DOI:
10.1039/D0RA10418C
(Paper)
RSC Adv., 2021,
11, 7369-7380
Electrochemically dealloyed nanoporous Fe40Ni20Co20P15C5 metallic glass for efficient and stable electrocatalytic hydrogen and oxygen generation†
Received
11th December 2020
, Accepted 7th February 2021
First published on 12th February 2021
Abstract
The anion exchange membrane (AEM) in fuel cells requires new, stable, and improved electrocatalysts for large scale commercial production of hydrogen fuel for efficient energy conversion. Fe40Ni20Co20P15C5, a novel metallic glass ribbon, was prepared by arc melting and melt spinning method. The metallic glass was evaluated as an efficient electrocatalyst in water-splitting reactions, namely hydrogen evolution reaction under acidic and alkaline conditions. In addition, oxygen evolution reaction in alkaline medium was also evaluated. In 0.5 M H2SO4, the metallic glass ribbons, after electrochemical dealloying, needed an overpotential of 128 mV for hydrogen evolution reaction, while in 1 M KOH they needed an overpotential of 236 mV for hydrogen evolution. For the oxygen evolution reaction, the overpotential was 278 mV. The electrochemical dealloying procedure significantly reduced the overpotential, and the overpotential remained constant over 20 hours of test conditions under acidic and alkaline conditions. The improved electrocatalytic activity was explained based on the metastable nature of metallic glass and the synergistic effect of metal hydroxo species and phosphates. Based on the excellent properties and free-standing nature of these metallic glass ribbons in electrolyte medium, we propose the current metallic glass for commercial, industrial electrocatalytic applications.
1. Introduction
There is an increased demand in the present world to find a potential and highly efficient substitute for the traditional fossil fuels that emit a lot of CO2.1–3 Hydrogen fuel is a potential candidate because of its substantial mass-energy density and environmentally friendly nature, and it is a renewable fuel too.4 The generation of hydrogen (H2) through electrochemical water splitting is highly efficient since water is freely abundant in the earth and free of carbon.5 The two standard methods to produce hydrogen fuel are electrochemical and photochemical water splitting. Here we focus on the electrochemical water splitting as the conversion efficiency of the photochemical splitting method is moderately very low.6 Water splitting involves hydrogen evolution reaction (HER) at the cathode and oxygen evolution reaction (OER) at the anode. The OER is a rate-determining process in water splitting electrolyzer and fuel cells, and Li-ion batteries.7 For the past three decades, researchers are exploring suitable catalytic materials by modifying the defect sites and improving the nanostructure architecture toward HER in acidic medium (2H+ + 2e− → H2), alkaline (2H2O + 2e− → H2 + 2OH−) medium, and OER in alkaline medium (4H2O → 4H+ + O2 + 4e−).8 In water-splitting reaction, the main hurdle is the extra potential when compared with the equilibrium condition otherwise termed overpotential for HER reaction and OER reaction. At equilibrium conditions, the hydrogen evolution reaction does not require an additional potential for initiating the response. But due to the practical resistance offered from the electrolyte and electrode–electrolyte interface, or capacitive double layer, an excess potential or overpotential is to be applied.9 For OER reaction, a potential of 1.23 V is needed for initiating reaction even in equilibrium condition in addition to the overpotential.10 Hence, electrochemical water splitting requires the development of a suitable, stable bifunctional electrocatalyst, which can reduce the overpotential needed for HER or OER.11 Currently, the reported electrocatalyst for HER and OER are based on precious noble metals like Pt, Ir, Ru, etc.12–14 The high cost and scarcity of these noble elements seriously hinder the extensive scale application of these catalysts in fuel cells. Thus many researchers have attempted to develop an alternative noble metal-free electrocatalyst for HER and OER such as transition metal oxides, carbides, sulphides, phosphides, and their additives with carbon nanostructures.15–17 Also, several attempts to enhance the efficiency of HER and OER activity of less expensive crystalline catalysts by alloying, modified nanostructures, and introducing lattice defects and strains; thus, a variety of nanoscale catalysts have been developed.18,19 The first-row transition metals like Ni, Fe, Co have low crystal field activation energies and are more attractive for HER and OER when compared to the expensive benchmark noble metal-based Pt, Ir, IrO2, RuO2 electrocatalysts.19–21 However, these nanoscale electrocatalysts are mainly powder form and are not free-standing in the electrolyte solution. Therefore, they are pasted on carbon supports with the help of polymer binders to act as the working electrode, and the improper contact through polymer binding lowers the conductivity and catalytic activity. Hence, these nanoscale electrocatalysts are challenging to industrialize.22 Amorphous alloys or metallic glasses (MG's) have attracted researchers' significant interest as a structural material due to their superior mechanical properties, and recently excellent catalytic activities of these alloys were also reported.23–25 Metallic glasses (MG's) with good intrinsic electrical conductivity are also suggested to be better electrocatalyst due to their corrosive resistant properties, free-standing nature in electrolyte solutions, metastable nature leading to high catalytic activity and industrially feasible production.26,27 The limitations of noble metal-based electrocatalysts led researchers to try transition metals like Fe, Co, Ni-based metallic glass as an alternative. Attempts to develop the bifunctional electrocatalyst for HER and OER reactions are of current interest to the research community intending to realize better water-splitting electrocatalyst.
