistic insights
Dinesh Kumar
 *a,
Pooja Sharma
ab,
Shabu
c,
Ramandeep Kaur
a,
Maloba M. M. Lobe
d,
Girish K. Gupta
e and
Fidele Ntie-Kang
*dfg
*a,
Pooja Sharma
ab,
Shabu
c,
Ramandeep Kaur
a,
Maloba M. M. Lobe
d,
Girish K. Gupta
e and
Fidele Ntie-Kang
*dfg
aSri Sai College of Pharmacy, Manawala, Amritsar-143001, Punjab, India. E-mail: dineshkumargndu@gmail.com; Tel: +91-9988902489
bDepartment of Pharmaceutical Sciences and Drug Research, Punjabi University, Patiala, India
cIndian Institute of Integrative Medicine (CSIR-IIIM), Canal Road, Jammu 180001, India
dDepartment of Chemistry, Faculty of Science, University of Buea, P. O. Box 63, Buea, Cameroon. E-mail: fidele.ntie-kang@ubuea.cm; Tel: +237 685625811
eDepartment of Pharmaceutical Chemistry, Sri Sai College of Pharmacy, Badhani, Pathankot-145001, Punjab, India
fInstitute for Pharmacy, Martin-Luther-Universität Halle-Wittenberg, Kurt-Mothes-Str. 3, 06120 Halle (Saale), Germany. E-mail: ntiekfidele@gmail.com; fidele.ntie-kang@pharmazie-uni-halle.de; Tel: +49 3455525043
gInstitute of Botany, Technical University of Dresden, Zellescher Weg 20b, 01062 Dresden, Germany. E-mail: fidele.ntie-kang@tu-dresden.de
First published on 18th May 2021
Abstract
The HIV/AIDS pandemic is a serious threat to the health and development of mankind, which has affected about 37.9 million people worldwide. The increasing negative health, economic and social impacts of this disease have led to the search for new therapeutic candidates for the mitigation of AIDS/HIV. However, to date, there is still no treatment that can cure this disease. Furthermore, the clinically available drugs have numerous severe side effects. Hence, the synthesis of novel agents from natural leads is one of the rational approaches to obtain new drugs in modern medicinal chemistry. This review article is an effort to summarize recent developments with regards to the discovery of novel analogs with promising biological potential against HIV/AIDS. Herein, we also aim to discuss prospective directions on the progress of more credible and specific analogues. Besides presenting design strategies, the present communication also highlights the structure–activity relationship together with the structural features of the most promising molecules, their IC50 values, mechanistic insights and some interesting key findings revealed during their biological evaluation. The interactions with the amino acid residues of the enzymes responsible for HIV-1 inhibition are also discussed. This collection will be of great interest for researchers working in this area.
 Dinesh Kumar | Dr Dinesh Kumar graduated in Pharmacy in 2005 from Punjab Technical University and achieved 4th position in the University. In 2008 he obtained his Master's Degree in Pharmaceutical Chemistry from the Department of Pharmaceutical Sciences, Guru Nanak Dev University, Amritsar, Punjab, India. He received his PhD in Pharmaceutical Sciences from Guru Nanak Dev University, Amritsar, where he worked on the Syntheses and Evaluation of Chalconoid Hybrids of Oxa, Aza and Thio Compounds as Potent Anti-proliferative Agents. In 2014, he joined the Department of Pharmaceutical Sciences, Guru Nanak Dev University, Amritsar as an Assistant Professor. Currently, he is working as Associate Professor, Director-Cum-Principal in the department of Pharmaceutical science, Sri Sai College of Pharmacy, Manawala, Amritsar, Punjab, India. He has more than 12 years of research experience in pharmaceutical field including academic, research and teaching. His research is focused on the synthesis and evaluation of novel heterocycles and their application in medicinal chemistry. He is also an editorial board member and reviewer of many reputed international and national journals. He has attended many national and international conferences. He has published his research in journals of international repute. |
 Pooja Sharma | Mrs Pooja Sharma graduated in Pharmacy from Punjab Technical University in 2005 and obtained her Master's Degree in the field of Pharmacognosy (Phytochemistry) in 2008 from the Department of Pharmaceutical Sciences, Guru Nanak Dev University, Amritsar. She achieved 2nd position in the University in her Master's programme. She is pursuing her PhD in the Department of Pharmaceutical Sciences and Drug Research, Punjabi University, Patiala. She also worked in the Council of Scientific & Industrial Research (CSIR) Laboratory of India at Jammu on the phytochemical investigation and standardization of natural products. Then she joined the Sri Sai College of Pharmacy, Manawala, Amritsar, Punjab, India as an Assistant Professor. Currently, she is working as the Head of Department at Sri Sai College of Pharmacy, Manawala, Amritsar, Punjab, India. She has more than 12 years of research experience in pharmaceutical field including academic, research and teaching. She is also a member of many pharmaceutical associations and societies. Her area of research is focused on the extraction and isolation of phytoconstituents of pharmaceutical interest. |
 Shabu | Ms. Sabu graduated with her BSc from Govt. College Dharamshala in 2016. In 2018, she obtained an MSc in Zoology from Himachal Pradesh University, Shimla. Currently, she is pursuing her PhD in Life Science, in Cancer Pharmacology Division from Indian Institute of Integrative Medicine, Jammu, India. She has attended many national and international conferences. |
 Ramandeep Kaur | Ms. Ramandeep Kaur graduated with a degree in Pharmacy in the first division from Sri Sai College of Pharmacy, Manawala, Amritsar, Punjab, India, affiliated with Punjab Technical University. Currently, she is working as a Pharmacy Officer at the Department of Health and Family Welfare, Punjab, India. She has attended many national and international conferences. |
 Maloba M. M. Lobe | Dr Maloba M. M. Lobe studied Chemistry at the University of Buea, where she obtained her BSc (2011), M.Sc. (2015) and PhD (2021) under the guidance of Prof. Simon M. N. Efange. Her research interest focuses on the design, synthesis and biological evaluation of novel small molecules against cancer and neglected tropical diseases. She was an Adolphe Monkedje Fellow 2016/2017 and received an award from the BMBF /DLR-DAAD Project (2018): Partnerships for Sustainable Solutions with Sub-Saharan Africa at TU Dortmund, where she obtained training on Natural Products: Chromatography, Spectroscopy and Biological Aspects. She is currently working on the development of novel spirooxindoles as potential pharmacologically active agents for the treatment of diseases, including cancer, malaria and tuberculosis, which has earned two original publications and two patents for her research group. |
 Girish K. Gupta | Dr Girish Kumar Gupta graduated in pharmacy in 2006 from Guru Jambheshwar University of Science and Technology, Hisar, India. In 2009 he obtained his Master's Degree in Pharmaceutical Chemistry from the University Institute of Pharmaceutical Sciences, Kurukshetra University, Kurukshetra, Haryana, India. Currently working as a Professor of Pharmaceutical Chemistry in Sri Sai of College of Pharmacy, Badhani, Pathankot, India. He has more than 12 years of research experience in the pharmaceutical field including academic and industrial research and teaching. He has over 80 publications in the field of heterocyclic chemistry in peer-reviewed high-impact journals. He is also a member of many pharmaceutical associations and societies. Moreover, he is also serving as the Director Research and Development at Sri Sai Group of Institutes, Pathankot, Punjab, India. He is also an editorial board member, editor, guest editor and reviewer of many reputed international and national journals. His area of research encompasses drug design, molecular modelling, and green synthesis related to nitrogen-containing heterocycles. |
 Fidele Ntie-Kang | Fidele Ntie-Kang heads the Molecular Simulations Laboratory, Chemistry Department, University of Buea. He studied Chemistry at the University of Douala (Cameroon) from 1999 to 2005, leading to Bachelor's and Master's Degrees. His PhD from the University of Douala (Cameroon) was based on molecular modeling of anti-tubercular drug targets to design novel inhibitors, followed by a Habilitation in Pharmaceutical Chemistry from Martin-Luther University Halle-Wittenberg (Germany) under the supervision of Prof. Wolfgang Sippl. His current focus is the discovery of bioactive natural products from African flora by the use of virtual screening followed by in vitro assays. A major contribution of his research team has been the development of the African natural products database. |
1. Introduction
The human immunodeficiency virus (HIV), which causes AIDS, is primarily responsible for the most severe public health challenges.1 However, there is a global promise to impede new HIV cases and ensure that everyone with HIV has access to HIV treatment. According to the worldwide statistics from UNAIDS, it was estimated that there were 37.9 million people suffering with HIV/AIDS in 2018. Clinically, more than thirty drugs targeting different steps of the viral life cycle have been approved to alleviate AIDS/HIV.2,3 Unfortunately, the use of drug cocktails is limited due to their severe noxious side effects and speedy emergence of resistant viral strains. Hence, new natural products can be considered as novel leads for the development of effective and selective potential therapeutic agents for AIDS. Thus, new anti-HIV agents that are less toxic and more effective in targeting HIV reservoirs in the body are still needed.4–7Traditionally, in the continuing search for natural products, the pedigrees of diverse medicinal agents are considered. Natural products play an important role in drug discovery and chemical biology.8–13 In drug discovery, numerous heterocycles such as pyrans, flavones, coumarins, pyrimidones, thiazoles, xanthenes, imidazoles, pyrazoles, isoxazolines and oxazolidines have been considered for the development of potential novel lead molecules.14–16 Typically, they are scaffolds that need to be decorated with selected substituents to exert the desired biological actions. These heterocycles have been used to construct diverse therapeutic drug candidates having an ample range of biological activities, viz, anticancer, anti-inflammatory, antimicrobial, antiviral, anti-HIV and anti-TB.17–23
Herein, we present a comprehensive summary of some recent advancements in the field of anti-AIDS by prominent scientists, researchers and scholars globally. Besides presenting the design strategies, this article also highlights the structure–activity relationship, IC50 values, and mechanistic insights revealed during the biological evaluation of compounds against HIV together with interesting key findings. The interactions with the amino acid residues of enzymes responsible for HIV-1 inhibition are also discussed diagrammatically.
To the best of our knowledge, this is the first comprehensive review of the recent advancements in the field of anti-HIV agents in the literature during the last decade.
The present assemblage will be of great interest to the scientific community to accelerate their further research for the development of novel heterocyclic compounds having potential against AIDS/HIV, thus helping them to adopt a focused and speedy target-oriented drug design. To present the rational approaches, we tried to classify the strategies based on the core functionalities of the chemical motifs. The classification is as follows:
(1) Andrographolide-based analogs
(2) Azaindole-based analogs
(3) Artemisinin-based analogs
(4) Maslinic acid-based analogs
(5) Calanolide-based analogs
(6) Labdane-based analogs
(7) Gomisin-G-based analogs
(8) Quinoline-based analogs
(9) 4-Hydroxypyrone-based analogs
(10) Ingenol-based analogs
(11) Lavendustin B-based analogs
(12) 3,7-Dihydroxytropolone-based analogs
(13) P1/P1′-substituted cyclic urea-based analogs
2. Rational approaches/design strategies
2.1. Andrographolide-based analogs
Andrographolide (1, Fig. 1a) is a diterpenoid lactone-based compound obtained from the plant Andrographis paniculata with potent anti-HIV activity.24–26 Andrographolide can also be used for the mitigation and management of several diseases including hepatitis, cancer, meningitis, hepatotoxicity, diabetes and other inflammatory disorders.27–32 3-Nitrobenzylidene, 2′6′-dichloro-nicotinoyl ester-type derivatives, 14-deoxy-11,12-didehydroandrographolide and 12-hydroxy-14-deoxy-13,14-dehydroandrographolide analogs of andrographolide have shown potent activity against the HIV virus. The diterpene lactone andrographolide 1 (IC50 (μM) = 0.59) shown in Fig. 1a possesses HIV-1 fusion inhibition properties and was evaluated in vitro using AZT (azidothymidine, also known as zidovudine) as a positive control.33–35 | ||
| Fig. 1 (a) Andrographolide-based (1–7) analogs. (b) Andrographolide analogs (8 and 9). (c) Structure of andrographolide-based analog (10). | ||
Reddy et al. reported the synthesis of andrographolide-based derivatives using different aromatic aldehydes together with Amberlyst-15 to obtain 3-nitrobenzylidene derivatives, which include 4′-hydroxyphenyl 2 (IC50 (μM) = 2.10), 3′,4′-methylenedioxy-phenyl 3 (IC50 (μM) = 2.75), 2′,4′-dihydroxy-phenyl 4 (IC50 (μM) = 1.09), 4′-chlorophenyl 5 (IC50 (μM) = 9.94), 4′-hydroxy-3′-methoxy-phenyl 6 (IC50 (μM) = 6.09) and 3′-nitro-phenyl 7 (IC50 (μM) = 0.51), as depicted in Fig. 1a. Upon treatment with acid chlorides, as presented in Fig. 1b, andrographolide afforded the andrographolide-substituted derivatives 2′,6′-dichloronicotinoyl 8 (IC50 (μM) = 0.83 and 2′-hydroxybenzoyl 9 (IC50 (μM) = 84.24). Further, upon treatment with p-toluene sulfonyl isocyanate, another derivative of p-toluene sulfonyl amino carbonyl andrographolide, 10 (IC50 (μM) = 2.08), was obtained, as depicted in Fig. 1c.36
2.2. Azaindole analogs/derivatives
The compound 1-(4-benzoylpiperazin-1-yl)-2-(4,7-dimethoxy-1H-pyrrolo[2,3-c]pyridin-3-yl)ethane-1,2-dione 16 was synthesized from azaindole.37–40 This derivative is considered to act on the HIV gp120 and inhibit the attachment of the viral glycoprotein with the CD4 molecule in the host cell, which is an essential step for viral entry.41–43 The synthesis of a potent azaindole-based derivative utilizing 5-bromo-2-chloro-3-nitropyridine 11 as the starting material is shown in Fig. 2. The reactant 11 was treated with vinyl magnesium bromide at very low temperature to give another compound, 4-bromo-7-chloro-6-azaindole 12 in 35% yield.44 | ||
| Fig. 2 Synthesis of azaindole derivatives together with important key findings. | ||
This intermediate is then catalyzed by copper metal, causing the replacement of bromide and chloride with sodium or potassium methoxide to give 4,7-dimethoxy-6-azaindole 13 in 75% yield.45,46 The latter compound was then treated with methyl chloro oxoacetate in the presence of aluminium chloride to produce ester 14. Further, this ester was hydrolyzed in the presence of aqueous methanolic potassium carbonate and converted into acid salt 15. The resulting acid salt underwent treatment with N-benzoyl piperazine in the presence of DEBPT, which is known as 3-(diethoxy-phosphoryloxy)-1,2,3-benzotriazin-4(3H)-one, to afford the desired compound 16, with an IC50 value of = ≥ 40 μM.47,48 These data reveal a safety profile consistent with exploring (16) in phase 1 clinical studies in normal healthy volunteers and proof-of-concept studies in HIV 1-infected subjects.37
2.3. Artemisinin analogs
Artemisinin 17 is a 1,2,4-trioxane sesquiterpene compound obtained from the plant Artemisia annua.49–51 Artemisinin 17 is significantly used towards chloroquinine-resistant and chloroquinine-sensitive strains of Plasmodium falciparum. However, its efficacy is limited because of its poor solubility.52,53 Dihydroartemisinin 18 is also produced from artemisinin via the reduction of its carbonyl group. Other potential reported derivatives of artemisinin are arteether 19, artemether 20 and artesunic acid 21. Artemisinin derivatives exhibit highly potent actions against malaria and cancer.54 Dihydroartemisinin 18 has attracted considerable interest because of its therapeutic activity, and being a versatile precursor, is also used for the synthesis of other artemisinin derivatives. Different analogs of dihydroartemisinin have been used in the treatment of bacterial, viral and autoimmune disorders, as depicted in Fig. 3a.55–62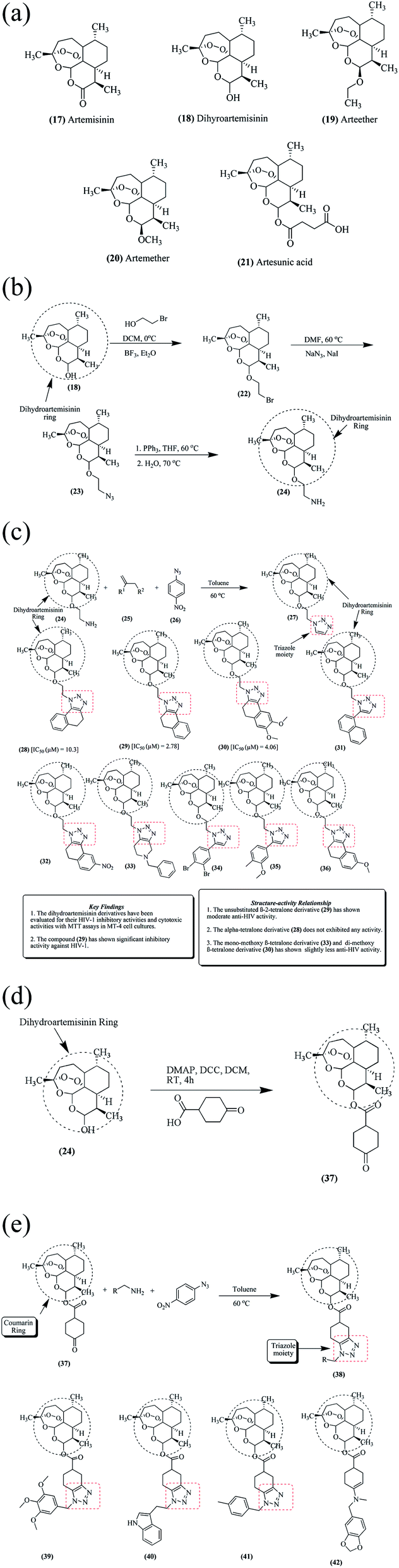 | ||
| Fig. 3 (a) Artemisinin analogs 17 to 21. (b) Synthetic route for the preparation of artemisinin analogs. (c) Artemisinin derivatives together with their SARs and key features. (d) Synthesis of a ketone precursor from dihydroartemisinin. (e) Triazole-based dihydroartemisinin ketone precursor analogs (38–42). | ||
Among the numerous derivatives from dihydroartemisinin 18, an amine precursor was synthesized and evaluated for its anti-HIV activity, as shown in Fig. 3b. Dihydroartemisinin 18 was reacted with 2-bromoethanol in the presence of catalysts, ethoxy ethane and boron trifluoride to convert it into 10-bromo ethoxy dihydroartemisinin 22. Compound 22 was then treated with sodium azide at 60 °C to form 2-(10β-dihydro artemisinoxy)ethyl azide 23 in 95% yield. The resultant compound 23 was finally converted into an amine precursor 2-(10β-dihydro artemisinoxy)ethyl amine 24 in 74% yield. From amine precursor 24, various potent derivatives have been synthesized and evaluated for their anti-HIV potential.49,63
The amino precursor 24 was further treated with α-tetralone 25 and 4-nitrophenyl azide 26 in the presence of toluene at 60 °C to yield another compound, 27, as depicted in Fig. 3c. Various dihydroartemisinin derivatives (28–36) have been synthesized and evaluated for their anti-HIV potential. It was found that compounds 28 and 29 exhibited significant activity with IC50 values of 2.78 μM and 4.06 μM against HIV-1, respectively.
