
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRapid synthesis of internal peptidyl α-ketoamides by on resin oxidation for the construction of rhomboid protease inhibitors†
Tim Van Kersavonda,
Raphael Konopatzkia,
Merel A. T. van der Plasscheb,
Jian Yangb and
Steven H. L. Verhelst *ab
*ab
aLeibniz Institute for Analytical Sciences ISAS, e.V., Otto-Hahn-Str. 6b, 44227 Dortmund, Germany
bKU Leuven, Department of Cellular and Molecular Medicine, Laboratory of Chemical Biology, Herestr. 49 box 802, 3000 Leuven, Belgium. E-mail: steven.verhelst@kuleuven.be
First published on 20th January 2021
Abstract
Rhomboid proteases are intramembrane serine proteases, which are involved in a wide variety of biological processes and have been implied in various human diseases. Recently, peptidyl α-ketoamides have been reported as rhomboid inhibitors with high potency and selectivity – owing to their interaction with both the primed and non-primed site of the target protease. However, their synthesis has been performed by solution phase chemistry. Here, we report a solid phase strategy towards ketoamides as rhomboid protease inhibitors, allowing rapid synthesis and optimization. We found that the primed site binding part of inhibitors is crucial for potency.
Introduction
Rhomboid proteases are one of the most widespread families of intramembrane proteases (IMPs). They were originally discovered in Drosophila melanogaster,1 but occur in virtually all sequenced organisms.2,3 Their roles are diverse and include EGFR signaling in the fruitfly,1 quorum sensing in specific bacteria4 and endoplasmatic reticulum associated degradation in mammalian cells.5 IMPs are potential drug targets,6 but the exact biomedical role and the druggability of rhomboids are still under investigation. One of the bottlenecks in rhomboid research has been the availability of potent and selective inhibitors.In the past decade, various electrophiles have been reported as scaffolds for the design and synthesis of rhomboid inhibitors.7 These include 4-chloro-isocoumarins,8,9 such as compound 1, β-lactams, e.g. compound 2,10,11 benzoxazinones including compound 3,12 and fluorophosphonates such as compound 4 (ref. 13 and 14) (Fig. 1A). All of these form a covalent intermediate with active site residues by alkylation, phosphorylation or acylation. Unfortunately, the aforementioned compounds are not highly selective. Peptidyl α-ketoamides (Fig. 1B), however, were recently reported as potent and highly selective rhomboid inhibitors.15 These compounds form a reversible covalent hemiketal intermediate, and elements at both sides of this electrophile contribute to interaction with the active site surroundings at the non-primed site and the primed site.