In the present work, the selected composition for metallic glass electrocatalyst is Fe40Ni20Co20P15C5. Even though many Fe–Co–Ni,28 Fe–Ni–P29 based metallic glasses were reported for electrocatalytic HER and OER reactions, the electrochemically dealloyed nanoporous amorphous surface structure for improved catalytic activity is not reported so far. For designing an electrocatalyst for water splitting reactions like HER and OER, thermoneutral elements need to be considered, i.e., elements with absolute Gibb's free energy (ΔGfree ∼ 0 eV) nearly zero at the same time with high exchange current density. When the free energy becomes high, the adsorption/desorption gets unbalanced, or in other words, when the adsorption is high, desorption will be low, and subsequently, HER reaction becomes sluggish.30 From the volcano plot, we may identify that Pt/Pd are close to the zero value of free energy and high exchange current density.31 Also, the transition metal elements like Fe, Co, and Ni are also closer next to Pt/Pd elements with optimum thermoneutral value for free energy and promising exchange current density. In the current metallic glass Fe40Ni20Co20P15C5, 40% is occupied by Fe since iron (Fe) is one of the most abundant transition metal on the earth and cheaper. Moreover, iron-phosphide (Fe–P) has an excellent activity for electrocatalytic hydrogen evolution reaction.32 Density functional theory (DFT) calculations show that the substitution of Co by Fe in Co–P has enhanced the electrocatalytic hydrogen adsorption free energy.33 Besides, Fe based MG's showed improved GFA and thermal stability.34 Amorphous Ni–P showed excellent HER catalytic performance in an alkaline medium with enhanced hydrophilicity.34 Even though chemically prepared FeCoNiP showed promising OER activity with an overpotential of 200 mV, there are many challenges associated with them, such as lack of stability and low electrical conductivity.35 The introduction of carbon (C) is reported to improve the stability of phosphide by preventing the oxidation of phosphorous (P) to high valance states in PdFePC MG ribbon catalyst.36 Inspired by the recent findings of electrocatalytically active metal phosphides like Fe–P,37,38 Co–P,39,40 and Ni–P40 and first-row transition element based metallic glasses, this typical composition Fe40Ni20Co20P15C5, is selected for the current research work. From the earlier reports, it is noted that the dealloying strategy leads to improved surface area through microstructural and morphological modification.41 Electrochemical dealloying of metallic glasses and the subsequent formation of a nanoporous structure with higher surface area is a more efficient and promising technique for improving the electrocatalyst performance in water splitting.42
2. Results and discussion
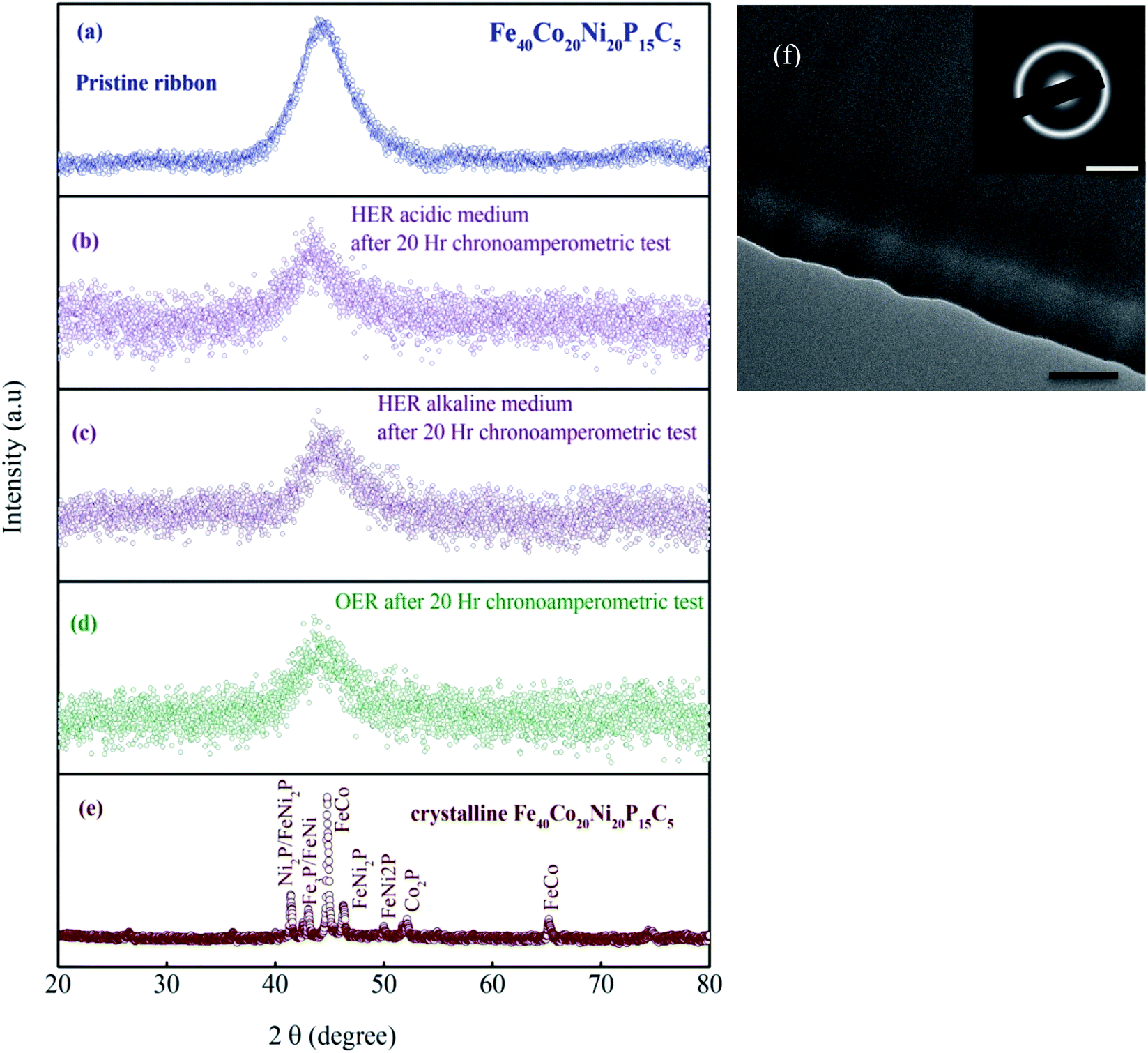
Fig. 1a and b shows the optical image of the as-prepared metallic glass Fe40Ni20Co20P15C5 electrocatalyst ribbon with approximately 2 mm wide and tens of centimeters long, and Fig. 1b shows that the MG in the form of ribbons can be cut or bent into different shapes for applications in electronic devices. Fig. 1c shows the evolution of H2/O2 in HER/OER reactions at the working MG electrode. The X-ray diffraction (XRD) spectra for the as-prepared ribbon, 20 hours tested samples for acidic HER, alkaline HER, alkaline OER, and crystalline Fe40Ni20Co20P15C5 is shown in Fig. 2a–e. From X-ray diffraction spectra, it may be noted that a broad halo is seen around 2 theta (θ) value of 44 degrees for the as-prepared MG ribbon as in Fig. 2a–d. The broad halo in the XRD spectrum is a characteristic feature of the amorphous state. Fig. 2f shows the TEM image of the current MG, and the inset shows the SAED diffraction pattern. The SAED pattern is a typical electron diffraction pattern of an amorphous structure with a diffused diffraction ring shape. The temperature-dependent heat flow measurements using differential scanning calorimeter (DSC) at a heating rate of 5 K min−1 (Fig. S1†) clearly showed amorphous glassy features like an endothermic heat flow peak around 620 K, and the glass transition temperature (Tg) is determined to be 620 K. The glass transition region is succeeded by a wide supercooled liquid region followed by a sharp exothermic peak of crystallization (Tx) at around 648 K. The MG ribbon was vacuum annealed at a temperature slightly above the glass transition temperature of 566 K for 60 minutes to convert the glassy metal to crystalline sample. The crystalline phase of the sample is confirmed from the XRD spectrum, as seen in Fig. 2e. The XRD spectrum of the crystalline sample showed sharp peaks, and different crystalline phases present in the sample were labeled. The electrocatalytic performance of the crystalline sample will also be compared with the MG electrocatalyst.
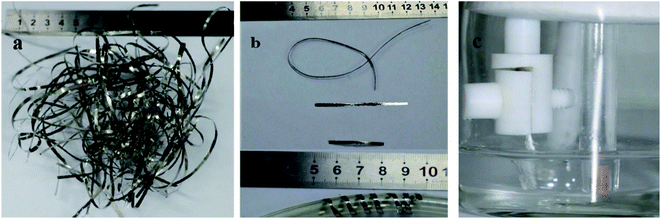 |
| | Fig. 1 (a) Optical image of the as-prepared MG ribbons, (b) ribbon sample twisted, cut, and wrapped around a tube, (c) evolution of H2/O2 gas bubbles at the MG working electrode toward HER/OER reactions. | |
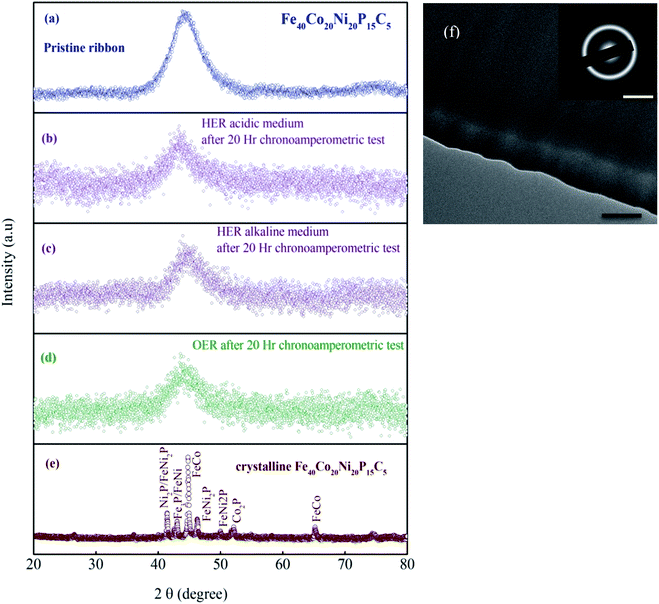 |
| | Fig. 2 XRD spectrum (a) of the MG ribbon in the as-prepared form. (b–d) after 20 h chronoamperometric tests. (e) The crystallized ribbon is composed mainly of crystalline phase FeCo, FeNi2P, Fe3P, Ni2P, and Co2P phases, which are confirmed from the PDF#65-4131, PDF#51-1367, PDF#19-0617, PDF#03-0953, PDF#89-3030, respectively. (f) TEM image of the as-prepared MG sample; scale bars: 100 nm and 5 nm−1 (inset). | |
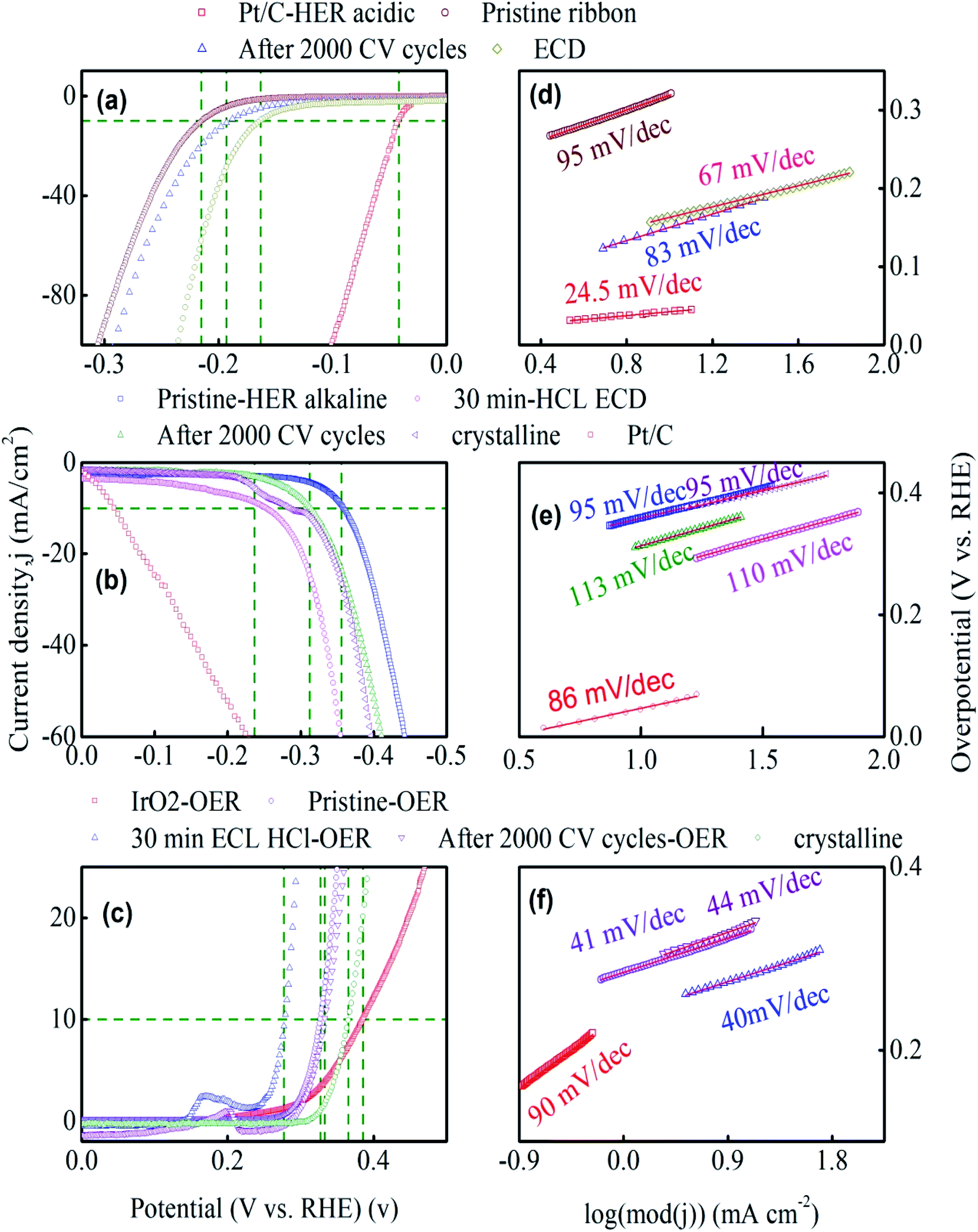
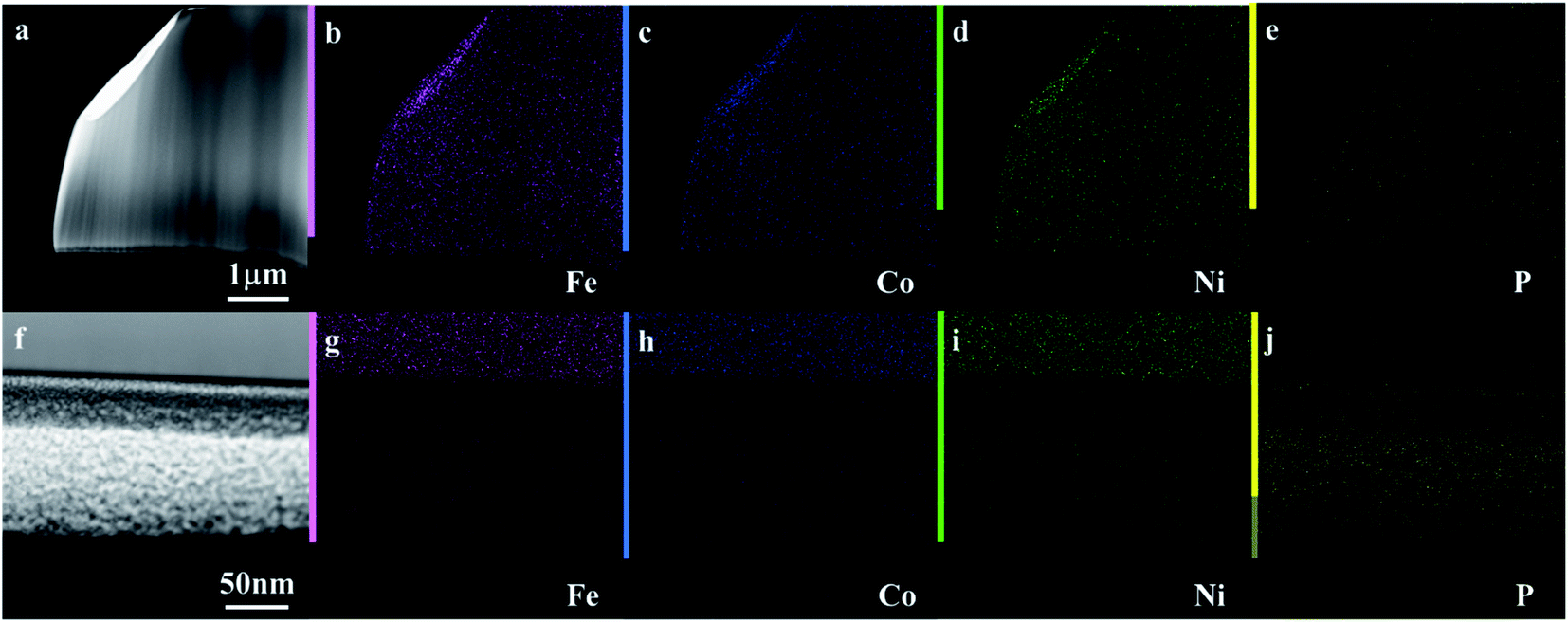