Ketone precursor 37 was also synthesized from dihydroartemisinin 24, as depicted in Fig. 3d. Treatment of 24 with cyclohexanone-4-carboxylic acid in the presence of DCC (N,N′-dicyclohexylcarbodiimide) and DMAP (4-dimethyl amino pyridine) for 4 h led to the formation of compound 37.49
Ketone derivative 37 of dihydroartemisinin was further reacted with several benzylamines to obtain active triazoles (38–42) with significant activity, as shown in Fig. 3e.49
2.4. Maslinic acid analogs
Maslinic acid 43 is an oleanene triterpenoid isolated from the Geum japonicum plant and evaluated for its anti-HIV potential.64,65 It is considered to inhibit the protease of HIV-1. Maslinic acid and its analogs also exhibited an array of therapeutic activities such as anti-tumor, antioxidant and antiviral.66,67 Maslinic acid 43 is generally condensed with α-amino acids (L-valine, glycine and L-alanine) and ω-amino acids (11-aminoundecanoic acid, γ-amino butyric acid and 6-amino hexanoic acid), as presented in Fig. 4a and b, respectively. | ||
| Fig. 4 (a) Synthesis of maslinic acid derivatives from α-amino acids. (b) Synthesis of maslinic acid analogs from ω-amino acids. | ||
The condensation of maslinic acid 43, with amino acids is accomplished in the presence of reagents like HOBt (N-hydroxy benzotriazole), TEA (triethylamine), DCM (dichloromethane) and DCC (N,N′-dicyclohexylcarbodiimide) at room temperature. Hence, different derivatives 44 and 46 were obtained using this approach. The resultant compounds upon saponification at room temperature in the presence of methanol, sodium hydroxide and THF yielded carboxylic acid derivatives 45 and 47 with 80% and 95% yield, respectively.64
Parra et al. reported that maslinic acid derivatives 45 and 47 exhibited potent inhibition of HIV replication and also found to induce apoptosis in HIV cells.64
2.5. Calanolides analogs
Calanolide A 48 and calanolide B 49 (Fig. 5) are two enantiomers isolated from the Calophyllum lanigerum plant and reported to have potent inhibitory activity against the reverse transcriptase enzyme of HIV-1.68–76 Several important racemic modifications were prepared from calanolides to achieve the desired objectives. Also, numerous synthetic derivatives of (+)-calanolide A and (−)-calanolide A have been prepared and evaluated for their activity. The anti-HIV activity has been attributed to (+)-calanolide A. Moreover, (+)-calanolide A has already been subjected to in vivo studies and up to phase II clinical trials in healthy, HIV-negative subjects.13,68,69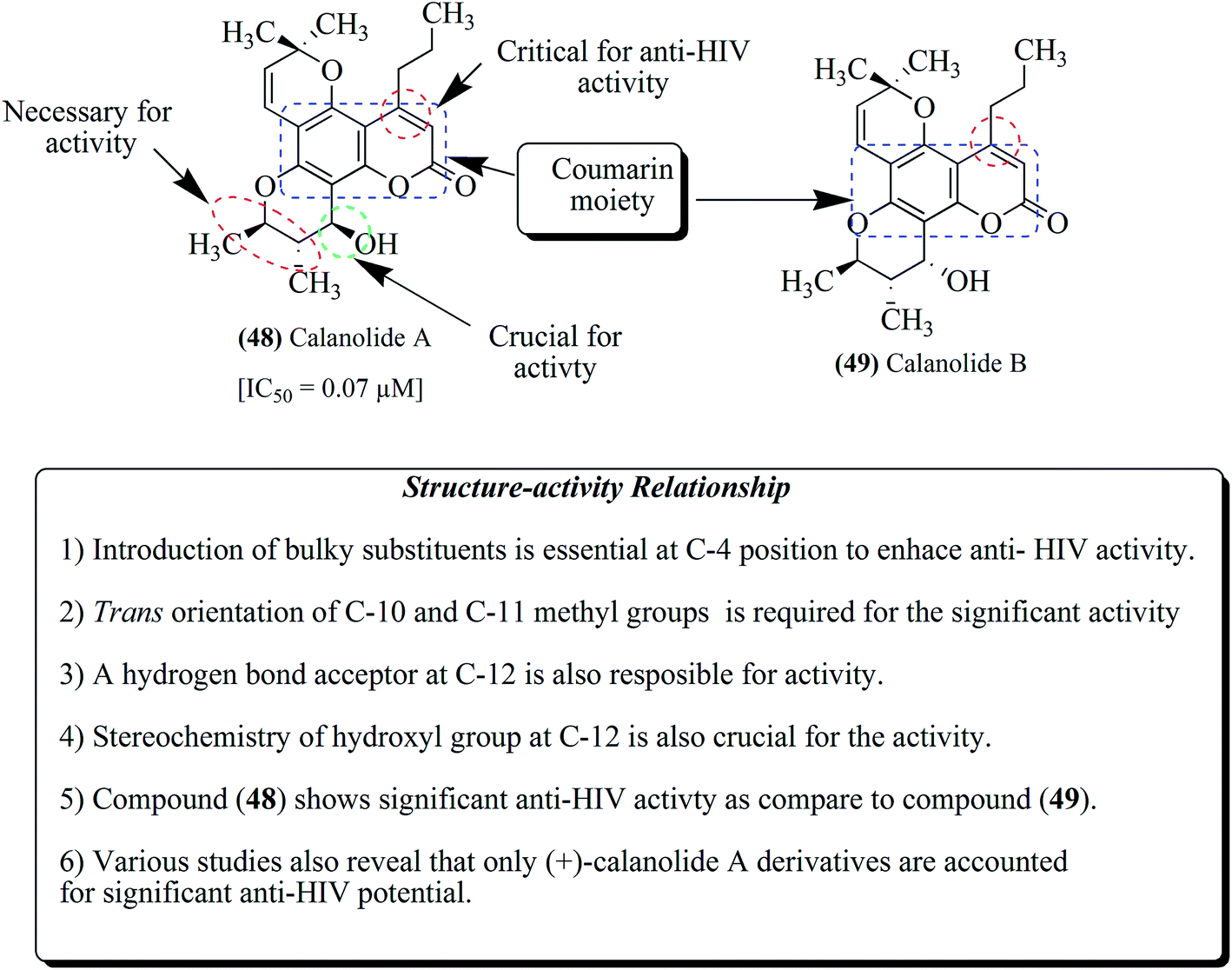 | ||
| Fig. 5 Structures of calanolide A and calanolide B. | ||
The synthesis utilizing coumarin compound 50 as a starting material is depicted in Fig. 6a. Compound 50 undergoes acetylation with propionic anhydride in the presence of aluminium chloride to produce an 8-propionyl derivative, which on further treatment with 1,1-dimethoxy-3-methylbutan-3-ol results in the formation of intermediate compounds 51 and 52.77,78
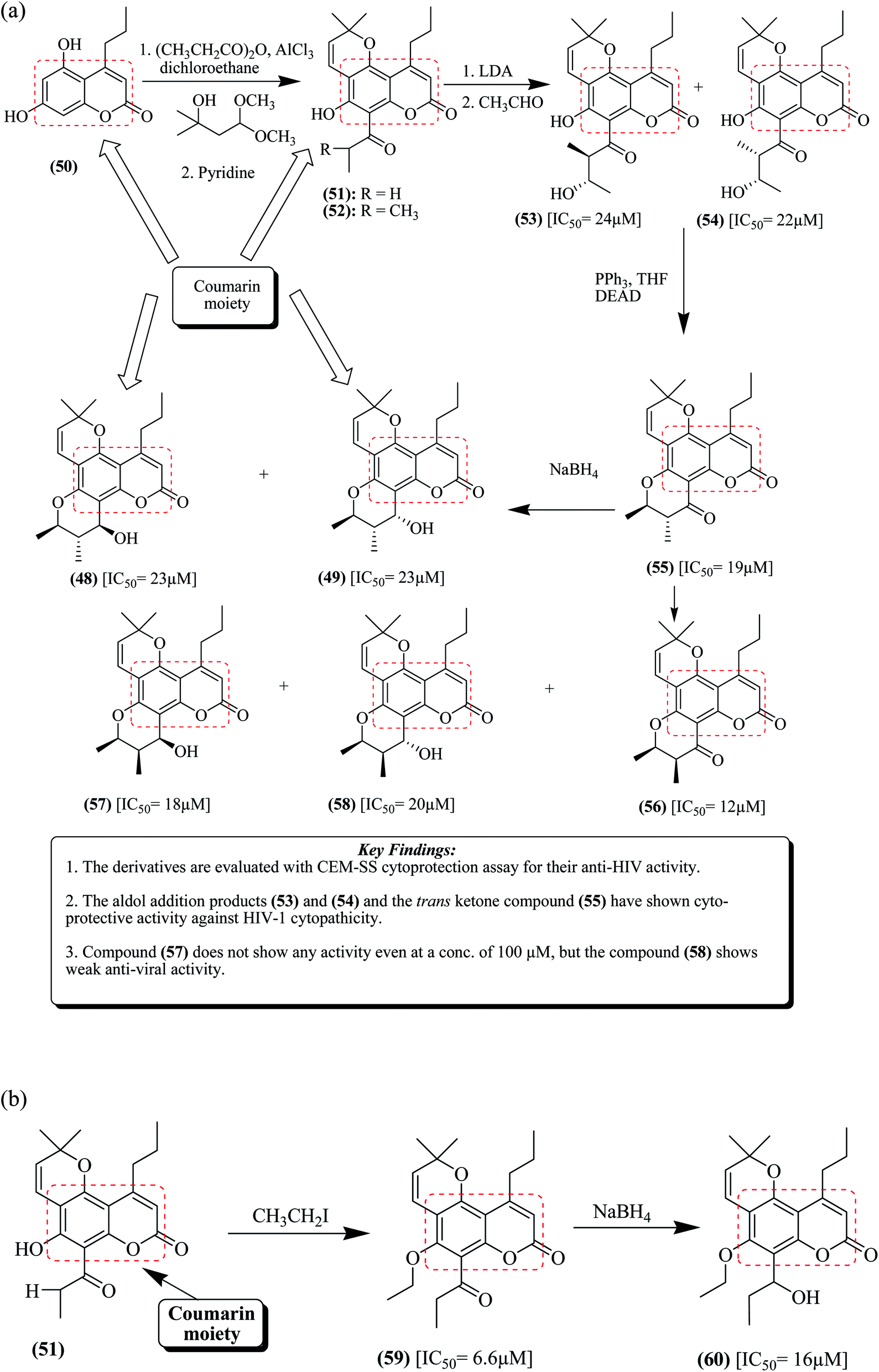 | ||
| Fig. 6 (a) Synthesis of racemic calanolide A together with important key findings. (b) Synthesis of calanolide A analogs. | ||
The latter compound was de-protonated by reacting it with LDA (lithium diisopropylamide) and acetaldehyde to produce a combination of erythro-aldol compound 53 and threo-aldol compound 54 with IC50 values of 24 μM and 22 μM, respectively. Further cyclization of compounds 53 or 54 was carried out to develop trans-ketone 55 and cis-ketone 56 with IC50 values of 19 μM and 12 μM, respectively. Compound 55 upon reduction with sodium borohydride in ethanol results in the formation of a combination of compounds calanolide A 48 and calanolide B 49 with almost equal IC50 values of 23 μM.68,79 Compound 49 is a racemic calanolide, which was resolved by HPLC to produce (+)-calanolide A and (−)-calanolide A. Compound 56 was reduced in the presence of sodium borohydride to produce a combination of compounds 57 and 58 with IC50 values of 18 μM and 20 μM, respectively. Compound 57 is a racemic calanolide C, which was separated via column chromatography.68,80,81
Zembower et al. reported a simple and direct modification of ring C of calanolides, which produced potent analogues, as depicted in Fig. 6b. Intermediate compound 51 underwent alkylation in the presence of iodo ethane to produce ketone intermediate 59 (IC50 = 6.6 μM). This intermediate was further reduced by sodium borohydride to afford compound 60 (IC50 = 16 μM). Compounds 53–55 exhibited significant anti-viral activity against HIV-1.68
2.6. Labdane analogs
Numerous labdane 61, analogs with different moieties such as hydroquinone, o-quinol and catechol have been synthesized and evaluated for their anti-HIV potential.82 Many natural terpenoidal compounds possessing catechol and o-quinol moieties also exhibit a broad spectrum of activity. The compound eremophilane, 62, is a sesquiterpenoid-based analog.83–86 Other compounds with o-keto–enol moieties are considered to act against the integrase enzyme of HIV, and thus exhibit anti-HIV activity. Cytosporic acid, 63, a fungal metabolite obtained from Cytospora sp., has shown inhibitory action towards HIV integrase.87,88 The quassinoid compound (64, Fig. 7) also exhibited many other therapeutic properties, viz, malaria, HIV, inflammation, allergies and tumors.86 | ||
| Fig. 7 Chemical structures of labdane analogs (61–64). | ||