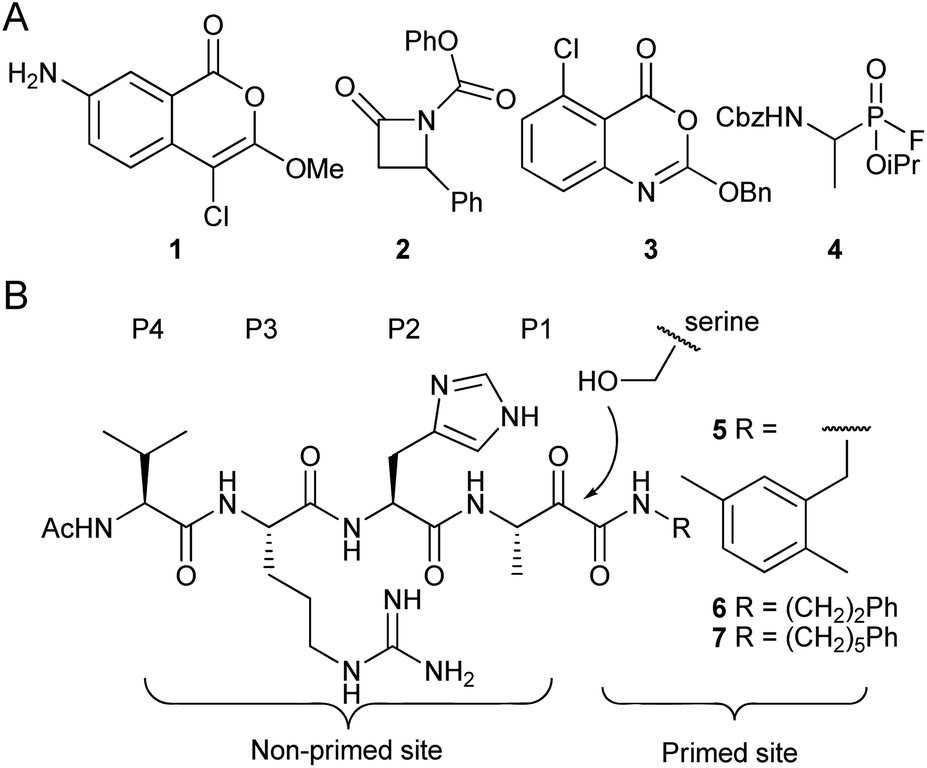 | ||
| Fig. 1 Examples of rhomboid inhibitors. (A) 4-chloro-isocoumarins (1), β-lactams (2), benzoxazinones (3) and fluorophosphonates (4). (B) α-Ketoamide rhomboid inhibitors (5–7). The peptidic element in the non-primed site is indicated with the P1–P4 position according to the Schechter and Berger protease substrate nomenclature.16 | ||
The peptidic nature of α-ketoamide rhomboid inhibitors makes it possible to utilize substrate preference information to rationally design effective rhomboid inhibitors. We therefore aimed at synthesizing α-ketoamides flanked with peptide sequences on each side by making use of solid phase peptide synthesis (SPPS). Various reports on the synthesis of internal peptidyl ketoamides by SPPS have been made in the past, for example by on-resin oxidation of α-hydroxyamides17,18 or by using Fmoc-protected building blocks containing as dithioketal-protected19 or acetal-protected ketoacids.20 We here report rhomboid substrate-derived ketoamides by Fmoc-based SPPS combined with an on-resin oxidation by IBX. Inhibition tests revealed that the precise nature of the primed site binding elements are crucial for effective rhomboid inhibition.
Results and discussion
Most rhomboid substrates contain an alanine at the P1 position. Consequently, the thusfar reported peptide-derived inhibitors for rhomboid proteases also display an alanine residue in this position.15,21,22 In order to explore a solid phase peptide synthesis of α-ketoamide inhibitors of rhomboid proteases, we first synthesized Fmoc-protected, alanine-derived α-keto-acid 13 with α-hydroxy-acid 12 as an intermediate (Scheme 1A). Known Fmoc L-alanine aldehyde 10 was generated from commercially available Fmoc-L-alanine (8) by conversion into Weinreb amide 9 and subsequent reduction under influence of LiAlH4. Cyanohydrin 11 was formed as a diastereomeric mixture by reaction of aldehyde 10 with hydrogen cyanide. Acidic hydrolysis of the nitrile function to a carboxylic acid furnished compound 12, which was oxidized to ketoacid 13 with Dess–Martin periodinane (Scheme 1A). With these building blocks in hand, we explored the synthesis of potential rhomboid inhibitors with peptide elements on both sides of the ketoamide moiety. In the primed site, a single alanine amino acid or an alanine–phenylalanine dipeptide was chosen. Coupling of keto-acid 13 to the free