Fig. 3a–c shows the compensated linear sweep voltammetry (LSV) curves for HER in acid medium, HER in alkaline medium, and OER in alkaline medium. Fig. 3d–f gives the corresponding Tafel plot with log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |J| on the x-axis and overpotential on the y-axis. LSV polarization curves for HER is quantified by two terms, namely the overpotential and Tafel slope. The overpotential is denoted by η10 and is defined as the excess voltage needed to drive a current density of −10 mA cm−2 for HER and +10 mA cm−2 for OER. The onset potential is the starting potential at which current density is just above zero value. HER/OER activity parameters of the current MG electrocatalyst are summarized in Table 1. For HER in acidic medium, as-prepared or pristine MG ribbon has an overpotential 215 mV. After 2000 CV cycles, the overpotential is improved to 163 mV. The improved electrocatalytic activity after 2000 CV cycles in 0.5 M H2SO4 is believed to be due to the increased number of active sites. For confirming this argument, SEM image (Fig. 4a and b) is taken and clearly showed the porous type structure developed. Since the MG ribbon sample was found to be dealloyed and a porous structure developed while measuring HER in an acid medium (0.5 M H2SO4), the ribbon sample is electrochemically dealloyed in 1 M HCl for different durations by applying an optimized voltage of 0.2 V for better activity.43 The electrochemical dealloying in 1 M HCl is optimized to 30 minutes, and the overpotential is reduced to 128 mV. Fig. 4c and d shows the SEM image of the electrochemically dealloyed MG ribbon and shows the nanoporous type structure on the surface. To evaluate the percentage of elements etched after dealloying, EDX spectrum is used and is shown in Fig. 4e. It may be seen that Fe, Co, Ni has been etched from the surface, and an oxide layer is formed, and an amorphous structure is retained as confirmed from XRD spectra (inset of Fig. 4e). The remaining average atomic percentage is Fe20Co10Ni10P15C5O40. The overpotential for the current material (η10 = 129 mV) is comparable to that reported for Ni40Fe40P20 MG (η10 = 193 mV) for HER in an acidic medium.29 To study the detailed structural morphology at the surface of the electrochemically dealloyed sample, TEM image at the interface with high resolution is measured (Fig. 5a and b). The high-resolution TEM images down to 5 nm nanometer scale (Fig. 5b) show the dealloyed part on the MG surface is amorphous without any evidence of crystalline particles. The HAADF-STEM image-based mapping of the constituent elements in the as-prepared MG ribbon is shown in Fig. 6a–e. To better understand the leaching of elements from the MG surface, the STEM image-based mapping of electrochemically dealloyed sample is measured and is shown in Fig. 6f–j. The mapping of elements for the as-prepared MG sample shows the uniformly homogenous distribution of all constituent elements in the MG. On the other hand, the electrochemically dealloyed sample showed a lower concentration of Fe, Co, and Ni and higher P in the dealloyed layer. The elemental mapping for C is not shown here since C present in the column can overlap and show error in result.
|J| on the x-axis and overpotential on the y-axis. LSV polarization curves for HER is quantified by two terms, namely the overpotential and Tafel slope. The overpotential is denoted by η10 and is defined as the excess voltage needed to drive a current density of −10 mA cm−2 for HER and +10 mA cm−2 for OER. The onset potential is the starting potential at which current density is just above zero value. HER/OER activity parameters of the current MG electrocatalyst are summarized in Table 1. For HER in acidic medium, as-prepared or pristine MG ribbon has an overpotential 215 mV. After 2000 CV cycles, the overpotential is improved to 163 mV. The improved electrocatalytic activity after 2000 CV cycles in 0.5 M H2SO4 is believed to be due to the increased number of active sites. For confirming this argument, SEM image (Fig. 4a and b) is taken and clearly showed the porous type structure developed. Since the MG ribbon sample was found to be dealloyed and a porous structure developed while measuring HER in an acid medium (0.5 M H2SO4), the ribbon sample is electrochemically dealloyed in 1 M HCl for different durations by applying an optimized voltage of 0.2 V for better activity.43 The electrochemical dealloying in 1 M HCl is optimized to 30 minutes, and the overpotential is reduced to 128 mV. Fig. 4c and d shows the SEM image of the electrochemically dealloyed MG ribbon and shows the nanoporous type structure on the surface. To evaluate the percentage of elements etched after dealloying, EDX spectrum is used and is shown in Fig. 4e. It may be seen that Fe, Co, Ni has been etched from the surface, and an oxide layer is formed, and an amorphous structure is retained as confirmed from XRD spectra (inset of Fig. 4e). The remaining average atomic percentage is Fe20Co10Ni10P15C5O40. The overpotential for the current material (η10 = 129 mV) is comparable to that reported for Ni40Fe40P20 MG (η10 = 193 mV) for HER in an acidic medium.29 To study the detailed structural morphology at the surface of the electrochemically dealloyed sample, TEM image at the interface with high resolution is measured (Fig. 5a and b). The high-resolution TEM images down to 5 nm nanometer scale (Fig. 5b) show the dealloyed part on the MG surface is amorphous without any evidence of crystalline particles. The HAADF-STEM image-based mapping of the constituent elements in the as-prepared MG ribbon is shown in Fig. 6a–e. To better understand the leaching of elements from the MG surface, the STEM image-based mapping of electrochemically dealloyed sample is measured and is shown in Fig. 6f–j. The mapping of elements for the as-prepared MG sample shows the uniformly homogenous distribution of all constituent elements in the MG. On the other hand, the electrochemically dealloyed sample showed a lower concentration of Fe, Co, and Ni and higher P in the dealloyed layer. The elemental mapping for C is not shown here since C present in the column can overlap and show error in result.