Labdane-based analogues having an o-quinol moiety are generally synthesized from compound 65 via Diels–Alder reaction with myrcene 66 and ocimene 70, respectively.82 Compound 65 treated with myrcene 66, as presented in Fig. 8, results in the formation of compound 67, a major para adduct. It was then subjected to treatment with selenium dioxide and tert-butyl peroxide to produce compounds 68 and 69. Similarly compound 65 upon treatment with ocimene 70 resulted in the formation of compounds 71–74, as depicted in Fig. 9. Furthermore, a Diels–Alder reaction was carried out by treatment of compound 75 with both myrcene 66 and ocimene 70 to give para adduct 76 and ortho adduct 77, respectively, as presented in Fig. 10. In another step, compound 78 was treated with both myrcene 66 and ocimene 70, resulting in the formation of ortho adduct 79 and para adduct 80, respectively. The resultants were evaluated against HIV and it was found that compounds 71, 76, 77 and 80 together with raltegravir as a positive control at 50 μM showed IC50 values of 5.0 μM, 12.3 μM, 4.6 μM, 12.4 μM and 0.5 μM for the inhibition of HIV-1, respectively, as depicted in Fig. 11.82
 | ||
| Fig. 8 Synthesis of labdane analogs via Diels–Alder reaction (67–69). | ||
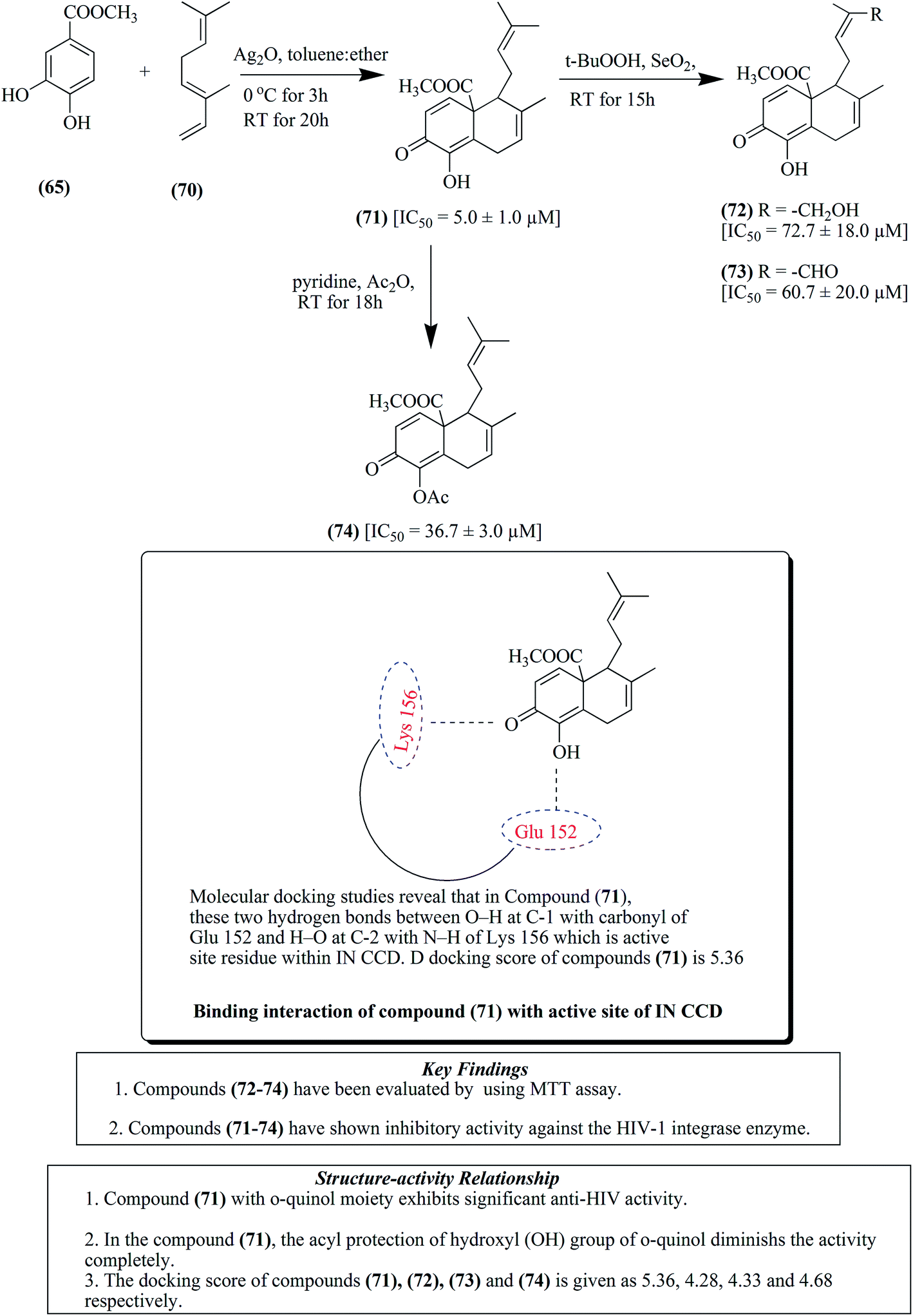 | ||
| Fig. 9 Labdane analogs by Diels–Alder reaction (71–74). | ||
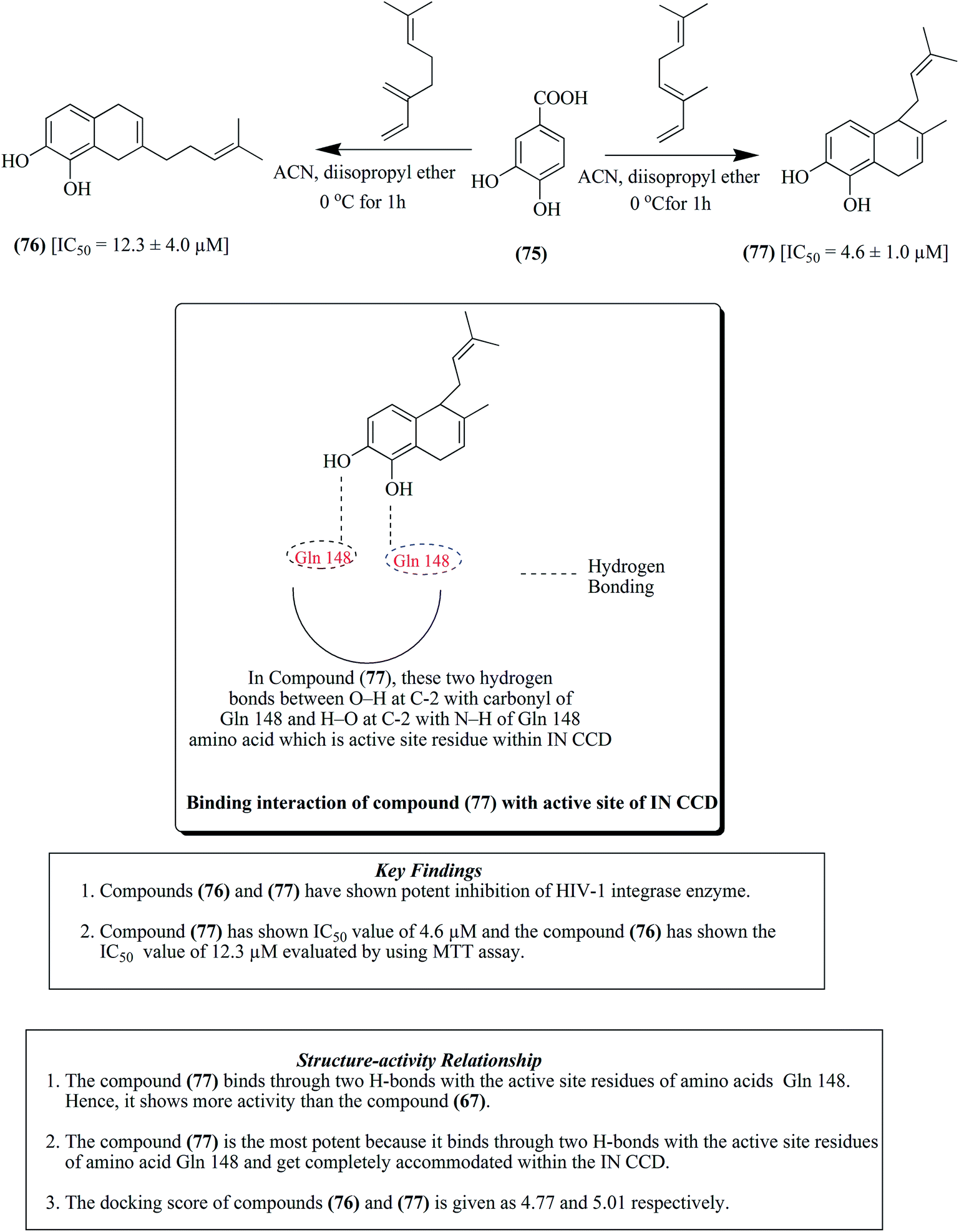 | ||
| Fig. 10 Labdane analogs by Diels–Alder reaction (76–77). | ||
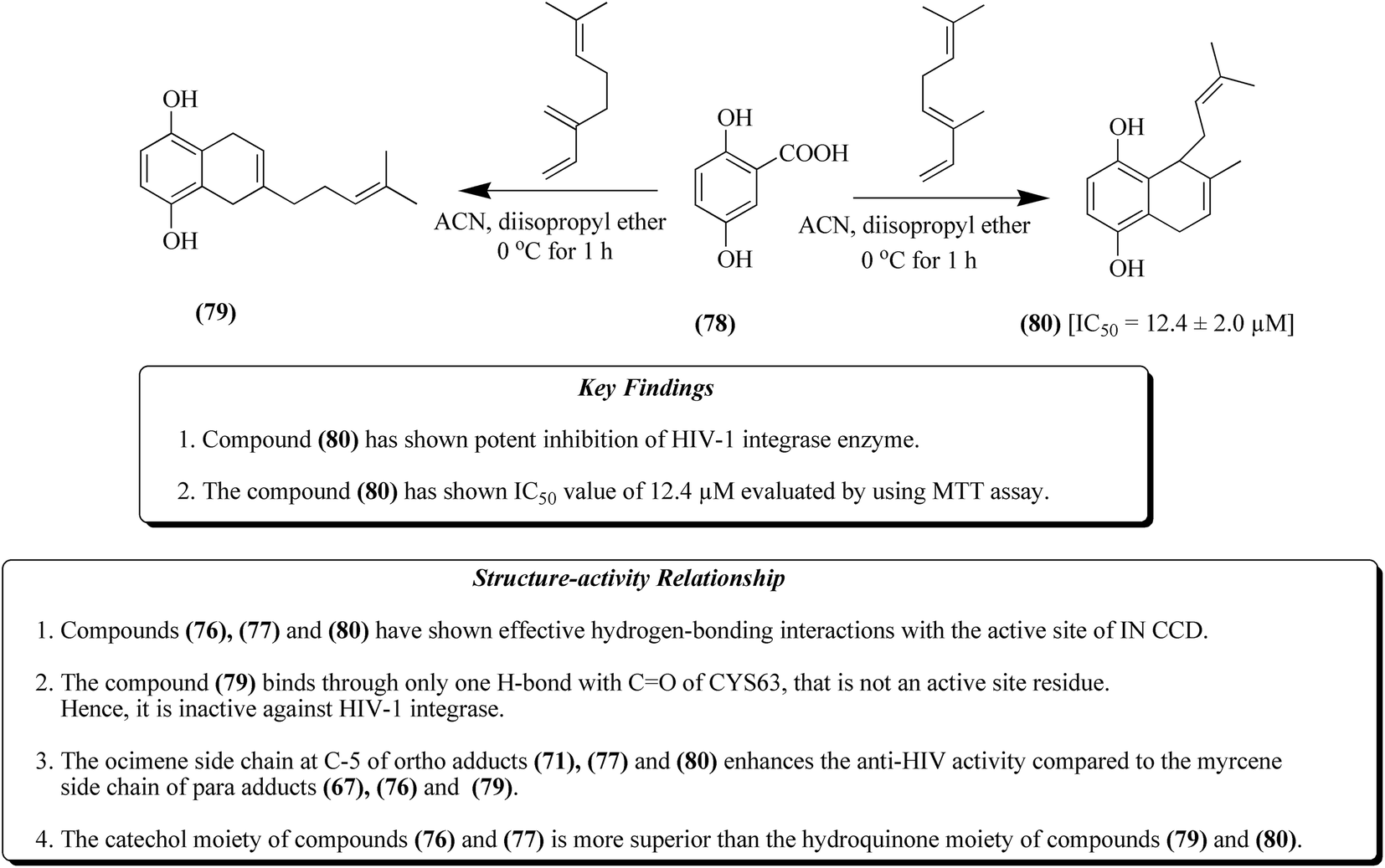 | ||
| Fig. 11 Synthesis of labdane analogues by Diels–Alder reaction (80) and their SAR features. | ||
2.7. Gomisin-G-based analogs
Gomisin-G 81 and some related lignans have been isolated from the ethanolic extract of Kadsura interior stems, exhibiting anti-HIV activity.89 Several other important natural lignans such as gomisin-A 82, schisantherin-B 83, deoxyschizandrin 84, schizandrin A 85 and schizandrin B 86, as depicted in Fig. 12a, also exhibited significant anti-HIV activity.90–96 From these natural compounds, various synthetic biphenyl compounds (87–96) have been designed by introducing different substitutions on the phenyl ring (Fig. 12b).96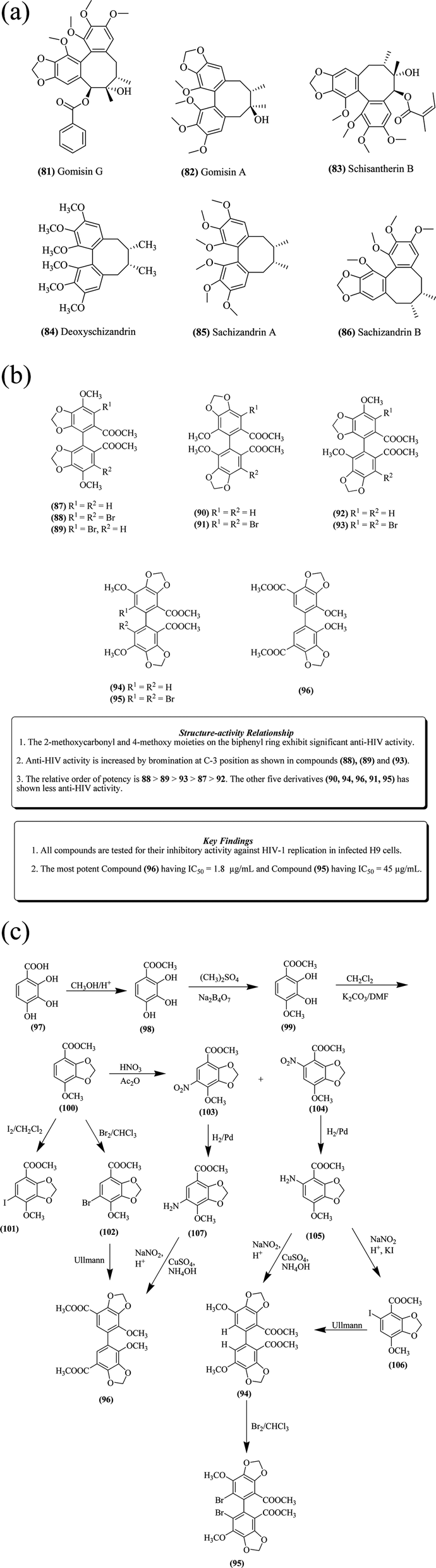 | ||
| Fig. 12 (a) Chemical structures of natural lignans. (b) Structures of synthetic biphenyl compounds (87 to 96). (c) Synthesis of anti-HIV biphenyl derivatives. | ||
Chen et al. reported the synthesis of 5,5′-dimethoxy-3,4,3′,4′-bis-methylene-dioxy-2,2-dimethoxy carbonyl biphenyl 94 and 2,2′-dimethoxy-3,4,3′,4′-bis-methylene-dioxy-5,5′-dimethoxy carbonyl biphenyl 96, which have reported to exhibit anti-HIV activity.89 In Fig. 12c, the esterification of 2,3,4-trihydroxybenzoic acid 97 in the presence of methanol results in the formation of methyl 2,3,4-trihydroxybenzoate 98. This compound 98 was subjected to treatment with dimethyl sulphate in the presence of sodium borate to produce methyl-2,3-dihydroxy-4-methoxybenzoate 99. The resultant intermediate 99 was further subjected to treatment in the presence of dichloromethane, potassium carbonate and DMF (N,N-dimethylformamide), resulting in the formation of methyl-2,3-methylene-dioxy-4-methoxybenzoate 100 in 91% yield. The latter compound then underwent bromination with dioxane dibromide or with dry bromine in trichloromethane to yield methyl-2,3-methylene-dioxy-4-methoxy-5-bromo-benzoate 102 in 80% yield. A small amount of diphenyl compound 96 could be obtained from compound 102 by reacting it with active copper powder via Ullmann reaction. To obtain a good yield of compound 96, compound 100 was iodinated with silver tri-fluoro acetate, giving a poor yield of halogenated precursor 101. The nitration of compound 100 was carried out with conc. HNO3 in acetic anhydride, leading to the formation of compounds 103 and 104. These two compounds were then reduced by Pd–C hydrogenation to produce amino acid derivatives 107 and 105, respectively. Furthermore, compound 105 underwent diazotization with sodium nitrite in the presence of sulphuric acid to produce a diazonium salt, which underwent treatment with KI and copper to form iodo derivative 106. The resulting compound subsequently underwent Ullmann reaction to give biphenyl derivative 94. Compound 94 was further subjected to treatment with bromine and trichloro methane to yield another derivative 95. Compound 94 can also be directly synthesized from compound 105 by diazotization reaction in the presence of alkaline medium. Simultaneously, compound 96 can also be formed directly from compound 107 via similar types of reaction conditions. The IC50 value of compounds 87, 88 and 90–92 is > 100 μg mL−1 towards HIV-1 inhibition, while compound 96 shows significant HIV-1 inhibition with an IC50 value of 1.8 μg mL−1.89
2.8. Quinoline-based analogs
Chemical scaffolds containing the quinoline pharmacophore exhibit an wide range of biological activities such as anti-HIV, anti-bacterial, anti-inflammatory, anti-fungal, immunosuppressive and anti-tumor activities.97–104 8-Hydroxyquinolines and 2-styrylquinolines are the two major categories among them that have been evaluated and considered to act on the integrase enzyme of HIV-1.105–107 Some natural quinolines such as, γ-fagarine 108 and haplopine 109 were extracted from the roots and barks of the Zanthoxylum ailanthoides plant and found to exert their anti-HIV actions through the inhibition of HIV replication.108 Various quinoline derivatives (110–116) have been synthesized with amide and styryl linkers and evaluated for inhibition of HIV-1 together with the positive control azidothymidine (AZT), as depicted in Fig. 13.97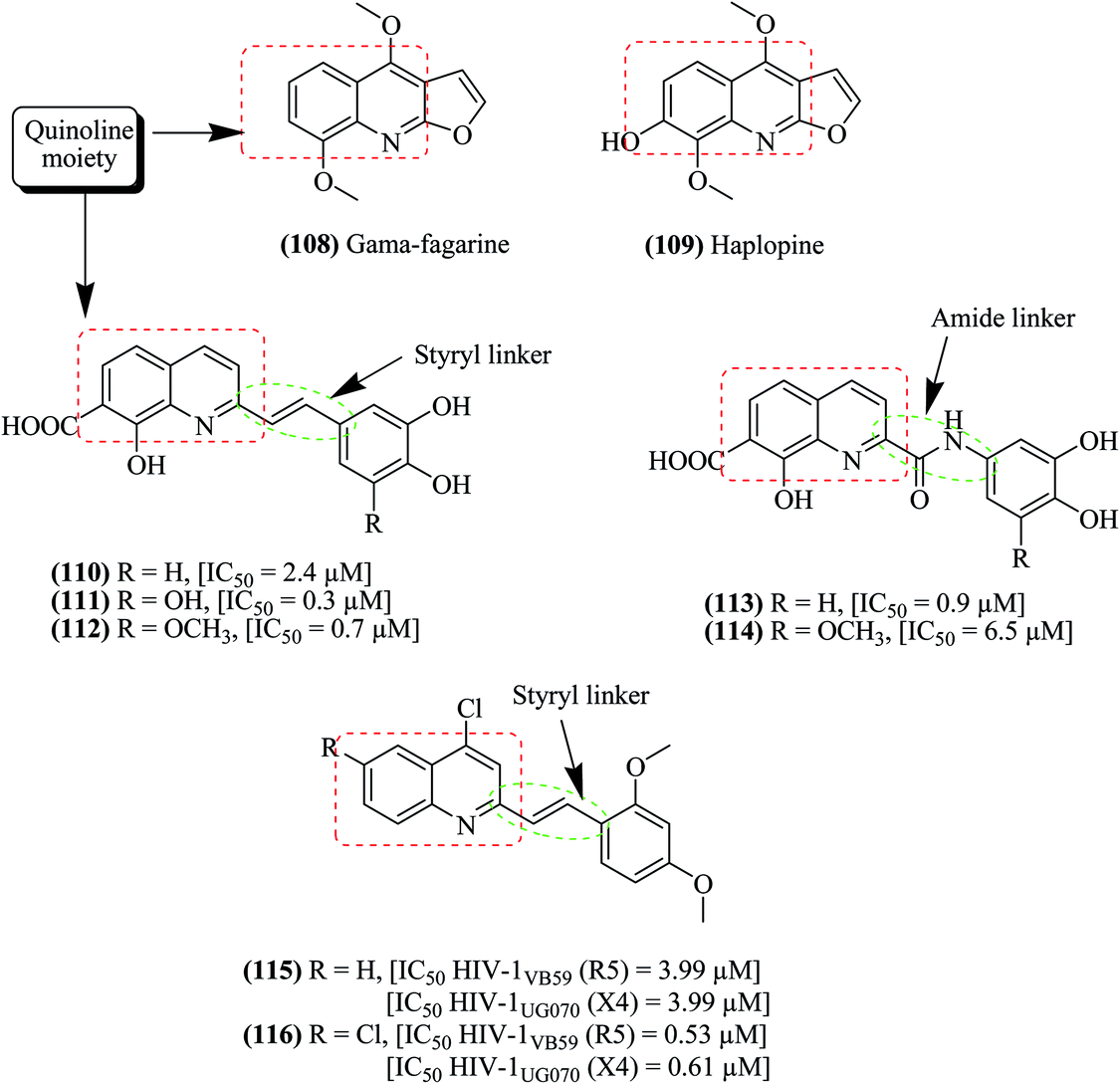 | ||
| Fig. 13 Quinoline analogs (108–116) possessing amide and styryl linkers with anti-HIV potential. | ||
Quinoline derivatives have been synthesized via two approaches, as depicted in Fig. 14. In the first step, the acetylation of aniline 117 by treatment with acetic anhydride at 40 °C for 10 min resulted in the formation of acetanilide 118 in 88% yield. Acetanilide was further refluxed with phosphoryl chloride in DMF at 80 °C for 8 h, which resulted the formation of another intermediate, 2-chloro-quinoline-3-carbaldehyde 119 using Vilsmeier–Haack reaction in 65% yield. From 2-chloro-quinoline-3-carbaldehyde 120, many 2,3-disubstituted quinoline derivative (121–127) were obtained in >60% yield using substituted anilines at 75 °C for 7 h.