amine of alanine on a Rink resin (14), followed by TFA cleavage, led to formation of alanine dimer 15 (Scheme 1B and Fig. S1†) by CO extrusion, as reported before.19 The elimination of CO has previously been attributed to a reaction under influence of TFA.19 However, we also observed CO extrusion when building block 13 was coupled in solution to alanine amide (16) (Scheme 1B and Fig. S2†). Although the mechanism of the CO extrusion during peptide coupling remains elusive, we reasoned that coupling of hydroxy-acid 12 would not lead to loss of CO during coupling. This could then be followed by on-resin oxidation of the α-hydroxyamide, which has been reported for products from an on-resin Passerini reaction,23 as well as for solution and solid phase approaches using various α-hydroxy-acids, followed by treatment with Dess–Martin periodinane.18,24 In these cases, CO extrusion did not take place after cleavage from the solid support. Hence, we reasoned that the proposed strategy may successfully lead to the formation of the desired products as potentially novel rhomboid inhibitors.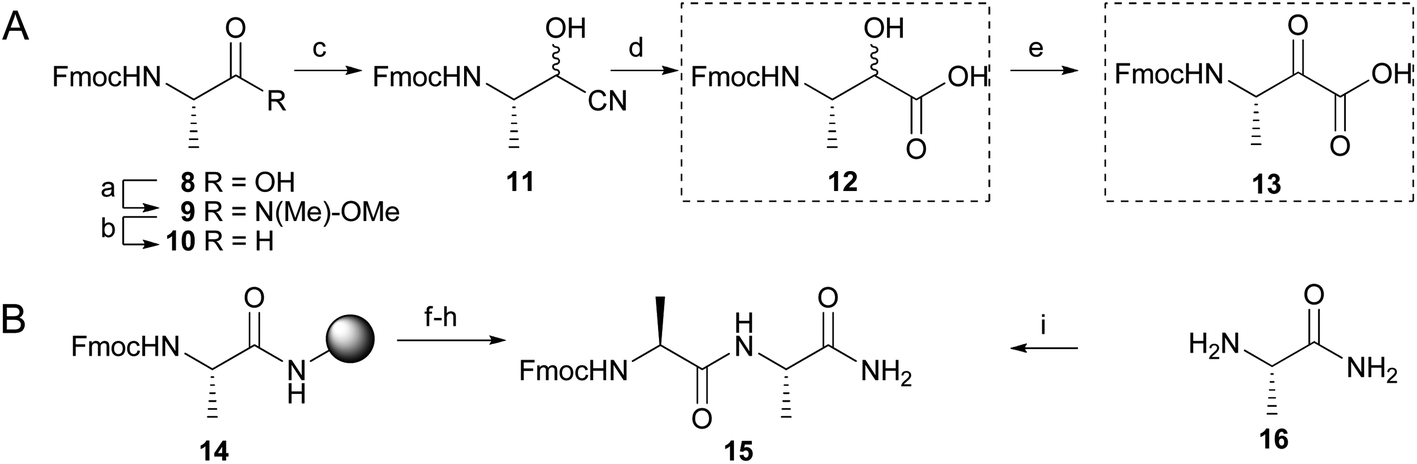 | ||
| Scheme 1 Synthesis and peptide coupling of building block 13, which leads to CO extrusion. (A) Synthesis of compound 13. (a) MeONHMe, HBTU, DIEA, DMF/DCM, 97%, (b) LiAlH4, THF, (c) NaCN, AcOH, DCM/MeOH, (d) conc HCl (aq)/dioxane 1/1, reflux, 48% (over three steps), (e) Dess-Martin reagent, DCM, 43%. (B) Peptide coupling with building block 13. (f) 20% piperidine in DMF, (g) 13 (3 eq.), HBTU (3 eq.), DIEA (6 eq.), DMF, (h) TFA/H2O/TIS 95/2.5/2.5, (i) 13 (1 eq.), HBTU (1 eq.), DIEA (2 eq), DMF. | ||
As P4–P1 residues, the desired compounds 18–20 contain the tetrapeptide element VRHA, as a most optimally binding sequence for GlpG15,21 or IATA, derived from the rhomboid substrate TatA.4 For the primed site, we chose an alanine or an alanine–phenylalanine dipeptide, as found in TatA (Scheme 2). The sequence was synthesized on a Rink amide resin to ensure a non-charged, C-terminal amide. After elongation of the peptide with the respective Fmoc amino acid building blocks, including building block 12, on resin oxidation was performed by using IBX in DMF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DMSO (1:1) (Scheme 2), which yielded target compounds 18–20 after cleavage from resin. Note that no CO extrusion was observed on the crude material (Fig. S3–S5†).
:DMSO (1:1) (Scheme 2), which yielded target compounds 18–20 after cleavage from resin. Note that no CO extrusion was observed on the crude material (Fig. S3–S5†).