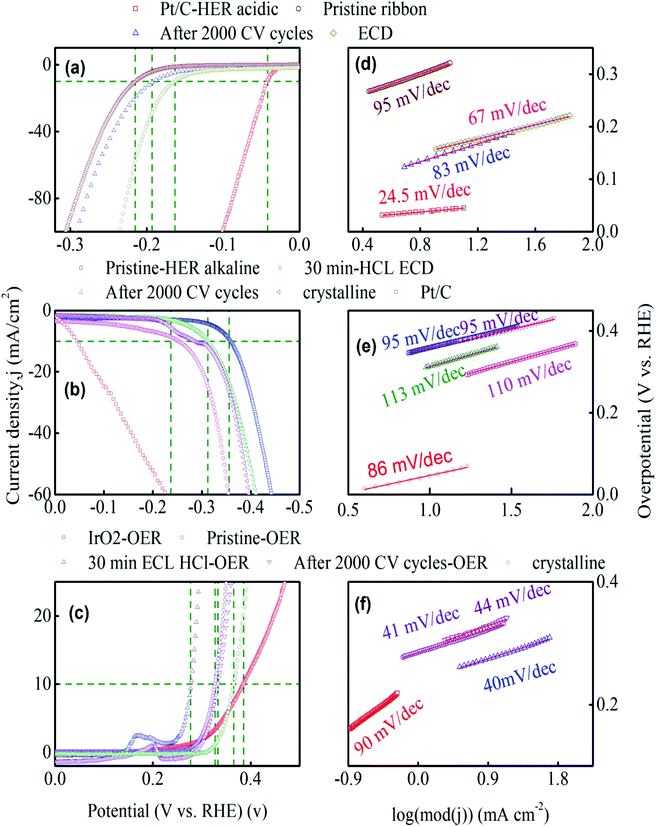 |
| | Fig. 3 HER and OER performance of Fe40Co20Ni20P15C5 MG. (a–c) Linear polarization curves of pristine glassy, after 2000 CV, electrochemically dealloyed and crystalline samples. (d–f) Tafel plots of their corresponding polarization curves. | |
Table 1 HER performance of Fe40Ni20Co20P15C5; overpotential (η10), Tafel slope, charge transfer resistance (Rct)
| Sample |
Overpotential, η (mV) |
Tafel slope mV dec−1 |
Charge transfer resistance, Rct (Ω) |
| Acidic |
HER pristine |
215 |
95 |
34.5 |
| HER after 2000 CV cycles |
163 |
83 |
31.3 |
| HER-ECD |
128 |
67 |
3.5 |
| Alkaline |
HER pristine |
355 |
95 |
83.1 |
| HER after 2000 CV cycles |
312 |
113 |
61.5 |
| HER-ECD |
236 |
110 |
28.7 |
| Alkaline |
OER pristine |
333 |
41 |
36.3 |
| OER after 2000 CV |
327 |
44 |
15.3 |
| OER-ECD |
278 |
40 |
8.4 |
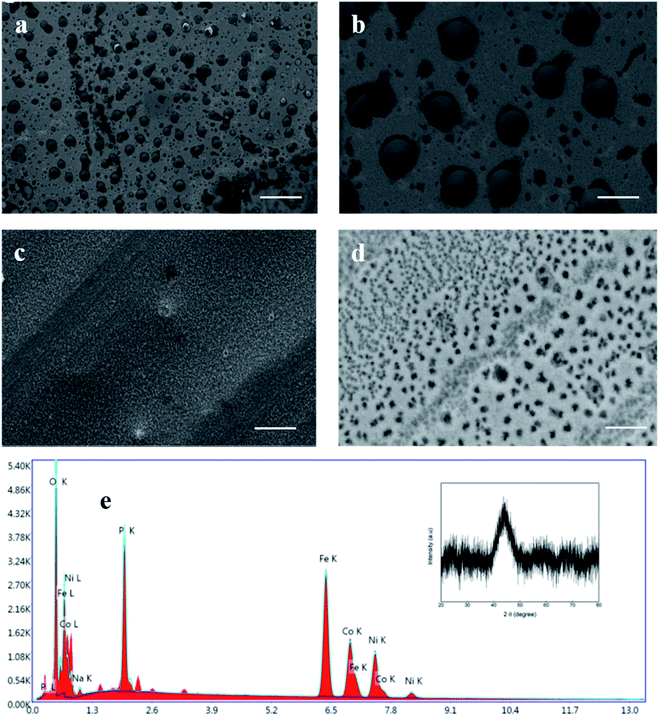 |
| | Fig. 4 (a and b) SEM image of electrochemically dealloyed MG ribbon in 0.5 M H2SO4 (c and d) MG ribbon electrochemically dealloyed in 1 M HCl. (e) EDX point-shoot spectrum for the porous structure after dealloying in 1 M HCl. Inset shows XRD spectra for electrochemically dealloyed in 1 M HCl; scale bars: (a) 40 μm, (b) 10 μm, (c) 5 μm, and (d) 1 μm. | |
 |
| | Fig. 5 TEM image of electrochemically dealloyed MG ribbon in 1 M HCl; scale bars: (a) 20 nm and (b) 5 nm. | |
 |
| | Fig. 6 (a–e) HAADF-STEM image and elemental mapping of Fe40Ni20Co20P15C5. (f–j) MG ribbon electrochemically dealloyed in 1 M HCl; (b and g) Fe, (c and h) Co, (d and i) Ni, and (e and j) P. | |
The onset potential for the as-prepared ribbon is 163 mV at a current density of 1.37 mA cm−2, and that for the dealloyed ribbon sample onset value is 115 mV. The overpotential for MG's and reported high-performance catalysts toward HER in the acid medium are given in Table S1.† The overpotential for standard Pt/C electrocatalyst is also measured for comparison and has an overpotential, η10 of 42 mV. Further innate features of the electrocatalyst for HER and OER are given by the Tafel slope, and the Tafel slope is an indicator to the reaction pathways. Tafel slope is obtained as the slope of the linear fit to the Tafel equation(η = a + blogj). The Tafel slope (Fig. 3d) for the as dealloyed MG ribbon toward HER in acidic medium is evaluated to be 67 mV dec−1, and it indicates that the HER reaction proceeds via Volmer–Heyrovsky mechanism (Volmer step: H+ + e− → Hads; Tafel slope: 120 mV dec−1 and Heyrovsky step: H+ + Hads + e− → H2(g); Tafel slope: 40 mV dec−1) with desorption as the rate-limiting step.36,44 The Tafel slope for Pt catalyst is measured to be 24.5 mV dec−1. Tafel slope for the current MG and reported electrocatalysts toward HER in the acid medium from other similar reports are given in Table S1.† Tan et al.,29 reported the bifunctional electrocatalyst Ni40Fe40P20 for both HER and OER in water splitting. They reported a working overpotential of 193 mV, exchange current density 0.024 mV cm−2, Tafel slope 65 mV dec−1 for HER in acidic medium, an overpotential of 270 mV for HER in alkaline medium, and OER overpotential 288 mV. Zhang et al.,28 reported Fe40Co40P13C7 to be an active catalyst for HER in an acidic medium and reported the overpotential to be 118 mV at a current density of 10 mA cm−2. Hu et al.,45 reported Ni40Fe40P20 for OER electrocatalyst and obtained an overpotential 219 mV with a Tafel slope 32 mV dec−1. Zhu et al.,46 reported the electrocatalytic hydrogen evolution catalyst of Ni61Zr36Mo3 metallic glass with better catalytic efficiency (over potential ∼71 mV at a current density of −20 mA cm−2, Tafel slope ∼57 mV dec−1) for HER in alkaline medium and the high catalytic activity is achieved by creating oxygen vacancies through chemical dealloying in HF acid. Xu et al.,43 reported a nanoporous phosphide bifunctional electrocatalyst for water splitting developed by dealloying NiFeP metallic glass ribbon and reported overpotentials of 245 mV and 120 mV at the current density of 10 mA cm−2 for OER and HER in 1 M KOH, respectively. The inherent activities of the electrocatalysts were further determined by exchange current density (jo) and are obtained by extrapolating the linear fit of the Tafel plot toward the current density axis. The exchange current density (jo) for as-prepared ribbon is 0.005 mA cm−2, and after 2000 CV cycles, jo is improved to 0.1 mA cm−2. For electrochemically dealloyed MG catalyst, an exchange current density of 0.1 mA cm−2 is observed. The standard Pt catalyst has an exchange current density of 0.15 mA cm−2. The largeness of the exchange current density value indicates high current towards electrocatalysis.47 For the HER reaction in alkaline medium, 1 M KOH, the pristine ribbons sample need an overpotential, η10 equal to 355 mV to increase the current density by 10 mA cm−2. After 2000 CV cycles, the overpotential is lowered to 312 mV. It is worthy to note that dealloying electrochemically in 1 M HCl for 30 minutes lowered the overpotential value to from 355 mV 235 mV for HER in alkaline medium. A reduction of 120 mV is a result of porous structure developed and a subsequent increase in active sites during the electrochemical dealloying procedure. The crystalline form of the Fe40Ni20Co20P15C5 sample showed an overpotential value 330 mV higher than pristine ribbon overpotential toward HER in alkaline medium. HER activity of standard Pt/C electrocatalyst is measured in alkaline medium and its overpotential, η10 is 44 mV. From the Tafel plots (Fig. 3e) for HER toward alkaline medium, the pristine MG ribbon has a Tafel slope 95 mV dec−1 while the electrochemically dealloyed sample shows a Tafel slope of 113 mV dec−1 and is comparable to Tafel slope for standard Pt catalyst which is 120 mV with Volmer or Heyrovsky step as the rate determining step (Volmer step: H2O + e− → Hads + OH−; Tafel slope: 120 mV dec−1, Heyrovsky step H2O + e− + Hads → H2(g) + OH−; Tafel slope: 40 mV dec−1).48 For the standard Pt/C, the measured Tafel slope is 86 mV dec−1 toward HER in the alkaline medium. For HER in an alkaline medium, the exchange current density of the crystalline sample is 0.0027 mA cm−2, while the electrochemically dealloyed sample showed an improved exchange current density value of 0.042 mA cm−2. The overpotential and Tafel slope values towards HER in the alkaline medium for the reported catalysts are given in Table S2.