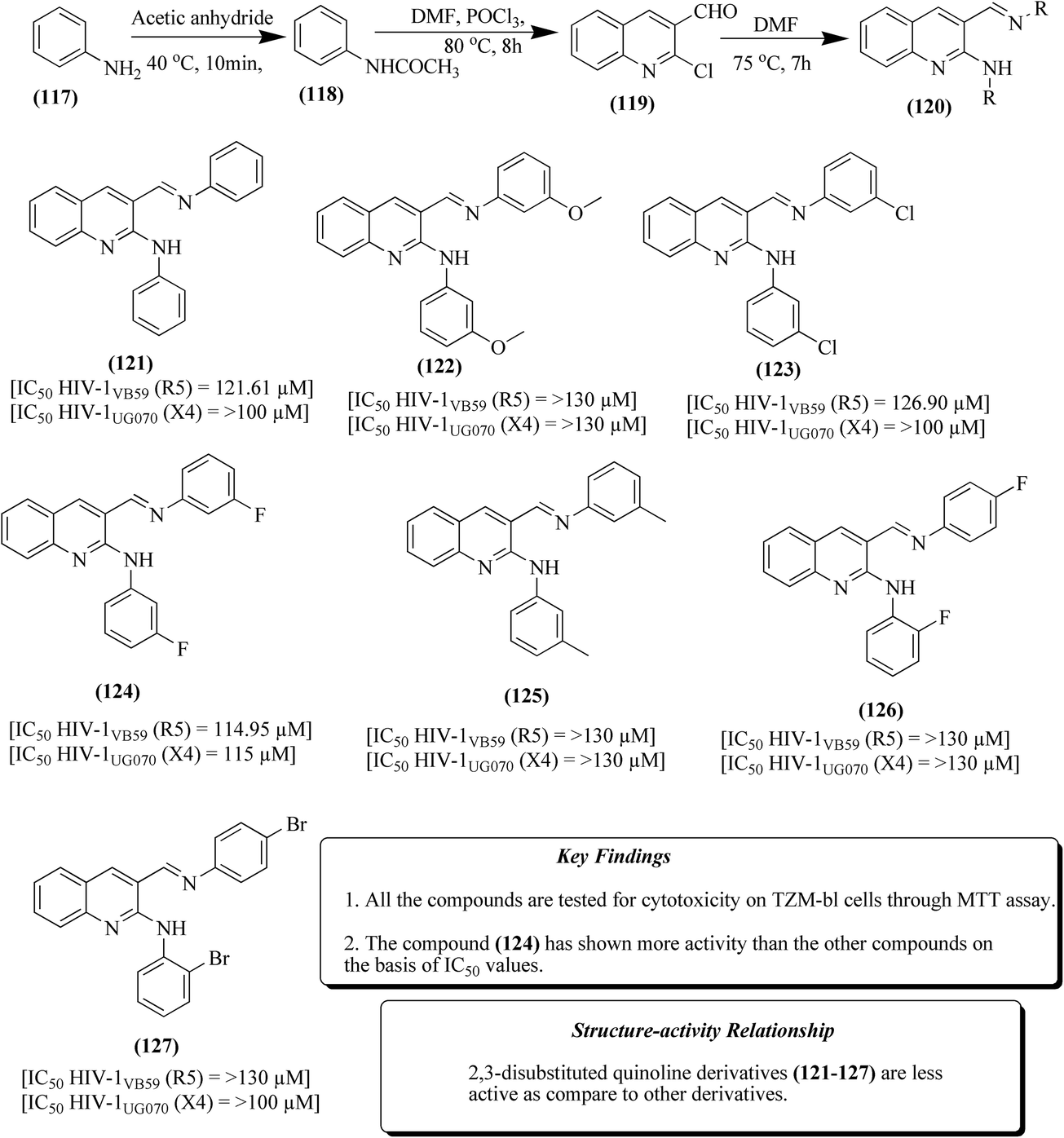 | ||
| Fig. 14 2,3-Disubstituted quinoline derivatives (121–127). | ||
Conversely, C-2-substituted 8-methoxyquinoline derivatives have been synthesized, as presented Fig. 15a, b, and c(i and ii). Compound o-anisidine 128 was used as the starting material and refluxed with 6 M HCl, toluene and crotonaldehyde at 110 °C for 3 h, resulting in the formation of intermediate 8-methoxy-2-quinaldine 129 in 61% yield via a modified Skraup synthesis. The resulting compound 129 was further treated with selenium dioxide and anhydrous 1,4-dioxane at 110 °C for 2 h to form 8-methoxyquinoline-2-carbaldehyde 130 in 81% yield. Compound 130 was reacted with pyridine and malonic acid at 70 °C for 2 h, giving compound (E)-3-(8-methoxyquinolin-2-yl) acrylic acid 133 in 65% total yield. Subsequently, compound 133 was reacted with substituted anilines in the presence of HOBt (hydroxylbenzotriazole), DMF and molecular nitrogen at 40 °C for 12 h to produce several α,β-unsaturated amide derivatives (134–139) in 60–70% yield. In another route, compound 130 was treated with acyl hydrazines in ethanol at 80 °C for 3 h, yielding 8-methoxyquinoline-2-carbaldehyde acyl hydrazine 131, which was then converted into compound, 132. The resulting hydrazines when treated with molecular iodine and potassium carbonate at 110 °C for 6–10 h produced 1,3,4-oxadiazole derivatives (140–157) in 70–90% in yield, as depicted in Fig. 15a, b, and c(i and ii) together with the important key findings.97
 | ||
| Fig. 15 (a) Synthesis of 2-substituted quinoline derivatives together with the structures of the most potent compounds (135–139). (b) 2-Substituted quinoline derivatives (140–154). (c(i)) 2-Substituted quinoline derivatives (155–157) together with their SAR and important key features. (c(ii)) 2-Substituted quinoline derivatives together with their important docking features. | ||
2.9. 4-Hydroxypyrone analogs
The 4-hydroxypyrone class of compounds was found to prevent HIV-1 replication by inhibiting the protease enzyme of this virus. The compound 4-hydroxycoumarin, 158, has been identified for its significant protease inhibition property.109–112 It was also found that compound 159 showed effective inhibition of the HIV-1 protease enzyme.112 4-Hydroxy-3-thio-substituted pyran-2-one derivative 160 was also found to be potent.113 Substitution on C-3 of compound 5,6-dihydro-4-hydroxy-pyran-2-one with a thio substituent resulted in the formation of a new derivative, 161, which was evaluated and found to possess potent HIV protease inhibitory actions. Another compound, 162, was screened and displayed potent anti-HIV activity with an EC50 value of 1.7 μM, as represented in Fig. 16.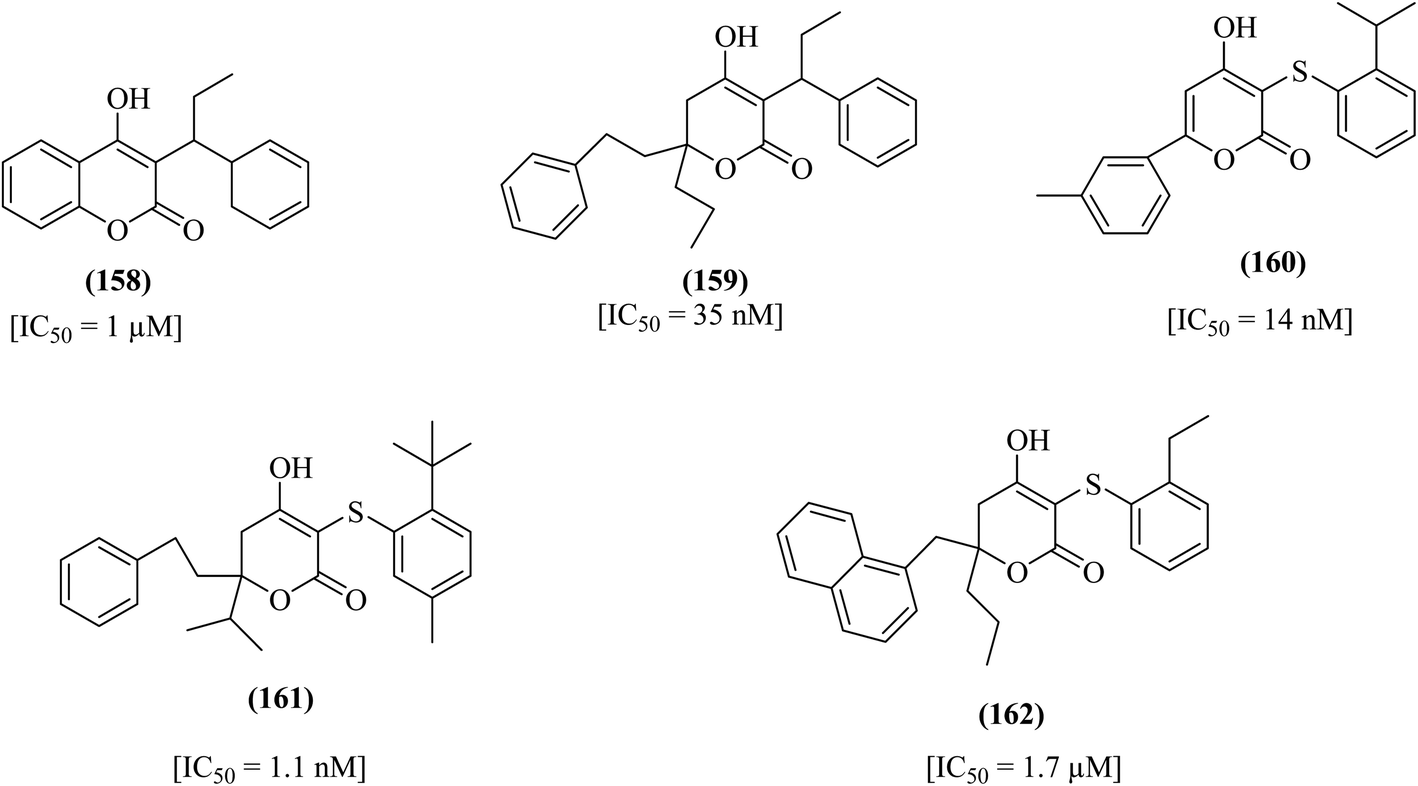 | ||
| Fig. 16 4-Hydroxypyrone HIV protease inhibitors (158–162). | ||
The synthesis of 4-hydroxypyrone derivatives has been described into two steps. In first approach (Fig. 17), the dihydro pyrone derivatives 163–170 were synthesized using different ketones. The ketones were treated with sodium hydride and butyl lithium in dry THF followed by the addition of ketone at 0 °C. Cyclization was achieved using sodium hydroxide and methanol, resulting in the formation of the substituted derivative in 50–65% yield.
 | ||
| Fig. 17 Structures of potent 4-hydroxypyrone derivatives (163–170) together with important key findings and their SAR features. | ||
Subsequently, the preparation of the compound 3-bromo-dihyropyrone was carried out with NBS (N-bromosuccinimide), and then the bromide atom was displaced by a suitable thiophenol substituent in the presence of thiophenol, dichloromethane and piperidine. Molecular modelling studies revealed that the naphthyl group at C-6 can possibly occupy the S2 pocket of the HIV protease and improve the anti-HIV potential, as depicted in Fig. 17. In the other step, a synthetic pathway has been described for the synthesis of the desired ketone derivatives 171–175, as depicted in Fig. 18.109
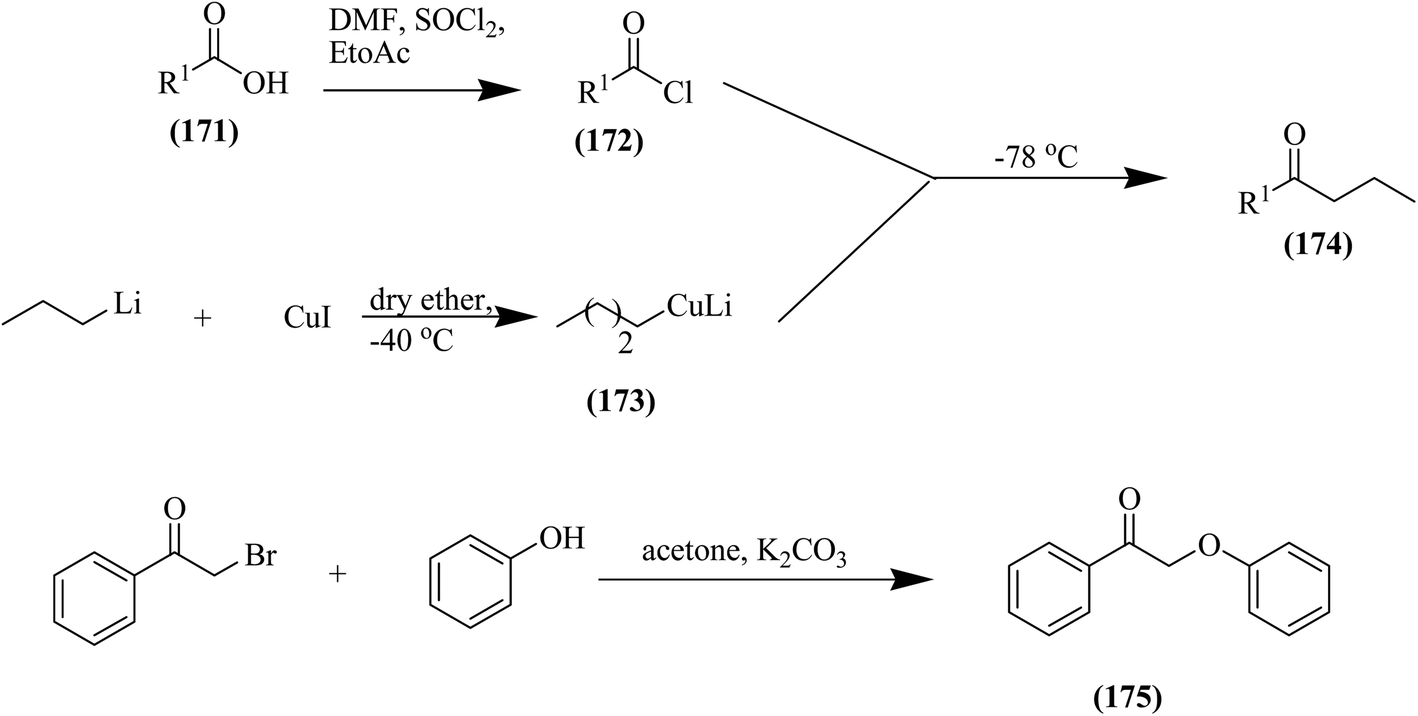 | ||
| Fig. 18 Synthesis of ketone derivatives. | ||
2.10. Ingenol-based analogs
Ingenol 176, is a diterpene compound isolated from plant genus Euphorbia. It is extracted from various species of Euphorbia like Euphorbia ingens, Euphorbia esula, Euphorbia sikkimensis, Euphorbia fischeriana and Euphorbia tirucalli.114–119 The isolated compounds from these species have been used for the treatment and management of various diseases such as cancer and keratosis.120–122 The synthetic modification of natural ingenol compounds has led to the formation of novel derivatives that are active against HIV-1. The modification of ingenol compound 176 at C-3 of its core ring with some significant ester chains led to the production of novel compounds such as ingenol-3-trans-cinnamate (ING-A) 177, ingenol-3-hexanoate (ING-B) 178 and ingenol-3-dodecanoate (ING-C) 179, as depicted in Fig. 19.114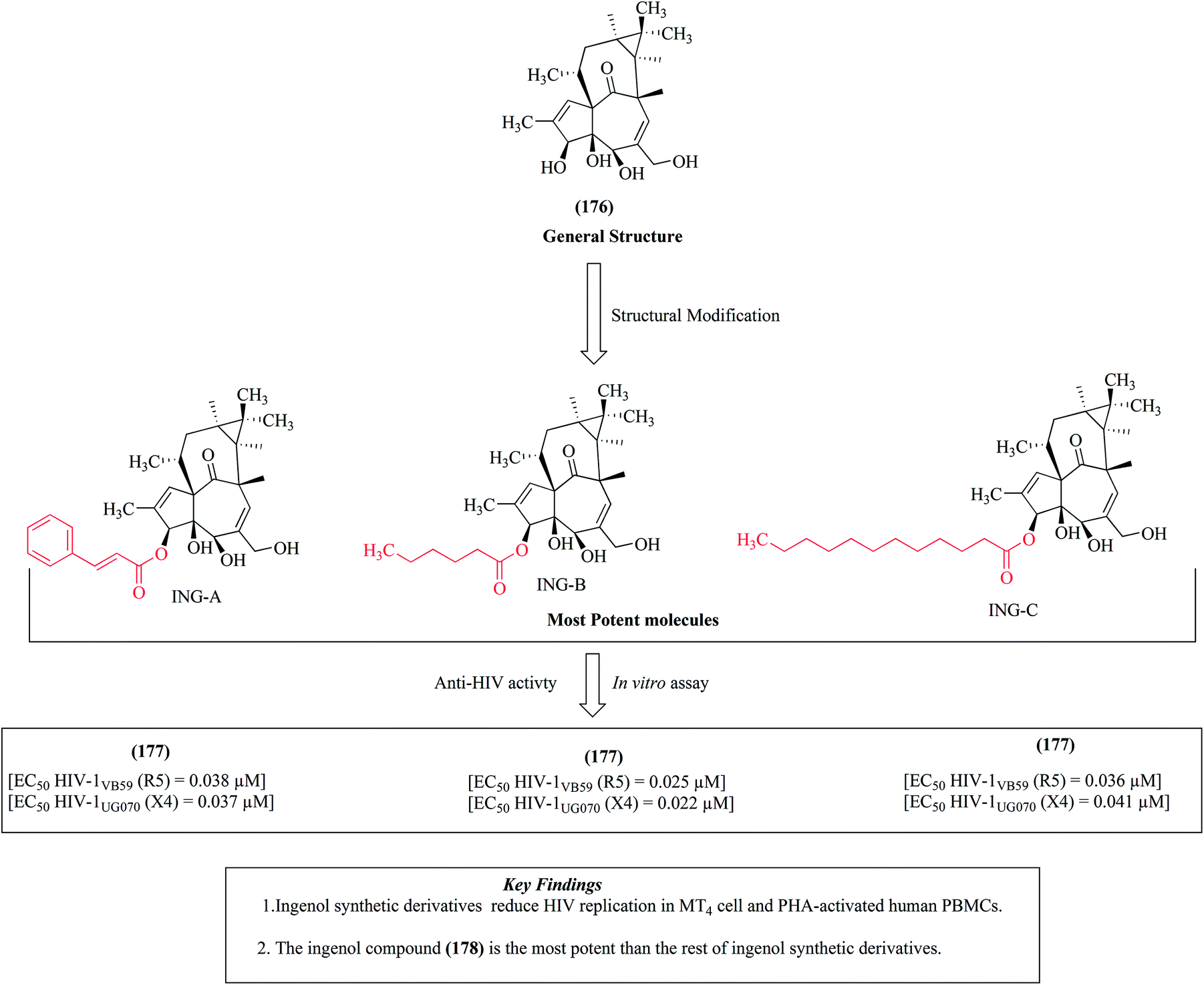 | ||
| Fig. 19 Structures of the most potent ingenol derivatives (176 to 179). | ||
2.11. Lavendustin B-based analogs
Lavendustin-B 180 is a natural product that has been identified as an allosteric inhibition inducer of the HIV-1 integrase enzyme.123 Integration is an important step for HIV replication, through which the viral genome gets inserted into the host genome for transcription of new viral proteins.124–133 Several other allosteric integrase inhibitors have been evolved, which act on the LEDGF/p75 binding site and inhibit the process of integration.134–138 Agharbaoui et al. reported the synthesis and evaluation of lavendustin-B and its derivatives. The structures of a few of the most potent molecules (181–190) having potential against HIV-1 are depicted in Fig. 20.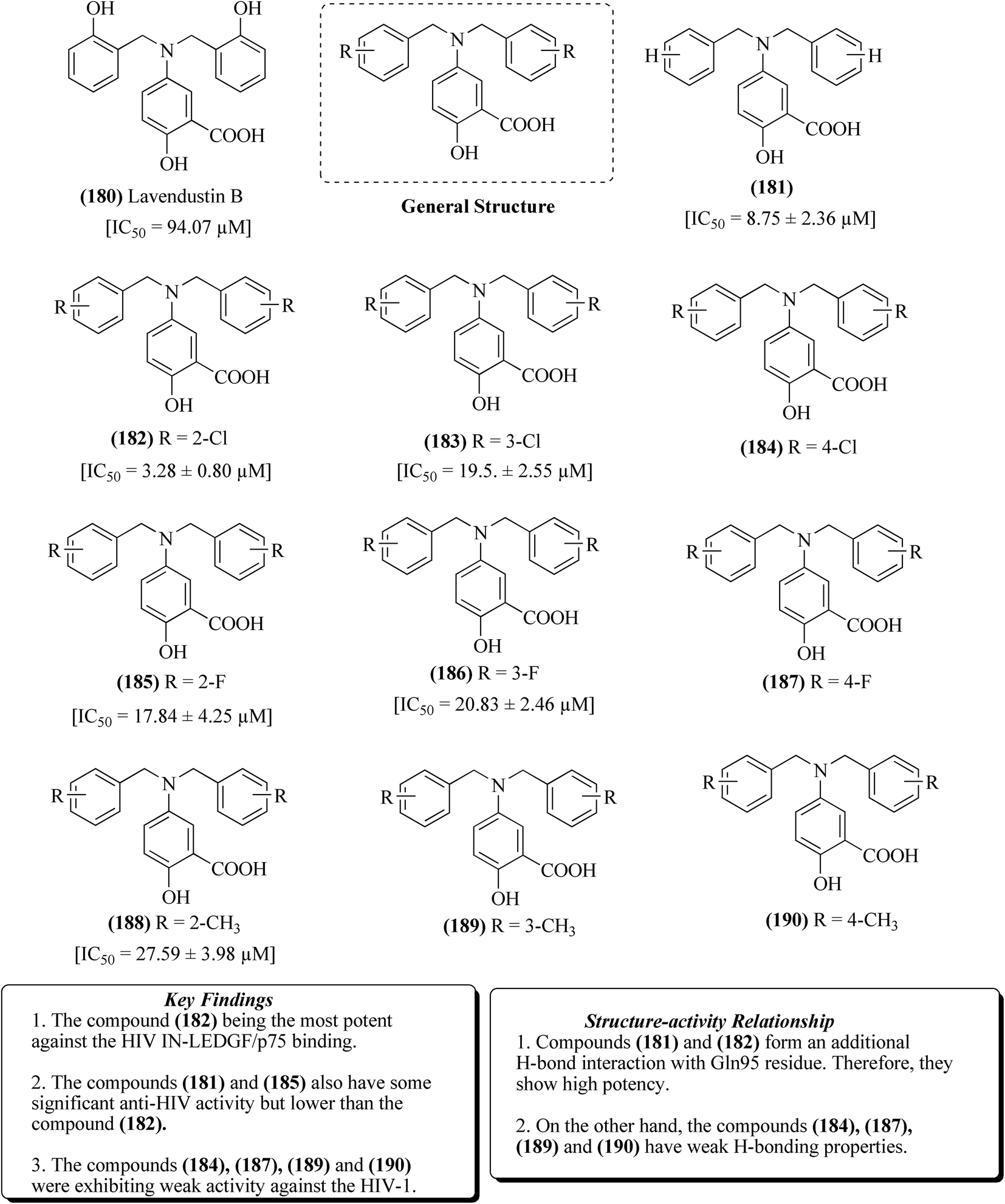 | ||
| Fig. 20 Lavendustin-B and its derivatives together with their SAR and key features. | ||