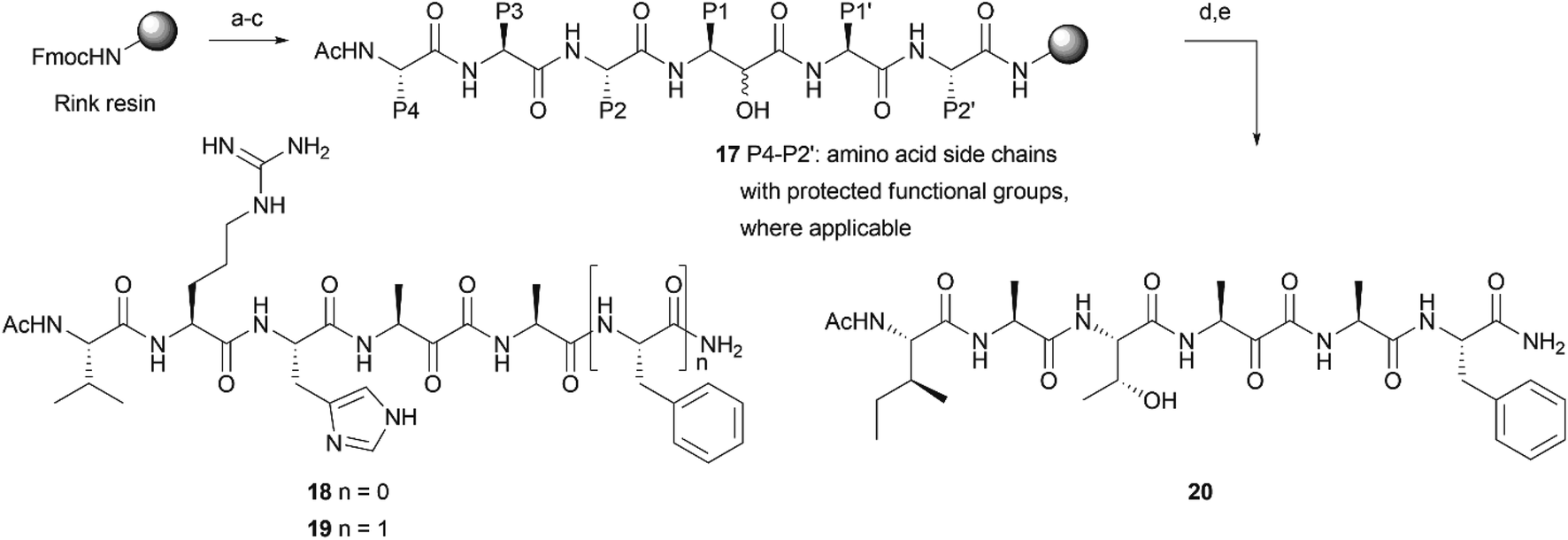 | ||
| Scheme 2 On resin synthesis of peptidyl α-ketoamides. (a) Fmoc-based solid phase peptide synthesis: (1) 20% piperidine in DMF, (2) Fmoc-amino acid (5 eq.), HBTU (5 eq.), DIEA (10 eq.), DMF (couple twice; except for building block 12, which was coupled in a single step); repeat from step 1 for every amino acid. Utilized amino acids: Fmoc-Phe-OH, Fmoc-Ala-OH, Fmoc-His(Trt)-OH, Fmoc-Arg(Pbf)-OH, Fmoc-Thr(tBu)-OH, Fmoc-Ile-OH (b) (1) 20% piperidine in DMF, (2) Ac2O (5 eq.), Et3N (5 eq.), pyridine (5 eq.), DMF, (c) 1 M KOH in MeOH (d) IBX (3 eq.), H2O (3 eq.) in DMF:DMSO (1:1) (e) TFA/H2O/TIS 95/2.5/2.5. | ||
We next performed an inhibition assay of E. coli rhomboid protease GlpG by using competitive activity-based protein profiling (competitive ABPP; Fig. 2A). To this end, GlpG was pretreated with 50 μM of compounds 18–20, known ketoamide inhibitors 5–7, or 100 μM dichloroisocoumarin (DCI) as a general rhomboid inhibitor control. Residually active GlpG was then labeled by the general serine hydrolase probe FP-Rh,25 which is also a general probe for rhomboid proteases.26 To our surprise, the ketoamide compounds 18–20 did not display any inhibition in the competitive ABPP assay, while the reference compounds 5–7 as well as DCI gave full inhibition.