† Linear polarization curves for OER are given in Fig. 3c. The as-prepared pristine ribbon sample showed an overpotential 333 mV for OER. It is noted that the electrochemically dealloyed MG electrocatalyst showed a low and a better overpotential of 277 mV which is even better than the benchmark IrO2 electrocatalyst which has an overpotential of 320 mV.49 The overpotential (η10) measured for IrO2 toward OER in the current work is 380 mV. The Tafel slope of the as-prepared ribbon MG is 41 mV dec−1 (Fig. 3f), while the electrochemically dealloyed sample shows a Tafel slope of 40 mV dec−1. It indicates that the OER step: MOH + OH− → MO + H2O + e−,50–52 (M = Ni, Fe, or Co; Tafel slope: 40 mV dec−1) is the rate-determining step. It is noted that the IrO2 sample in the present work has a Tafel slope 90 mV dec−1, whose rate determining step will be the OER step: M + OH− → MOH + e− (Tafel slope: 120 mV dec−1). The overpotentials and Tafel slopes for the similar reported OER electrocatalysts reported are given in Table S3.† The cyclic voltammograms (CVs) in the non-faradaic region at different scan rates (200 mV s−1–10 mV s−1) for HER acidic, HER alkaline and OER alkaline medium (Fig. S2a–c†) is analyzed for double layer capacitance (Cdl). The slope of linear fit to the plot of current density differences (Δj/2, Δj = Ja − Jc) vs. scan rate (Fig. S2d–f†) gives the Cdl value. The double layer capacitance for the electrochemically dealloyed MG electrocatalyst towards HER acidic, HER alkaline, and OER alkaline is 8.63 mF cm−2, 33 μF cm−2 and 550 μF cm−2, respectively. The electrochemical double layer capacitance (Cdl) thus determined implies that the current MG electrocatalyst affords a large electrochemically active surface area (ECSA) compared to amorphous nanosheets.53 The difference in double layer capacitance (Cdl) in different electrolyte arises due to different cell potential applied while measuring CV for HER in acidic and alkaline medium.
The stability of the electrocatalyst toward HER and OER is a critical area of attention. To evaluate the stability of the MG ribbon electrocatalyst, a chronoamperometric test is performed at constant overpotential for 20 hours, and the chronoamperometric spectrum is shown in Fig. 7a–c. LSV curves before and after chronoamperometric tests in HER acidic, HER alkaline, and OER alkaline is given in Fig. 7d–f. Linear polarization curves are measured before and after the chronoamperometric test to check whether the catalytic efficiency has been affected after 20 h HER and OER tests.
 |
| | Fig. 7 Chronoamperometric durability tests for 20 h (a) HER @ 0.5 M H2SO4 at overpotential 214 mV, (b) HER @ 1 M KOH at overpotential 319 mV, (c) OER @ 1 M KOH at overpotential 330 mV. (d–f) LSV curves before and after chronoamperometric tests. | |
The chronoamperometric test data shows that the MG electrocatalyst maintained a constant current density of 10 mA cm−2 for 20 h. This shows the superior stability of the current MG catalyst in the both acidic and alkaline medium for HER and OER experiments. The LSV curves show unchanged overpotential values before and after chronoamperometric tests indicating excellent electrocatalytic stability for a long period. In the case of HER in the alkaline medium, the present MG decreased overpotential value from 360 mV to 320 mV after the chronoamperometry test. Thus the superior durability of the current electrocatalyst is confirmed from the unchanged catalytic parameters after 20 hours chronoamperometric test and after 2000 CV cycles. Therefore, it is to be ascertained that the structure, mechanical stability, and catalytic performance were unaffected even for 20 hours prolonged HER and OER chronoamperometric tests.
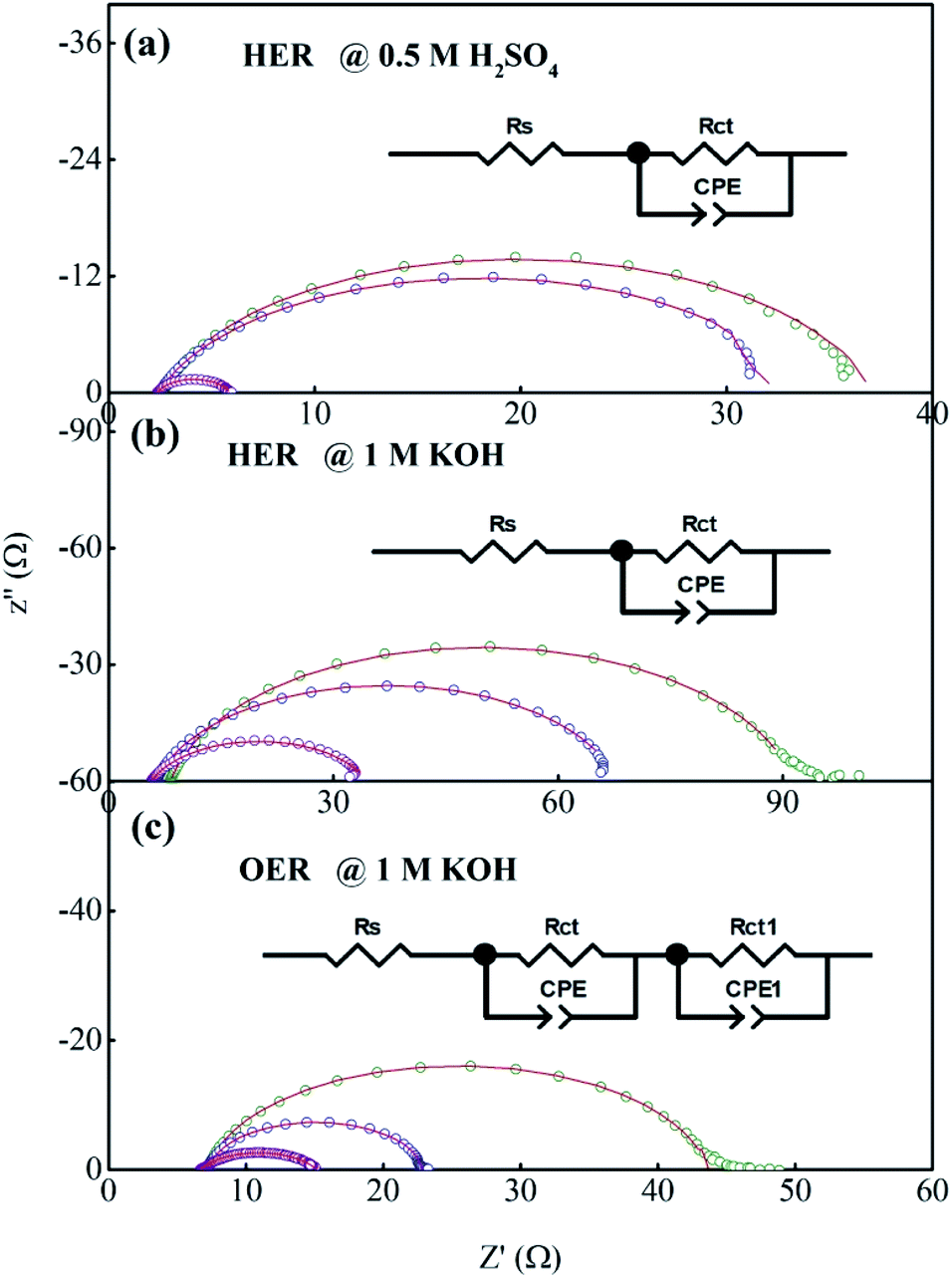
Fig. 8a–c shows the electrochemical impedance spectra (EIS) for the MG electrocatalyst toward HER acidic, HER alkaline, and OER alkaline. The impedance Cole–Cole plot exhibited different depressed semicircles corresponding to impedance relaxation from the as-prepared ribbon sample, after 2000 CV tested samples, and electrochemically dealloyed (ECD) catalysts. In an EIS spectrum, the relaxation arises due to mass transfer relaxation, which is seen as a semicircle in the intermediate frequency region, and charge transfer relaxation, which is seen as a semicircle in the high frequency region separated by different time constants.54 The Cole–Cole plot for the current material shows a depressed semicircle corresponding to the charge transfer relaxation. The charge transfer resistance (Rct) is given by the high frequency intercept of the Cole–Cole plot and the real impedance (Z′) axis.55 Rct value is deduced by fitting the impedance Cole–Cole plot using a series resistance, Rs with a parallel combination of Rct and a theoretical constant phase element (CPE), i.e., Rct‖CPE.56 The CPE is associated with the surface heterogeneities, and its impedance is given by 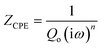 where Qo is a pseudo-capacitance arising from the double layer for the adsorbed hydrogen or adsorbed oxygen (Hads or Oads), ω is the angular frequency (ω = 2πf, f is the linear frequency),
where Qo is a pseudo-capacitance arising from the double layer for the adsorbed hydrogen or adsorbed oxygen (Hads or Oads), ω is the angular frequency (ω = 2πf, f is the linear frequency),  , and ‘n’ is an empirical constant between 0 and 1. CPE is a pure capacitor when n = 1 and is a pure resistor when n = 0.57 It may be noted that the faster kinetics toward HER and OER is given by the low value of charge transfer resistance as well as the diameter of the semicircle in the impedance Cole–Cole plot. The charge transfer resistance of the as-prepared MG ribbon toward HER acidic reaction is 34.5 Ω. After electrochemical dealloying, the Rct value is reduced to 3.5 Ω, indicating the faster HER kinetics as confirmed from linear polarization data. For the HER alkaline reaction, the as-prepared ribbon has a charge transfer resistance of 83 Ω while the electrochemically dealloyed sample has a reduced value of 28.7 Ω. For the OER reaction, the pristine sample has an Rct value of 36.3 Ω, while the electrochemically dealloyed sample has an Rct value of 8.4 Ω. The reduced value of charge transfer resistance emphasizes the increased catalytic activity in dealloyed samples and after 2000 CV cycle tested samples.58