As shown in Fig. 21, 2-hydroxy-5-nitrobenzoic acid, 191 was reduced with zinc dust in the presence of conc. HCl and subjected to refluxing for 4 h at 0–5 °C to produce 5-amino-2-hydroxy-benzoic acid 192. Compound 192 underwent esterification by treating with methanol in the presence of sulphuric acid to form 5-amino-2-hydroxybenzoate 193. Intermediate compound 193 formed in the previous step was treated with different substituted benzaldehydes in the presence of NaCNBH3 (sodium cyanoborohydride), affording methyl-5-(benzylamino)-2-hydroxybenzoates 194–199. These intermediates 194–199 were further subjected to treatment with dimethyl formamide together with sodium hydride and benzyl bromide under an argon atmosphere, resulting in 5-(dibenzyl amino)-2-hydroxybenzoates 200–205. In the last step, the desired lavendustin B derivatives 181–183, 185, 186, and 188 were formed by treating compounds 200–205 with sodium hydroxide and methanol.123 The anti-HIV potential of all the lavendustin B derivatives was evaluated for their inhibitory activity against IN-LEDGF/p75 binding using HTRF (homogeneous time-resolved fluorescence)-based assays. Lavendustin-B 180 and compounds 181 and 182 displayed IC50 values of 94.07 μM, 8.75 μM and 3.28 μM against IN-LEDGF/p75 binding, respectively. The most potent compound 182 together with its docking features are also presented in Fig. 21.123
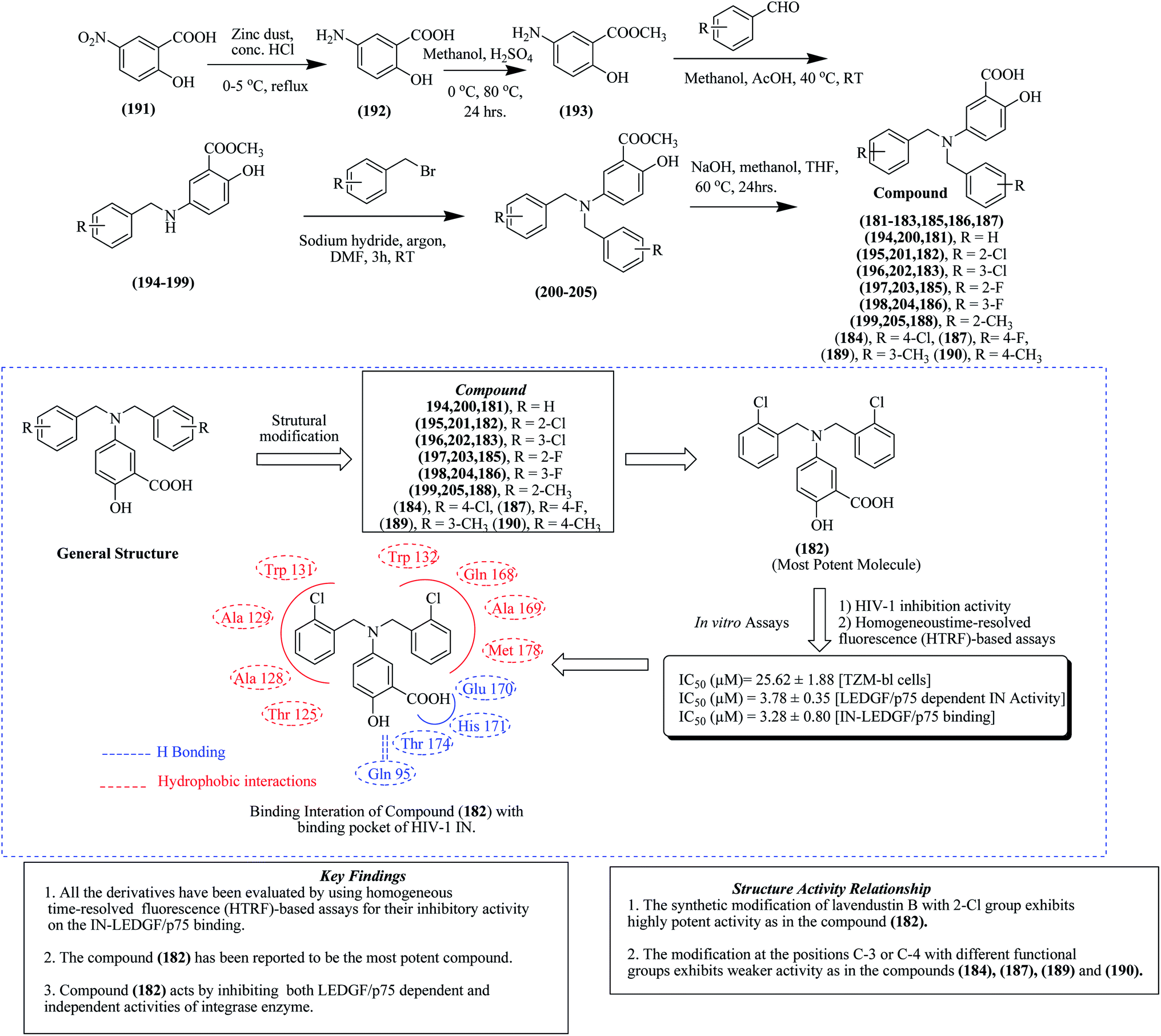 | ||
| Fig. 21 Structures of lavendustin B analogs together with their docking features. | ||
2.12. 3,7-Dihydroxytropolone-based analogs
3,7-Dihydroxytropolones are oxygenated troponoid compounds, which have been used as potential agents against several human disorders including viral infections.139 7-Hydroxytropolones (α-HTs) 206 are troponoid compounds containing three oxygen atoms, whereas 3,7-dihydroxytropolones 207 are troponoid compounds containing four oxygen atoms, as shown in Fig. 22.140,141 | ||
| Fig. 22 Hydroxylated tropolones. | ||
The synthesis of various 3,7-dihydroxytropolone derivatives has been described using the following approaches. Fig. 23 demonstrates that in the first approach, D-(+)-galactose 208 was utilized as a substrate for the synthesis of new 7-hydroxytropolone and 3,7-dihydroxytropolone derivatives.139,142 In another approach, kojic acid 209 was used as a starting material for the synthesis of two types of hydroxytropolone derivatives.139
 | ||
| Fig. 23 Synthesis of tropolone derivatives through D-(+)-galactose route and through kojic acid route. | ||
The cyclo addition of kojic acid with (oxido)pyrylium triflate salt 210a and iodo alkynes 211 and 212 resulted in the formation of iodo bicycles 213a/213b, respectively. These iodo bicycles were further subjected to methanolysis in the presence of DMAP (4-dimethylaminopyridine) to form methoxy bicycles 214a and 214b, respectively. The resultant iodo bicycles were subsequently treated with various reagents to afford compounds 215a and 215b and 216a and 216b. Compound 213b was treated with DMAP in the presence of CDCl3 (deuterated chloroform) to obtain compound 217 and further treatment of compound 213b with potassium carbonate and methanol yielded another compound, 218, as depicted in Fig. 24. Furthermore, compound 213b was treated with MsOH (methane sulfonic acid) to produce (oxido)pyrylium dimer compound 219a, as presented in Fig. 25.
 | ||
| Fig. 24 3,7-Dihydroxytropolone and tropolone derivatives. | ||
 | ||
| Fig. 25 Conversion of oxabicyclic intermediates into (oxido)pyrylium dimer compound (219a). | ||
As shown in Fig. 26, the methyl ester of compound 215a when treated with hydrobromic acid in the presence of acetic acid yielded 3,7-dihydroxytropolone 216c at 120 °C.139 Puberulonic acid 222 and puberulic acid 224 are two important 3,7-dihydrotropolones obtained from the Penicillium puberulum fungus.143 These two compounds are considered to be very similar to stipitatonic acid 221 and stipitatic acid 223, which are derivatives of 6-hydroxytropolone compounds.144,145 Also, lactone stipitalide 220 was used for the synthesis of stipitatonic acid 221, which was further utilized for the synthesis of stipitatic acid 223, as depicted in Fig. 27. Puberulonic acid 222 and puberulic acid 224 can also be synthesized from 3,7-dihyroxytropolone. Also, compound 216d has been utilized for the biosynthesis of different 3,7-dihydroxytropolones. Fig. 28 also demonstrates another strategy for the synthesis of compound 216d.139
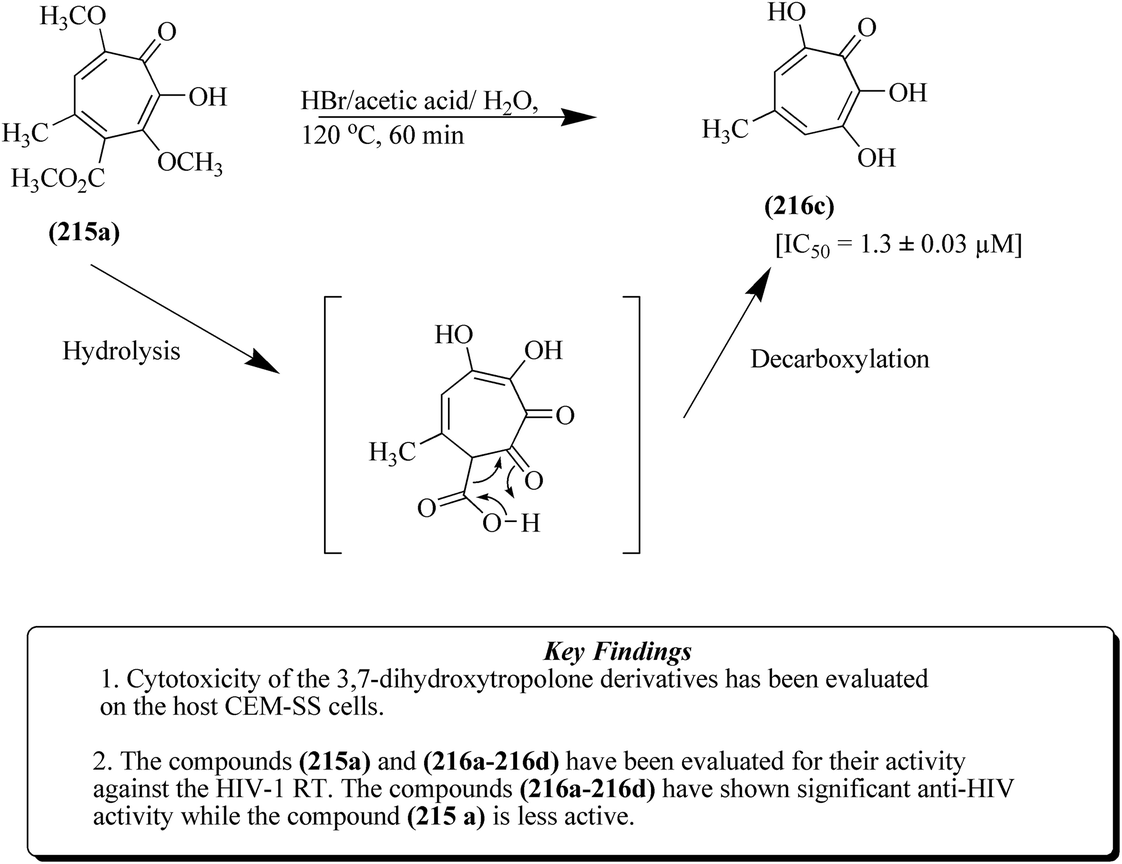 | ||
| Fig. 26 Acid-mediated hydrolysis and decarboxylation of compound (215a). | ||
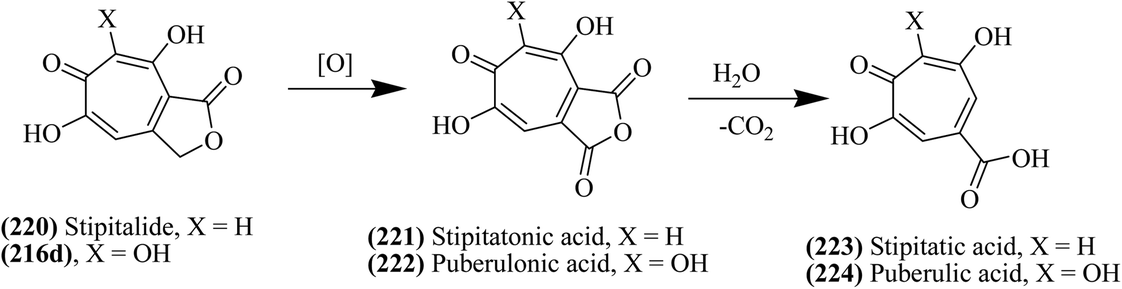 | ||
| Fig. 27 Synthesis of compounds (221–224) from stipitalide and other 3,7-DHTs. | ||
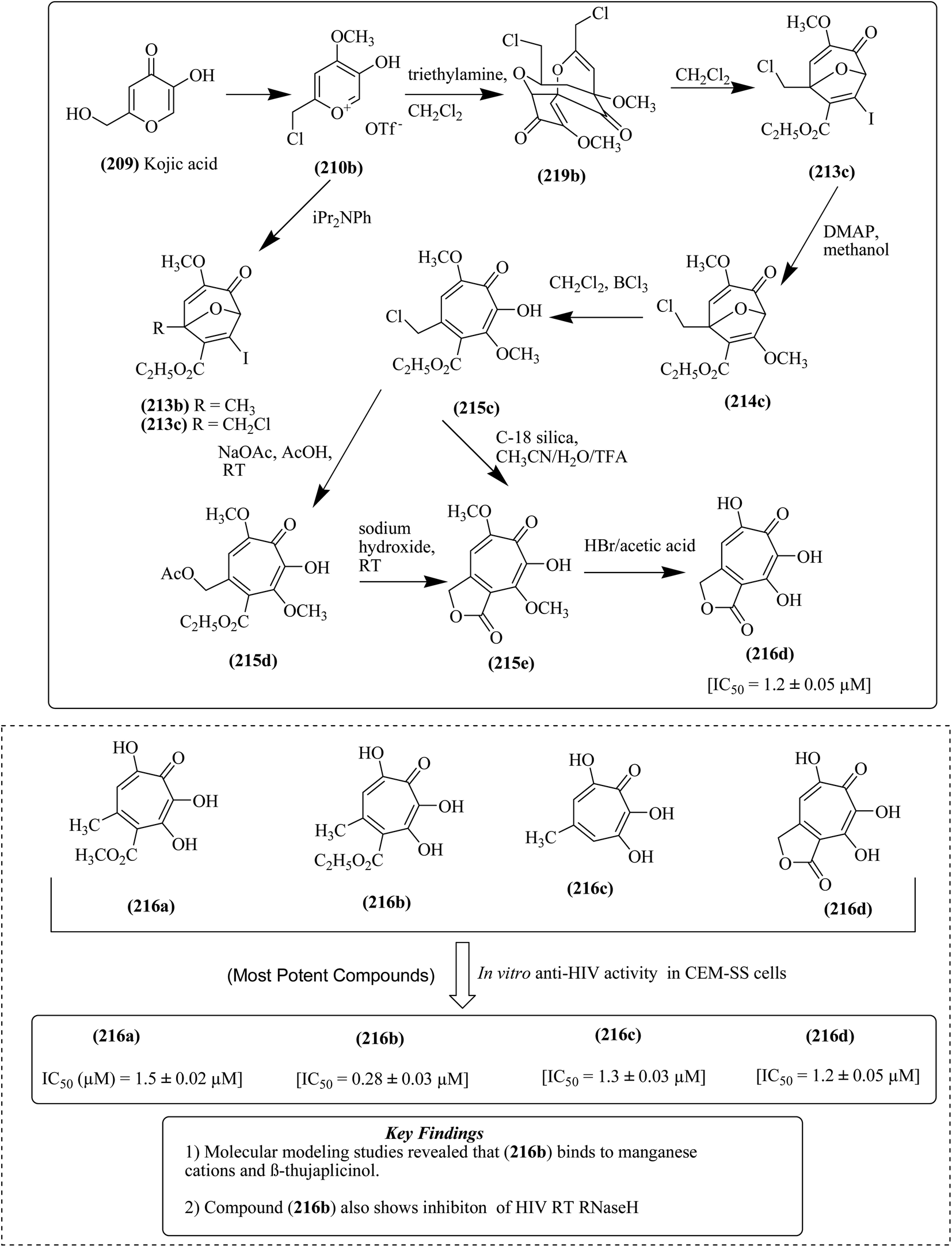 | ||
| Fig. 28 Synthesis of 3,7-dihydroxytropolone analogs together with important key findings of the potent compounds. | ||
In a different approach, compounds 213b/213c were synthesized from compound, 210b together with the compound 219b, which was formed using TEA (triethyl amine) and dichloromethane. Subsequently, methoxy tropolone compound 215c was formed, and then converted into compound 216d through various reaction steps such as acetolysis, lactonization and demethylation. The 3,7-dihydroxytropolone compounds 216a and 216b displayed IC50 values of 1.5 μM and 1.1 μM, respectively, against HIV-RT, as depicted in Fig. 28.139
2.13. P1/P1′-substituted cyclic urea-based analogs
P1/P1′-substituted cyclic urea-based compounds have been reported to exhibit potent activity against the protease enzyme of HIV-1.146 In recent years, several compounds have been identified that act as protease inhibitors and exhibit potent anti-viral activities.147,148 However, it has been established that the modification at P1/P1′ binding is necessary to enhance the potency of protease inhibitors as potential agents.149 It has been shown that the potency has about an 8–15 fold enhancement by introducing 4-(3-hydroxypropyl) at the P1′ phenyl ring.The introduction of a new (hydroxyl ethyl) urea isostere-based protease inhibitor permits deviations of the P1 substituent.150 DMP 323 225 is a 7-membered cyclic urea-based HIV-1 protease inhibitor, which showed poor binding affinity towards the protease enzyme of HIV-1.151 Oppositely, the DMP 450 226 molecule exhibited good pharmacokinetic properties together with significant inhibition and recently completed phase I clinical trials, as depicted in Fig. 29.152
 | ||
| Fig. 29 Chemical structures of P1/P1′-substituted cyclic urea compounds. | ||
There are three different strategies adopted for the synthesis of P1/P1′-substituted cyclic ureas. DMP 323 225 has been synthesized from amino acids.153 The commercially available N-Cbz-D-alanine 227 upon treatment with NMM (N-methylmorpholine) and N,O-dimethoxy hydroxyl amine resulted in the formation of amide compound 228, as presented in (Fig. 30). Subsequent reduction with LAH (lithium aluminium hydride) yielded an amino aldehyde in 96% yield. The formed amino aldehyde was further added to a slurry of [V2Cl3(THF)6]2 [Zn2Cl6] in the presence of dichloromethane to produce compound 229 in 66% yield.