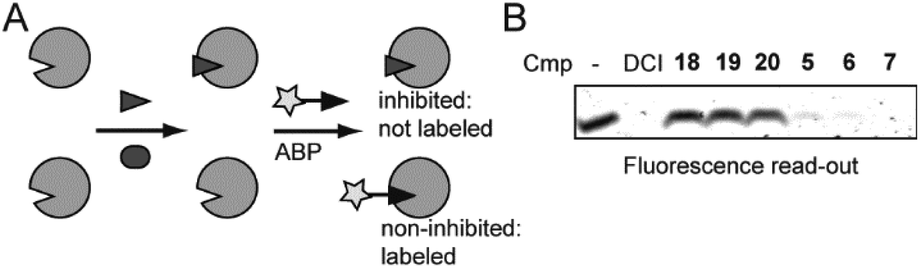 | ||
| Fig. 2 Evaluation of peptide α-ketoamides as inhibitors of the E. coli rhomboid GlpG. (A) Schematic representation of competitive ABPP. Pretreatment of a serine protease with a small molecule may lead to inhibition (above) or not (below). Subsequent addition of an activity-based probe (ABP) will lead to fluorescent labeling of the residually active enzyme, which can be measured by in-gel fluorescent scanning. (B) E. coli GlpG was incubated with the indicated ketoamides, the pan rhomboid inhibitor 3,4-dichloroisocoumarin (DCI) or dmso control for 30 min. Residually active GlpG was determined by incubation with the general serine hydrolase activity-based probe FP-Rh. Disappearance of fluorescent bands indicate full inhibition. | ||
Conclusions
In this paper, we have described a synthesis of peptides with an internal α-ketoamide electrophile that can be fully executed on solid support. Crucial to the synthesis is an α-hydroxy-β-amino acid derivative, exemplified by compound 12, which is oxidized on resin after elongation of the peptide sequence. Importantly, this enables a synthesis of peptidyl α-ketoamides that can be fully executed on resin, and circumvents the need for building blocks with protected ketone function.19,20 The synthesized α-ketoamides, despite close resemblance of rhomboid substrate sequences and previously reported inhibitors, turned out to be inactive. This indicates that the structure located at the C-terminal side of the α-ketoamide is crucial for potent inhibition of rhomboids. It also underlines the difficulty of using specificity information from both primed and non-primed sites of rhomboid protease substrates, which may complicate the design of inhibitors based on substrate sequences. Since α-ketoamides show a broad range of biological activities against proteases and other targets, we expect that the presented solid support strategy will find application in the future synthesis of biologically active peptidyl ketoamide derivatives.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Kvido Strisovsky for providing ketoamides 5–7. We acknowledge funding from the German Research Foundation DFG (grant VE 502-4/1 to SHLV), the China Scholarship Council (PhD fellowship to JY), the FWO (grant G0E3617N), the Ministerium für Kultur und Wissenschaft des Landes Nordrhein-Westfalen, the Regierende Bürgermeister von Berlin–inkl. Wissenschaft und Forschung, and the Bundesministerium für Bildung und Forschung.Notes and references
- S. Urban, J. R. Lee and M. Freeman, Cell, 2001, 107, 173–182 CrossRef CAS
.