, and ‘n’ is an empirical constant between 0 and 1. CPE is a pure capacitor when n = 1 and is a pure resistor when n = 0.57 It may be noted that the faster kinetics toward HER and OER is given by the low value of charge transfer resistance as well as the diameter of the semicircle in the impedance Cole–Cole plot. The charge transfer resistance of the as-prepared MG ribbon toward HER acidic reaction is 34.5 Ω. After electrochemical dealloying, the Rct value is reduced to 3.5 Ω, indicating the faster HER kinetics as confirmed from linear polarization data. For the HER alkaline reaction, the as-prepared ribbon has a charge transfer resistance of 83 Ω while the electrochemically dealloyed sample has a reduced value of 28.7 Ω. For the OER reaction, the pristine sample has an Rct value of 36.3 Ω, while the electrochemically dealloyed sample has an Rct value of 8.4 Ω. The reduced value of charge transfer resistance emphasizes the increased catalytic activity in dealloyed samples and after 2000 CV cycle tested samples.58
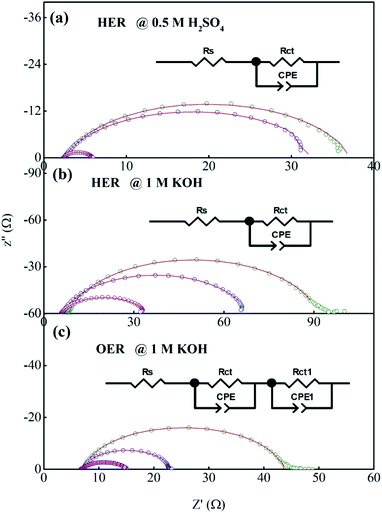 |
| | Fig. 8 (a–c) Impedance Cole–Cole plots for the Fe40Co20Ni20P15C5 MG during HER in acidic medium and alkaline medium, and OER in alkaline medium. | |
The chemical states of the constituent elements present in the as-prepared MG catalyst and, after 20 hours, chronoamperometric tests were examined by high-resolution X-ray photoelectron spectroscopy (XPS). The deconvoluted Fe XPS spectra for as-prepared, 20 h tested samples are shown for the Fig. 9a–d. The deconvoluted Fe 2p XPS spectra for the as-prepared sample shows two main peaks at 710.4 eV and 723.5 eV, two satellite peaks at 713.3 eV and 716.5 eV corresponding to Fe 2P3/2. For Fe 2P1/2, two main peaks were observed at 719.6 eV and 723.5 eV, two satellite peaks at 727.4 eV, and 732.8 eV with Fe metal peak at 706.8 eV. The peak from elemental Fe is not present after the 20 h chronoamperometry test toward OER (Fig. 9b). Also, satellite peaks corresponding to Fe 2P3/2 at 716.5 eV and 727.4 eV in the as-prepared sample XPS spectrum are absent for OER 20 h tested MG sample. It may also be noted that the main peaks corresponding to Fe 2P1/2 at 710.4 eV and 723.5 eV in the as-prepared sample shift to higher binding energies 711.4 eV and 724.9 eV after 20 hours OER chronoamperometric tested samples. These new peak positions may be assigned to iron hydroxo species.59 The XPS spectrum for Co 2p for the as-prepared ribbon (Fig. 9e) showed a Co metal peak corresponding at binding energy position 777.9 eV. Co 2P3/2 main peak is observed at peak position 780.9 eV and two satellite peaks at 785.9 eV and 792.4 eV. Co 2P1/2 showed two main peaks at 796.9 eV and 802 eV for the as-prepared tested samples. After 20 h OER chronoamperometric test, XPS spectra for Co 2p (Fig. 9f) shows the following notable features like the disappearance of the metal peak corresponding to Co, the disappearance of Co 2P3/2 satellite peak originally found at 792.4 eV, and a satellite peak of Co 2P1/2 at 807.4 eV in the as-prepared MG electrocatalyst.
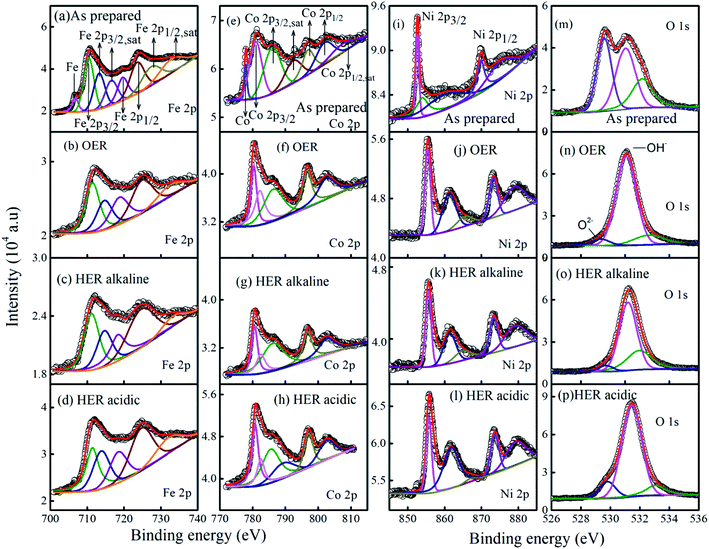 |
| | Fig. 9 XPS spectra for as prepared and chronoamperometric tested MG samples. (a–d) Fe 2p, (e–h) Co 2p, (i–l) Ni 2p, and (m–p) O 1s. | |
After the 20 h OER test, the main peak of Co 2P3/2 showed multiple splitting feature with a new peak formed at 782.2 eV. The multiple splitting features signify the oxide formation in the surface, enhancing the OER activity.60,61 Fig. 9j–l shows the XPS spectra for Ni 2p after 20 h chronoamperometric tests for OER alkaline, HER alkaline, and HER acidic reactions. In Ni 2p XPS spectra, the shifting of main peaks (Ni 2P3/2 and Ni 2P1/2) to slightly higher binding energies and the satellite peaks become sharper. The new shifted main peaks for Ni 2P3/2 at 855.6 eV, and 873.2 eV may be assigned to nickel hydroxo species.62,63 Fig. 9m–p shows the O 1s spectra of as-prepared and chronoamperometric tested MG samples. For the as-prepared MG sample, O 1s showed a main peak at 529.4 eV corresponding to surface oxides present. Another major peak at 531.2 eV corresponds to oxygen from OH− ions. It is similar to H2PO4− species observed at 531.7 eV as in phosphorous doped NiFe2O4.64 Another peak at 533 eV arises from phosphate species (PO3−). In the 20 h tested samples toward OER and HER, the peak at 529.4 eV, corresponding to surface oxides, is seen as a minor peak while the hydroxyl ion peak at 531.2 eV becomes the dominant contribution (Fig. 9n–p). Thus it is understood that there is a reassembly of the metallic glass Fe40Ni20Co20P15C5 surface during electrocatalysis for HER and OER. Further checking the P 2p spectrum (Fig. S3a–d†), it is observed that a peak corresponding to phosphate species is present at position 133.2 eV in addition to phosphide components P 2P3/2 at 129.2 eV and a shoulder P 2P1/2 at 130.1 eV. After 20 h HER test, the phosphate peak becomes dominant and other phosphide components significantly reduces. After 20 h chronoamperometric tests for HER and OER in alkaline medium the phosphide peaks are absent while the phosphate peaks remains as a dominant contribution. Here it is to be noted that the enhanced HER/OER activity arises due to transfer of electrons from metal atoms to P atoms and the P atoms are stabilized by C atoms.65 Electronic structure modification due to P on FeCoNi is not true. It was observed in nickel phosphates (Ni2P), electron transfer is from nickel to phosphorous, and in nickel borides (Ni2B), electron transfer is from boron to nickel, confirming electronic structure modification is not sufficient to explain the enhanced OER activity in phosphorus-containing catalysts.66 Moreover, it was found that the geometric changes induced by B and P on the lattice structure of Ni, notably, the Ni–Ni bond distances, as well as the Ni–P and Ni–B bond distances and is found to be the crucial factors for enhanced OER activity in Ni2B and Ni2P electrocatalysts. Similarly, in the current metallic glass electrocatalyst Fe40Ni20Co20P15C5 phosphorous is believed to play an important role in enhancing the electrocatalytic activity, and the synergistic effect from the nickel–iron cobalt oxo/hydroxo species and phosphate species contribute towards the improved catalytic adsorption/desorption in HER and OER.