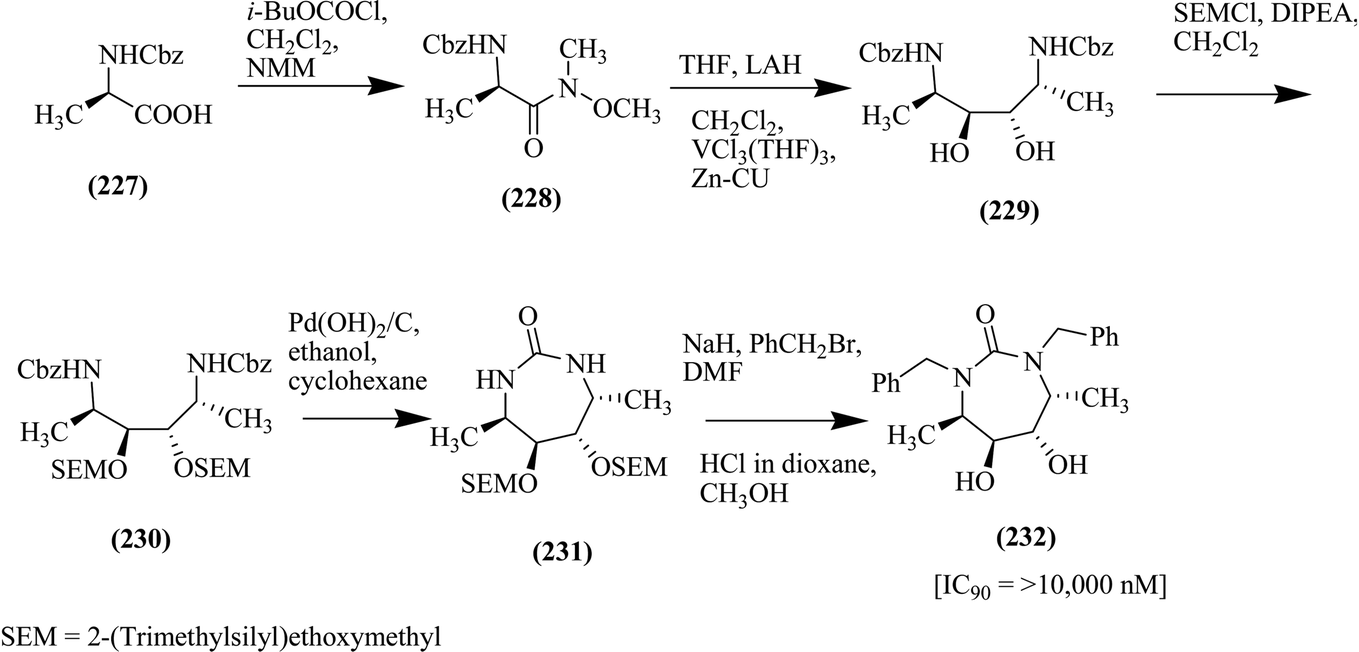 | ||
| Fig. 30 Synthesis of P1/P1′-substituted cyclic ureas. | ||
Compound 229 was treated with 2-(trimethylsilyl) ethoxy methyl chloride (SEM-Cl) to yield ether compound 230 in 87% yield. Subsequent reactions of compound 230 in the presence of catalytic Pd(OH)2 and CDI (1,1-carbonyldiimidazole) produced urea-based scaffold 231 in 81% yield. N-alkylation of the urea in the presence of benzyl bromide and NaH afforded the P2/P2′ substituent-based analogs. Acid-catalysed cyclization of the latter in methanol resulted in the target cyclic urea-based compound 232 in 54% yield.146,154
In the second approach, L-tartaric acid is used as the starting material for the synthesis of P1/P1′-substituted cyclic ureas. (−)-Dimethyl 2,3-O-isopropylidene-L-tartrate 233 was treated with trimethyl aluminium and N,O-dimethyl hydroxylamine hydrochloride to afford a new bis-Weinreb amide compound 234. It was further treated with p-isopropyl benzyl magnesium chloride in THF to form diketone scaffold 235 in 95% yield. The E:Z oxime isomer 236 was obtained together with the E:Z isomer by reacting compound 235 with hydroxylamine hydrochloride. The resulting oxime mixture was further reduced with diisobutyl aluminium hydride (DIBAL) in the presence of toluene to produce a di-amine compound, 237. Di-amine 237 was subsequently reacted with 1,1′-carbonyl-diimidazole to produce an imidazolide intermediate, which was further cyclized in the presence of tetrachloroethane to produce cyclic urea-based compound 238 in 62% yield. Compound 238 further underwent alkylation in the presence of benzyl bromide and potassium tert-butoxide. The resultant compound was treated with HCl and methanol, yielding the target urea-based compound 239 in 67% yield, as presented in Fig. 31.146,155,156
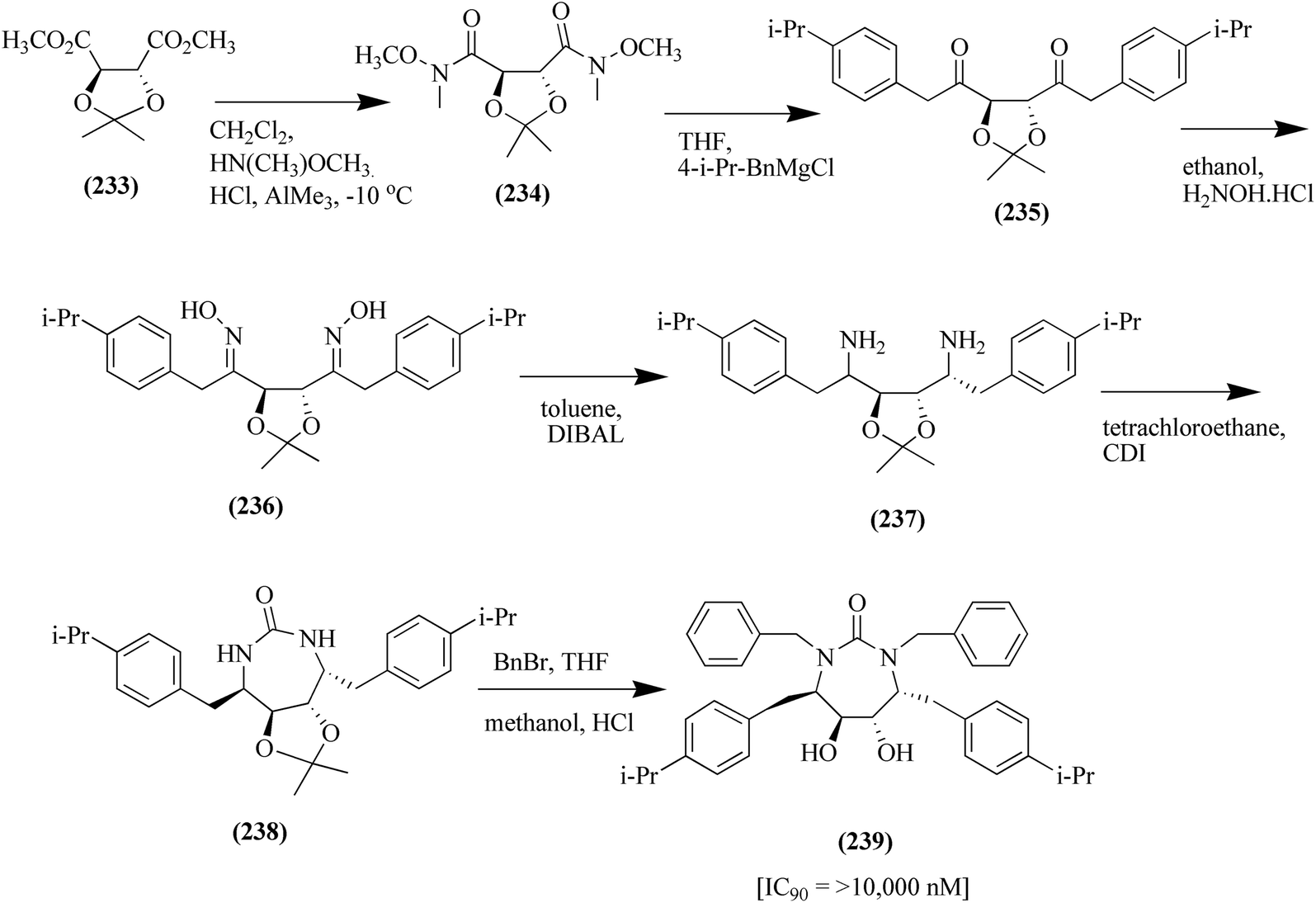 | ||
| Fig. 31 Synthesis of P1/P1′-substituted cyclic urea derivatives (239). | ||
In the third strategy, lactone 240 was reduced with lithium borohydride in the presence of methanol to form a crude L-mannitol. The reaction of the latter with 2,2-dimethoxypropane and hydrochloric acid in acetone resulted in the formation of a new compound triacetonide 241 in 80% yield. Further treatment with acetic acid yielded a tetraol intermediate in 80% yield. This tetraol intermediate was subsequently reacted with triphenyl phosphine and diethyl azo dicarboxylate (DEAD) in the presence of toluene for the production of volatile diepoxide 242 in 80% yield. Diepoxide 242 was reacted in the presence of butyl lithium and 4-CH3SC6H4Br to obtain diol compound 243.
The reaction of diol compound 243 in the presence of DEAD, triphenyl phosphine and diphenyl phosphorazidate resulted in the formation of diazide scaffold 244 together with significant amounts of monoazide compound 245. The diazide compound was further reduced using LAH to form a diamine, which was further cyclized to form urea-based compound 246. The cyclic urea-based compound 246 was alkylated in the presence of benzyl bromide and potassium tert-butoxide. In the last step, treatment with HCl afforded the target urea-based compound 247 in 60–70% yield, as presented in Fig. 32 together with the important key findings of the most potent compounds 248–250.146,157
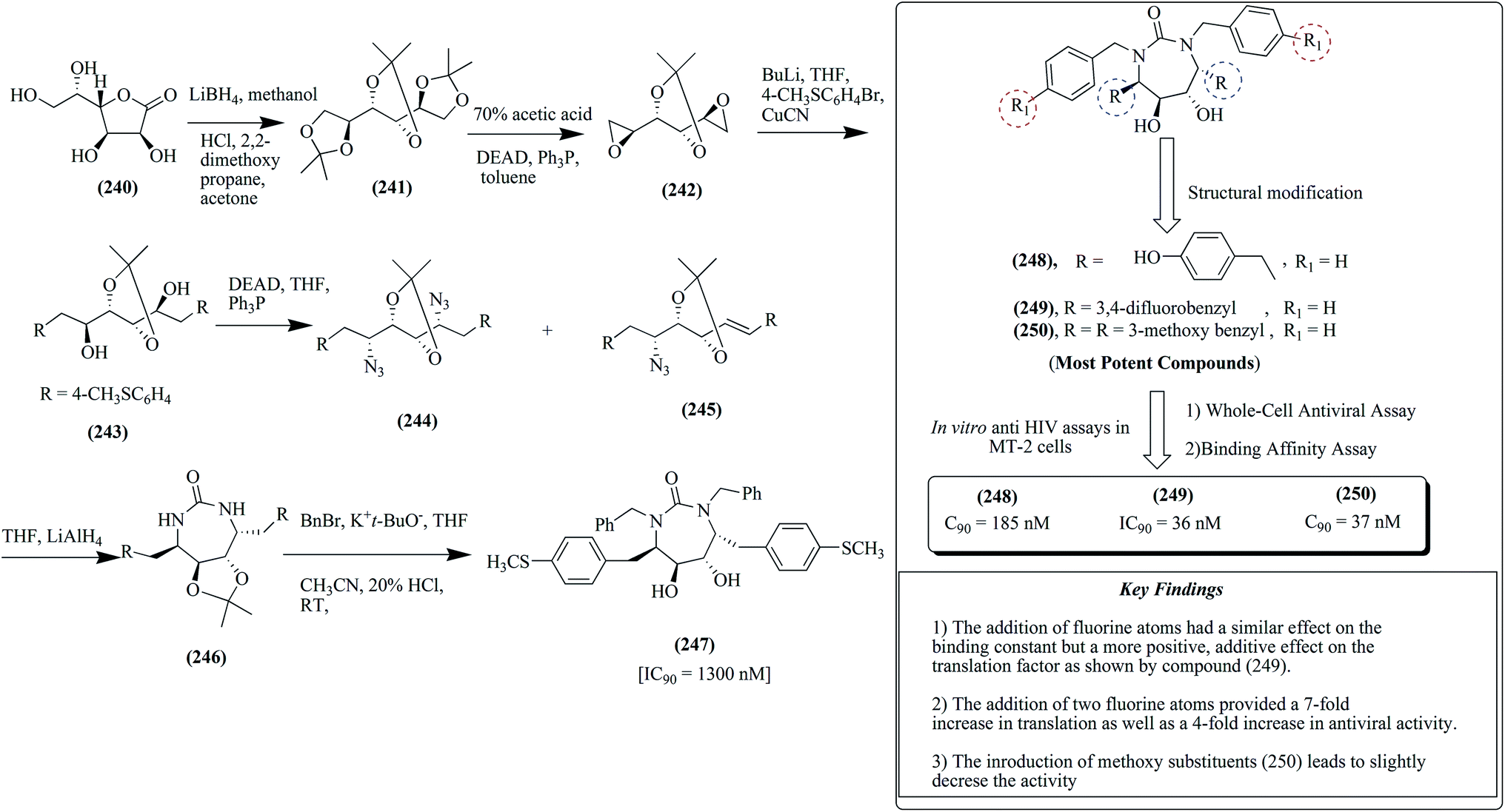 | ||
| Fig. 32 Synthesis of P1/P1′-substituted cyclic urea-based derivatives together with important key findings of most potent compounds (248–250). | ||
3. Conclusion
Numerous efforts have been made in the recent decades for the synthesis of anti-HIV agents. The literature survey clearly indicated that several chemical entities have been clinically approved and recommended for the management of AIDS. However, there is still great demand to search novel scaffolds because the existing drugs exhibit severe noxious harmful effects. This review article presented some interesting rational approaches behind the design of potential therapeutic candidates employed by researchers, prominent scientists, academicians and scholars globally.In the present communication, we summarized the design strategies, structure of the most potent molecules, their IC50 values, structure–activity relationships, most important key findings, binding interactions with the amino acid residues of the enzymes responsible for HIV 1 inhibition and mechanistic insights into plant-based analogs such as andrographolide, azaindole, artemisinin, maslinic acid, calanolides, labdane, gomisin-G, ingenol, lavendustin-B, dihydroxy tropolone, and cyclic urea-based analogs in the context of their action against HIV/AIDS. The synthesized compounds display modified pharmacological profiles together with increased potency against different strains of HIV-1. The present assemblage can be immensely beneficial for medicinal chemists focusing on the design of therapeutic candidates having significant anti-HIV potential.