- E. V. Koonin, K. S. Makarova, I. B. Rogozin, L. Davidovic, M. C. Letellier and L. Pellegrini, Genome Biol., 2003, 4, R19 CrossRef
- M. K. Lemberg and M. Freeman, Genome Res., 2007, 17, 1634–1646 CrossRef CAS
- L. G. Stevenson, K. Strisovsky, K. M. Clemmer, S. Bhatt, M. Freeman and P. N. Rather, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 1003–1008 CrossRef CAS
- L. Fleig, N. Bergbold, P. Sahasrabudhe, B. Geiger, L. Kaltak and M. K. Lemberg, Mol. Cell, 2012, 47, 558–569 CrossRef CAS
- S. H. L. Verhelst, FEBS J., 2017, 284, 1489–1502 CrossRef CAS
- E. V. Wolf and S. H. Verhelst, Biochimie, 2016, 122, 38–47 CrossRef CAS
- K. R. Vinothkumar, K. Strisovsky, A. Andreeva, Y. Christova, S. Verhelst and M. Freeman, EMBO J., 2010, 29, 3797–3809 CrossRef CAS
- O. Vosyka, K. R. Vinothkumar, E. V. Wolf, A. J. Brouwer, R. M. Liskamp and S. H. L. Verhelst, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 2472–2477 CrossRef CAS
- O. A. Pierrat, K. Strisovsky, Y. Christova, J. Large, K. Ansell, N. Bouloc, E. Smiljanic and M. Freeman, ACS Chem. Biol., 2011, 6, 325–335 CrossRef CAS
- K. R. Vinothkumar, O. A. Pierrat, J. M. Large and M. Freeman, Structure, 2013, 21, 1051–1058 CrossRef CAS
- J. Yang, M. Barniol-Xicota, M. T. N. Nguyen, A. Ticha, K. Strisovsky and S. H. L. Verhelst, Bioorg. Med. Chem. Lett., 2018, 28, 1423–1427 CrossRef CAS
- Y. Xue, S. Chowdhury, X. Liu, Y. Akiyama, J. Ellman and Y. Ha, Biochemistry, 2012, 51, 3723–3731 CrossRef CAS
- Y. Xue and Y. Ha, J. Biol. Chem., 2012, 287, 3099–3107 CrossRef CAS
- A. Ticha, S. Stanchev, K. R. Vinothkumar, D. C. Mikles, P. Pachl, J. Began, J. Skerle, K. Svehlova, M. T. N. Nguyen, S. H. L. Verhelst, D. C. Johnson, D. A. Bachovchin, M. Lepsik, P. Majer and K. Strisovsky, Cell Chem. Biol., 2017, 24, 1523–1536 e1524 CrossRef CAS
- I. Schechter and A. Berger, Biochem. Biophys. Res. Commun., 1967, 27, 157–162 CrossRef CAS
- A. Arasappan, F. G. Njoroge, T. Y. Chan, F. Bennett, S. L. Bogen, K. Chen, H. Gu, L. Hong, E. Jao, Y. T. Liu, R. G. Lovey, T. Parekh, R. E. Pike, P. Pinto, B. Santhanam, S. Venkatraman, H. Vaccaro, H. Wang, X. Yang, Z. Zhu, B. McKittrick, A. K. Saksena, V. Girijavallabhan, J. Pichardo, N. Butkiewicz, R. Ingram, B. Malcolm, A. Prongay, N. Yao, B. Marten, V. Madison, S. Kemp, O. Levy, M. Lim-Wilby, S. Tamura and A. K. Ganguly, Bioorg. Med. Chem. Lett., 2005, 15, 4180–4184 CrossRef CAS
- Y. Liu, V. S. Stoll, P. L. Richardson, A. Saldivar, J. L. Klaus, A. Molla, W. Kohlbrenner and W. M. Kati, Arch. Biochem. Biophys., 2004, 421, 207–216 CrossRef CAS
- A. Papanikos and M. Meldal, J. Comb. Chem., 2004, 6, 181–195 CrossRef CAS
- F. Rohrbacher, A. Zwicky and J. W. Bode, Helv. Chim. Acta, 2018, 101 Search PubMed
- S. Zoll, S. Stanchev, J. Began, J. Skerle, M. Lepsik, L. Peclinovska, P. Majer and K. Strisovsky, EMBO J., 2014, 33, 2408–2421 CrossRef CAS
- S. Cho, S. W. Dickey and S. Urban, Mol. Cell, 2016, 61, 329–340 CrossRef CAS
- A. Basso, L. Banfi, R. Riva, P. Piaggio and G. Guanti, Tetrahedron Lett., 2003, 44, 2367–2370 CrossRef CAS
- G. K. Newton, T. R. Perrior, K. Jenkins, M. R. Major, R. E. Key, M. R. Stewart, S. Firth-Clark, S. M. Lloyd, J. Zhang, N. J. Francis-Newton, J. P. Richardson, J. Chen, P. Lai, D. R. Garrod and C. Robinson, J. Med. Chem., 2014, 57, 9447–9462 CrossRef CAS
- M. P. Patricelli, D. K. Giang, L. M. Stamp and J. J. Burbaum, Proteomics, 2001, 1, 1067–1071 CrossRef CAS
- E. V. Wolf, A. Zeissler and S. H. Verhelst, ACS Chem. Biol., 2015, 10, 2325–2333 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra10614c |
| This journal is © The Royal Society of Chemistry 2021 |