3. Conclusions
In summary, a new class of Fe40Ni20Co20P15C5 metallic glass ribbons is prepared through the conventional industrial melt spinning technique. The amorphous ribbon can directly act as electrodes for electrochemical water splitting without using any polymer binders. The free-standing nature of these glassy ribbons in electrolyte solutions and its improved catalytic properties with stability and less cost of production than standard noble metal-based electrocatalysts are the desirable features of the current electrocatalyst. The amorphous MG electrocatalyst gives an OER activity very close to the standard catalyst. Facile electrochemical dealloying improved the electrocatalytic HER and OER activity significantly. The improved and stable values of electrocatalytic activity of the multicomponent Fe40Ni20Co20P15C5 arises from the metastable state, the synergistic effect of metal hydroxo species, and phosphate species. Hence the current metallic glass is an attractive candidate in electrochemical water splitting, and this glassy alloy has potential for industrial applications. This research suggests an efficient approach to identifying new electrocatalysts and designing low-cost, high-performance electrocatalysts for electrochemical water splitting based on metallic glasses.
Funding sources
This work is supported by China National Natural Science Foundation (No. 52071217) and China National Key Research and Development Program (No. 2018YFA0703605).
Conflicts of interest
The authors declare no competing financial interest.
References
- J. W. F. To, J. W. D. Ng, S. Siahrostami, A. L. Koh, Y. Lee, Z. Chen, K. D. Fong, S. Chen, J. He, W.-G. Bae, J. Wilcox, H. Y. Jeong, K. Kim, F. Studt, J. K. Nørskov, T. F. Jaramillo and Z. Bao, Nano Res., 2017, 10, 1163–1177 CrossRef CAS
 .
. - Y. Zheng, Y. Jiao, Y. Zhu, L. H. Li, Y. Han, Y. Chen, A. Du, M. Jaroniec and S. Z. Qiao, Nat. Commun., 2014, 5, 3783 CrossRef .
- R. C. Sekol, G. Kumar, M. Carmo, F. Gittleson, N. Hardesty-Dyck, S. Mukherjee, J. Schroers and A. D. Taylor, Small, 2013, 9, 2081–2085 CrossRef CAS .
- H. T. Hwang and A. Varma, Curr. Opin. Chem. Eng., 2014, 5, 42–48 CrossRef .
- I. Roger, M. A. Shipman and M. D. Symes, Nat. Rev. Chem., 2017, 1, 3 CrossRef CAS .
- T. J. Jacobsson, Energy Environ. Sci., 2018, 11, 1977–1979 RSC .
- F.-T. Tsai, Y.-T. Deng, C.-W. Pao, J.-L. Chen, J.-F. Lee, K.-T. Lai and W.-F. Liaw, J. Mater. Chem. A, 2020, 8, 9939–9950 RSC .
- S. Y. Tee, K. Y. Win, W. S. Teo, L.-D. Koh, S. Liu, C. P. Teng and M.-Y. Han, Adv. Sci., 2017, 4, 1600337 CrossRef .
- N. Mahmood, Y. Yao, J.-W. Zhang, L. Pan, X. Zhang and J.-J. Zou, Adv. Sci., 2018, 5, 1700464 CrossRef .
- M. Gong and H. Dai, Nano Res., 2015, 8, 23–39 CrossRef CAS .
- H. Qian, K. Li, X. Mu, J. Zou, S. Xie, X. Xiong and X. Zeng, Int. J. Hydrogen Energy, 2020, 45, 16447–16457 CrossRef CAS .
- W. T. Hong, M. Risch, K. A. Stoerzinger, A. Grimaud, J. Suntivich and Y. Shao-Horn, Energy Environ. Sci., 2015, 8, 1404–1427 RSC .
- Y. Jiao, Y. Zheng, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2015, 44, 2060–2086 RSC .
- L. Yang, W. Zhou, J. Lu, D. Hou, Y. Ke, G. Li, Z. Tang, X. Kang and S. Chen, Nano Energy, 2016, 22, 490–498 CrossRef CAS .
- A. Eftekhari, Int. J. Hydrogen Energy, 2017, 42, 11053–11077 CrossRef CAS .
- S. Anantharaj, S. R. Ede, K. Sakthikumar, K. Karthick, S. Mishra and S. Kundu, ACS Catal., 2016, 6, 8069–8097 CrossRef CAS .
- F. Lu, M. Zhou, Y. Zhou and X. Zeng, Small, 2017, 13, 1701931 CrossRef .
- H. Wang, S. Xu, C. Tsai, Y. Li, C. Liu, J. Zhao, Y. Liu, H. Yuan, F. Abild-Pedersen, F. B. Prinz, J. K. Nørskov and Y. Cui, Science, 2016, 354, 1031–1036 CrossRef CAS .
- Y.-J. Lee, Y.-S. Lee, J. Y. Cha, Y. S. Jo, H. Jeong, H. Sohn, C. W. Yoon, Y. Kim, K.-B. Kim and S. W. Nam, Int. J. Hydrogen Energy, 2020, 45, 19181–19191 CrossRef CAS .
- A. Li, Y. Sun, T. Yao and H. Han, Chem.–Eur. J., 2018, 24, 18334–18355 CrossRef CAS .
- L. Han, S. Dong and E. Wang, Adv. Mater., 2016, 28, 9266–9291 CrossRef CAS .
- M. W. Glasscott, A. D. Pendergast, S. Goines, A. R. Bishop, A. T. Hoang, C. Renault and J. E. Dick, Nat. Commun., 2019, 10, 2650 CrossRef .
- Y.-C. Hu, C. Sun and C. Sun, ChemCatChem, 2019, 11, 2401–2414 CrossRef CAS .
- Z. Li, J. Shen, J. Q. Wang, D. Wang, Y. Huang and J. Zou, CrystEngComm, 2012, 14, 1874–1880 RSC .
- Z.-J. Wang, M.-X. Li, J.-H. Yu, X.-B. Ge, Y.-H. Liu and W.-H. Wang, Adv. Mater., 2020, 32, 1906384 CrossRef CAS .
- M. Carmo, R. C. Sekol, S. Ding, G. Kumar, J. Schroers and A. D. Taylor, ACS Nano, 2011, 5, 2979–2983 CrossRef CAS .
- W. E. Brower, M. S. Matyjaszczyk, T. L. Pettit and G. V Smith, Nature, 1983, 301, 497–499 CrossRef CAS .
- F. Zhang, J. Wu, W. Jiang, Q. Hu and B. Zhang, ACS Appl. Mater. Interfaces, 2017, 9, 31340–31344 CrossRef CAS .
- Y. Tan, F. Zhu, H. Wang, Y. Tian, A. Hirata, T. Fujita and M. Chen, Adv. Mater. Interfaces, 2017, 4, 1601086 CrossRef .
- S. Sarkar and S. C. Peter, Inorg. Chem. Front., 2018, 5, 2060–2080 RSC .
- M. Zeng and Y. Li, J. Mater. Chem. A, 2015, 3, 14942–14962 RSC .
- Y. Tan, H. Wang, P. Liu, C. Cheng, F. Zhu, A. Hirata and M. Chen, Adv. Mater., 2016, 28, 2951–2955 CrossRef CAS .
- Y. Lin, K. Sun, X. Chen, C. Chen, Y. Pan, X. Li and J. Zhang, J. Energy Chem., 2021, 55, 92–101 CrossRef .