Abbreviations
| AIDS | Acquired immunodeficiency syndrome |
| AZT | Azidothymidine |
| CDI | Carbonyl diimidazole |
| DCC | Dicyclohexylcarbodiimide |
| DCM | Dichloromethane |
| DIBAL | Diisobutyl aluminium hydride |
| DMAP | Dimethyl amino pyridine |
| DMF | Dimethylformamide |
| DNA | Deoxyribonucleic acid |
| HCl | Hydrochloric acid |
| HIV | Human immunodeficiency virus |
| HOBt | Hydroxybenzotriazole |
| HPLC | High performance liquid chromatography |
| HTRF | Homogeneous time-resolved fluorescence |
| LAH | Lithium aluminium hydride |
| LDA | Lithium diisopropyl amide |
| μM | Micromolar |
| NBS | N-Bromosuccinimide |
| NMM | N-Methyl morpholine |
| SARs | Structure activity-relationships |
| TEA | Triethylamine |
| THF | Tetrahydrofuran |
| UNAIDS | Joint United Nations Programme on AIDS/HIV |
Author contributions
Conceptualization, G. K. G, F. N. K and D. K.; methodology, R. K., P. S. and D. K.; software, data curation, R. K., P. S., S., F. N. K and D. K.; writing—original draft preparation, D. K., P. S., and R. K., writing—review and editing, R. K., P. S., S., G. K. G, F. N. K, and D. K.; visualization, X. X.; supervision, F. N. K., and D. K.; project administration, F. N. K., G. K. G, and D. K.; funding acquisition, F. N. K. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
F. N. K. acknowledges financial support from the German Academic Exchange Services (DAAD) through the guest professorship program.References
- B. Salehi, N. V. A. Kumar, B. Sener, M. Sharifi-Rad, M. Kılıç, G. B. Mahady, S. Vlaisavljevic, M. Iriti, F. Kobarfard, W. N. Setzer, S. A. Ayatollahi, A. Ata and J. Sharifi-Rad, Int. J. Mol. Sci., 2018, 19, 1459 CrossRef PubMed.
- C. Reynolds, C. B. de Koning, S. C. Pelly, W. A. L. Otterlo and M. L. Bode, Chem. Soc. Rev., 2012, 41, 4657–4670 RSC.
- www.hiv.gov, accessed on 16 December 2020.
- D. Y. Lu, H. Y. Wu, N. S. Yarla, B. Xu, J. Ding and T. R. Lu, Infect. Disord.: Drug Targets, 2018, 18, 15–22 CAS.
- M. S. Cohen, Y. Q. Chen, M. McCauley, T. Gamble, M. C. Hosseinipour, N. Kumarasamy, J. G. Hakim, J. Kumwenda, B. Grinsztejn and J. H. Pilotto, N. Engl. J. Med., 2011, 365, 493–505 CrossRef CAS PubMed.
- WHO, Bull. W. H. O., 1989, 87, 613–618 Search PubMed.
- P. Cos, L. Maes, D. V. Berghe, N. Hermans, L. Pieters and A. Vlietinck, J. Nat. Prod., 2004, 67, 284–293 CrossRef CAS PubMed.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2012, 75, 311e335 CrossRef PubMed.
- Y. W. Chin, M. J. Balunas, H. B. Chai and A. D. Kinghorn, AAPS J., 2006, 8, 239–253 CrossRef PubMed.
- D. Kumar and S. K. Jain, Curr. Med. Chem., 2016, 23, 4338–4394 CrossRef CAS PubMed.
- V. Kumar, K. Kaur, G. K. Gupta and A. K. Sharma, Eur. J. Med. Chem., 2013, 69, 735–753 CrossRef CAS PubMed.
- K. Kaur, V. Kumar, G. K. Gupta and A. K. Sharma, Eur. J. Med. Chem., 2014, 77, 121–133 CrossRef CAS PubMed.
- R. Kaur, P. Sharma, G. K. Gupta, F. Ntie-Kang and D. Kumar, Molecules, 2020, 25, 2070 CrossRef CAS PubMed.
- D. Kumar, P. Sharma, H. Singh, K. Nepali, G. K. Gupta, S. K. Jain and F. Ntie-Kang, RSC Adv., 2017, 7, 36977–36999 RSC.
- K. Nepali, S. Sharma, D. Kumar, A. Budiraja and K. L. Dhar, Recent Pat. Anti-Cancer Drug Discovery, 2014, 9, 303–339 CrossRef CAS PubMed.
- D. Kumar, K. Nepali, P. M. S. Bedi, S. Kumar, F. Malik and S. Jain, Anti-Cancer Agents Med. Chem., 2015, 15, 793–803 CrossRef CAS PubMed.
- D. Kumar, O. Singh, K. Nepali, P. M. S. Bedi, A. Qayum, S. Singh and S. K. Jain, Anti-Cancer Agents Med. Chem., 2016, 16, 881–890 CrossRef CAS PubMed.
- D. Kumar, F. Malik, P. M. S. Bedi and S. K. Jain, Chem. Pharm. Bull., 2016, 64, 399–409 CrossRef CAS PubMed.
- D. Kumar, G. Singh, P. Sharma, A. Qayum, G. Mahajan, M. J. Mintoo, S. K. Singh, D. M. Mondhe, P. M. S. Bedi, S. K. Jain and G. K. Gupta, Anti-Cancer Agents Med. Chem., 2018, 18, 57–73 CrossRef CAS PubMed.
- D. Kumar, P. Sharma, K. Nepali, G. Mahajan, M. J. Mintoo, A. Singh, G. Singh, D. M. Mondhe, G. Singh, S. K. Jain, G. K. Gupta and F. Ntie-Kang, Heliyon, 2018, 4, e00661 CrossRef PubMed.
- P. Sharma, R. Sharma, H. S. Rao and D. Kumar, Int. J. Pharma Sci. Res., 2015, 6, 505–513 CrossRef.
- D. Kumar and P. M. S. Bedi, Indian Drugs, 2009, 46, 675–681 Search PubMed.
- T. Kaur, P. Sharma, G. K. Gupta, F. N. Kang and D. Kumar, Plant Arch., 2019, 19(2), 2168–2176 Search PubMed.
- C. Calabrese, S. H. Berman, J. G. Badish, X. Ma, L. Shinto, M. Dorr, K. Well, C. A. Wenner and L. J. Standish, Phytother. Res., 2000, 14, 333–338 CrossRef CAS PubMed.
- W. Li, X. Xu, H. Zhang, C. Ma, H. Fong, V. Richard and F. John, Chem. Pharm. Bull., 2007, 55, 455–458 CrossRef CAS PubMed.
- R. S. Chang, L. Ding, G. Q. Chen, Q. C. Pan, J. L. Zhao and K. M. Smith, Proc. Soc. Exp. Biol. Med., 1991, 197, 59–66 CrossRef CAS PubMed.
- V. Rajpal, Standardization of Botanicals, Estern Publishers, New Delhi, 1st edn, 2002, pp. 29–37 Search PubMed.
- S. Vetriselvan and U. Subasini, Asian J. Phytomed. Clin. Res., 2013, 1, 57–63 Search PubMed.
- R. Kumar, K. Sridevi, N. Kumar, N. Srinivas and S. Rajagopal, J. Ethnopharmacol., 2004, 92, 291–295 CrossRef CAS PubMed.
- S. Jada, G. Subur, C. Matthews, A. Hamzah, N. Lajis and M. Saad, Phytochemistry, 2007, 68, 904–912 CrossRef CAS PubMed.
- V. Menon and S. V. Bhat, Nat. Prod. Commun., 2010, 5, 717–720 CrossRef CAS PubMed.
- H. W. Xu, F. D. Gui, Z. L. Gai, F. W. Jun and M. L. Hong, Bioorg. Med. Chem., 2007, 15, 4247–4255 CrossRef CAS PubMed.
- V. L. N. Reddy, S. M. Reddy, V. Ravikanth, P. Krishnaiah, T. V. Goud, T. P. Rao, T. S. Ram, R. G. Gonnade, M. Bhadbhade and Y. Venkateswarlu, Nat. Prod. Res., 2005, 19, 223–230 CrossRef CAS PubMed.
- B. Wang, L. Ge, W. Huang, H. Zhang, H. Qian, J. Li and Y. Zheng, Med. Chem., 2010, 6, 252–258 CrossRef CAS PubMed.
- B. Wang, J. Li, W. L. Haung, H. B. Zhang, H. Qian and Y. T. Zheng, Chin. Chem. Lett., 2011, 22, 781–784 CrossRef CAS.
- M. M. Uttekar, T. Das, R. S. Pawar, B. Bhandari, V. Menon, Nutan, S. K. Gupta and S. V. Bhat, Eur. J. Med. Chem., 2012, 56, 368–374 CrossRef CAS PubMed.
- T. Wang, Z. Yin, Z. Zhang, J. A. Bender, Z. Yang, G. Johnson, Z. Yang, L. M. Zadjura, C. J. D'Arienzo, D. D. G. Parker, C. Gesenberg, G. A. Yamanaka, Y. F. Gong, H. T. Ho, H. Fang, N. Zhou, B. V. McAuliffe, B. J. Eggers, L. N. Fan, B. Sans, I. B. Dicker, Q. Gao, R. J. Colonno, P. F. Lin, N. A. Meanwell and J. F. Kadow, J. Med. Chem., 2009, 52, 7778–7787 CrossRef CAS PubMed.
- T. Wang, Z. Zhang, O. B. Wallace, M. Deshpande, H. Fang, Z. Yang, L. M. Zadjura, D. L. Tweedie, S. Huang, F. Zhao, S. Ranadive, B. S. Robinson, Y. F. Gong, K. Ricarrdi, T. P. Spicer, C. Deminie, R. Rose, H. G. H. Wang, W. S. Blair, P. Y. Shi, P. F. Lin, R. J. Colonno and N. A. Meanwell, J. Med. Chem., 2003, 46, 4236–4239 CrossRef CAS PubMed.
- P. F. Lin, W. S. Blair, T. Wang, T. P. Spicer, Q. Guo, N. Zhou, Y. F. Gong, H. G. H. Wang, R. Rose, G. Yamanaka, B. Robinson, C. B. Li, R. Fridell, C. Deminie, G. Demers, Z. Yang, L. Zadjura, N. A. Meanwell and R. J. Colonno, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 11013–11018 CrossRef CAS PubMed.
- H. T. Ho, L. Fan, B. Nowicka-Sans, B. McAuliffe, C. B. Li, G. Yamanaka, N. Zhou, H. Fang, I. Dicker, R. Dalterio, Y. F. Gong, T. Wang, Z. Yin, Y. Ueda, J. Matiskella, J. F. Kadow, P. Clapham, J. Robinson, R. J. Colonno and P. F. Lin, J. Virol., 2006, 80, 4017–4025 CrossRef CAS PubMed.
- N. Madani, A. L. Perdigoto, K. Srinivasan, J. M. Cox, J. J. Chruma, J. LaLonde, M. Head, A. B. Smith and J. G. Sodroski, J. Virol., 2004, 78, 3742–3752 CrossRef CAS PubMed.
- Z. Si, N. Madani, J. M. Cox, J. J. Chruma, J. C. Klein, A. Schon, N. Phan, W. Wang, A. C. Biorn, S. Cocklin, I. Chaiken, E. Freire, A. B. Smith and J. G. Sodroski, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5036–5041 CrossRef CAS PubMed.
- G. Hanna, J. Lalezari, J. Hellinger, D. Wohl, T. Masterson, W. Fiske, J. F. Kadow, P. F. Lin, M. Giordano, R. J. Colonno and D. Grasela, Antiviral Activity, Safety, and Tolerability of a Novel, Oral Small-Molecule HIV-1 Attachment Inhibitor, BMS-488043, in HIV-1-Infected Subjects, San Francisco, CA, 2004 Search PubMed.
- Z. Zhang, Z. Yang, N. A. Meanwell, J. F. Kadow and T. Wang, J. Org. Chem., 2002, 67, 2345–2347 CrossRef CAS PubMed.
- T. Yamato, C. Hideshima and M. Tashiro, Chem. Express, 1990, 5, 845–848 CAS.
- H. Van de Poel, G. Guillaumet and M. C. Viaud-Massuard, Heterocycles, 2002, 57, 55–71 CrossRef CAS.
- H. Li, X. Jiang, Y. H. Ye, C. Fan, T. Romoff and M. Goodman, Org. Lett., 1999, 1, 91–93 CrossRef CAS PubMed.
- Z. Zhang, Z. Yang, H. Wong, J. Zhu, N. A. Meanwell, J. F. Kadow and T. Wang, J. Org. Chem., 2002, 67, 6226–6227 CrossRef CAS PubMed.
- S. Jana, S. Iram, J. Thomas, M. Q. Hayat, C. Pannecouque and W. Dehaen, Molecules, 2017, 22, 303 CrossRef PubMed.
- Y. Tu, Nat. Med., 2011, 17, 1217–1220 CrossRef CAS PubMed.
- I. S. Lubbe, T. Klimkait and F. V. D. Kooy, J. Ethnopharmacol., 2012, 141, 854–859 CrossRef PubMed.
- D. L. Klayman, Science, 1985, 228, 1049–1055 CrossRef CAS PubMed.
- D. Chaturvedi, A. Goswami, P. P. Saikia, N. C. Barua and P. G. Rao, Chem. Soc. Rev., 2010, 39, 435–454 RSC.
- M. Jung, K. Lee, H. Kim and M. Park, Curr. Med. Chem., 2004, 11, 1265–1284 CrossRef CAS PubMed.
- J. J. Lu, S. M. Chen, X. W. Zhang, J. Ding and L. H. Meng, Invest. New Drugs, 2011, 29, 1276–1283 CrossRef CAS PubMed.
- H. Chen, B. Sun, S. Pan, H. Jiang and X. Sun, Anticancer Drugs, 2009, 20, 131–140 CrossRef CAS PubMed.
- J. J. Lu, L. H. Meng, Y. J. Cai, Q. Chen, L. J. Tong, L. P. Lin and J. Ding, Cancer Biol. Ther., 2008, 7, 1017–1023 CrossRef CAS PubMed.
- Y. Li, J. M. Wu, F. Shan, G. S. Wu, J. Ding, D. Xiao, J. X. Han, G. Atassi, S. Leonce and D. H. Caignard, Bioorg. Med. Chem., 2003, 11, 977–984 CrossRef CAS PubMed.
- Z. S. Yang, W. L. Zhou, Y. Sui, J. X. Wang, J. M. Wu, Y. Zhou, Y. Zhang, P. L. He, J. Y. Han and W. Tang, J. Med. Chem., 2005, 48, 4608–4617 CrossRef CAS PubMed.
- D. Mu, W. Zhang, D. Chu, T. Liu, Y. Xie, E. Fu and F. Jin, Cancer Chemother. Pharmacol., 2008, 61, 639–645 CrossRef CAS PubMed.
- T. Chen, M. Li, R. Zhang and H. Wang, J. Cell. Mol. Med., 2009, 13, 1358–1370 CrossRef CAS PubMed.
- C. Wu, J. Liu, X. Pan, W. Xian, B. Li, W. Peng, J. Wang, D. Yang and H. Zhou, Molecules, 2013, 18, 6866–6882 CrossRef CAS PubMed.
- Y. Liu, Z. Liu, J. Shi, H. Chen, B. Mi, P. Li and P. Gong, Molecules, 2013, 18, 2864–2877 CrossRef CAS PubMed.
- A. Parra, F. Rivas, P. L. Lopez, A. G. Granados, A. Martinez, F. Albericio, N. Marquez and E. Munoz, Bioorg. Med. Chem., 2009, 17, 1139–1145 CrossRef CAS PubMed.
- H. X. Xu, F. Q. Zeng, M. Wan and K. Y. Sim, J. Nat. Prod., 1996, 59, 643 CrossRef CAS PubMed.
- R. Mukherjee, V. Kumar, S. K. Srivastava, S. K. Agarwal and A. C. Burman, Anti-Cancer Agents Med. Chem., 2006, 6, 271–279 CrossRef CAS PubMed.
- M. P. Montilla, A. Agil, M. C. Navarro, M. I. Jimenez, A. G. Garcia, A. Parra and M. M. Cabo, Planta Med., 2003, 69, 472–474 CrossRef CAS PubMed.
- D. E. Zembower, S. Liao, M. T. Flavin, Z. Q. Xu, T. L. Stup, R. W. Buckheit Jr, A. Khilevich, A. A. Mar and A. K. Sheinkman, J. Med. Chem., 1997, 40, 1005–1017 CrossRef CAS PubMed.
- T. Creagh, J. L. Ruckle, D. T. Tolbert, J. Giltner, D. A. Eiznhamer, B. Dutta, M. T. Flavin and Z. Q. Xu, Antimicrob. Agents Chemother., 2001, 45, 1379–1386 CrossRef CAS PubMed.
- Z. Q. Xu, E. R. Kern, L. Westbrook, L. B. Allen, R. W. Buckheit, C. K. Tseng, T. Jenta and M. T. Flavin, Antiviral Chem. Chemother., 2000, 11, 23–29 CrossRef CAS PubMed.
- A. D. Patil, A. J. Freyer, D. S. Eggleston, R. C. Haltwanger, M. F. Bean, P. B. Taylor, M. J. Caranfa, A. L. Breen, H. R. Bartus, R. K. Johnson, R. P. Hertzberg and J. W. Westley, J. Med. Chem., 1993, 36, 4131–4138 CrossRef CAS PubMed.
- Y. Kashman, K. R. Gustafson, R. W. Fuller, J. H. Cardellina, J. B. McMahon, M. J. Currens, R. W. Buckheit Jr, S. H. Hughes, G. M. Cragg and M. R. Boyd, J. Med. Chem., 1992, 35, 2735–2743 CrossRef CAS PubMed.
- J. H. Cardellina, H. R. Bokesch, T. C. McKee and M. R. Boyd, Bioorg. Med. Chem. Lett., 1995, 5, 1011–1014 CrossRef CAS.
- M. T. Flavin, J. D. Rizzo, A. Khilevich, A. Kucherenko, A. K. Sheinkman, V. Vilaychack, L. Lin, W. Chen, E. M. Greenwood, T. Pengsuparp, J. M. Pezzuto, S. H. Hughes, T. M. Flavin, M. Cibulski, W. A. Boulanger, R. L. Shone and Z. Q. Xu, J. Med. Chem., 1996, 39, 1303–1313 CrossRef CAS PubMed.
- A. Kucherenko, M. T. Flavin, W. A. Boulanger, A. Khilevich, R. L. Shone, J. D. Rizzo, A. K. Sheinkman and Z. Q. Xu, Tetrahedron Lett., 1995, 36, 5475–5478 CrossRef CAS.