- H. X. Li, Z. C. Lu, S. L. Wang, Y. Wu and Z. P. Lu, Prog. Mater. Sci., 2019, 103, 235–318 CrossRef CAS .
- J. Lian, Y. Wu, H. Zhang, S. Gu, Z. Zeng and X. Ye, Int. J. Hydrogen Energy, 2018, 43, 12929–12938 CrossRef CAS .
- R. Jiang, Z. Cui, W. Xu, S. Zhu, Y. Liang, Z. Li, S. Wu, C. Chang and A. Inoue, Electrochim. Acta, 2019, 328, 135082 CrossRef CAS .
- D. Y. Chung, S. W. Jun, G. Yoon, H. Kim, J. M. Yoo, K.-S. Lee, T. Kim, H. Shin, A. K. Sinha, S. G. Kwon, K. Kang, T. Hyeon and Y.-E. Sung, J. Am. Chem. Soc., 2017, 139, 6669–6674 CrossRef CAS .
- P. Jiang, Q. Liu, Y. Liang, J. Tian, A. M. Asiri and X. Sun, Angew. Chem., Int. Ed., 2014, 53, 12855–12859 CrossRef CAS .
- C. Tang, R. Zhang, W. Lu, L. He, X. Jiang, A. M. Asiri and X. Sun, Adv. Mater., 2017, 29, 1602441 CrossRef .
- Q. Liu, J. Tian, W. Cui, P. Jiang, N. Cheng, A. M. Asiri and X. Sun, Angew. Chem., Int. Ed., 2014, 53, 6710–6714 CrossRef CAS .
- S.-X. Liang, W. Zhang, W. Wang, G. Jia, W. Yang and L.-C. Zhang, J. Phys. Chem. Solids, 2019, 132, 89–98 CrossRef CAS .
- J. Li, G. Doubek, L. McMillon-Brown and A. D. Taylor, Adv. Mater., 2019, 31, 1802120 CrossRef .
- W. Xu, S. Zhu, Y. Liang, Z. Cui, X. Yang and A. Inoue, J. Mater. Chem. A, 2018, 6, 5574–5579 RSC .
- D. B. Kayan, D. Koçak, M. İlhan and A. Koca, Int. J. Hydrogen Energy, 2017, 42, 2457–2463 CrossRef CAS .
- F. Hu, S. Zhu, S. Chen, Y. Li, L. Ma, T. Wu, Y. Zhang, C. Wang, C. Liu, X. Yang, L. Song, X. Yang and Y. Xiong, Adv. Mater., 2017, 29, 1606570 CrossRef .
- Y. Zhu, Y. Pan, W. Dai and T. Lu, ACS Appl. Energy Mater., 2020, 3, 1319–1327 CrossRef CAS .
- S. Xue, Z. Liu, C. Ma, H.-M. Cheng and W. Ren, Sci. Bull., 2020, 65, 123–130 CrossRef CAS .
- T. Shinagawa, A. T. Garcia-Esparza and K. Takanabe, Sci. Rep., 2015, 5, 13801 CrossRef .
- C. C. L. McCrory, S. Jung, I. M. Ferrer, S. M. Chatman, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2015, 137, 4347–4357 CrossRef CAS .
- A. Grimaud, O. Diaz-Morales, B. Han, W. T. Hong, Y.-L. Lee, L. Giordano, K. A. Stoerzinger, M. T. M. Koper and Y. Shao-Horn, Nat. Chem., 2017, 9, 457–465 CrossRef CAS .
- N.-T. Suen, S.-F. Hung, Q. Quan, N. Zhang, Y.-J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337–365 RSC .
- J. Song, C. Wei, Z.-F. Huang, C. Liu, L. Zeng, X. Wang and Z. J. Xu, Chem. Soc. Rev., 2020, 49, 2196–2214 RSC .
- J. Zhang, Y. Hu, D. Liu, Y. Yu and B. Zhang, Adv. Sci., 2017, 4, 1600343 CrossRef .
- M. Vasić, M. Čebela, I. Pašti, L. Amaral, R. Hercigonja, D. M. F. Santos and B. Šljukić, Electrochim. Acta, 2018, 259, 882–892 CrossRef .
- K. Krukiewicz, Electrochem. Commun., 2020, 116, 106742 CrossRef CAS .
- R. N. Bhowmik and K. S. Aneesh Kumar, Mater. Chem. Phys., 2016, 177, 417–428 CrossRef CAS .
- R. Martinez, A. Kumar, R. Palai, J. F. Scott and R. S. Katiyar, J. Phys. D: Appl. Phys., 2011, 44, 105302 CrossRef .
- Y. Liu, X. Liang, L. Gu, Y. Zhang, G.-D. Li, X. Zou and J.-S. Chen, Nat. Commun., 2018, 9, 2609 CrossRef .
- A. N. Mansour and R. A. Brizzolara, Surf. Sci. Spectra, 1996, 4, 357–362 CrossRef CAS .
- A. Jena, T. R. Penki, N. Munichandraiah and S. A. Shivashankar, J. Electroanal. Chem., 2016, 761, 21–27 CrossRef CAS .
- Y. Tang, L. Dong, S. Mao, H. Gu, T. Malkoske and B. Chen, ACS Appl. Energy Mater., 2018, 1, 2698–2708 CrossRef CAS .
- I. G. Casella, M. R. Guascito and M. G. Sannazzaro, J. Electroanal. Chem., 1999, 462, 202–210 CrossRef CAS .
- L. Trotochaud, J. K. Ranney, K. N. Williams and S. W. Boettcher, J. Am. Chem. Soc., 2012, 134, 17253–17261 CrossRef CAS .
- Q. Chen, R. Wang, F. Lu, X. Kuang, Y. Tong and X. Lu, ACS Omega, 2019, 4, 3493–3499 CrossRef CAS .
- S. Li, X. Jian, J. Liu, S. Guo, C. Zhou, P. Zhang, Y. Yang and L. Chen, Int. J. Hydrogen Energy, 2020, 45, 4435–4443 CrossRef CAS .
- J. Masa, C. Andronescu, H. Antoni, I. Sinev, S. Seisel, K. Elumeeva, S. Barwe, S. Marti-Sanchez, J. Arbiol, B. Roldan Cuenya, M. Muhler and W. Schuhmann, ChemElectroChem, 2019, 6, 235–240 CrossRef CAS .
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra10418c |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Jinsen Tian
a,
Jinsen Tian



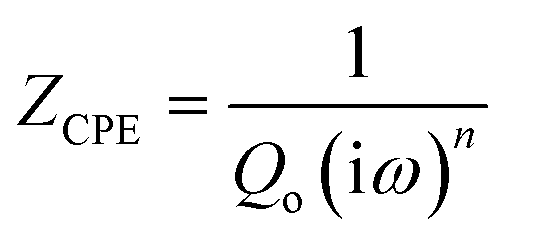 where Qo is a pseudo-capacitance arising from the double layer for the adsorbed hydrogen or adsorbed oxygen (Hads or Oads), ω is the angular frequency (ω = 2πf, f is the linear frequency),
where Qo is a pseudo-capacitance arising from the double layer for the adsorbed hydrogen or adsorbed oxygen (Hads or Oads), ω is the angular frequency (ω = 2πf, f is the linear frequency), 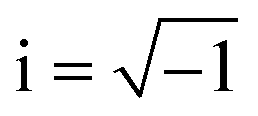 , and ‘n’ is an empirical constant between 0 and 1. CPE is a pure capacitor when n = 1 and is a pure resistor when n = 0.57 It may be noted that the faster kinetics toward HER and OER is given by the low value of charge transfer resistance as well as the diameter of the semicircle in the impedance Cole–Cole plot. The charge transfer resistance of the as-prepared MG ribbon toward HER acidic reaction is 34.5 Ω. After electrochemical dealloying, the Rct value is reduced to 3.5 Ω, indicating the faster HER kinetics as confirmed from linear polarization data. For the HER alkaline reaction, the as-prepared ribbon has a charge transfer resistance of 83 Ω while the electrochemically dealloyed sample has a reduced value of 28.7 Ω. For the OER reaction, the pristine sample has an Rct value of 36.3 Ω, while the electrochemically dealloyed sample has an Rct value of 8.4 Ω. The reduced value of charge transfer resistance emphasizes the increased catalytic activity in dealloyed samples and after 2000 CV cycle tested samples.58
, and ‘n’ is an empirical constant between 0 and 1. CPE is a pure capacitor when n = 1 and is a pure resistor when n = 0.57 It may be noted that the faster kinetics toward HER and OER is given by the low value of charge transfer resistance as well as the diameter of the semicircle in the impedance Cole–Cole plot. The charge transfer resistance of the as-prepared MG ribbon toward HER acidic reaction is 34.5 Ω. After electrochemical dealloying, the Rct value is reduced to 3.5 Ω, indicating the faster HER kinetics as confirmed from linear polarization data. For the HER alkaline reaction, the as-prepared ribbon has a charge transfer resistance of 83 Ω while the electrochemically dealloyed sample has a reduced value of 28.7 Ω. For the OER reaction, the pristine sample has an Rct value of 36.3 Ω, while the electrochemically dealloyed sample has an Rct value of 8.4 Ω. The reduced value of charge transfer resistance emphasizes the increased catalytic activity in dealloyed samples and after 2000 CV cycle tested samples.58