- A. Khilevich, J. D. Rizzo, M. T. Flavin, A. K. Sheinkman, A. Mar, A. Kucherenko, C. Yan, S. Dzekhtser, D. Brankovic, L. Lin, J. Liu, T. M. Rizzo and Z. Q. Xu, Synth. Commun., 1996, 20, 3757–3771 CrossRef.
- B. Chenera, M. L. West, J. A. Finkelstein and G. B. Dreyer, J. Org. Chem., 1993, 58, 5605–5606 CrossRef CAS.
- A. L. Gemal and J. L. Luche, J. Am. Chem. Soc., 1981, 103, 5454–5459 CrossRef CAS.
- L. Crombie, R. C. F. Jones and C. J. Palmer, J. Chem. Soc., Perkin Trans. 1, 1987, 317–331 RSC.
- C. J. Palmer and J. L. Josephs, J. Chem. Soc., Perkin Trans. 1, 1995, 3135–3151 RSC.
- T. C. McKee, J. H. Cardellina, G. B. Dreyer and M. R. Boyd, J. Nat. Prod., 1995, 58, 916–920 CrossRef CAS PubMed.
- R. Pawar, T. Das, S. Mishra, Nutan, B. Pancholi, S. K. Gupta and S. V. Bhat, Bioorg. Med. Chem. Lett., 2014, 24, 302–307 CrossRef CAS PubMed.
- T. Konishi, M. Azuma, R. Itoga, S. Kiyosawa, Y. Fujiwara and Y. Shimada, Chem. Pharm. Bull., 1996, 44, 229–231 CrossRef CAS.
- D. Kenifield, G. Bunkers, G. Strobel and F. Sugawara, In: Ref., 1989, 26, 319–335.
- M. F. Braulio, Nat. Prod. Rep., 2001, 18, 650 RSC.
- S. V. Bhat, Life Chem. Rep., 1994, 12, 137 CAS.
- D. J. Lee and W. E. Robinson Jr, Antimicrob. Agents Chemother., 2006, 50, 134–142 CrossRef CAS PubMed.
- H. Jayasuriya, Z. Guan, J. D. Polishook, A. W. Dombrowski, P. J. Felock, D. J. Hazuda and S. B. Singh, J. Nat. Prod., 2003, 66, 551–553 CrossRef CAS PubMed.
- D. F. Chen, S. X. Zhang, L. Xie, J. X. Xie, K. Chen, Y. Kashiwada, B. N. Zhou, P. Wang, L. M. Cosentino and K. H. Lee, Bioorg. Med. Chem., 1997, 5, 1715–1723 CrossRef CAS PubMed.
- T. Fujihashi, H. Hara, T. Sakata, K. Mori, H. Higuchi, A. Tanaka, H. Kaji and A. Kaji, Antimicrob. Agents Chemother., 1995, 39, 2000–2007 CrossRef CAS PubMed.
- X. W. Yang, H. Miyanshiro, M. Hattori, T. Namba, Y. Tezuka, T. Kikuchi, D. F. Chen, G. J. Xu, T. Hori, M. Extine and H. Mizuno, Chem. Pharm. Bull., 1992, 40, 1510–1516 CrossRef CAS PubMed.
- Y. Ikeya, H. Taguchi, I. Yosioka and H. Kobayashi, Chem. Pharm. Bull., 1979, 27, 1583 CrossRef CAS.
- R. S. Coleman and S. R. Gurrala, Org. Lett., 2005, 7, 1849–1852 CrossRef CAS PubMed.
- Y. Y. Chen, Z. B. Shu and L. N. Li, Sci. Sin., 1976, 19, 276–290 CAS.
- Y. Ikeya, H. Taguchi and I. Yosioka, Chem. Pharm. Bull., 1982, 30, 3207 CrossRef CAS.
- J. X. Xie, J. Zhou, C. Z. Zhang, J. H. Yang and J. X. Chen, Sci. Sin., 1983, 26, 1291 CAS.
- P. Shah, D. Naik, N. Jariwala, D. Bhadane, S. Kumar, S. Kulkarni, K. K. Bhutani and I. P. Singh, Bioorg. Chem., 2018, 80, 591–601 CrossRef CAS PubMed.
- J. L. McCormick, T. C. Mckee, J. H. Cardellina and M. R. Boyd, J. Nat. Prod., 1996, 59, 469–471 CrossRef CAS PubMed.
- C. Benard, F. Zouhiri, M. Normand-Bayle, M. Danet, D. Desmaele, H. Leh, J. F. Mouscadet, G. Mbemba, C. M. Thomas, S. Bonnenfant, M. L. Bret and J. D'Angelo, Bioorg. Med. Chem. Lett., 2004, 14, 2473–2476 CrossRef CAS PubMed.
- Q. Q. He, S. X. Gu, J. Liu, H. Q. Wu, X. Zhang, L. M. Yang, Y. T. Zheng and F. E. Chen, Bioorg. Med. Chem., 2011, 19, 5039–5045 CrossRef CAS PubMed.
- S. Mahajan, S. Gupta, N. Jariwala, D. Bhadane, K. K. Bhutani, S. Kulkarni and I. P. Singh, Lett. Drug Des. Discovery, 2018, 15, 937–944 CrossRef CAS.
- S. Chen, R. Chen, M. He, R. Pang, Z. Tan and M. Yang, Bioorg. Med. Chem., 2009, 17, 1948–1956 CrossRef CAS PubMed.
- R. Musiol, Curr. Pharm. Des., 2013, 19, 1835–1849 CrossRef CAS PubMed.
- A. Marella, O. P. Tanwar, R. Saha, M. R. Ali, S. Srivastava, M. Akhter, M. Shaquiquzzaman and M. M. Alam, Saudi Pharm. J., 2013, 21, 1–12 CrossRef PubMed.
- M. Billamboz, V. Suchaud, F. Bailly, C. Lion, J. Demeulemeester, C. Calmels, M. L. Andréola, F. Christ, Z. Debyser and P. Cotelle, ACS Med. Chem. Lett., 2013, 4, 606–611 CrossRef CAS PubMed.
- E. Serrao, B. Debnath, H. Otake, Y. Kuang, F. Christ, Z. Debyser and N. Neamati, J. Med. Chem., 2013, 56, 2311–2322 CrossRef CAS PubMed.
- J. F. Mouscadet and D. Desmaele, Molecules, 2010, 15, 3048–3078 CrossRef CAS PubMed.
- M. J. Cheng, K. H. Lee, I. L. Tsai and I. S. Chen, Bioorg. Med. Chem., 2005, 13, 5915–5920 CrossRef CAS PubMed.
- C. L. Sun, R. F. Pang, H. Zhang and M. Yang, Bioorg. Med. Chem. Lett., 2005, 15, 3257–3262 CrossRef CAS PubMed.
- K. R. Romines and R. A. Chrusciel, Curr. Med. Chem., 1995, 2, 825 CAS.
- J. V. Prasad, F. E. Boyer, J. M. Domagala, E. L. Ellsworth, C. Gajda, H. W. Hamilton, S. E. Hagen, L. J. Markoski, B. A. Steinbaugh, B. D. Tait, C. Humblet, E. A. Lunney, A. Pavlovsky, J. R. Rubin, D. Ferguson, N. Graham, T. Holler, D. Hupe, C. Nouhan, P. J. Tummino, A. Urumov, E. Zeikus, G. Zeikus, S. J. Gracheck and J. W. Erickson, Bioorg. Med. Chem., 1999, 7, 2775–2800 CrossRef CAS.
- S. Thaisrivongs, D. L. Romero, R. A. Tommasi, M. N. Janakiraman, J. W. Strohbach, S. R. Tuner, C. Biles, R. R. Morge, P. D. Johnson, P. A. Aristoff, P. K. Tomich, J. C. Lynn, M. M. Horng, K. T. Chong, R. R. Hinshaw, W. J. Howe, B. C. Finzel and K. D. Watenpaugh, J. Med. Chem., 1996, 39, 4630–4642 CrossRef CAS.
- H. I. Skulnick, P. D. Johnson, W. J. Howe, P. K. Tomich, K. T. Chong, K. D. Watenpaugh, M. N. Janakiraman, L. A. Dolak, J. P. McGrath and J. C. Lynn, J. Med. Chem., 1995, 38, 4968–4971 CrossRef CAS.
- C. M. Abreu, S. L. Price, E. N. Shirk, R. D. Cunha, L. F. Pianowski, J. E. Clements, A. Tanuri and L. Gama, PLoS One, 2014, 9, e97257 CrossRef PubMed.
- C. Jiang, P. Luo, Y. Zhao, J. Hong, S. L. Morris-Natschke, J. Xu, C. H. Chen, K. H. Lee and Q. Gu, J. Nat. Prod., 2016, 79, 578–583 CrossRef CAS PubMed.
- M. Zhou, L. Deng and V. Lacoste, J. Virol., 2004, 78, 13522–13533 CrossRef CAS PubMed.
- A. R. Jassbi, Phytochemistry, 2006, 67, 1977–1984 CrossRef CAS PubMed.
- P. Hampson, K. Wang and J. M. Lord, Drugs Future, 2005, 30, 1003–1005 CrossRef CAS.
- R. Barrero, B. Chapman, Y. Yang, P. Moolhuijzen and G. Keeble-Gagne`re, BMC Genomics, 2011, 12, 600 CrossRef CAS PubMed.
- R. Norman, JAMA Dermatol., 2013, 149, 666–670 CrossRef PubMed.
- L. Jorgensen, S. J. McKerrall, C. A. Kuttruff, F. Ungeheuer and J. Felding, Science, 2013, 341, 878–882 CrossRef CAS PubMed.
- M. Nirmala, A. Samundeeswari and P. Sankar, Res Plant Biol., 2011, 1, 1–14 Search PubMed.
- F. E. Agharbaoui, A. C. Hoyte, S. Ferro, R. Gitto, M. R. Buemi, J. R. Fuchs, M. Kvaratskhelia and L. D. Luca, Eur. J. Med. Chem., 2016, 123, 673–683 CrossRef CAS PubMed.
- L. Feng, R. C. Larue, A. Slaughter, J. J. Kessl and M. Kvaratskhelia, Curr. Top. Microbiol. Immunol., 2015, 389, 93–119 CAS.
- M. Jaskolski, J. N. Alexandratos, G. Bujacz and A. Wlodawer, FEBS J., 2009, 276, 2926–2946 CrossRef CAS PubMed.
- M. Li, M. Mizuuchi, T. R. Burke Jr and R. Craigie, EMBO J., 2006, 25, 1295–1304 CrossRef CAS PubMed.
- A. Engelman and P. Cherepanov, Nat. Rev. Microbiol., 2012, 10, 279–290 CrossRef CAS PubMed.
- M. Llano, D. T. Saenz, A. Meehan, P. Wongthida, M. Peretz, W. H. Walker, W. Teo and E. M. Poeschla, Science, 2006, 314, 461–464 CrossRef CAS PubMed.
- K. Busschots, J. Vercammen, S. Emiliani, R. Benarous, Y. Engelborghs, F. Christ and Z. Debyser, J. Biol. Chem., 2005, 280, 17841–17847 CrossRef CAS PubMed.
- P. Cherepanov, G. Maertens, P. Proost, B. Devreese, J. V. Beeumen, Y. Engelborghs, E. D. Clercq and Z. Debyser, J. Biol. Chem., 2003, 278, 372–381 CrossRef CAS PubMed.
- L. Q. Al-Mawsawi and N. Neamati, Trends Pharmacol. Sci., 2007, 28, 526–535 CrossRef CAS PubMed.
- M. C. Shun, N. K. Raghavendra, N. Vandegraaff, J. E. Daigle, S. Hughes, P. Kellam, P. Cherepanov and A. Engelman, Genes Dev., 2007, 21, 1767–1778 CrossRef CAS PubMed.
- P. Cherepanov, E. Devroe, P. A. Silver and A. Engelman, J. Biol. Chem., 2004, 279, 48883–48892 CrossRef CAS PubMed.
- F. Christ, A. Voet, A. Marchand, S. Nicolet, B. A. Desimmie, D. Marchand, D. Bardiot, N. J. Van der Veken, B. Van Remoortel, S. V. Strelkov, M. D. Maeyer, P. Chaltin and Z. Debyser, Nat. Chem. Biol., 2010, 6, 442–448 CrossRef CAS PubMed.
- A. Sharma, A. Slaughter, N. Jena, L. Feng, J. J. Kessl, H. J. Fadel, N. Malani, F. Male, L. Wu, E. Poeschla, F. D. Bushman, J. R. Fuchs and M. Kvaratskhelia, PLoS Pathog., 2014, 10, e1004171 CrossRef PubMed.
- L. D. Fader, E. Malenfant, M. Parisien, R. Carson, F. Bilodeau, S. Landry, M. Pesant, C. Brochu, S. Morin, C. Chabot, T. Halmos, Y. Bousquet, M. D. Bailey, S. H. Kawai, R. Coulombe, S. LaPlante, A. Jakalian, P. K. Bhardwaj, D. Wernic, P. Schroeder, M. Amad, P. Edwards, M. Garneau, J. Duan, M. Cordingley, R. Bethell, S. W. Mason, M. Bos, P. Bonneau, M. A. Poupart, A. M. Faucher, B. Simoneau, C. Fenwick, C. Yoakim and Y. Tsantrizos, ACS Med. Chem. Lett., 2014, 5, 422–427 CrossRef CAS PubMed.
- N. Van Bel, Y. van der Velden, D. Bonnard, E. L. Rouzic, A. T. Das, R. Benarous and B. Berkhout, PLoS One, 2014, 9, e103552 CrossRef PubMed.
- L. D. Luca, F. Morreale, F. Christ, Z. Debyser, S. Ferro and R. Gitto, Eur. J. Med. Chem., 2013, 68, 405–411 CrossRef PubMed.
- D. R. Hirsch, D. V. Schiavone, A. J. Berkowitz, L. A. Morrison, T. Masaoka, J. A. Wilson, E. Lomonosova, H. Zhao, B. S. Patel, S. H. Datla, S. G. Hoft, S. J. Majidi, R. K. Pal, E. Gallicchio, L. Tang, J. E. Tavis, S. F. J. Le Grice, J. A. Beutler and R. P. Murelli, Org. Biomol. Chem., 2018, 16, 62 RSC.
- C. Meck, M. P. D'Erasmo, D. R. Hirsch and R. P. Murelli, Med. Chem. Commun., 2014, 5, 842–852 RSC.
- S. R. Budihas, I. Gorshkova, S. Gaidamakov, A. Wamiru, M. K. Bona, M. M. A. Parniak, R. J. Crouch, J. B. McMahon, J. A. Beutler and S. F. J. L'Grice, Nucleic Acids Res., 2005, 33, 1249–1256 CrossRef CAS PubMed.
- G. Sennari, T. Hirose, M. Iwatsuki, S. Omura and T. Sunazuka, Chem. Commun., 2014, 50, 8715–8718 RSC.
- L. D. Ferretti and J. H. Richards, Proc. Natl. Acad. Sci. U. S. A., 1960, 46, 1438–1444 CrossRef CAS PubMed.
- J. Davison, A. al. Fahad, M. Cai, Z. Song, S. Y. Yehia, C. M. Lazarus, A. M. Bailey, T. J. Simpson and R. J. Cox, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 7642–7647 CrossRef CAS PubMed.
- J. H. Birkinshaw, A. H. Chambers and H. Raistrick, Biochem. J., 1942, 36, 242–251 CrossRef CAS PubMed.
- D. A. Nugiel, K. Jacobs, T. Worley, M. Patel, R. F. Kaltenbach, D. F. Meyer, P. K. Jadhav, G. V. D'Lucca, T. E. Smyser, R. M. Klabe, L. T. Bacheler, M. M. Rayner and S. P. Seitz, J. Med. Chem., 1996, 39, 2156–2169 CrossRef CAS PubMed.
- D. D. Ho, A. U. Neumann, A. S. Perelson, W. Chen, J. M. Leonard and M. Markowitz, Nature, 1995, 373, 123–126 CrossRef CAS PubMed.
- K. E. B. Parkes, D. J. Bushnell, P. H. Crackett, S. J. Dunsdon, A. C. Freeman, M. P. Gunn, R. A. Hopkins, R. W. Lambert, J. A. Martin, J. H. Merrett, S. Redshaw, W. C. Spurden and G. J. Thomas, J. Org. Chem., 1994, 59, 3656–3664 CrossRef CAS.
- S. Thaisrivongs, Annu. Rep. Med. Chem., 1994, 29, 133–144 CAS.
- D. P. Getman, G. A. DeCrescenzo, R. M. Heintz, K. L. Reed, J. J. Talley, M. L. Bryant, M. Clare, K. A. Houseman, J. J. Marr, R. A. Mueller, M. L. Vazquez, H. S. Shieh, W. C. Stallings and R. A. Stegeman, J. Med. Chem., 1993, 36, 288–291 CrossRef CAS PubMed.
- C. H. Chang, M. C. Schadt, F. A. Lewandowski, R. DeLoskey, J. Duke, J. C. Calabrese, P. Y. S. Lam, P. K. Jadhav, C. J. Eyerman, C. N. Hodge and P. C. Weber, J. Biol. Chem., 1998, 273, 12325–12398 CrossRef PubMed.
- C. N. Hodge, P. E. Aldrich, L. T. Bacheler, C. H. Chang, C. J. Eyerman, M. Grubb, D. A. Jackson, P. K. Jadhav, B. Korant, P. Y. S. Lam, M. B. Maurin, J. L. Meek, M. J. Otto, M. M. Rayner, T. R. Sharpe, L. Shum, D. L. Winslow and S. EricksonViitanen, Chem. Biol., 1996, 3, 301–314 CrossRef CAS PubMed.
- P. Y. S. Lam, Y. Ru, P. K. Jadhav, P. E. Aldrich, G. V. De Lucca, C. J. Eyermann, C. H. Chang, G. Emmett, E. R. Holler, W. F. Daneker, L. Li, P. N. Confalone, R. J. McHugh, Q. Han, R. Li, J. A. Markwalder, S. P. Seitz, T. R. Sharpe, L. T. Bacheler, M. M. Rayner, R. M. Klabe, L. Shum, D. L. Winslow, D. M. Kornhauser, D. A. Jackson, S. Erickson-Viitanen and C. N. Hodge, J. Med. Chem., 1996, 39, 3514–3525 CrossRef CAS PubMed.
- A. W. Konradi and S. F. Pederson, J. Org. Chem., 1992, 57, 28–32 CrossRef CAS.
- S. Sasatani, T. Miyazaki, K. Maruoka and H. Yamamoto, Tetrahedron Lett., 1983, 24, 4711–4712 CrossRef CAS.
- J. I. Levin, E. Turos and S. M. Weinreb, Synth. Commun., 1982, 12, 989–993 CrossRef CAS.
- O. Mitsunobu, Synthesis, 1981, 1–28 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |